

Abstract
Objective
The current standard treatment for ovarian carcinoma, consisting of surgery followed by chemotherapy with carboplatin and paclitaxel, is fraught with a high rate of recurrences. We hypothesized that targeted inhibition of specific signaling pathways in combination with conventional drugs may increase chemotherapeutic efficacy.
Methods
We analyzed the expression and activation profiles of various signaling pathways in nine established ovarian cancer cell lines (CAOV-3, ES2, PA-1, SKOV-3, NIHOVCAR3, OV90, TOV112D, A1847, A2780) and 24 freshly procured human ovarian tumors. The PI3 kinase pathway component Akt was frequently overexpressed and/or activated in tumor cells. The effect of several PI3K pathway inhibitors (rapamycin, LY294002, SH-6) and rapamycin in combination with carboplatin on various tumor cell growth characteristics was tested in cell lines and fresh tumor-derived transient monolayer and organ cultures.
Results
Rapamycin by itself and additively with carboplatin inhibited the growth and invasion, and increased the sensitivity to anoikis of most of the ovarian cancer cell lines and fresh tumors. The additive inhibitory effect may be due to enhanced apoptosis as demonstrated by Poly-ADP-Ribose Polymerase (PARP) cleavage and Annexin V staining in cells treated with both rapamycin and carboplatin.
Conclusions
Rapamycin in combination with standard chemotherapeutic agents may improve the efficiency of ovarian cancer treatment.
Keywords: Ovarian carcinoma, Rapamycin, PI3 kinase, Carboplatin, Chemotherapy
Introduction
The mortality rate of ovarian cancer, the second most frequent malignancy of the female genital tract in the western world, is higher than that of all other gynecologic malignancies combined. Serous carcinoma accounts for almost 60% of all ovarian malignancies. Of these, more than 70% present at an advanced stage with widespread disease in the peritoneal cavity and/or distant metastases.[1] The standard treatment of ovarian carcinoma consists of cytoreductive surgery with subsequent chemotherapy including carboplatin and paclitaxel. Despite initially high response rates to this treatment, most patients develop recurrent disease within few years.[1] Second line treatment regimens are less standardized and include platinum-based agents, gemcitabine, topotecan, etoposide, 5-fluorouracil, doxorubicin and combinations thereof.[1, 2] Median survival has improved with these regimens, but long-term survival and disease mortality have remained largely unchanged due to drug-resistant recurrent tumor. Therefore, more effective treatment options for ovarian carcinoma are needed. The molecular mechanisms of ovarian oncogenesis are poorly understood. Among cell growth and survival controlling mechanisms, the phosphatidylinositol-3 kinase (PI3K) signaling pathway is often activated. Aberrations of its signaling molecules are frequently found in ovarian cancer cells. This includes overexpression of the upstream receptor protein kinases (RPTKs),[3–6] mutations/amplifications of the PI3K catalytic (PIK3CA, p110) and regulatory (p85) subunits, [7–9] Akt activation,[10] and Akt 2 amplification[11]. Loss of the negative regulator PTEN due to deletion, inactivating mutations or epigenetic silencing has been associated with ovarian carcinomas of endometrioid histology.[12–14] Aside from uncontrolled growth, the capacity of invasion and metastatic spread are pathogenic features of cancer cells. Numerous reports suggest a role of PI3K signaling in invasion and metastasis.[15–21] Therefore, the component molecules of the PI3K signaling pathway are logical targets for new anti-cancer drug development.
Rapamycin, a macrolide produced by Streptomyces hygroscopicus, leads to cell cycle arrest in the G1 phase by inhibition of the mammalian target of rapamycin complex 1 (mTORC1). mTORC1 controls protein translation and several cytokine-driven signaling pathways involved in cell cycle progression.[22, 23] Rapamycin and its analogues have been in clinical use for about a decade as immunosuppressants. Recently they have been tested as potential anti-cancer drugs against breast, gastrointestinal, head and neck, renal and other solid tumors.[22, 24–27] Temserolimus was FDA approved for the treatment of advanced renal cell carcinoma in 2007 and is currently being evaluated in several clinical trials for its use in the treatment of gynecologic malignancies, including ovarian carcinoma.[28] Cell culture studies [10, 29–31] and mouse models [32–37] also suggested that PI3K pathway inhibitors are efficient in suppressing ovarian cancer cell growth. We analyzed the effect of the PI3K, AKT and mTORC1 inhibitors LY294002, SH-6 and rapamycin, respectively, on ovarian cancer cell growth using established cell lines and fresh human tumor tissue. Since the biologic targets of PI3K pathway inhibitors and carboplatin are different, we hypothesized that combination treatment may result in an additive effect. Our results show that rapamycin alone and additively in combination with carboplatin effectively inhibits various oncogenic properties of ovarian cancer cells.
Materials and Methods
Cell lines and cultures
The ovarian cancer cell lines CAOV-3, ES-2, PA-1, SKOV-3, OV-90, NIHOVCAR3, TOV-112D (obtained from ATCC), A1847 and A2780 (obtained from Dr. Stuart Aaronson, Mount Sinai School of Medicine) were cultured in DMEM with 10% fetal calf serum (FCS).
Reagents
LY294002 is a specific inhibitor (IC50 = 1.40 µM) of PI3 kinase. Rapamycin (sirolimus) binds to FK506 binding protein 12 (FKBP12) and selectively inhibits the catalytic activity of the mammalian target of rapamycin (mTOR) when complexed to the regulatory associated protein of mTOR (raptor) in the mTOR-complex 1 (mTORC1). SH-6 (Akt inhibitor III) is a phosphatidylinositol analog that inhibits the activation of Akt without decreasing phosphorylation of other kinases downstream of Ras. Carboplatin (cis-Diamine[1,1-cyclobutanedicarboxylato]platinum[II]), an analog of Cisplatin with reduced nephrotoxicity, is an alkylating agent that cross-links DNA strands and induces apoptosis in cancer cells, possibly via caspase-3 activation, and is used in the standard chemotherapy for ovarian carcinoma. LY294002 and rapamycin were purchased from L.C. Laboratories, Woburn, MA. SH-6 and carboplatin from Calbiochem, San Diego, CA. The dosage of these reagents was based on our previous experience.[38] For SH-6, we empirically determined to use 20ng/ml to effectively inhibit more than 80% of the phospho-AKT. For drug combination experiments, we targeted 50% inhibition by either drug.
