

Abstract
This paper reports the biochemical characterization of a purified and reconstituted two-component 3-ketosteroid 9α-hydroxylase (KSH). KSH of Rhodococcus rhodochrous DSM 43269, consisting of a ferredoxin reductase (KshB) and a terminal oxygenase (KshA), was heterologously expressed in Escherichia coli. E. coli cell cultures, expressing both KshA and KshB, converted 4-androstene-3,17-dione (AD) into 9α-hydroxy-4-AD (9OHAD) with a >60% molar yield over 48 h of incubation. Coexpression and copurification were critical to successfully obtain pure and active KSH. Biochemical analysis revealed that the flavoprotein KshB is an NADH-dependent reductase using flavin adenine dinucleotide as a cofactor. Reconstitution experiments confirmed that KshA, KshB, and NADH are essential for KSH activity with steroid substrates. KSH hydroxylation activity was inhibited by several divalent metal ions, especially by zinc. The reconstituted KSH displayed subtle steroid substrate specificity; a range of 3-ketosteroids, i.e., 5α-Η, 5β-Η, Δ1, and Δ4 steroids, could act as KSH substrates, provided that they had a short side chain. The formation of 9OHAD from AD by KSH was confirmed by liquid chromatography-mass spectrometry analysis and by the specific enzymatic conversion of 9OHAD into 3-hydroxy-9,10-secoandrost-1,3,5(10)-triene-9,17-dione using 3-ketosteroid Δ1-dehydrogenase. Only a single KSH is encoded in the genome of the human pathogen Mycobacterium tuberculosis H37Rv, shown to be important for survival in macrophages. Since no human KSH homolog exists, the M. tuberculosis enzyme may provide a novel target for treatment of tuberculosis. Detailed knowledge about the biochemical properties of KSH thus is highly relevant in the research fields of biotechnology and medicine.
Hydroxylated steroids are pharmaceutically very interesting bioactive compounds. 9α-Hydroxylated steroids are of particular importance for the synthesis of corticoids such as 9α-fluorohydrocortisone. Microorganisms are widely used for the stereo-specific hydroxylation of steroids, but little is known about the enzymes involved, and current processes suffer from low conversion rates and yields (12, 18, 23).
Rhodococcus species are well known for their broad catabolic potential and ability to degrade sterols and steroids (14, 21, 25, 39). In this paper, we focus on 3-ketosteroid 9α-hydroxylase (KSH), which is essential for the growth of Rhodococcus strains on steroids (38). KSH acts on the B-ring of 3-keto-Δ4 steroids, e.g., 4-androstene-3,17-dione (AD), introducing a 9α-hydroxyl moiety (Fig. 1). Subsequent Δ1-dehydrogenation of 9α-hydroxy-AD (9OHAD) by 3-ketosteroid Δ1-dehydrogenase (Δ1-KSTD) initiates the opening of the B-ring through formation of a chemically unstable intermediate that spontaneously hydrolyzes, forming 3-hydroxy-9,10-secoandrost-1,3,5(10)-triene-9,17-dione (3-HSA). KSH activity has been observed in various actinobacterial genera, e.g., Mycobacterium (1, 3, 6), Nocardia (35), Arthrobacter (11), and Rhodococcus (38). In view of their amino acid sequences, KSH enzymes are predicted to belong to the class IA monooxygenases (5, 38). Monooxygenases, or mixed-function oxidases, incorporate one atom of O2 into the substrate (oxygenase function). The second atom of O2 is reduced to H2O (oxidase function) (17, 24). By definition, class IA monooxygenases are two-component systems consisting of a terminal oxygenase (KshA of KSH) and a ferredoxin reductase (KshB of KSH). The reductase component is a flavoprotein containing an NAD-binding domain and a plant-type iron-sulfur cluster, [Fe2S2Cys4]. A Rieske-type iron-sulfur cluster, [Fe2S2Cys2His2], and a non-heme Fe2+-binding domain are characteristic for the terminal oxygenase (5, 38). The two protein subunits are linked by an electron transport chain. NAD(P)H donates electrons to the flavin of the reductase; these electrons are transferred via the iron-sulfur clusters to the oxygenase, eventually leading to hydroxylation of the substrate (Fig. 1). The non-heme Fe2+ is involved in the binding and activation of O2 and substrate hydroxylation (4, 10, 13, 36).
FIG. 1.
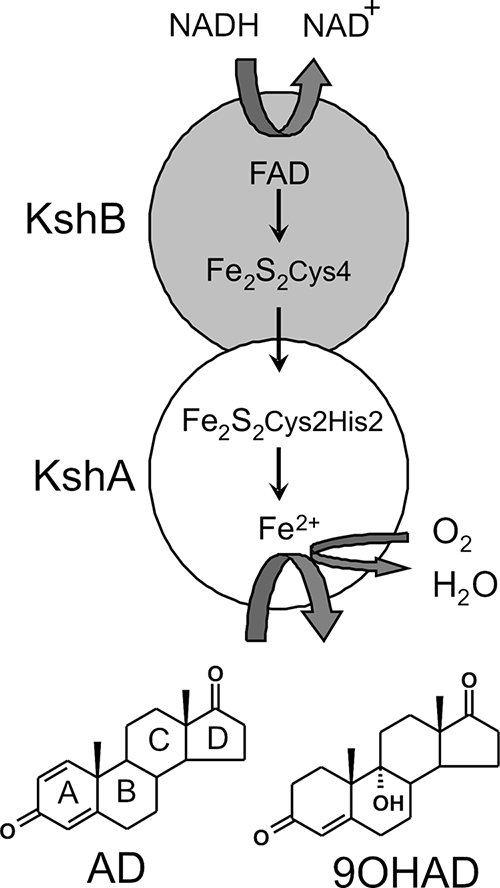
Schematic representation of the Rhodococcus two-component iron-sulfur monooxygenase KSH comprised of KshA and KshB. Small arrows indicate electron transfer from NADH to the FAD cofactor of KshB and via the Fe2S2 clusters to KshA, where substrate hydroxylation occurs at the Fe2+ binding domain. The substrate depicted is AD. The product is 9OHAD.
