

Abstract
AIM: To investigate the microsatellite alterations in phenotypically normal esophageal squamous epithelium and metaplasia-dysplasia-adenocarcinoma sequence.
METHODS: Forty-one specimens were obtained from esophageal cancer (EC) patients. Histopathological assessment identified 23 squamous cell carcinomas (SCC) and 18 adenocarcinomas (ADC), including only 8 ADC with Barrett esophageal columnar epithelium (metaplasia) and dysplasia adjacent to ADC. Paraffin-embedded normal squamous epithelium, Barrett esophageal columnar epithelium (metaplasia), dysplasia and esophageal tumor tissues were dissected from the surrounding tissues under microscopic guidance. DNA was extracted using proteinase K digestion buffer, and DNA was diluted at 1:100, 1:1000, 1:5000, 1:10 000 and 1:50 000, respectively. Seven microsatellite markers (D2S123, D3S1616, D3S1300, D5S346, D17S787, D18S58 and BATRII loci) were used in this study. Un-dilution and dilution polymerase chain reactions (PCR) were performed, and microsatellite analysis was carried out.
RESULTS: No statistically significant difference was found in microsatellite instability (MSI) and loss of heterozygosity (LOH) of un-diluted DNA between SCC and ADC. The levels of MSI and LOH were high in the metaplasia-dysplasia-adenocarcinoma sequence of diluted DNA. The more the diluted DNA was, the higher the rates of MSI and LOH were at the above 7 loci, especially at D3S1616, D5S346, D2S123, D3S1300 and D18S58 loci.
CONCLUSION: The sequence of metaplasia-dysplasia-adenocarcinoma is associated with microsatellite alterations, including MSI and LOH. The MSI and LOH may be the early genetic events during esophageal carcinogenesis, and genetic alterations at the D3S1616, D5S346 and D3S123 loci may play a role in the progress of microsatellite alterations.
Keywords: Microsatellite alteration, Dilution PCR, Metaplasia-dysplasia-adenocarcinoma sequence, Esophageal squamous epithelium, Squamous cell carcinoma
INTRODUCTION
Barrett esophagus, thought to be primarily due to uncontrolled gastric acid and biliopancreatic reflux into the esophagus, is a precancerous condition[1,2]. It is estimated that the risk of neoplasia in Barrett esophagus, through the intermediate step of dysplastic transformation of the columnar epithelium, is 125-fold higher than that in general population[1]. There is evidence that adenocarcinoma (ADC) of the esophagus arises in similar metaplastic epithelium, which has been termed “short segment Barrett esophagus” by some investigators[3,4]. However, this etiology alone is insufficient to explain the rising incidence of this highly lethal form of cancer[5,6]. Although several studies focusing on the squamous cell carcinoma (SCC) and the advanced stages of progression from dysplasia to ADC have investigated the role of genetic alterations in esophageal cancer (EC)[7–10], scarce data are available on genetic alterations occurring in metaplasia-dysplasia-adenocarcinoma sequence of Barrett esophagus. To better understand the pathogenesis of esophageal ADC, we investigated the microsatellite alterations in these lesions. We dissected Barrett esophageal columnar epithelium (metaplasia) adjacent to dysplastic and neoplastic changes, dysplasia, and ADC, from the formalin-fixed, paraffin-embedded tissue blocks. Microsatellite alterations were evaluated at D2S123, D3S1616, D3S1300, D5S346, D17S787, D18S58 and BATRII loci, using the technique of dilution polymerase chain reaction (PCR). Our aim was to assess the early microsatellite alterations characterizing Barrett esophagus.
MATERIALS AND METHODS
Tissue specimens
Forty-one specimens were obtained from EC patients identified from Xiamen First Hospital affiliated to Fujian Medical University, Fujian Province, China, during 2002-2005. All the patients did not receive chemotherapy or radiation before surgery. Formalin-fixed, paraffin-embedded archival esophageal tissue specimens from 41 EC patients were accrued according to the guidelines established by the Ethics Committee of Xiamen First Hospital Affiliated to Fujian Medical University. Histopathological assessment identified 23 SCC and 18 ADC, including only 8 ADC with metaplasia and dysplasia adjacent to ADC. Strict criteria were used to diagnose ADC of primary esophageal origin as previously described[11]. Briefly, these included the presence of Barrett epithelium and dysplasia adjacent to ADC, at least 75% of tumor mass located in the distal one third of esophagus, invasion of periesophageal tissue, minimal gastric involvement, and clinical symptoms of dysphagia due to esophageal obstruction. Tissue histopathology was reviewed independently by 2 gastrointestinal pathologists.