Fresh tumor cells and organ culture
This study was approved by the Institutional Review Board. A waiver of informed consent was obtained for studies of human tissue. Between April 2003 and June 2005, de-identified fresh tumor and, when possible, matched normal tissue samples were harvested from 24 different ovarian carcinoma patients. In order to prevent confounding results due to the great variety of histologic subtypes of ovarian malignancies, we focused on primary ovarian papillary serous carcinomas (OPSC), which constitute the vast majority of our study cohort (Supplementary table 1). The histopathologic diagnoses, tumor grade and stage were verified according to the World Health Organization (WHO) classification and the American Joint Committee on Cancer (AJCC) staging system.[39, 40] Due to limited tissue availability, not all tissue samples could be subjected to all tests. Monolayer and organ cultures were prepared as follows: Fresh tissue was harvested within 15 minutes of surgical removal, placed in DMEM containing 10% FCS and kept at 4° C. The specimen was divided into 3 pieces, which were used for protein extraction, preparation of monolayer cell culture and organ culture, respectively. For monolayer culture, the normal or tumor tissue was minced and treated with trypsin (0.5%) and collagenase (1 mg/ml) in phosphate buffered saline containing 0.2% EDTA at room temperature for 30 minutes with constant stirring. After sedimentation for 5 minutes, the supernatant was harvested, while the sediment was subjected to a second cycle of trypsin and collagenase treatment. The combined supernatant cell suspension was centrifuged, and the collected cells were resuspended in DMEM and seeded in culture dishes. In some cases, the cells were cultured in DMEM containing 1.3% methylcellulose for 48 hours to enrich for anoikis resistant tumor cells before seeding on the culture dishes.[41] With this protocol, about 50% of the initial cells survived. Significant growth of monolayer tumor cells could be observed during the initial 3 to 4 passages. The monolayer tumor cell cultures with or without preselection in methylcellulose medium could be maintained for 3 to 6 weeks (Supplementary Fig. 1A). The epithelial origin was confirmed by immunostaining for cytokeratins (Supplementary Fig. 2).
For transient organ culture, 1–2 mm3 tumor pieces were placed on a nylon mesh platform floating on DMEM and cultured in the air-medium interphase. Changing the medium every three days, the tumor tissue remained viable for 3 to 4 weeks. Cell growth could be observed around the periphery of the tissue fragments (Supplementary Fig. 1B) and was assessed after one week by 3H-thymidine incorporation assay.
Protein analysis
Growth factor receptors and signaling molecules were analyzed by Western blots as described.[16] Total lysates from either cell cultures or freshly frozen tumor tissues were prepared using RIPA buffer.[16, 18, 41] The frozen tissues were minced and dounce homogenized prior to RIPA extraction. Protein lysates were resolved by SDS-PAGE, probed with primary antibodies against ErbB2, EGFR (Calbiochem, San Diego, CA), IGF1R [42], IR, AKT-1, AKT-2, phospho-AKT(S473) (Santa Cruz Biotechnology, Santa Cruz, CA), MAP kinase, phospho-MAP kinase, phospho- AKT(T308) ( Cell Signaling, Danvers, MA), Stat3 (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-Stat3 (Cell Signaling, Danvers, MA), PARP (Beckton Dickinson, Franklin Lakes, NJ), GAPDH (Chemicon International, Temecula, CA) and β-tubulin (Sigma, St Louis, MO), and visualized by chemiluminescence (PerkinElmer, Boston, MA, and Roche, Nutley, NJ).
Monolayer growth assay
105 cells were seeded in 6cm Petri dishes and maintained in the growth medium with or without rapamycin (5 or 10 ng/ml), carboplatin (1 or 2 µg/ml, as indicated in the figure legends) or both. 72 to 96 hours later the cells were trypsinized and counted.
Colony assay
105 cells were seeded in a 6cm Petri dish and cultured in agar containing DMEM without or with LY294002 (10 µM), SH-6 (20 ng/ml), rapamycin (5ng/ml), carboplatin (2µg/ml), or rapamycin (5ng/ml) and carboplatin (2µg/ml) combined. After 14 days, colonies were quantified as described previously.[18]
Invasion assay
105 cells were plated in a Boyden chamber, and the number of invaded cells was quantified as described [18], except that the insert was precoated with 100µl of 0.5µg/ml growth factor reduced Matrigel (Beckton Dickinson, Franklin Lakes, NJ) in DMEM containing 0.1% bovine serum albumin (BSA) and incubated at 37ºC for 30 minutes before use. The observation time was 6 to 24h.
Anoikis assay
106 cells per ml were suspended in growth medium containing 1.3% methylcellulose as described.[41] 48 to 72 hours later cells were recovered and subjected to trypan blue exclusion viability assay.[41]
DNA synthesis assay
Organ cultures were washed three times with DMEM containing 10% dialyzed FCS and then incubated in 4 ml of the same medium containing 100 µCi of 3H-thymidine (20 Ci/mM, PerkinElmer Life and Analytical Sciences, Boston, MA) with or without rapamycin (5 ng/ml), carboplatin (2 µg/ml) or both. After 96 hours, the tissues were washed with PBS and treated with Western extraction buffer as described.[16] A small aliquot was taken for assaying protein concentration (Bio-Rad reagent). DNA of the remaining sample was extracted by adjusting to 0.1 N KOH, neutralized with HCl and deproteinized with phenol/chloroform. DNA was precipitated by adjusting to 70% ethanol and 0.1 N NaCl and kept at −20ºC overnight. The DNA was pelleted by centrifugation and redissolved in 100 µl water. Radioactivity was measured by liquid scintillation. The DNA synthesis, as measured by the 3H-thymidine incorporation, was normalized to the protein amount.