The kshA and kshB genes, encoding KshA and KshB, respectively, were first identified in Rhodococcus erythropolis SQ1 (38). Gene deletion studies have shown that both kshA and kshB of R. erythropolis SQ1 were required for KSH activity, suggesting that these two proteins constitute KSH. Biochemical evidence for this interaction, however, had been lacking. In fact, knowledge of KSH at the biochemical level has been extremely limited. Steroid 9α-hydroxylation has been reported to occur in cell extracts of Nocardia restricta (7), and a partly purified three-component enzyme system of Nocardia species M117 has been described (35). Heterologous expression of kshA of M. smegmatis mc2155 in Escherichia coli has recently been reported (1, 3). Upon purification, however, KSH activities were lost or became too low for biochemical characterization of the enzyme. Recently, the heterologous expression and characterization of KSH of Mycobacterium tuberculosis H37Rv was reported and a crystal structure of KshA was described (6). Addition of Fe2+-chelating agents to cell cultures of Rhodococcus rhodochrous DSM 43269 (= IFO3338) incubated with cholesterol was shown to chemically inactivate KSH, resulting in the accumulation of pharmaceutically interesting steroid pathway intermediates, i.e., 1,4-androstadiene-3,17-dione (ADD) and 23,24-bisnorcholesta-1,4-diene-22-oic acid (2). A single kshA ortholog (locus tag rv3526) has been identified in the genome of the human pathogen M. tuberculosis H37Rv (9, 40). Intriguingly, H37Rv genome-wide studies have revealed that rv3526 is specifically upregulated in macrophages and that rv3526 is important for the survival of M. tuberculosis in macrophages (29, 32, 34). Since no human homolog exists, KSH may provide a novel target for the treatment of a devastating disease, tuberculosis. Detailed knowledge about the biochemical properties of KSH thus is highly relevant in the research fields of biotechnology and medicine.
Here, we report the characterization of purified and reconstituted KSH and show at the biochemical level that both KshA and KshB are essential components of the two-component, iron-sulfur-containing KSH of R. rhodochrous.
MATERIALS AND METHODS
Steroids.
Structures of steroids used in this study are shown in Fig. S1 in the supplemental material. AD, ADD, 19-nor-AD (nordion), 3α-hydroxy-5α-pregnane-20-one, 5α-androstan-17β-ol-3-one (stanolon), 3β-hydroxy-5α-androstane-17-one, and 9α-hydroxy-AD were obtained from Schering-Plough. 17β-Hydroxy-4-androstene-3-one (testosterone), 11β-hydrocortisone, 4-cholestene-3-one (cholestenone), and 5-cholestene-3β-ol (cholesterol) were obtained from Sigma-Aldrich. 4-Pregnene-3,20-dione (progesterone) was obtained from ICN Biomedicals Inc. 1-(5α)-Androstene-3,17-dione, 5α-androstane-3,17-dione, 5β-androstane-3,17-dione, 5α-androstane-17-one, and 4-pregnene-3-one-20β-carboxylic acid were obtained from Steraloids.
Cloning of kshA and kshB from R. rhodochrous DSM 43269.
Chromosomal DNA of R. rhodochrous DSM 43269 (DSMZ culture collection, Braunschweig, Germany) grown in LB (Luria-Bertani) broth (Sigma-Aldrich) was isolated using a genomic DNA isolation kit (Sigma-Aldrich), digested by Sau3A, sized by sucrose gradient centrifugation (6 to 10 kb), and ligated into BglII-digested pRESQ (38). Transformation of E. coli DH5α (Bethesda Research Laboratories) with the ligation mixture generated a genomic library of approximately 16,000 transformants, in which approximately 90% of the constructs contained an insert. The sizes of the inserts varied considerably (6 to 20 kb).
The kshA gene was identified and isolated from the genomic library of R. rhodochrous DSM 43269 by PCR using forward primer KshAType-F [5′-TG(C/T)CCITTCCA(C/T)GA(C/T)TGGCGITGGGGIGG] and reverse primer KshAType-R [5′-TGIACGTAAAGAAG(A/G)TGIGCCAT(A/G)TC] (41). Identification and isolation of kshB were performed with forward primer KshBcon-F [5′-G(G/T/C)CTC(G/C)AACTGG(C/T)TGTGCGA-3′] and reverse primer KshBcon-R [5′-TCGCG(A/G)TT(G/C)GCGTAGA(A/C/G/T)CAGC-3′]. PCR was performed in a reaction mixture (25 μl) consisting of Tris-HCl (10 mM, pH 8), polymerase buffer, the deoxyribonucleotide triphosphates (0.2 mM), dimethyl sulfoxide (2%), primers (10 ng/μl), and Taq polymerase (0.1 U; Fermentas) under the following conditions: 5 min at 95°C; 30 cycles of 1 min at 95°C, 45 s at 60°C, and 1 min at 72°C; and 5 min at 72°C.
Heterologous kshA and kshB expression and protein purification.
The kshA gene was amplified by PCR (Expand long-template PCR system polymerase [0.1 U/25 μl]; Roche) from chromosomal DNA of R. rhodochrous DSM 43269 using kshA-specific primers (forward primer KshARho-4-F, 5′-CATATGACCGTCCCTCAGGAGCG-3′, and reverse primer KshARho-4-R, 5′-GCGTCACTCCGCCGTACCGGCGA-3′). The PCR product was cloned into an EcoRV-digested pZero2.1 (Invitrogen) vector (pKSH804). A 1,176-bp NdeI-BamHI fragment of pKSH804 was ligated into pET15b (Novagen), which had been digested with NdeI-BamHI (pKSH808). The kshB gene was amplified by PCR (0.02 U Vent polymerase; New England Biolabs Inc.) from chromosomal DNA of R. rhodochrous DSM 43269 with kshB-specific primers (forward primer KshBrho-F, 5′-CATATGACGACTGTCGAGGTG-3′, and reverse primer KshBrho-R, 5′-GGTTCAGAACTCGATCTTGAGG-3′). The PCR product was cloned into EcoRV-digested pZero2.1 (pBrho4). The 1,119-bp NdeI-HindIII fragment of pBrho4 was ligated into pET15b, digested with NdeI-HindIII (pBrho5). The construct for coexpression was made by ligation of a 1,216-bp XbaI-HindIII fragment of pBrho5 into pKSH808, digested with SpeI-HindIII (pA4rho4) (Fig. 2). Expression plasmids were used to transform E. coli BL21(DE3) (Invitrogen) or E. coli C41(DE3) (26). Cultures of E. coli harboring expression plasmids were grown (37°C, 200 rpm) in LB broth supplemented with ampicillin (100 μg/ml) to an optical density at 660 nm (OD660) approximately equal to 0.2. Isopropyl-β-d-thiogalactopyranoside (IPTG; 1 mM) was added for induction, and the cultures were incubated at 30°C and 200 rpm for an additional 4 h. The cells were washed in 50 mM Tris-HCl buffer, pH 7.0, disrupted with a French press, and centrifuged at 20,000 × g for 30 min at 4°C (Sorvall SS-34). All subsequent purification steps were performed at 4°C. Cell extracts were loaded on a Ni-nitrilotriacetic acid column (Sigma-Aldrich) for His tag purification and washed with buffer (50 mM Tris-HCl, pH 7.0, 500 mM NaCl) containing 0, 5, 15, and 30 mM imidazole. KshA and KshB were eluted from the column with 1 to 2 ml of elution buffer (50 mM Tris-HCl, pH 7.0, 500 mM NaCl, 100 mM imidazole).