Tissue cellularity, DNA isolation and DNA dilution
Paraffin-embedded normal squamous epithelium, Barrett esophageal columnar epithelium (metaplasia), dysplasia and esophageal tumor tissues were dissected from the surrounding tissues under microscopic guidance in order to enrich the appropriate cell population for subsequent DNA extraction (Figure 1). All the hematoxylin-eosin (HE) stained sections of the above tissues from each EC patient were reviewed and cellularity was determined by estimating the relative density of neoplastic cells to normal tissue components (infiltrating stroma, muscle cells, white blood cells) per microscopic field. For the majority of cases, carcinoma specimens with at least 75% neoplastic cellularity were selected for microsatellite analyses. A series of unstained 10 μm-thick sections of the above tissues were prepared from the corresponding paraffin-embedded tissue block with the microtone appropriately cleaned between cases in order to prevent tissue contamination. Previously selected areas on HE stained sections were then properly oriented and traced carefully on the corresponding unstained sections using a mark pen. Paraffin shaving from the unstained 10 μm-thick sections was collected and pooled together in 0.5 mL Eppendorf tubes and DNA was extracted using proteinase K digestion buffer (10 mmol/L Tris-HCl, pH 8.0, 100 mmol/L KCl, 2.5 mmol/L MgCl2, 0.45% Tween20 and 2.5 mg/mL proteinase K) as previously described[12]. Briefly, samples were deparaffinized at 95°C for 10 min, cooled 1 h to room temperature and incubated at 65°C for 1 h with the proteinase K digestion buffer (100-200 μL). Samples were then denatured at 95°C for 10 min to remove excess proteinase K. The mixture was centrifuged at 5 000 r/min for 5 min and the supernatant was carefully transferred by aspiration to a clean microcentrifuge tube. The quality of DNA was 1.07-1.24 (A260/A280), the quantity of DNA was 255.4-407.0 ng/μL. To increase its sensitivity, DNA samples were diluted at 1:100, 1:1000, 1:5000, 1:10 000 and 1:50 000, respectively.
Figure 1.
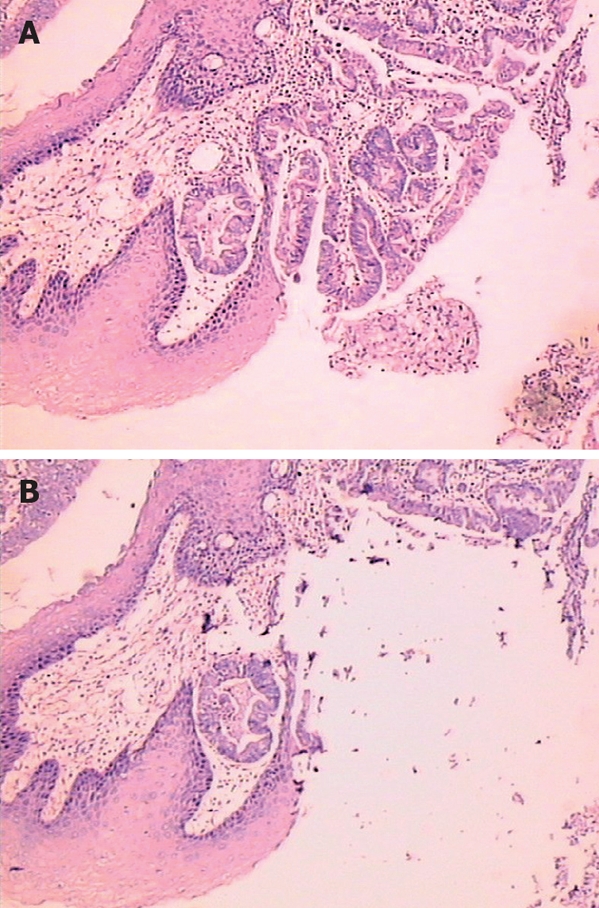
Photomicrograph of epithelial dysplasia (HE, × 200) in Barrett esophagus (A) and after dissected under microscopic guidance (B).