Annexin V staining assay for apoptotic cells
A2780, PA-1, ES2, and OVT24 cells were treated with various concentrations of rapamycin, carboplatin, or both for 48 or 72 h. Cells were collected and stained with Annexin V-FITC according to the manufacturer’s instruction (BD Biosciences). 1×104 cells were collected, and the percentage of Annexin V-FITC positive cells was considered as the percentage of apoptotic cells.
Statistical analysis
For every test three experiments were conducted in duplicates, except for the 3H-thymidine incorporation test, which was performed with two duplicate experiments. Histograms shown in the figures represent mean values and standard deviation.
Protein expression (Supplementary Fig. 3) was graded visually on a four tier-scale ranging from 0 (no expression) to 3 (strong expression). To detect associations between tumor stage, grade, histologic subtype or age, and expression level of the PI3K pathway-related signaling molecules, a one tail Student’s t test was used. Two-tailed t tests were used to assess the statistical significance of growth inhibition under rapamycin, carboplatin or combined therapy.
Results
Expression and activation of PI3K pathway mediators
Cell lines expressed ErbB2, IGF-1 receptor (IGF-1R), EGF receptor (EGFR), or insulin receptor (IR) to varying degrees (Supplementary Fig. 3A). ES2 overexpressed IGF-1R and EGFR, and PA-1 overexpressed IGF-1R and IR, whereas OV90, TOV112D and A1847 did not show significant expression of these RTPKs. ES2, SKOV-3 and OV90 had relatively higher levels of activated MAP kinase, and all except OV90 and A2780 displayed activated Akt. Except for A2780, all lines with overexpression of RPTK displayed activation of MAP kinase, Akt or both.
Nineteen of the 24 tumor samples and 9 matched normal tissues were examined for protein expression. The tumor tissues had higher expression levels of ErbB2, IGF-1R, EGFR, IR, Akt2 or Stat3, and displayed higher levels of phosphorylated MAP kinase, Akt and Stat3 than the normal tissues (Supplementary Fig. 3B). We were unable to identify a statistically significant correlation between protein expression patterns and tumor type or stage.
Effect of PI3K pathway inhibitors and carboplatin on ovarian cancer cell lines
The six lines with robust colony forming ability (ES2, NIHOVCAR3, SKOV-3, OV-90, TOV-112D, and A2780, Fig. 1A) were treated with LY294002, SH-6 and rapamycin,. Except for ES2 and A2780, LY294002 reduced the colony formation ability to 30 – 70% of the controls (Fig. 2A). SH-6 inhibited 30 to 60%, while rapamycin inhibited 50 to 80% of the colony formation. All lines tested were inhibited more than 45% by rapamycin. Of the five lines that were further tested for combined drug inhibition, all were inhibited by rapamycin alone, and four showed a significant further colony formation reduction under combined rapamycin/carboplatin treatment. Colony formation ability of SKOV3 was so strongly inhibited by rapamycin alone that a further reduction could not be demonstrated by the addition of carboplatin (Fig. 2B).
Fig. 1. Colony and invasion activity of various ovarian cancer lines.

A Colony assays, photographed 14 days after plating. B Boyden chamber invasion assays: invaded cells photographed after 24 hours.
Fig. 2. Effect of PI3K pathway inhibitors and carboplatin on growth and invasion of ovarian cancer cells.

All histograms except those in C represent the average values with standard errors of 3 independent experiments. The results in C represent the average of two experiments. P-values indicate the statistical significance between the effects of carboplatin or rapamycin as single agents versus their combined use.
A Colony assays using DMSO solvent, 10µM LY294002, 20ng/ml AKT inhibitor (SH-6) or 10ng/ml rapamycin (Ra). The value for DMSO treated control cells was set as 100%. B Colony assays using 5ng/ml rapamycin (Ra), 2µg/ml carboplatin (Carbo), or both. C Boyden chamber invasion assays. D Growth assay was performed by plating 1 × 105 cells in duplicate 6-cm dishes in DMEM with 10% FCS. The following day the medium was changed with addition of DMSO solvent, 5 ng/ml rapamycin, 2 µg/ml carboplatin, or both. Cells were counted 72 hours later.
Similarly, six cell lines with higher invasion activity (Fig. 1B) were tested for inhibition by LY294002, SH-6 and rapamycin. PA-1 was relatively resistant to all three inhibitors. NIHOVCAR3 and CAOV3 were relatively resistant to rapamycin, but were sensitive to LY294002 and SH-6. The rest were inhibited 60 to 80% by rapamycin (Fig. 2C).
In monolayer growth assays, OV90, SKOV3 and NIH OVCAR3 were relatively resistant to carboplatin, but most lines were sensitive to rapamycin. In most cases, combination of rapamycin and carboplatin significantly enhanced growth inhibition compared to either drug alone (Fig. 2D).
Effect of rapamycin and carboplatin on fresh tumor-derived monolayer and organ cultures
Cells were immunohistochemically analyzed for cytokeratin expression to assure their epithelial origin. The majority of cultured cells were positive for cytokeratins (Supplementary Fig. 2).
Fifteen of the 24 tumors grew monolayer cultures for at least two transfers. Organ cultures were prepared from OVT8 onwards and could be maintained for one to four weeks in 11 of 16 tumors (Supplementary table 1).
Rapamycin and carboplatin individually showed different degrees (20 to 80%) of growth inhibition on the tumor cells in monolayer or in organ culture as measured by cell count and 3H-thymidine incorporation, respectively. Combined treatment resulted in significantly enhanced inhibition in most cases (Fig. 3A–B). Interestingly, OVT22 had been subjected to neoadjuvant chemotherapy with carboplatin and paclitaxel (Supplementary table 1), and the lower sensitivity to carboplatin may reflect an emerging drug resistance.