FIG. 2.

Construction of the pA4rho4 vector used for coexpression of KshA and KshB in E. coli C41(DE3). Both proteins are expressed with a His tag at their N termini. Abbreviations: amp, ampicillin resistance marker; lacI, lactose repressor gene; RBS, ribosomal binding site; pT7 (filled squares), T7 promoter for overexpression in E. coli.
Whole-cell steroid bioconversion by recombinant E. coli cells.
Cell cultures of E. coli BL21(DE3) harboring the expression vector pKSH808 (kshA) and cell cultures of E. coli C41(DE3) harboring the expression vector pBrho5 (kshB) or pA4rho4 (kshA and kshB) were grown in triplicate in 25 ml LB broth at 37°C to an OD660 of approximately 0.2. At this point, IPTG (1 mM) and AD (1 g/liter) were added to the cultures and the incubation was continued at 30°C. Cultures of E. coli C41(DE3) harboring pET15b were used as a negative control.
Samples were taken 4, 6, 7.5, 24, and 48 h after IPTG induction and diluted fivefold in 80% methanol-H2O prior to analysis by high-performance liquid chromatography with UV detection (HPLC-UV) (see below). Product formation (9OHAD) was quantified using a calibration curve of authentic 9OHAD (200 to 800 μM).
Analysis of the KshB flavin cofactor.
The flavin cofactor KshB was extracted in quadruplicate by acid treatment (27). Perchloric acid (70 to 72%) was added at a 3% (vol/vol) final concentration to purified KshB (0.6 to 1.5 mg/ml protein), and the mixture was incubated for 1 h at 4°C and centrifuged at 10,000 × g for 10 min at 4°C (Sorvall SS-34). The supernatant containing the released flavin was analyzed by HPLC-UV (375 nm) (reversed-phase C18 column [250 by 4.6 mm], 35°C) using a mobile phase consisting of 20 mM ammonium acetate (pH 6.0) in 9% acetonitrile at a flow rate of 1 ml/min. Under these conditions, the retention times of flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) were 6.8 min and 11.2 min, respectively. The flavin cofactor was quantified using a calibration curve of authentic FAD (7 to 38 μM). Measurements were performed in quadruplet with different batches of enzymes.
Noncovalent binding of the flavin cofactor was determined as follows. Purified KshB (15 μg of protein) was run on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel. The gel was treated with 5% acetic acid solution and subjected to UV (254 nm) to detect and visualize separated flavin from KshB.
Standard KshB reductase activity assay.
The reductase activity of KshB was measured spectrophotometrically (600 nm) in an assay with NAD(P)H (0.25 mM) as the electron donor and 2,6-dichlorophenolindophenol (DCPIP; 0.1 mM) as the electron acceptor (ɛ = 21 mM/cm). KshB (1 to 2 μg) was added to the assay. Assays were performed at room temperature (22°C) in Tris-HCl (50 mM; pH 7.0) buffer.
Determination of the content of iron and acid-labile sulfur in KshB and KSH.
The iron content of KshB and KSH was determined using the method described by Fischer and Price (15), as modified by Zabinski et al. (43). FeCl3 was used as a standard. The acid-labile sulfur contents of these proteins were determined using the method described by Chen and Mortenson (8).
Standard KSH enzyme activity assay.
The KSH activity assay mixture (total volume, 500 μl) consisted of 50 mM Tris-HCl buffer (pH 7.0), NADH (105 μM), and purified, coexpressed KshA and KshB (20 to 25 μg). The reaction was started by the addition of 250 μM steroid substrate dissolved in 2-propanol (100%). All assays were performed at 35°C, unless stated otherwise. Soft-max PRO4 (Life Science edition) program was used to continuously record NADH oxidation at 340 nm (ɛ = 6.22 mM/cm). Sigmaplot version 10.0 was used to process the data using the Michaelis-Menten formula y = (a × x)/(b + x), where y is the reaction velocity, a is the maximum velocity, x is the substrate concentration, and b is the half-saturation constant. The relation between enzyme activity and pH was measured using citric acid-phosphate buffers (pHs 5 to 7) and Tris-HCl buffers (pHs 7 to 9). The low solubility of steroids may affect the measurement. Therefore, the kinetic parameters do not represent absolute but rather relative values, providing a strong indication of the substrate preference of KSH.
Product formation was analyzed by HPLC-UV on an Alltima C18 column (250 by 4.6 mm; particle size, 5 μm) at 35°C using methanol-water (80:20) as a mobile phase (1 ml/min) at 254 nm. Samples were diluted fivefold with 80% methanol in water prior to analysis.
Confirmation of 9α-hydroxy-AD product formation by KSH.
A KSH activity assay was performed using AD as the steroid substrate. The assay mixture consisted of KSH (184 μg/ml), NADH (320 μM), AD (200 μM), and Tris-HCl (50 mM, pH 7.0). The mixture was incubated for 16 h at 30°C and 220 rpm. Steroids were extracted from the assay mixture with dichloromethane and dried by N2 evaporation.
The extracted steroids (200 μM) were analyzed by LC-mass spectrometry (MS) analysis. HPLC was performed on an Alltima C18 column (250 by 4.6 mm; particle size, 5 μm) at 35°C using methanol-water (80:20) as a mobile phase (0.5 ml/min). MS was performed using atmospheric-pressure chemical ionization.
The extracted steroids were also used as a substrate in an assay with the Δ1-KSTD enzyme of R. erythropolis SQ1 (20, 37) and analyzed for the formation of 3-HSA. The Δ1-KSTD dehydrogenation reaction mixture consisted of KSH steroid extract (200 μM), DCPIP (50 μM), and Δ1-KSTD enzyme (74.8 μg/ml) in Tris-HCl buffer (50 mM pH 7.0). HPLC analysis with diode array detection was performed using a Lichrosorb reversed-phase C18 column (250 by 4.6 mm) at 35°C with methanol-water (70:30) as the mobile phase (1 ml/min). Samples were diluted fivefold in 70% methanol prior to analysis. Authentic 3-HSA (Schering-Plough, Oss, The Netherlands) was used as a standard.
Influence of metal ions on KSH and KshB activities.
Divalent metal ions or Fe3+ ions were added at a final concentration of 100 μM to standard KSH or KshB enzyme assays. Activities measured in a standard assay mixture were set to 100%. No effect on KSH or KshB activity was observed following the addition of 0.1 M HCl (1:100, vol/vol) (the control). The following compounds, prepared in 10 mM stock solutions in 0.1 M HCl, were tested: CaCl2·2H2O, CoSO4·7H2O, CuCl2·2H2O, FeSO4·7H2O, FeCl3, MgCl2·6H2O, MnSO4·H2O, NiSO4·6H2O, and ZnSO4·7H2O (4).