Dillution PCR and microsatellite analysis
Seven microsatellite markers (D2S123, D3S1616, D3S1300, D5S346, D17S787, D18S58 and BATRII loci) were used in this study. The oligonucleotide primers used for PCR amplifications were obtained from Research Genetics (Huntsville, AL). Each PCR reaction contained 1 μL of DNA samples each diluted at 1:100, 1:1000, 1:5 000, 1:10 000 or 1:50 000, respectively, 0.4 μCi [r-32p] ATP-radiolabeled microsatellite primer, 0.2 mmol/L dNTP, 10 mmol/L Tris-HCl (pH 8.3), 1.5 mmol/L MgCl2, 50 mmol/L KCl, and 0.4 units of Taq polymerase in a total reaction volume of 10 μL (that was called dilution PCR). The hot start PCR was used to eliminate nonspecific amplification. The samples underwent 35 cycles of PCR amplification (95°C for 30 s, 58°C for 40 s, and 72°C for 60 s) followed by an extension at 72°C for 2 min. Several PCR amplifications were performed for each DNA dilution. PCR products were diluted at 1:1 in a formalin loading buffer containing 95% formalin, 0.05% bromophend blue, 0.05% xylene cyanol and 20 mmol/L EDTA, and 5 μL aliquots of PCR products was electrophoresed on 7% denaturing polyacrylamide gels (19:1 acrylamide:bisacrylamide) containing 5.6 mol/Lurea, 32% formamide and 1 × TBE [0.089 mol/L Tris (pH 8.3)], 0.089 mol/L borata and 0.002 mol/L EDTA] at 75 W for 2-4 h. Gels were vacuum-dried at 80°C and exposed to a Kodak MR film (Kodak, Xiamen, China) for 6-24 h. Microsatellite analysis was carried out. A sample was scored positive for microsatellite instability (MSI) if additional new alleles were observed in Barrett esophageal epithelium or dysplasia or tumor DNA compared to control DNA from the same individual. Loss of heterozygosity (LOH) was defined as a visible reduction (at least 2 folds or more) in the ratio of signal intensities of the 2 alleles in metaplasia or dysplasia or tumor DNA relative to that observed in the corresponding normal DNA of the same individual. In all positive results analyzed, since losses were readily visible or obvious, quantification was unnecessary. To ensure that the allele in intensities was identified in microsatellite analysis, multiple exposures of each autoradiograph were carried out. The auto-radiographs were independently scored by JCC and HPZ.
Statistical analysis
Fisher’s exact test was performed to assess any statisti-cally significant correlation of the frequency of the 2 histological subtypes (SCC vs ADC) or 2 subgroups (un-dilution vs dilution) by SPSS 10.0. P < 0.05 was considered statistically significant.
RESULTS
Microsatellite alterations in undiluted DNA of SCC and ADC
A varying level of MSI, ranging from 0% (0/23) to 28.6% (6/23) at the 7 loci tested, was detected in 23 SCC. The rate of MSI was 25% (3/18) at the D2S123 or D3S1616 or D5S346 locus in 18 ADC. The rate of MSI was 0 at the D3S1300, D17S787, D18S58 and BAT-RII loci in 18 ADC. Only informative cases (heterozygous for the microsatellite markers used) were considered for LOH analysis. The rate of LOH was 23.5% (4/17 informative cases) at the D3S1300 locus, and very low for the other loci in 23 SCC. The rate of LOH was 10.0% (1/10 informative cases) at the D2S123 or D3S1300 or D3S1616 locus, the others were 0 in 18 ADC. The results of the microsatellite analysis are summarized in Tables 1 and 2. No statistically significant difference was found in the rate of MSI and LOH at one or more positive loci, or at two or more positive loci between SCC and ADC.
Table 1.
Microsatellite alterations of un-diluted and diluted DNA in different esophageal tissues
| Tissue DNA | Number studied |
MSI |
LOH |
||||
| Negative | At ≥ 1 locus | At ≥ 2 loci | Negative | At ≥ 1 locus | At ≥ 2 loci | ||
| Classification | |||||||
| SCC | 23 | 12 | 5 | 6 | 17 | 3 | 3 |
| ADC | 18 | 11 | 4 | 31 | 15 | 2 | 12 |
| Normal | |||||||
| Undilution | 8 | 8 | 0 | 0 | 8 | 0 | 0 |
| Dilution | 8 | 3 | 2 | 33 | 4 | 2 | 24 |
| Metaplasia | |||||||
| Undilution | 8 | 7 | 1 | 0 | 7 | 1 | 0 |
| Dilution | 8 | 1 | 4 | 35 | 2 | 3 | 36 |
| Dysplasia | |||||||
| Undilution | 8 | 7 | 1 | 0 | 7 | 1 | 0 |
| Dilution | 8 | 1 | 3 | 47 | 2 | 2 | 48 |
| Carcinoma | |||||||
| Undilution | 8 | 6 | 2 | 0 | 6 | 2 | 0 |
| Dilution | 8 | 0 | 3 | 59 | 0 | 4 | 410 |
Normal: Normal esophageal squamous epithelium DNA; Metaplasia: Barrett esophageal columnar epithelium DNA; Dysplasia: Dysplastic esophageal epithelium DNA; Carcinoma: Esophageal adenocarcinoma DNA;
P = 0.758,
P = 0.696,
P = 0.026,
P = 0.069,
P = 0.009, 6P = 0.034,
P = 0.009,
P = 0.029,
P = 0.004,
P = 0.004 vs other groups.