Fig. 3. Fresh tumor-derived cells under rapamycin and carboplatin.

A Monolayer culture growth. B DNA synthesis of transient organ cultures. Bars represent the average of duplicate cultures for each treatment. C Invasion assay. D Colony assay. E Anoikis assay. For A, C, D and E, bars represent average values with standard errors of three independent experiments.
The effect of rapamycin and carboplatin on invasion, colony formation and sensitivity to anoikis of fresh tumor-derived cells is illustrated in Fig. 3C–E. Rapamycin and carboplatin individually inhibited these three oncogenic properties. Combined treatment resulted in an enhanced inhibition in anchorage independent growth and resistance to anoikis, but less apparent effect on invasion activity. Among our tumor cases, only OVT24 displayed a significant and robust colony forming ability and continuous growth for over 20 passages.
Induction of apoptosis by rapamycin and carboplatin
To explore the mechanism of the enhanced antiproliferative effect under carboplatin/rapamycin combination treatment, apoptosis assays using PARP cleavage and Annexin V staining were conducted. PARP expression and cleavage was assessed in four representative cell lines and one fresh tumor-derived culture. Rapamycin induced PARP cleavage in all cell lines, and addition of carboplatin resulted in an increased PARP cleavage in PA-1 and ES2 cells (Fig. 4A). In OVT24 cells, carboplatin induced PARP cleavage and addition of rapamycin led to an enhanced cleavage.
Fig. 4. PARP cleavage and apoptosis assay under rapamycin and carboplatin.
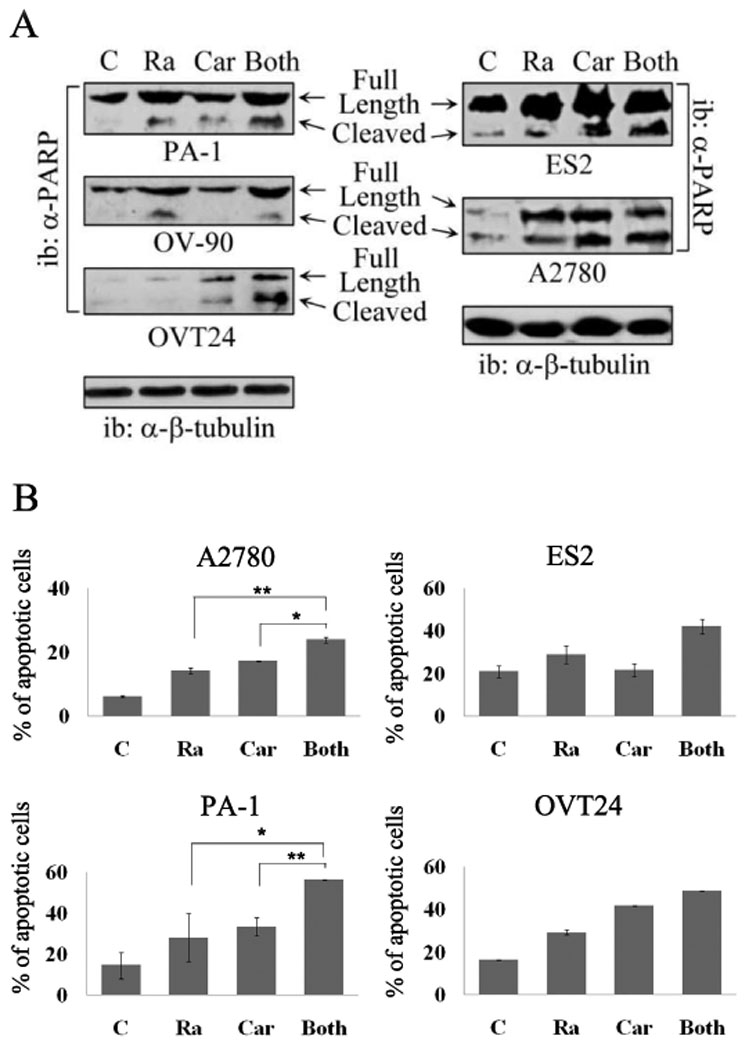
A PARP cleavage. Upper band represents total PARP, lower band represents cleavage product. B Annexin assay. Four ovarian cell lines, A2780, PA-1, ES2, and OVT24 were treated with rapamycin, carboplatin, or both for 48 h (PA-1) or 72 h (A2780, ES2, and OVT24). For PA-1 and A2780 cells, 10 ng/ml rapamycin, 2 µg/ml carboplatin or their combination was added. For ES2 and OVT24 cells, 50 ng/ml rapamycin, 10 µg/ml carboplatin or their combination was added. Cells were then stained with Annexin V-FITC. FACS analysis was performed to quantify the percentage of cells undergoing apoptosis with Annexin V positive staining. (*, p<0.05; **, p<0.01). C – control; Ra – rapamycin; Car – carboplatin.
Annexin V staining indicated increased apoptosis in all four cell lines tested. The difference between either rapamycin or carboplatin alone and their combined application was statistically significant in two cell lines (A2780 and PA-1, Fig. 4B). These findings suggest that the combination of rapamycin and carboplatin can lead to increased apoptosis.