Nucleotide sequence accession numbers.
DNA nucleotide sequencing was performed by AGOWA, Berlin, Germany. The sequence data have been submitted to the DDBJ/EMBL/GenBank databases under accession numbers FJ238095 (kshA) and FJ238096 (kshB).
RESULTS AND DISCUSSION
Cloning of the kshA and kshB genes from R. rhodochrous DSM 43269.
The kshA gene was identified on a clone from the genomic library of R. rhodochrous DSM 43269 and isolated by PCR using primers KshAType-F and KshAType-R. Nucleotide sequencing analysis of the insert (3.2 kb) revealed the presence of an open reading frame (kshA) of 1,137 nucleotides, encoding a protein of 378 amino acids with high sequence similarities to KshA from R. erythropolis SQ1 (58% identity) and KshA (Rv3526) from Mycobacterium tuberculosis H37Rv (56% identity). In these three KshA proteins, the Rieske [Fe2S2Cys2His2] motif (CXHX16CX2H, corresponding to residues 67 to 89 of KshA of R. rhodochrous) and the non-heme Fe2+ motif (DX3DX2Hx4H, residues 174 to 186) were fully conserved. The kshB gene from R. rhodochrous DSM 43269 was isolated from the genomic library using degenerate primers (KshBcon-F, KshBcon-R) developed on conserved regions (SNWLCD, residues 91 to 96, and LXYANRD, residues 151 to 157) identified by alignment of KshB of R. erythropolis SQ1 with orthologous protein sequences found in databases. A clone carrying kshB (1,056 nucleotides) on a ±15-kb insert was isolated. R. rhodochrous KshB has 74% amino acid sequence identity with KshB of R. erythropolis SQ1. The typical class IA monooxygenase reductase domains previously identified in KshB of R. erythropolis, a flavin-binding domain (RXYSL, corresponding to residues 64 to 69 of KshB of R. rhodochrous), an NAD-binding domain (GSGITP, residues 128 to 133), and a C-terminal, plant-type iron-sulfur cluster [Fe2S2Cys4] domain (CX4CX2CX29C, residues 300 to 338), were fully conserved in KshB of R. rhodochrous.
Heterologous expression of the KshA and KshB proteins in E. coli.
The kshA and kshB genes were ligated into the pET15b vector for heterologous expression in E. coli. SDS-PAGE analysis of proteins in cell extracts showed that expression of kshA (calculated molecular mass of the His-tagged protein, 44.3 kDa) was achieved in E. coli BL21(DE3) but that expression of kshB (calculated molecular mass of the His-tagged protein, 38.7 kDa) was obtained only with E. coli C41(DE3). Standard KSH enzyme activity assays, using E. coli extracts with the KshA and KshB components expressed separately or jointly plus NADH and AD as substrates, resulted in a clear disappearance of AD and the appearance of a new product, identified by HPLC-UV as 9OHAD, using an authentic reference sample (Fig. 3). The time point of IPTG induction for KshA expression proved crucial for high KSH hydroxylation activity; specific enzyme activity was considerably higher when IPTG induction was started at an OD660 of 0.2 instead of an OD660 of 0.5.
FIG. 3.

HPLC elution profiles confirming the AD 9α-hydroxylation activity of E. coli-expressed KshA and KshB components. Profile a, Authentic reference sample of 9OHAD; profile b, authentic reference sample of AD; profile c, product analysis following incubation of KshA, KshB, NADH (140 μM), and AD (200 μM) in Tris-HCl buffer (50 mM, pH 7.0).
Whole-cell steroid bioconversion by recombinant E. coli cells.
Cell cultures of E. coli C41(DE3) coexpressing KshA and KshB displayed excellent steroid bioconversion potential, converting AD (1 g/liter) into 9OHAD within 48 h of incubation after IPTG induction in a >60% molar yield (Fig. 4). No AD conversion occurred with E. coli cells expressing only KshA or KshB, indicating that both KshA and KshB are essential components of KSH activity. Conversion of AD to 9OHAD was also not detected in cultures of E. coli C41(DE3) harboring the pET15b vector as a negative control.
FIG. 4.
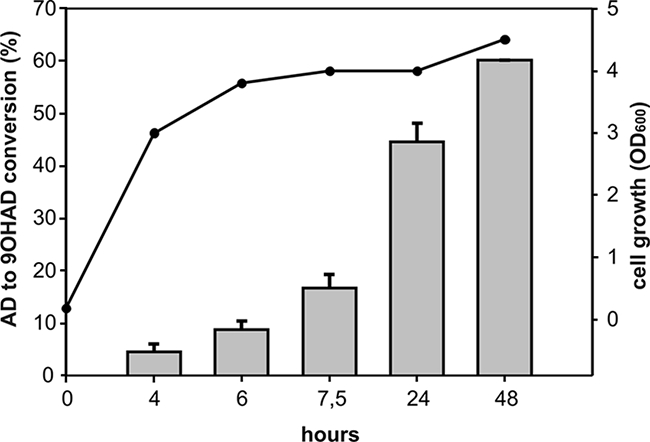
Bioconversion of AD into 9-OHAD by E. coli C41(DE3) cell cultures coexpressing KshA and KshB. E. coli BL21(DE3) cells expressing KshA alone and E. coli C41(DE3) cells harboring pET15b showed no conversion of AD into 9OHAD at 48 h after IPTG induction. Error bars indicate standard errors of the means (from three experiments). Time points are numbers of hours after IPTG induction. Filled circles indicate increases in the biomass of E. coli C41(DE3) coexpressing KshA and KshB, measured as turbidity of the cell culture at the OD660.
(Co)Purification of KshA and KshB proteins.
Both KSH subunits KshA and KshB were His tag purified separately. Addition of the purified KSH subunits separately to a standard KSH assay mixture with AD as a substrate, however, did not result in detectable KSH activity. Purified KshB was still active in the reductase assay with DCPIP, indicating either that the KshA component lost activity upon purification or that reconstitution of a functional KshAB enzyme complex failed. The purified R. rhodochrous KshB (0.5 to 2 mg/ml protein) could be stored (in 20% glycerol) at −20°C for several weeks without significant loss of activity. In contrast, rapid loss of KSH activity was observed in cell extracts stored at −20°C (in 20% glycerol). These observations indicate that the R. rhodochrous KshA protein was unstable under these conditions.