Table 2.
Microsatellite alterations of undiluted and diluted DNA at 7 loci in different esophageal tissues
| Tissue DNA | Number studied |
D2S123 |
D3S1616 |
D3S1300 |
BATRII |
D5S346 |
D17S787 |
D18S61 |
|||||||
| MSI | LOH | MSI | LOH | MSI | LOH | MSI | LOH | MSI | LOH | MSI | LOH | MSI | LOH | ||
| Classification | |||||||||||||||
| SCC | 23 | 3 | 1 | 6 | 2 | 3 | 4 | 0 | 0 | 2 | 1 | 2 | 1 | 2 | 0 |
| ADC | 18 | 4 | 1 | 3 | 1 | 2 | 1 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 0 |
| Normal | |||||||||||||||
| Undilution | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dilution | 8 | 2 | 1 | 1 | 2 | 2 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 |
| Metaplasia | |||||||||||||||
| Undilution | 8 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dilution | 8 | 2 | 2 | 2 | 3 | 1 | 2 | 1 | 0 | 1 | 1 | 2 | 1 | 1 | 0 |
| Dysplasia | |||||||||||||||
| Undilution | 8 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dilution | 8 | 4 | 3 | 4 | 3 | 4 | 3 | 1 | 0 | 1 | 1 | 4 | 2 | 1 | 1 |
| Carcinoma | |||||||||||||||
| Undilution | 8 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dilution | 8 | 6 | 4 | 6 | 7 | 6 | 4 | 2 | 0 | 4 | 0 | 6 | 3 | 3 | 2 |
Normal: Normal esophageal squamous epithelium DNA; Metaplasia: Barrett esophageal columnar epithelium DNA; Dysplasia: Dysplastic esophageal epithelium DNA; Carcinoma: Esophageal adenocarcinoma DNA.
Microsatellite alterations in diluted DNA of phenotypically normal esophageal squamous epithelium and metaplasia-dysplasia-ademocarcinoma sequence
Dilution PCR showed that the more sensitive strategy for analyzing microsatellite changes was developed, reasoning that if a rare cell in a population harbored microsatellite alterations, the new microsatellite alleles would not be detected amid the large background of normal alleles[13,14]. High levels of MSI and LOH were very commonly found in phenotypically normal esophageal squamous epithelium and metaplasia-dysplasia-adenocarcinoma sequence of diluted DNA. The more the dilution of DNA (up to 1:5 000) was, the higher the rates of MSI and LOH were at the above 7 loci, especially at D3S1616, D5S346, D3S123, D2S1300 and D18S58 loci. However, the rates of MSI and LOH were low when DNA was diluted at 1:10 000 and up, because most PCR products were not available. The data described here could document a profound microsatellite alteration in phenotypically normal esophageal squamous epithelium and metaplasia-dysplasia-ademocarcinoma sequence, although un-diluted DNA was not discovered. The results of microsatellite analysis are summarized in Tables 1, 2 and Figure 2.
Figure 2.

Microsatellite alterations in diluted DNA. Arrow and arrowhead indicate MSI and LOH, respectively. N: Normal esophageal squamous epithelium DNA; BE: Barrett esophageal columnar epithelium (metaplasia) DNA; Undil: Un-diluted; 1/100-1/50 000: Dilution fold.
DISSCUSSION
Tumor of Barrett esophagus occurs via a multi-step pathway, and is histopathologically recognized as a metaplasia-dysplasia-adenocarcinoma sequence[15]. DNA and certain molecular abnormalities under the form of proto-oncogene alterations, LOH and mutations of tumor suppressor genes are thought to represent the genetic background of metaplastic and dysplastic changes[16–19]. In recent years, frequent or infrequent MSI at multiple microsatellite loci corresponding to diverse tumor suppressor genes has been demonstrated in ADC of colon and rectum, small intestine, stomach, gallbladder and esophagus[20–24]. However, few data are available on the correlation between microsatellite alterations and metaplasia-dysplasia-adenocarcinoma sequence in Barrett esophagus, especially in normal squamous epithelium and metaplastic tissue adjacent to dysplasia and neoplasm. Our study was to assess the microsatellite alterations in metaplasia-dysplasia-adenocarcinoma sequence of Barrett esophagus, as well as in normal squamous epithelium adjacent to metaplastic tissue. Tissue was dissected under microscopic guidance with a new technique which ensures enrichment and purity of the tissues to be studied and the specificity of the molecular event encountered.