Discussion
Our results show that inhibition of the PI3K pathway is highly effective in suppressing proliferation and invasion of ovarian cancer cells. Due to limitations in specificity and potential side effects of LY294002 [43, 44], and the lack of clinical experience with SH-6, we focused on rapamycin in the test of combination therapy. Rapamycin is already established as an immunosuppressive agent clinically, and its toxicity profile is known. We used rapamycin at concentrations of 5–10 ng/ml, which is similar to that in the blood of transplant patients treated for graft rejection prophylaxis.[45]
Over the past years a substantial body of evidence has been accumulated regarding the therapeutic usefulness of PI3 kinase pathway inhibitors for various malignancies including breast, gastrointestinal, head and neck, renal and other solid tumors.[22, 25, 26] Several phase I/II clinical trials using PI3 kinase inhibitors for a variety of solid tumors are under way.[46] After its FDA approval for the treatment of advanced renal cell carcinoma, the rapamycin derivative temserolimus is currently being tested as single agent and in combination with other drugs for the treatment of gynecologic malignancies, including ovarian carcinoma.[28] Although other PI3K pathway inhibitors have been tested in combination with cisplatin or carboplatin on ovarian carcinoma cells [47, 48], this is the first report about the combined use of rapamycin and carboplatin in human ovarian cancer.
Carboplatin, the single most important chemotherapeutic agent used in the treatment of ovarian carcinoma, is an alkylating agent that cross-links DNA strands, thereby interfering with DNA replication and leading to apoptosis.[49] The action mechanism of rapamycin as an inhibitor of the PI3K pathway is principally different, which may explain the enhanced antiproliferative effect under combination treatment. Furthermore, the PI3K pathway has been implicated in the development of drug resistance. The X-linked inhibitor of apoptosis (XIAP) protein and Her-2/neu can induce resistance to chemotherapeutic agents and tumor necrosis factor (TNF) through PI3K pathway-mediated inhibition of the caspase cascade.[6, 50, 51] AKT has been implicated in mediating resistance to cicplatin and paclitaxel in ovarian cancer cells.[52, 53] The PI3K inhibitor LY294002 was able to revert PI3K-driven drug resistance to Paclitaxel in ovarian cancer models.[54] Therefore, the addition of a PI3K pathway inhibitor to the standard cytotoxic chemotherapy might not only target tumor cell growth, but also antagonize the development of drug resistance.
Relatively few published reports have shown the antitumoral effect of PI3K pathway inhibitors in combination with other chemotherapeutic agents. Growth assays on human carcinoma cells and tumor models using athymic mice inoculated with human ovarian carcinoma cell lines showed enhanced tumor sensitivity to platinum-based chemotherapy under inhibition of the PI3K pathway. In a mouse model, combination treatment with the mTORC1 inhibitor RAD001 (everolimus, a rapamycin derivative) and cisplatin resulted in prolonged survival.[36] The mechanism of this phenomenon is thought to be an increased efficacy of platinum-induced apoptosis.[47, 48] Consistent with these reports, we observed increased PARP cleavage under rapamycin/carboplatin combination treatment. In head and neck cancer cells, rapamycin as a single agent has been shown to reduce cell proliferation and induce apoptosis, while an additive synergistic effect was observed when combined with carboplatin and paclitaxel.[55]
Recent observations suggest that inhibition of mTORC1 may lead to secondary activation of not only the PI3 kinase pathway, but also the MAP kinase pathway by relieving a feedback mechanism mediated by the negative regulator S6K1.[23, 56] The effect of selective mTORC1 inhibition may vary considerably from tumor to tumor due to the numerous mTOR-independent substrates of Akt, abundant cross-talk between PI3 kinase signaling and other signaling pathways, and the specific cell signaling aberrations in any given tumor. Drugs or drug combinations targeting multiple signaling molecules simultaneously (e.g. mTORC1, Akt and ERK) may overcome the drawbacks of mTORC1 inhibition monotherapy. Here we show that rapamycin by itself has a strong antiproliferative effect on ovarian carcinoma cells, which is further enhanced in combination with carboplatin.
In addition to uncontrolled proliferation, malignant tumor cells are characterized by the ability to destructively invade surrounding tissue and to form metastases. Our results and previous reports support a role of the PI3K pathway in invasion and metastasis formation.[15–19] Thus, inhibition of the PI3K pathway might also interfere with these crucial properties of malignant cells.
A transient organ culture assay as described in this study may be helpful in the clinical setting to determine which patients might benefit from a PI3K pathway inhibitor added to the standard chemotherapeutic regimen. Development and standardization of the culturing and assaying protocol could be of considerable clinical value.
In conclusion, we present evidence that rapamycin, especially in combination with carboplatin, may be useful in the treatment of human ovarian carcinoma. As an inhibitor of the PI3K pathway, rapamycin targets tumor cell growth, invasion and metastasis, antagonizes drug resistance and enhances the efficiency of apoptosis induced by platinum-based agents. While adding these new dimensions to tumor therapy, rapamycin may increase the efficiency of standard chemotherapy and/or allow lower doses of cytotoxic agents to reduce side effects.
Supplementary Material
Numbers indicate individual tumor cases. Monolayer (A) and organ (B) cultures were maintained in DMEM containing 10% FCS. The photographs were taken one week later.
Immunofluorescent stain for cytokeratins and phase contrast images. Original magnification: 100×.
Equivalent amounts of protein lysates from various ovarian cancer cell lines, fresh ovarian tumors (OVT) and matched normal tissues (N) were resolved by SDS-PAGE and blotted with the indicated antibodies. For the detection of phospho-AKT, a mixture of antibodies against both the T308 and S473 epitopes was used.
Acknowledgements
This study was supported by NIH grant CA 29339 and DOD Army grant DAMD17-02-1-0504 (LHW).
List of abbreviations
- PI3K
Phosphatidylinositol-3 kinase
- PARP
Poly-adenosine diphosphate-ribose Polymerase
- RPTK
Receptor protein tyrosine kinase
- mTOR
Mammalian target of rapamycin
- mTORC1
Mammalian target of rapamycin complex 1
- EGFR
Epithelial growth factor receptor
- IGFR
Insulin-like growth factor receptor
- IR
Insulin receptor
- MAP kinase
Mitogen activated protein kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Parts of this work were presented as a poster presentation at the 37th annual meeting of the Society of Gynecologic Oncologists, March 22–26, 2006, Palm Springs, CA.