Interestingly, active KshA could be purified when E. coli extract with KshA was mixed with E. coli extract with KshB, prior to the loading of these extracts onto the Ni-nitrilotriacetic acid column. This resulted in coelution of both proteins and detection of KSH activity in the standard assay. As an alternative, plasmid pA4rho4 (Fig. 2) was constructed to attempt coexpression of kshA and kshB in E. coli C41(DE3) and subsequent copurification of KshA and KshB proteins (Fig. 5). Coexpression and copurification of KshA and KshB dramatically enhanced the in vitro KSH enzyme activity. This marked effect of coexpression and copurification on KSH activity strongly suggests that protein-protein interactions between the KshA and KshB subunits are critically important for maintaining KSH activity. KshA may be kept in a reduced state by the presence of KshB during purification, mimicking purification in the presence of reducing agents or under anaerobic conditions, as has been reported for several other related oxygenases (4, 6, 22, 30, 42). The molar ratio of KshA and KshB protein in purified KSH was determined as 1:(0.83 ± 0.058). The iron and acid-labile sulfur content of KSH was determined to be 3.94 ± 0.08 mol and 4.3 ± 1 mol per mol of KSH, respectively. The spectra of purified and dithionite-reduced KSH were characteristic for the presence of an [Fe2S2] cluster (Fig. 6A). The activity of purified KSH could now be determined spectrophotometrically by measuring substrate (e.g., AD)-dependent NADH oxidation at 340 nm, which was not possible in cell extracts because of high background NADH oxidation activity. Dialysis of the purified sample (3 h at 4°C) to remove imidazole and NaCl reduced KSH activity to about 30% of the original activity, which could have resulted from the loss of non-heme Fe2+ present in KshA during dialysis. However, the loss of activity could not be restored by the addition of Fe2+ and thus might have resulted from protein unfolding or loss of protein-protein interactions between KshA and KshB following dialysis. Undialyzed, purified KSH (1 to 2 mg/ml) could be stored for at least 2 weeks in 20% glycerol at −20°C without significant loss of activity and was used for all subsequent experiments.
FIG. 5.

Lanes: a, SDS-PAGE analysis of protein markers; b, E. coli cell extract coexpressing KshA and KshB; c, E. coli cell extract expressing KshB; d, His tag-copurified KshA (45.4 kDa)/KshB (40 kDa) (10 μg); e, His tag-purified KshB (10 μg).
FIG. 6.

Absorption spectra of KSH (57.5 μM) (A) and KshB (33 μM) (B) as purified (thin line) and after dithionite (10 mM) treatment (thick line).
Biochemical characterization of KshB as an NADH-dependent oxygenase-reductase.
In the reductase assay with DCPIP, purified KshB was strictly NADH dependent for activity (specific activity = 4.1 μmol min−1 mg−1). No activity was found in a reductase assay with NADPH as the electron donor. Purified KshB had an orange color, which is in agreement with the presence of a flavin and a plant-type iron-sulfur cluster (4). Cofactor extraction from KshB and subsequent HPLC analysis of this extract identified the cofactor as FAD (0.72 ± 0.14 mol FAD per mol KshB) (Fig. 7). The spectrum of KshB was consistent with the presence of an [Fe2S2] cluster (Fig. 6B). The purified material contained 1.75 ± 0.25 mol iron and 2.03 ± 0.37 mol acid-labile sulfur per mol of KshB. The flavin cofactor of KshB is clearly noncovalently bound, running separately from KshB on an SDS-PAGE gel, visualized by acetic acid treatment of an SDS-PAGE gel and UV (254 nm) irradiation.
FIG. 7.

HPLC elution profiles identifying FAD as the flavin cofactor of KshB. Profile a, flavin cofactor extracted from KshB by acid treatment; profile b, authentic FAD; profile c, authentic FMN.
The amino acid sequences of KshA and KshB predict that Rhodococcus KSH belongs to the class IA monooxygenases (38; this study). By definition, oxygenase reductases of class IA monooxygenases contain FMN as a cofactor, whereas class IB oxygenase reductases contain FAD (5). Our finding in the present study that FAD acts as a KshB cofactor thus is not consistent with the classification of this protein in class IA. Several other examples of enzymes that do not fit the Batie classification of ring-hydroxylating oxygenases have been reported (19, 31, 33). Another classification system has been proposed for oxygenase components involved in ring-hydroxylating oxygenations (28). In this scheme, based on amino acid sequences of terminal oxygenases, KshA of KSH belongs to group I. Enzymes of group I vary greatly, however, in the amino acid sequence of the oxygenase component, contain either two or three components, and include both monooxygenases and dioxygenases. Further biochemical characteristics thus appear required for proper classification of oxygenases.
Characteristics of the reconstituted KSH enzyme system.
To confirm that KSH activity is dependent on KshA, KshB, and NADH, we performed standard steroid hydroxylation activity assays using AD as the substrate and analyzed product formation by HPLC. Purified KshB and E. coli cell extracts containing active KshA were used, instead of copurified KshA and KshB, enabling us to omit each of the components of the KSH system. Omitting KshA, KshB, or NADH separately from the reaction mixture resulted in no detectable KSH activity, indicating that these are all essential components. Interestingly, the KSH-specific activity of copurified KshA and KshB (16 μg), using AD (100 μM) as the substrate, could be maximally enhanced nearly twofold by the addition of purified KshB (15 μg) (510 ± 8 nmol min−1 mg−1 KSH). At saturated KshB levels (4 μg copurified KshAB with 34 μg KshB), however, the KSH-specific activity was significantly lower (150 ± 18 nmol min−1 mg−1 KSH). Since the increase in KSH-specific activity was less than twofold, we decided to perform all subsequent experiments with copurified KSH without the addition of purified KshB.
Next, the optimal temperature and pH of KSH were determined using copurified KshA/KshB. Activity measurements at different temperatures and pH values revealed a rather narrow pH range with an optimum of pH 7.0 and a temperature optimum of 33°C. No decrease in the initial KSH enzyme activity was observed after a 5-min preincubation at 35°C, indicating that the KSH enzyme was stable during the assay. Initial activity was reduced by 25% following a 30-min preincubation at 35°C.
The KSH-dependent introduction of a 9α-hydroxyl moiety into AD was also investigated. The hydroxylated product formed during an overnight incubation of AD (200 μM) with copurified and reconstituted KSH was extracted and analyzed by LC-MS. The MS spectrum of the product of the KSH activity assay was identical to the spectrum of authentic 9OHAD, confirming C9α-hydroxylation by KSH.
The steroid extract from the KSH reaction mixture was additionally used in a Δ1-KSTD enzyme assay. Δ1-Dehydrogenation of 9OHAD results in a chemically unstable compound (9OHADD), leading to a nonenzymatic B-ring opening and the formation of 3-HSA. HPLC analysis with diode array detection of the KSTD reaction mixture indeed confirmed the disappearance of 9OHAD and the formation of 3-HSA.