In this study, the frequency of MSI was investigated in 23 SCC and 18 ADC, including 8 ADC with Barrett esophageal columnar epithelium and dysplasia. MSI of at least one microsatellite locus was detected in 10 out of 23 SCC (47.6%) and 5 out of 18 ADC (41.7%). MSI of two or more microsatellite loci was found in 5 out of 23 SCC (23.8%) and 3 out of 18 ADC (25.0%). MSI was found in 1 out of 8 Barrett esophageal columnar epithelium tissue samples, 2 out of 8 dysplasia tissue samples adjacent to ADC only at one microsatellite locus. Although no significant difference was found in the frequency of MSI between SCC and ADC in this study, the results are in agreement with the findings of some previous studies showing frequent MSI in EC[25–28], but not consistent with some recent reports showing infrequent MSI in EC[24,29].
It was reported that tumors develop via a process of clonal expansion resulting from the selection of increasingly abnormal sub-populations[10]. According to this theory, microscopic tumor usually represents the clonal expansion of one or a few cells, indicating that cells of a given neoplasm should share some commom genetic abnormalities[10]. In order to find whether microsatellite alterations are an early event in esophageal ADC, we diluted DNA of metaplasia, dysplasia, ADC and normal esophageal squamous epithelium from the same 8 patients, and performed dilution PCR which showed surprisingly high levels of MSI and LOH expression at the early stage of metaplasia-dysplasia-adenocarcinoma sequence.
However, some studies also suggested that PCR artifacts are found when paraffin-embedded tissues are used, especially when the number of cells analyzed is small[30,31]. With a larger number of target cells, enough non-damaged templates dominate the amplification process. With a smaller amount of cells, only fragmented DNA presents and requires a few PCR cycles to achieve an in vitro repaired template that would yield an exponential amplification[30,31]. Thus, to minimize the potential for artificial microsatellite alterations resulting from the diluted DNA, DNA was diluted at 1:100 to 1:50 000 for the dilution PCR. The concentration was estimated to be 25.5-40.7 pg/μL DNA when diluted at 1:10 000. PCR amplification was performed for each diluted DNA. For example, in patient 1, a concentration of 34.3-40.7 pg/μL DNA diluted at 1:10 000 was used.
Interestingly, microsatellite alterations were detected in all the eight patients having the “sequence” of metaplasia-dysplasia-adenocarcinoma at each representative DNA dilution. Similarly, microsatellite alterations were detected both in metaplasia and in dysplasia adjacent to ADC at the 7 microsatellite loci. The more the dilution of DNA was, the higher the rate of microsatellite alterations was at the D3S1616, D5S346, D2S123, D3S1300 and D18S58 loci depending on development of the metaplasia-dysplasia-adenocarcinoma sequence, clearly revealing that there are extensive microsatellite alterations at the D3S1616, D5S346, D2S123, D3S1300 and D18S58 loci in the metaplasia-dysplasia-adenocarcinoma sequence of Barrett esophagus, even in the histopathologically normal esophageal squamous epithelium. The observations of shared molecular abnormalities in metaplasia, dysplasia and ADC suggested a process of colonal expansion in the proposed histological pathway of tumor development, which may be partly interpreted by the fact that only partial cells undergo molecular alterations under a similar circumstance.
In conclusion, high levels of MSI and LOH are frequently found in metaplasia, dysplasia and adenocarcinoma. Development of esophageal ADC is associated with microsatellite alterations, including MSI and LOH. These alterations occurring in recognized precursor lesions provide evidence for support of the proposed metaplasia-dysplasia-adenocarcinoma sequence in Barrett esophagus. MSI and LOH may be the early genetic events during esophageal carcinogenesis. Additional genetic alterations at the D3S1616, D5S346 and D3S123 loci probably play a role in the progression of these diseases. D3S1616, D5S346, D2S123, D3S1300 and D18S58 loci may be used as predictive markers for the early detection of esophageal ADC.