Conflict of interest statement:
The authors declare that there are no conflicts of interest
References
- 1.Ozols RF, Bookman MA, Connolly DC, Daly MB, Godwin AK, Schilder RJ, Xu X, Hamilton TC. Focus on epithelial ovarian cancer. Cancer. Cell. 2004;5:19–24. doi: 10.1016/s1535-6108(04)00002-9. [DOI] [PubMed] [Google Scholar]
- 2.Sundar S, Symonds RP, Decatris MP, Kumar DM, Osman A, Vasanthan S, O'byrne KJ. Phase II trial of oxaliplatin and 5-Fluorouracil/Leucovorin combination in epithelial ovarian carcinoma relapsing within 2 years of platinum-based therapy. Gynecol. Oncol. 2004;94:502–508. doi: 10.1016/j.ygyno.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 3.Lafky JM, Wilken JA, Baron AT, Maihle NJ. Clinical implications of the ErbB/epidermal growth factor (EGF) receptor family and its ligands in ovarian cancer. Biochim. Biophys. Acta. 2008;1785:232–265. doi: 10.1016/j.bbcan.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Steffensen KD, Waldstrom M, Andersen RF, Olsen DA, Jeppesen U, Knudsen HJ, Brandslund I, Jakobsen A. Protein levels and gene expressions of the epidermal growth factor receptors, HER1, HER2, HER3 and HER4 in benign and malignant ovarian tumors. Int. J. Oncol. 2008;33:195–204. [PubMed] [Google Scholar]
- 5.Ross JS, Yang F, Kallakury BV, Sheehan CE, Ambros RA, Muraca PJ. HER-2/neu oncogene amplification by fluorescence in situ hybridization in epithelial tumors of the ovary. Am. J. Clin. Pathol. 1999;111:311–316. doi: 10.1093/ajcp/111.3.311. [DOI] [PubMed] [Google Scholar]
- 6.Zhou BP, Hu MC, Miller SA, Yu Z, Xia W, Lin SY, Hung MC. HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-kappaB pathway. J. Biol. Chem. 2000;275:8027–8031. doi: 10.1074/jbc.275.11.8027. [DOI] [PubMed] [Google Scholar]
- 7.Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB, Phillips WA. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64:7678–7681. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- 8.Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, Boyd J. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin. Cancer Res. 2005;11:2875–2878. doi: 10.1158/1078-0432.CCR-04-2142. [DOI] [PubMed] [Google Scholar]
- 9.Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, Thomas RJ, Phillips WA. The phosphatidylinositol 3'-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61:7426–7429. [PubMed] [Google Scholar]
- 10.Altomare DA, Wang HQ, Skele KL, De Rienzo A, Klein-Szanto AJ, Godwin AK, Testa JR. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene. 2004;23:5853–5857. doi: 10.1038/sj.onc.1207721. [DOI] [PubMed] [Google Scholar]
- 11.Nakayama K, Nakayama N, Kurman RJ, Cope L, Pohl G, Samuels Y, Velculescu VE, Wang TL, Shih I. Sequence mutations and amplification of PIK3CA and AKT2 genes in purified ovarian serous neoplasms. Cancer. Biol. Ther. 2006;5:779–785. doi: 10.4161/cbt.5.7.2751. [DOI] [PubMed] [Google Scholar]
- 12.Willner J, Wurz K, Allison KH, Galic V, Garcia RL, Goff BA, Swisher EM. Alternate molecular genetic pathways in ovarian carcinomas of common histological types. Hum. Pathol. 2007;38:607–613. doi: 10.1016/j.humpath.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Kolasa IK, Rembiszewska A, Janiec-Jankowska A, Dansonka-Mieszkowska A, Lewandowska AM, Konopka B, Kupryjanczyk J. PTEN mutation, expression and LOH at its locus in ovarian carcinomas. relation to TP53, K-RAS and BRCA1 mutations. Gynecol. Oncol. 2006;103:692–697. doi: 10.1016/j.ygyno.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 14.Obata K, Morland SJ, Watson RH, Hitchcock A, Chenevix-Trench G, Thomas EJ, Campbell IG. Frequent PTEN/MMAC mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer Res. 1998;58:2095–2097. [PubMed] [Google Scholar]
- 15.Alessandro R, Kohn EC. Signal transduction targets in invasion. Clin. Exp. Metastasis. 2002;19:265–273. doi: 10.1023/a:1015547804511. [DOI] [PubMed] [Google Scholar]
- 16.Sachdev P, Jiang YX, Li W, Miki T, Maruta H, Nur-E-Kamal MS, Wang LH. Differential requirement for rho family GTPases in an oncogenic insulin-like growth factor-I receptor-induced cell transformation. J. Biol. Chem. 2001;276:26461–26471. doi: 10.1074/jbc.M010995200. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen KT, Wang WJ, Chan JL, Wang LH. Differential requirements of the MAP kinase and PI3 kinase signaling pathways in src- versus insulin and IGF-1 receptors-induced growth and transformation of rat intestinal epithelial cells. Oncogene. 2000;19:5385–5397. doi: 10.1038/sj.onc.1203911. [DOI] [PubMed] [Google Scholar]
- 18.Sachdev P, Zeng L, Wang LH. Distinct role of phosphatidylinositol 3-kinase and rho family GTPases in Vav3-induced cell transformation, cell motility, and morphological changes. J. Biol. Chem. 2002;277:17638–17648. doi: 10.1074/jbc.M111575200. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen KT, Zong CS, Uttamsingh S, Sachdev P, Bhanot M, Le MT, Chan JL, Wang LH. The role of phosphatidylinositol 3-kinase, rho family GTPases, and STAT3 in ros-induced cell transformation. J. Biol. Chem. 2002;277:11107–11115. doi: 10.1074/jbc.M108166200. [DOI] [PubMed] [Google Scholar]