The non-heme Fe2+ binding domain of KshA is predicted to be important in catalysis (10). Indeed, addition of o-phenanthroline (100 μM), a highly specific Fe2+ chelator, to the standard KSH enzyme assay resulted in an 80% reduction of KSH activity. The activity of KshB in the reductase assay was not reduced by the same treatment, confirming that Fe2+ bound in the non-heme Fe2+ domain in KshA is important for KSH activity.
Influence of divalent metal ions on KshB reductase and KSH activities.
The influence of metal ions on KshB reductase and KSH activities in a standard enzyme assay was studied (Table 1). AD (100 μM) was added as the substrate to the KSH activity assay. None of the metal ions added enhanced the activity of KSH or KshB substantially. Addition of Fe2+, Ca2+, Mn2+, or Mg2+ to the enzyme mixture had a significant effect neither on KSH activity nor on KshB reductase activity. Several metals, however, were shown to inhibit the hydroxylation activity. Zn2+ was the strongest inhibitor, completely abolishing KSH activity. The inhibitory effects of Fe3+, Co2+, Zn2+, and Ni2+ on KSH activity clearly are KshA specific, since the reductase activity of KshB was not inhibited by these metal ions. KshB was inhibited only by copper. It has been reported that copper can disrupt the binding of FAD (16). In view of the inhibitory effect of Ni2+ on KSH activity, the nickel affinity column chromatography step used for purification of His-tagged proteins in hindsight appears less suited. This may at least partly explain the KshA activity loss.
TABLE 1.
Effect of metal ions on the activities of KshB and KSH, displayed as percentages of control activities
| Assay | Activity with indicated metal ion relative to control activity (%)a
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| None | Fe2+ | Fe3+ | Cu2+ | Ca2+ | Co2+ | Zn2+ | Mn2+ | Mg2+ | Ni2+ | |
| KshB | 100 | —b | 111 | 8 | 116 | 102 | 80 | 98 | 103 | 78 |
| KSH | 100 | 98 | 40 | 18 | 95 | 36 | 0 | 103 | 91 | 23 |
Control activity is the activity measured in a standard assay, which was set at 100%.
The influence of Fe2+ on the KshB reductase activity could not be determined because of interference with the assay.
KSH acts on 3-ketosteroids with a range of A-ring configurations.
To determine the substrate range of KSH, standard enzyme assays were performed with a range of steroid substrate concentrations (10 to 250 μM) with copurified KshA/KshB, and NADH oxidation was measured at 340 nm. No NADH oxidation was observed in a KSH standard enzyme assay without the addition of steroid substrate. Steroid substrates were tested with variations in A-ring configuration and side chain length (Table 2). The enzyme followed Michaelis-Menten kinetics, and maximal reaction rates were observed with several substrates in the range of 10 to 100 μM. NADH oxidation was well coupled to steroid substrate utilization as determined by HPLC or gas chromatography. The highest KSH activity was observed with AD, ADD, 4-androstene-17β-ol-3-one (testosterone) and 4-pregnene-3,20-dione (progesterone) (Table 2). Km values for these substrates, however, were below 10 μM and could not be determined accurately, since KSH reaction rates at substrate concentrations below 10 μM resulted in minimal differences in absorbance at 340 nm and inaccurate Michaelis-Menten curves. On the other hand, Km values for 19-nor-AD (10 ± 1 μM), 5α-androstane-3,17-dione (23 ± 2 μM), and 5β-androstane-3,17-dione (33 ± 5 μM) could be accurately determined.
TABLE 2.
Steroid substrate range of the KSH enzyme of R. rhodochrous DSM 43269a
| Steroid substrate | Exptl Vmax (nmol min−1 mg−1) | Relative activity (%) |
|---|---|---|
| AD | 276 ± 20 | 100 |
| ADD | 246 ± 20 | 89 |
| 4-Androstene-17β-ol-3-one (testosterone) | 279 ± 29 | 101 |
| 4-Pregnene-3,20-dione (progesterone) | 272 ± 38 | 99 |
| 19-Nor-4-androstene-3,17-dione (nordion) | 216 ± 25 | 78 |
| 1-(5α)-Androstene-3,17-dione | 188 ± 20 | 68 |
| 5α-Androstane-3,17-dioneb | 177 ± 19 | 64 |
| 5β-Androstane-3,17-dioneb | 160 ± 22 | 58 |
| 5-Cholestene-3β-ol (cholesterol)c | ND | ND |
| 5α-Androstane-17β-ol-3-one (stanolon) | ND | ND |
| 5α-Androstane-17-one | ND | ND |
| 3α-Hydroxy-5α-pregnane-20-onec | ND | ND |
| 11β-Hydrocortisone | ND | ND |
| 3β-Hydroxy-5α-androstane-17-one | ND | ND |
| 4-Cholestene-3-onec | ND | ND |
| 4-Pregnene-3-one-20β-carboxylic acidc | ND | ND |
| 9OHAD (negative control) | ND | ND |
Experimental enzyme activities (Exptl Vmax) at a 100 μM steroid substrate concentration were calculated in nmol−1·min−1·mg−1 KSH enzyme. The relative activities are expressed as percentages of the activity with AD, which was set at 100%. Errors were calculated as standard errors of the means (from three experiments). Steroid structures are shown in Fig. S1 in the supplemental material. ND, no detectable activity.
Steroid substrate concentration, 200 μM.
Steroid substrate concentration, 25 μM, due to the low solubility of the substrate.
KSH accepts a variety of steroid substrates with different A-ring confirmations. The highest KSH activity was observed with 3-keto-Δ4 steroid substrates, e.g., AD. The presence of a 3-keto moiety appears essential for KSH activity: no activity was found with 3α- or 3β-hydroxysteroids or steroids without a hydroxyl moiety at this position. The configuration of the A-ring does not appear to play an important role in determining substrate specificity, provided that a 3-keto moiety is present. Activity was observed with saturated 3-ketosteroids having either a 5α-H or a 5β-H configuration, as well as with unsaturated Δ1, Δ4, and Δ1,4 3-ketosteroids (Table 2). Striking is the difference in substrate affinity observed for 1-(5α)-androstene-3,17-dione and 5α-androstane-3,17-dione. The Δ1 unsaturated bond in the former appears to make the substrate more favorable for the enzyme, suggesting that in vivo 9α-hydroxylation might be preceded by Δ1-dehydrogenation. The ability of KSH to use 4-androstene-17β-ol-3-one (testosterone) but not 5α-androstane-17β-ol-3-one (stanolon) as a substrate is remarkable, since activity was observed with 5α-androstane-3,17-dione. Apparently, the active site of KshA cannot accommodate steroids with a 17β-hydroxyl group and a 5α-H-configuration simultaneously. The substituent present at the C-17 position of the steroid molecule appears to be an important determinant of KSH substrate specificity. Unlike with progesterone, a C21 steroid with a relatively small side chain, no KSH activity was observed with 4-pregnene-3-one-20β-carboxylic acid (C22) or 4-cholestene-3-one (C27), with more bulky side chains (Table 2).