COMMENTS
Background
It is estimated that the risk of neoplasia in Barrett esophagus is 125-fold higher than that in the general population, through the intermediate step of dysplastic transformation of the columnar epithelium. There is evidence that adenocarcinoma (ADC) of the esophagus arises in metaplastic epithelium, termed “short segment Barrett esophagus”. However, this etiology alone is insufficient to explain the rising incidence of this highly lethal form of cancer. Although several studies focusing on the squamous cell carcinoma (SCC) and the advanced stages of progression from dysplasia to ADC, have investigated the role of genetic alterations in esophageal cancer (EC), scarce data are available on genetic alterations occurring in metaplasia-dysplasia-adenocarcinoma sequence of Barrett esophagus.
Research frontiers
Tumor occurring in Barrett esophagus via a multi-step pathway is histopathologically recognized as a metaplasia-dysplasia-adenocarcinoma sequence. Certain molecular abnormalities in the form of proto-oncogene alterations, loss of heterozygosity (LOH) and mutations of tumor suppressor genes are thought to represent the genetic background of metaplastic and dysplastic changes. In recent years, frequent or infrequent microsatellite instability (MSI) at multiple microsatellite loci corresponding to the diverse tumor suppressor genes has been found in ADC of colon and rectun, small intestine, stomach and esophagus. However, few data are available on the correlation between microsatellite alterations and metaplasia-dysplasia-adenocarcinoma sequence in Barrett esophagus, especially in normal squamous epithelium and metaplastic tissue adjacent to dysplasia and neoplasm.
Innovations and breakthroughs
Dilution polymerase chain reaction (PCR), the more sensitive strategy for analyzing microsatellite changes, was developed, reasoning that if a rare cell in a population harbored microsatellite alterations, the new microsatellite alleles would not be detectable amid the large number of normal alleles. High levels of MSI and LOH are frequently found in phenotypically normal esophageal squamous epithelium and metaplasia-dysplasia-adenocarcinoma sequence of diluted DNA in this study.
Applications
MSI and LOH might be the early genetic events during esophageal carcinogenesis. Additional genetic alterations at the D3S1616, D5S346 and D3S123 loci may play a role in progression of ADC. The microsatellite markers (D3S1616, D5S346, D2S123, D3S1300 and D18S58 loci) may be used as predictive markers in the early detection of esophageal ADC.
Terminology
Dilution PCR refers to diluted DNA of metaplasia, dysplasia, ADC and normal esophageal squamous epithelium in the same patient before PCR is performed.
Peer review
The study was well designed and provided a great finding at metaplasia-dysplasia-adenocarcinoma sequence in Barrett esophagus. It is interesting.
Acknowledgments
The authors thank Professor Bapat B and Professor Chen WY for their technical support and invaluable advice.
Supported by by The Xiamen Science and Technology Founda-tion (No. 3502Z20052018), and Xiamen Healthy Bureau Research Foundation (No. WSK0301)
Peer reviewer: Alvaro Montiel-Jarquin, PhD, Department of General Surgery, Instituto Mexicano Del Seguro Social, Ave Cue Merlo 802-2 San Baltazar Campeche, Puebla, Puebla 72550, Mexico
S- Editor Li DL L- Editor Wang XL E- Editor Lin YP
References
- 1.Romagnoli S, Roncalli M, Graziani D, Cassani B, Roz E, Bonavina L, Peracchia A, Bosari S, Coggi G. Molecular alterations of Barrett's esophagus on microdissected endoscopic biopsies. Lab Invest. 2001;81:241–247. doi: 10.1038/labinvest.3780232. [DOI] [PubMed] [Google Scholar]