- 20.Ellerbroek SM, Halbleib JM, Benavidez M, Warmka JK, Wattenberg EV, Stack MS, Hudson LG. Phosphatidylinositol 3-kinase activity in epidermal growth factor-stimulated matrix metalloproteinase-9 production and cell surface association. Cancer Res. 2001;61:1855–1861. [PubMed] [Google Scholar]
- 21.Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, Danino M, Karlan BY, Slamon DJ. Overexpression of AKT2/protein kinase bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003;63:196–206. [PubMed] [Google Scholar]
- 22.Mita MM, Mita A, Rowinsky EK. Mammalian target of rapamycin: A new molecular target for breast cancer. Clin. Breast Cancer. 2003;4:126–237. doi: 10.3816/cbc.2003.n.018. [DOI] [PubMed] [Google Scholar]
- 23.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2008 doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Punt CJ, Boni J, Bruntsch U, Peters M, Thielert C. Phase I and pharmacokinetic study of CCI-779, a novel cytostatic cell-cycle inhibitor, in combination with 5-fluorouracil and leucovorin in patients with advanced solid tumors. Ann. Oncol. 2003;14:931–937. doi: 10.1093/annonc/mdg248. [DOI] [PubMed] [Google Scholar]
- 25.Raymond E, Alexandre J, Faivre S, Vera K, Materman E, Boni J, Leister C, Korth-Bradley J, Hanauske A, Armand JP. Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J. Clin. Oncol. 2004;22:2336–2347. doi: 10.1200/JCO.2004.08.116. [DOI] [PubMed] [Google Scholar]
- 26.Atkins MB, Hidalgo M, Stadler WM, Logan TF, Dutcher JP, Hudes GR, Park Y, Liou SH, Marshall B, Boni JP, Dukart G, Sherman ML. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J. Clin. Oncol. 2004;22:909–918. doi: 10.1200/JCO.2004.08.185. [DOI] [PubMed] [Google Scholar]
- 27.O'Donnell A, Faivre S, Burris HA, 3rd, Rea D, Papadimitrakopoulou V, Shand N, Lane HA, Hazell K, Zoellner U, Kovarik JM, Brock C, Jones S, Raymond E, Judson I. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J. Clin. Oncol. 2008;26:1588–1595. doi: 10.1200/JCO.2007.14.0988. [DOI] [PubMed] [Google Scholar]
- 28.Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer. 2009;9:167–181. doi: 10.1038/nrc2583. [DOI] [PubMed] [Google Scholar]
- 29.Aguirre D, Boya P, Bellet D, Faivre S, Troalen F, Benard J, Saulnier P, Hopkins-Donaldson S, Zangemeister-Wittke U, Kroemer G, Raymond E. Bcl-2 and CCND1/CDK4 expression levels predict the cellular effects of mTOR inhibitors in human ovarian carcinoma. Apoptosis. 2004;9:797–805. doi: 10.1023/B:APPT.0000045781.46314.e2. [DOI] [PubMed] [Google Scholar]
- 30.Gao N, Flynn DC, Zhang Z, Zhong XS, Walker V, Liu KJ, Shi X, Jiang BH. G1 cell cycle progression and the expression of G1 cyclins are regulated by PI3K/AKT/mTOR/p70S6K1 signaling in human ovarian cancer cells. Am. J. Physiol. Cell. Physiol. 2004;287:C281–C291. doi: 10.1152/ajpcell.00422.2003. [DOI] [PubMed] [Google Scholar]
- 31.Shi Y, Frankel A, Radvanyi LG, Penn LZ, Miller RG, Mills GB. Rapamycin enhances apoptosis and increases sensitivity to cisplatin in vitro. Cancer Res. 1995;55:1982–1988. [PubMed] [Google Scholar]
- 32.Hu P, Margolis B, Skolnik EY, Lammers R, Ullrich A, Schlessinger J. Interaction of phosphatidylinositol 3-kinase-associated p85 with epidermal growth factor and platelet-derived growth factor receptors. Mol. Cell. Biol. 1992;12:981–990. doi: 10.1128/mcb.12.3.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang HY, Feng YJ. Inhibition of hypoxia-inducible factor 1alpha expression and tumor growth in SKOV3 ovarian cancer model by sirolimus. Zhonghua Fu Chan Ke Za Zhi. 2004;39:474–477. [PubMed] [Google Scholar]
- 34.Xing D, Orsulic SA. genetically defined mouse ovarian carcinoma model for the molecular characterization of pathway-targeted therapy and tumor resistance. Proc. Natl. Acad. Sci. U. S. A. 2005;102:6936–6941. doi: 10.1073/pnas.0502256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huynh H, Teo CC, Soo KC. Bevacizumab and rapamycin inhibit tumor growth in peritoneal model of human ovarian cancer. Mol. Cancer. Ther. 2007;6:2959–2966. doi: 10.1158/1535-7163.MCT-07-0237. [DOI] [PubMed] [Google Scholar]
- 36.Mabuchi S, Altomare DA, Cheung M, Zhang L, Poulikakos PI, Hensley HH, Schilder RJ, Ozols RF, Testa JR. RAD001 inhibits human ovarian cancer cell proliferation, enhances cisplatin-induced apoptosis, and prolongs survival in an ovarian cancer model. Clin. Cancer Res. 2007;13:4261–4270. doi: 10.1158/1078-0432.CCR-06-2770. [DOI] [PubMed] [Google Scholar]
- 37.Mabuchi S, Altomare DA, Connolly DC, Klein-Szanto A, Litwin S, Hoelzle MK, Hensley HH, Hamilton TC, Testa JR. RAD001 (everolimus) delays tumor onset and progression in a transgenic mouse model of ovarian cancer. Cancer Res. 2007;67:2408–2413. doi: 10.1158/0008-5472.CAN-06-4490. [DOI] [PubMed] [Google Scholar]
- 38.Wang LH, Chan JL, Li W. Rapamycin together with herceptin significantly increased anti-tumor efficacy compared to either alone in ErbB2 over expressing breast cancer cells. Int. J. Cancer. 2007;121:157–164. doi: 10.1002/ijc.22606. [DOI] [PubMed] [Google Scholar]