The stereo-specific steroid hydroxylation catalyzed by the KSH system, its subtle substrate specificity for various 3-ketosteroids, and the efficient use of AD in E. coli whole-cell bioconversions make this R. rhodochrous DSM 43269 enzyme an interesting candidate for industrial applications.
Supplementary Material
Acknowledgments
This project is financially supported by The Netherlands Ministry of Economic Affairs and B-Basic partner organizations (http://www.b-basic.nl) through B-Basic, a public/private NWO (Netherlands Organization for Scientific Research)-ACTS (Advanced Chemical Technologies for Sustainability) program. Schering-Plough (Oss, The Netherlands) also supported this project.
We gratefully acknowledge Jan Knol (University of Groningen) for supplying Δ1-KSTD enzyme and the Department of Biochemistry (University of Groningen) for use of equipment and assistance with LC-MS.
Footnotes
Published ahead of print on 26 June 2009.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Andor, A., A. Jekkel, D. A. Hopwood, F. Jeanplong, E. Ilkoy, A. Konya, I. Kurucz, and G. Ambrus. 2006. Generation of useful insertionally blocked sterol degradation pathway mutants of fast-growing mycobacteria and cloning, characterization, and expression of the terminal oxygenase of the 3-ketosteroid 9α-hydroxylase in Mycobacterium smegmatis mc2155. Appl. Environ. Microbiol. 72:6554-6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arima, K., W. Nakamatsu, and T. Beppu. 1978. Microbial production of 3-oxobisnorchola-1,4-dien-22-oic acid. Agric. Biol. Chem. 42:411-416. [Google Scholar]
- 3.Arnell, R., R. Johannisson, J. Lindholm, T. Fornstedt, B. Ersson, A. Ballagi, and K. Caldwell. 2007. Biotechnological approach to the synthesis of 9alpha-hydroxylated steroids. Prep. Biochem. Biotechnol. 37:309-321. [DOI] [PubMed] [Google Scholar]
- 4.Batie, C. J., E. LaHaie, and D. P. Ballou. 1987. Purification and characterization of phthalate oxygenase and phthalate oxygenase reductase from Pseudomonas cepacia. J. Biol. Chem. 262:1510-1518. [PubMed] [Google Scholar]
- 5.Batie, C. J., D. P. Ballou, and C. J. Correll. 1991. Phthalate dioxygenase reductase and related flavin-iron-sulfur containing electron transferases, p. 543-556. In F. Müller (ed.), Chemistry and biochemistry of flavoenzymes. CRC Press, Boca Raton, FL.
- 6.Capyk, J. K., I. D'Angelo, N. C. Strynadka, and L. D. Eltis. 2009. Characterization of 3 ketosteroid 9α-hydroxylase, a Rieske oxygenase in the cholesterol degradation pathway of Mycobacterium tuberculosis. J. Biol. Chem. 284:9937-9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang, F. N., and C. J. Sih. 1964. Mechanisms of steroid oxidation by microorganisms. VII. Properties of the 9α-hydroxylase. Biochemistry 3:1551-1557. [DOI] [PubMed] [Google Scholar]
- 8.Chen, J.-S., and L. E. Mortenson. 1977. Inhibition of methylene blue formation during determination of the acid-labile sulfide of iron-sulfur protein samples containing dithionite. Anal. Biochem. 79:157-165. [DOI] [PubMed] [Google Scholar]
- 9.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry III, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quail, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead, and B. G. Barrell. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. [DOI] [PubMed] [Google Scholar]
- 10.Coulter, E. D., N. Moon, C. J. Batie, W. R. Dunham, and D. P. Ballou. 1999. Electron paramagnetic resonance measurements of the ferrous mononuclear site of phthalate dioxygenase substituted with alternate metal ions: direct evidence for ligation of two histidines in the copper(II)-reconstituted protein. Biochemistry 38:11062-11072. [DOI] [PubMed] [Google Scholar]
- 11.Dutta, R. K., M. K. Roy, and H. D. Singh. 1992. Role of plasmid pJL1 of Arthrobacter oxydans 317 in the degradation of β-sitosterol. J. Basic Microbiol. 32:317-324. [DOI] [PubMed] [Google Scholar]
- 12.Fernandes, P., A. Cruz, B. Angelova, H. M. Pinheiro, and J. M. S. Cabral. 2003. Microbial conversion of steroid compounds: recent developments. Enzyme Microb. Technol. 32:688-705. [Google Scholar]
- 13.Ferraro, D. J., L. Gakhar, and S. Ramaswamy. 2005. Rieske business: structure-function of Rieske non-heme oxygenases. Biochem. Biophys. Res. Commun. 338:175-190. [DOI] [PubMed] [Google Scholar]
- 14.Finnerty, W. R. 1992. The biology and genetics of the genus Rhodococcus. Annu. Rev. Microbiol. 46:193-218. [DOI] [PubMed] [Google Scholar]
- 15.Fischer, D. S., and D. C. Price. 1964. A simple serum iron method using the new sensitive chromogen tripyridyl-s-triazine. Clin. Chem. 10:21-31. [PubMed] [Google Scholar]
- 16.Green, J., S. D. Prior, and H. Dalton. 1985. Copper ions as inhibitors of protein C of soluble methane monooxygenase of Methylococcus capsulatus (Bath). Eur. J. Biochem. 153:137-144. [DOI] [PubMed] [Google Scholar]
- 17.Harayama, S., M. Kok, and E. L. Neidle. 1992. Functional and evolutionary relationships among diverse oxygenases. Annu. Rev. Microbiol. 46:565-601. [DOI] [PubMed] [Google Scholar]
- 18.Holland, H. L. 1999. Recent advances in applied and mechanistic aspects of the enzymatic hydroxylation of steroids by whole-cell biocatalysts. Steroids 64:178-186. [DOI] [PubMed] [Google Scholar]
- 19.Jouanneau, Y., C. Meyer, J. Jakoncic, V. Stojanoff, and J. Gaillard. 2006. Characterization of a ring-hydroxylating dioxygenase able to oxidize a wide range of polycyclic aromatic hydrocarbons including four and five ring carcinogens. Biochemistry 45:12380-12391. [DOI] [PubMed] [Google Scholar]
- 20.Knol, J., K. Bodewits, G. I. Hessels, L. Dijkhuizen, and R. van der Geize. 2008. 3-Keto-5alpha-steroid delta(1)-dehydrogenase from Rhodococcus erythropolis SQ1 and its orthologue in Mycobacterium tuberculosis H37Rv are highly specific enzymes that function in cholesterol catabolism. Biochem. J. 410:339-346. [DOI] [PubMed] [Google Scholar]