- 2.Rossi M, Barreca M, de Bortoli N, Renzi C, Santi S, Gennai A, Bellini M, Costa F, Conio M, Marchi S. Efficacy of Nissen fundoplication versus medical therapy in the regression of low-grade dysplasia in patients with Barrett esophagus: a prospective study. Ann Surg. 2006;243:58–63. doi: 10.1097/01.sla.0000194085.56699.db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oberg S, Wenner J, Johansson J, Walther B, Willen R. Barrett esophagus: risk factors for progression to dysplasia and adenocarcinoma. Ann Surg. 2005;242:49–54. doi: 10.1097/01.sla.0000167864.46462.9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rumpel CA, Powell SM, Moskaluk CA. Mapping of genetic deletions on the long arm of chromosome 4 in human esophageal adenocarcinomas. Am J Pathol. 1999;154:1329–1334. doi: 10.1016/S0002-9440(10)65386-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nair KS, Naidoo R, Chetty R. Microsatellite analysis of the APC gene and immunoexpression of E-cadherin, catenin, and tubulin in esophageal squamous cell carcinoma. Hum Pathol. 2006;37:125–134. doi: 10.1016/j.humpath.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 6.Devesa SS, Blot WJ, Fraumeni JF Jr. Changing patterns in the incidence of esophageal and gastric carcinoma in the United States. Cancer. 1998;83:2049–2053. [PubMed] [Google Scholar]
- 7.Mashimo H, Wagh MS, Goyal RK. Surveillance and screening for Barrett esophagus and adenocarcinoma. J Clin Gastroenterol. 2005;39:S33–S41. doi: 10.1097/01.mcg.0000155859.26557.45. [DOI] [PubMed] [Google Scholar]
- 8.Jankowski JA, Wright NA, Meltzer SJ, Triadafilopoulos G, Geboes K, Casson AG, Kerr D, Young LS. Molecular evolution of the metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Pathol. 1999;154:965–973. doi: 10.1016/S0002-9440(10)65346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walch AK, Zitzelsberger HF, Bruch J, Keller G, Angermeier D, Aubele MM, Mueller J, Stein H, Braselmann H, Siewert JR, et al. Chromosomal imbalances in Barrett's adenocarcinoma and the metaplasia-dysplasia-carcinoma sequence. Am J Pathol. 2000;156:555–566. doi: 10.1016/S0002-9440(10)64760-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujiki T, Haraoka S, Yoshioka S, Ohshima K, Iwashita A, Kikuchi M. p53 Gene mutation and genetic instability in superficial multifocal esophageal squamous cell carcinoma. Int J Oncol. 2002;20:669–679. [PubMed] [Google Scholar]
- 11.Casson AG, Mukhopadhyay T, Cleary KR, Ro JY, Levin B, Roth JA. p53 gene mutations in Barrett's epithelium and esophageal cancer. Cancer Res. 1991;51:4495–4499. [PubMed] [Google Scholar]
- 12.Reitmair AH, Cai JC, Bjerknes M, Redston M, Cheng H, Pind MT, Hay K, Mitri A, Bapat BV, Mak TW, et al. MSH2 deficiency contributes to accelerated APC-mediated intestinal tumorigenesis. Cancer Res. 1996;56:2922–2926. [PubMed] [Google Scholar]
- 13.Parsons R, Li GM, Longley M, Modrich P, Liu B, Berk T, Hamilton SR, Kinzler KW, Vogelstein B. Mismatch repair deficiency in phenotypically normal human cells. Science. 1995;268:738–740. doi: 10.1126/science.7632227. [DOI] [PubMed] [Google Scholar]
- 14.Mao L, Lee DJ, Tockman MS, Erozan YS, Askin F, Sidransky D. Microsatellite alterations as clonal markers for the detection of human cancer. Proc Natl Acad Sci USA. 1994;91:9871–9875. doi: 10.1073/pnas.91.21.9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gleeson CM, Sloan JM, McGuigan JA, Ritchie AJ, Weber JL, Russell SE. Barrett's oesophagus: microsatellite analysis provides evidence to support the proposed metaplasia-dysplasia-carcinoma sequence. Genes Chromosomes Cancer. 1998;21:49–60. [PubMed] [Google Scholar]
- 16.Fang M, Lew E, Klein M, Sebo T, Su Y, Goyal R. DNA abnormalities as marker of risk for progression of Barrett's esophagus to adenocarcinoma: image cytometric DNA analysis in formalin-fixed tissues. Am J Gastroenterol. 2004;99:1887–1894. doi: 10.1111/j.1572-0241.2004.30886.x. [DOI] [PubMed] [Google Scholar]
- 17.Roth MJ, Hu N, Emmert-Buck MR, Wang QH, Dawsey SM, Li G, Guo WJ, Zhang YZ, Taylor PR. Genetic progression and heterogeneity associated with the development of esophageal squamous cell carcinoma. Cancer Res. 2001;61:4098–4104. [PubMed] [Google Scholar]