- 39.Scully RF, Young RH, Clement PB. Tumors of the ovary, mal-developed gonads, fallopian tube, and broad ligament. Washington, D.C.: Armed Forces Institute of Pathology; 1998. [Google Scholar]
- 40.AJCC cancer staging handbook. New York: Springer Verlag; 2002. [Google Scholar]
- 41.Uttamsingh S, Zong CS, Wang LH. Matrix-independent activation of phosphatidylinositol 3-kinase, Stat3, and cyclin A-associated Cdk2 is essential for anchorage-independent growth of v-ros-transformed chicken embryo fibroblasts. J. Biol. Chem. 2003;278:18798–18810. doi: 10.1074/jbc.M211522200. [DOI] [PubMed] [Google Scholar]
- 42.Liu D, Rutter WJ, Wang LH. Enhancement of transforming potential of human insulinlike growth factor 1 receptor by N-terminal truncation and fusion to avian sarcoma virus UR2 gag sequence. J. Virol. 1992;66:374–385. doi: 10.1128/jvi.66.1.374-385.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo M, Joiakim A, Reiners JJ., Jr Suppression of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-mediated aryl hydrocarbon receptor transformation and CYP1A1 induction by the phosphatidylinositol 3-kinase inhibitor 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) Biochem. Pharmacol. 2000;60:635–642. doi: 10.1016/s0006-2952(00)00379-8. [DOI] [PubMed] [Google Scholar]
- 44.Hu L, Zaloudek C, Mills GB, Gray J, Jaffe RB. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002) Clin. Cancer Res. 2000;6:880–886. [PubMed] [Google Scholar]
- 45.Physicians' desk reference. Oradell, N.J.: Medical Economics Co; 2007. [Google Scholar]
- 46.Maira SM, Stauffer F, Schnell C, Garcia-Echeverria C. PI3K inhibitors for cancer treatment: Where do we stand? Biochem. Soc. Trans. 2009;37:265–272. doi: 10.1042/BST0370265. [DOI] [PubMed] [Google Scholar]
- 47.Ohta T, Ohmichi M, Hayasaka T, Mabuchi S, Saitoh M, Kawagoe J, Takahashi K, Igarashi H, Du B, Doshida M, Mirei IG, Motoyama T, Tasaka K, Kurachi H. Inhibition of phosphatidylinositol 3-kinase increases efficacy of cisplatin in in vivo ovarian cancer models. Endocrinology. 2006;147:1761–1769. doi: 10.1210/en.2005-1450. [DOI] [PubMed] [Google Scholar]
- 48.Westfall SD, Skinner MK. Inhibition of phosphatidylinositol 3-kinase sensitizes ovarian cancer cells to carboplatin and allows adjunct chemotherapy treatment. Mol. Cancer. Ther. 2005;4:1764–1771. doi: 10.1158/1535-7163.MCT-05-0192. [DOI] [PubMed] [Google Scholar]
- 49.Havrilesky LJ, Elbendary A, Hurteau JA, Whitaker RS, Rodriguez GC, Berchuck A. Chemotherapy-induced apoptosis in epithelial ovarian cancers. Obstet. Gynecol. 1995;85:1007–1010. doi: 10.1016/0029-7844(95)00058-y. [DOI] [PubMed] [Google Scholar]
- 50.Cheng JQ, Jiang X, Fraser M, Li M, Dan HC, Sun M, Tsang BK. Role of X-linked inhibitor of apoptosis protein in chemoresistance in ovarian cancer: Possible involvement of the phosphoinositide-3 kinase/Akt pathway. Drug Resist Updat. 2002;5:131–146. doi: 10.1016/s1368-7646(02)00003-1. [DOI] [PubMed] [Google Scholar]
- 51.Asselin E, Mills GB, Tsang BK. XIAP regulates akt activity and caspase-3-dependent cleavage during cisplatin-induced apoptosis in human ovarian epithelial cancer cells. Cancer Res. 2001;61:1862–1868. [PubMed] [Google Scholar]
- 52.Page C, Lin HJ, Jin Y, Castle VP, Nunez G, Huang M, Lin J. Overexpression of Akt/AKT can modulate chemotherapy-induced apoptosis. Anticancer Res. 2000;20:407–416. [PubMed] [Google Scholar]
- 53.Yang X, Fraser M, Moll UM, Basak A, Tsang BK. Akt-mediated cisplatin resistance in ovarian cancer: Modulation of p53 action on caspase-dependent mitochondrial death pathway. Cancer Res. 2006;66:3126–3136. doi: 10.1158/0008-5472.CAN-05-0425. [DOI] [PubMed] [Google Scholar]
- 54.Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3'-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002;62:1087–1092. [PubMed] [Google Scholar]
- 55.Aissat N, Le Tourneau C, Ghoul A, Serova M, Bieche I, Lokiec F, Raymond E, Faivre S. Antiproliferative effects of rapamycin as a single agent and in combination with carboplatin and paclitaxel in head and neck cancer cell lines. Cancer Chemother. Pharmacol. 2008;62:305–313. doi: 10.1007/s00280-007-0609-2. [DOI] [PubMed] [Google Scholar]
- 56.Grant S. Cotargeting survival signaling pathways in cancer. J. Clin. Invest. 2008;118:3003–3006. doi: 10.1172/JCI36898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Numbers indicate individual tumor cases. Monolayer (A) and organ (B) cultures were maintained in DMEM containing 10% FCS. The photographs were taken one week later.
Immunofluorescent stain for cytokeratins and phase contrast images. Original magnification: 100×.
Equivalent amounts of protein lysates from various ovarian cancer cell lines, fresh ovarian tumors (OVT) and matched normal tissues (N) were resolved by SDS-PAGE and blotted with the indicated antibodies. For the detection of phospho-AKT, a mixture of antibodies against both the T308 and S473 epitopes was used.