- 21.Larkin, M. J., L. A. Kulakov, and C. C. Allen. 2005. Biodegradation and Rhodococcus—masters of catabolic versatility. Curr. Opin. Biotechnol. 16:282-290. [DOI] [PubMed] [Google Scholar]
- 22.Locher, H. H., T. Leisinger, and A. M. Cook. 1991. 4-Sulphobenzoate 3,4-dioxygenase: purification and properties of a desulphonative two-component enzyme system from Comamonas testosterone T-2. Biochem. J. 274:833-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahato, S. B., and S. Garai. 1997. Advances in microbial steroid biotransformation. Steroids 62:332-345. [DOI] [PubMed] [Google Scholar]
- 24.Mason, J. R., and R. Cammack. 1992. The electron transport proteins of hydroxylating bacterial dioxygenases. Annu. Rev. Microbiol. 46:277-305. [DOI] [PubMed] [Google Scholar]
- 25.McLeod, M. P., R. L. Warren, W. W. Hsiao, N. Araki, M. Myhre, C. Fernandes, D. Miyazawa, W. Wong, A. L. Lillquist, D. Wang, M. Dosanjh, H. Hara, A. Petrescu, R. D. Morin, G. Yang, J. M. Stott, J. E. Schein, H. Shin, D. Smailus, A. S. Siddiqui, M. A. Marra, S. J. Jones, R. Holt, F. S. Brinkman, K. Miyauchi, M. Fukuda, J. E. Davies, W. W. Mohn, and L. D. Eltis. 2006. The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc. Natl. Acad. Sci. USA 103:15582-15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miroux, B., and J. E. Walker. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260:289-298. [DOI] [PubMed] [Google Scholar]
- 27.Mohamed, M. E.-S., A. Zaar, C. Ebenau-Jehle, and G. Fuchs. 2001. Reinvestigation of a new type of aerobic benzoate metabolism in the proteobacterium Azoarcus evansii. J. Bacteriol. 183:1899-1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nam, J. W., H. Nojiri, T. Yoshida, H. Habe, H. Yamane, and T. Omori. 2001. New classification system for oxygenase components involved in ring-hydroxylating oxygenations. Biosci. Biotechnol. Biochem. 65:254-263. [DOI] [PubMed] [Google Scholar]
- 29.Rengarajan, J., B. R. Bloom, and E. J. Rubin. 2005. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. USA 102:8327-8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosche, B., B. Tsisuaka, S. Fetzner, and F. Lingens. 1995. 2-Oxo-1,2-dihydroquinoline 8-monooxygenase, a two-component enzyme system from Pseudomonas putida 86. J. Biol. Chem. 270:17836-17842. [DOI] [PubMed] [Google Scholar]
- 31.Rosche, B., B. Tshisuaka, B. Hauer, F. Lingens, and S. Fetzner. 1997. 2-Oxo-1,2-dihydroquinoline 8-monooxygenase: phylogenetic relationship to other multicomponent nonheme iron oxygenases. J. Bacteriol. 179:3549-3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sassetti, C. M., and E. J. Rubin. 2003. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 100:12989-12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato, S., J. W. Nam, K. Kasuga, H. Nojiri, H. Yamane, and T. Omori. 1997. Identification and characterization of genes encoding carbazole 1,9α-dioxygenase in Pseudomonas sp. strain CA10. J. Bacteriol. 179:4850-4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schnappinger, D., S. Ehrt, M. I. Voskuil, Y. Liu, J. A. Mangan, I. M. Monahan, G. Dolganov, B. Efron, P. D. Butcher, C. Nathan, and G. K. Schoolnik. 2003. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198:693-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strijewski, A. 1982. The steroid-9 alpha-hydroxylation system from Nocardia species. Eur. J. Biochem. 128:125-135. [DOI] [PubMed] [Google Scholar]
- 36.Tarasev, M., and D. P. Ballou. 2005. Chemistry of the catalytic conversion of phthalate into its cis-dihydrodiol during the reaction of oxygen with the reduced form of phthalate dioxygenase. Biochemistry 44:6197-6207. [DOI] [PubMed] [Google Scholar]
- 37.van der Geize, R., G. I. Hessels, R. van Gerwen, P. van der Meijden, and L. Dijkhuizen. 2001. Unmarked gene deletion mutagenesis of kstD, encoding 3-ketosteroid delta1-dehydrogenase, in Rhodococcus erythropolis SQ1 using sacB as counter-selectable marker. FEMS Microbiol. Lett. 205:197-202. [DOI] [PubMed] [Google Scholar]
- 38.van der Geize, R., G. I. Hessels, R. Van Gerwen, P. Van der Meijden, and L. Dijkhuizen. 2002. Molecular and functional characterization of kshA and kshB, encoding two components of 3-ketosteroid 9α-hydroxylase, a class IA monooxygenase, in Rhodococcus erythropolis strain SQ1. Mol. Microbiol. 45:1007-1018. [DOI] [PubMed] [Google Scholar]
- 39.van der Geize, R., and L. Dijkhuizen. 2004. Harnessing the catabolic diversity of rhodococci for environmental and biotechnological applications. Curr. Opin. Microbiol. 7:255-261. [DOI] [PubMed] [Google Scholar]
- 40.Van der Geize, R., K. Yam, T. Heuser, M. H. Wilbrink, H. Hara, M. C. Anderton, E. Sim, L. Dijkhuizen, J. E. Davies, W. W. Mohn, and L. D. Eltis. 2007. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl. Acad. Sci. USA 104:1947-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Geize, R., G. I. Hessels, M. Nienhuis-Kuiper, and L. Dijkhuizen. 2008. Characterization of a second 3-ketosteroid 9α-hydroxylase activity in Rhodococcus erythropolis SQ1 comprised of a terminal oxygenase homologue, KshA2, active with oxygenase-reductase component KshB. Appl. Environ. Microbiol. 74:7197-7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolfe, M. D., D. J. Altier, A. Stubna, C. V. Popescu, E. Münck, and J. D. Lipscomb. 2002. Benzoate 1,2-dioxygenase from Pseudomonas putida: single turnover kinetics and regulation of a two-component Rieske dioxygenase. Biochemistry 41:9611-9626. [DOI] [PubMed] [Google Scholar]
- 43.Zabinski, R., E. Münck, P. M. Champion, and J. M. Wood. 1972. Kinetic and Mössbauer studies on the mechanism of protocatechuic acid 4,5-oxygenase. Biochemistry 11:3212-3219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.