- 18.Barrett MT, Sanchez CA, Prevo LJ, Wong DJ, Galipeau PC, Paulson TG, Rabinovitch PS, Reid BJ. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat Genet. 1999;22:106–109. doi: 10.1038/8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walch AK, Zitzelsberger HF, Bink K, Hutzler P, Bruch J, Braselmann H, Aubele MM, Mueller J, Stein H, Siewert JR, et al. Molecular genetic changes in metastatic primary Barrett's adenocarcinoma and related lymph node metastases: comparison with nonmetastatic Barrett's adenocarcinoma. Mod Pathol. 2000;13:814–824. doi: 10.1038/modpathol.3880143. [DOI] [PubMed] [Google Scholar]
- 20.Sawhney MS, Farrar WD, Gudiseva S, Nelson DB, Lederle FA, Rector TS, Bond JH. Microsatellite instability in interval colon cancers. Gastroenterology. 2006;131:1700–1705. doi: 10.1053/j.gastro.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe T, Kobunai T, Toda E, Yamamoto Y, Kanazawa T, Kazama Y, Tanaka J, Tanaka T, Konishi T, Okayama Y, et al. Distal colorectal cancers with microsatellite instability (MSI) display distinct gene expression profiles that are different from proximal MSI cancers. Cancer Res. 2006;66:9804–9808. doi: 10.1158/0008-5472.CAN-06-1163. [DOI] [PubMed] [Google Scholar]
- 22.Hasuo T, Semba S, Li D, Omori Y, Shirasaka D, Aoyama N, Yokozaki H. Assessment of microsatellite instability status for the prediction of metachronous recurrence after initial endoscopic submucosal dissection for early gastric cancer. Br J Cancer. 2007;96:89–94. doi: 10.1038/sj.bjc.6603532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagahashi M, Ajioka Y, Lang I, Szentirmay Z, Kasler M, Nakadaira H, Yokoyama N, Watanabe G, Nishikura K, Wakai T, et al. Genetic changes of p53, K-ras, and microsatellite instability in gallbladder carcinoma in high-incidence areas of Japan and Hungary. World J Gastroenterol. 2008;14:70–75. doi: 10.3748/wjg.14.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yanagi M, Keller G, Mueller J, Walch A, Werner M, Stein HJ, Siewert JR, Hofler H. Comparison of loss of heterozygosity and microsatellite instability in adenocarcinomas of the distal esophagus and proximal stomach. Virchows Arch. 2000;437:605–610. doi: 10.1007/s004280000322. [DOI] [PubMed] [Google Scholar]
- 25.Kagawa Y, Yoshida K, Hirai T, Toge T, Yokozaki H, Yasui W, Tahara E. Microsatellite instability in squamous cell carcinomas and dysplasias of the esophagus. Anticancer Res. 2000;20:213–217. [PubMed] [Google Scholar]
- 26.Kulke MH, Thakore KS, Thomas G, Wang H, Loda M, Eng C, Odze RD. Microsatellite instability and hMLH1/hMSH2 expression in Barrett esophagus-associated adenocarcinoma. Cancer. 2001;91:1451–1457. doi: 10.1002/1097-0142(20010415)91:8<1451::aid-cncr1152>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi M, Tamura G, Jin Z, Kato I, Sato M, Shibuya Y, Yang S, Motoyama T. Microsatellite instability in esophageal squamous cell carcinoma is not associated with hMLH1 promoter hypermethylation. Pathol Int. 2003;53:270–276. doi: 10.1046/j.1440-1827.2003.01478.x. [DOI] [PubMed] [Google Scholar]
- 28.Mathew R, Arora S, Mathur M, Chattopadhyay TK, Ralhan R. Esophageal squamous cell carcinomas with DNA replication errors (RER+) are associated with p16/pRb loss and wild-type p53. J Cancer Res Clin Oncol. 2001;127:603–612. doi: 10.1007/s004320100249. [DOI] [PubMed] [Google Scholar]
- 29.Kubo N, Yashiro M, Ohira M, Hori T, Fujiwara I, Hirakawa K. Frequent microsatellite instability in primary esophageal carcinoma associated with extraesophageal primary carcinoma. Int J Cancer. 2005;114:166–173. doi: 10.1002/ijc.20725. [DOI] [PubMed] [Google Scholar]
- 30.Yamano M, Fujii H, Takagaki T, Kadowaki N, Watanabe H, Shirai T. Genetic progression and divergence in pancreatic carcinoma. Am J Pathol. 2000;156:2123–2133. doi: 10.1016/S0002-9440(10)65083-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu N, Hu N, Li WJ, Roth MJ, Wang C, Su H, Wang QH, Taylor PR, Dawsey SM. Microsatellite alterations in esophageal dysplasia and squamous cell carcinoma from laser capture microdissected endoscopic biopsies. Cancer Lett. 2003;189:137–145. doi: 10.1016/s0304-3835(02)00555-4. [DOI] [PubMed] [Google Scholar]