

Abstract
A gene that codes for a novel intracellular poly(3-hydroxybutyrate) (PHB) depolymerase, designated PhaZ1, has been identified in the genome of Bacillus megaterium. A native PHB (nPHB) granule-binding assay showed that purified soluble PhaZ1 had strong affinity for nPHB granules. Turbidimetric analyses revealed that PhaZ1 could rapidly degrade nPHB granules in vitro without the need for protease pretreatment of the granules to remove surface proteins. Notably, almost all the final hydrolytic products produced from the in vitro degradation of nPHB granules by PhaZ1 were 3-hydroxybutyric acid (3HB) monomers. Unexpectedly, PhaZ1 could also hydrolyze denatured semicrystalline PHB, with the generation of 3HB monomers. The disruption of the phaZ1 gene significantly affected intracellular PHB mobilization during the PHB-degrading stage in B. megaterium, as demonstrated by transmission electron microscopy and the measurement of the PHB content. These results indicate that PhaZ1 is functional in intracellular PHB mobilization in vivo. Some of these features, which are in striking contrast with those of other known nPHB granule-degrading PhaZs, may provide an advantage for B. megaterium PhaZ1 in fermentative production of the biotechnologically valuable chiral compound (R)-3HB.
Polyhydroxyalkanoates (PHAs) are a group of polyesters that are produced by numerous bacteria as carbon and energy storage materials in response to nutritional stress (13, 27, 29). Poly(3-hydroxybutyrate) (PHB) is the most common and intensively studied PHA. Intracellular native PHB (nPHB) granules are composed of a hydrophobic PHB core and a surface layer consisting of proteins and phospholipids (13). The PHB of intracellular nPHB granules is in an amorphous state. When intracellular nPHB granules are exposed to extracellular environments due to cell death and lysis, the amorphous PHB is transformed into a denatured semicrystalline state. nPHB granules subjected to physical damage or solvent extraction to remove the surface layer can also crystallize into denatured PHB (dPHB) (13, 15). Artificial PHB (aPHB) granules, in which PHB is in an amorphous state, can be prepared from semicrystalline dPHB and detergents (1, 11, 23, 31).
Various extracellular PHB depolymerases (PhaZs) that are secreted by many PHB-degrading bacteria have been demonstrated to specifically degrade dPHB (13, 14, 37). One exception is that PhaZ7, an extracellular PHB depolymerase secreted by Paucimonas lemoignei, displays unusual substrate specificity for amorphous PHB, with 3-hydroxybutyrate (3HB) oligomers as the main products of enzymatic hydrolysis (7). PhaZ7 exhibits no enzymatic activity toward dPHB. So far, a growing number of intracellular PHB depolymerases have been characterized. The intracellular PHB depolymerase PhaZa1 of Ralstonia eutropha (also called Cupriavidus necator) H16 has recently been established to be especially important for the intracellular mobilization of accumulated PHB (42). The main in vitro hydrolytic products of PhaZa1 degradation of amorphous aPHB are 3HB oligomers (31). PhaZd1, another intracellular PHB depolymerase of R. eutropha H16, shows no significant amino acid similarity to PhaZa1. The in vitro hydrolytic products of PhaZd1 degradation of amorphous aPHB are also 3HB oligomers. A 3HB monomer is rarely detected as a hydrolytic product (1). The intracellular PHB depolymerase PhaZ of Paracoccus denitrificans was reported previously to degrade protease-treated nPHB granules in vitro, with the release of 3HB dimers and oligomers as the main hydrolytic products (6). Recently, we have identified a novel intracellular PHB depolymerase from Bacillus thuringiensis serovar “israelensis” (39). The B. thuringiensis PhaZ shows no significant amino acid similarity to any known PHB depolymerase. This PhaZ has strong amorphous PHB-hydrolyzing activity and can release a considerable amount of 3HB monomers by the hydrolysis of trypsin-treated nPHB granules (39). It is of note that purified PhaZd1 from R. eutropha, PhaZ from P. denitrificans, and PhaZ from B. thuringiensis need pretreatment of nPHB granules with protease to remove surface proteins for PHB degradation (1, 6, 39). They show only very little or no activity toward nPHB granules without trypsin pretreatment. It has been demonstrated previously that these intracellular PHB depolymerases cannot hydrolyze dPHB (1, 31, 39).
(R)-3HB, a biotechnologically valuable chiral compound, has been widely used for syntheses of antibiotics, vitamins, and pheromones (3, 30, 38). One way to produce (R)-3HB is heterologous coexpression of a PHB synthetic operon and a gene encoding an amorphous PHB-degrading PhaZ in Escherichia coli (3, 18, 25, 33, 38). A common problem encountered by this method is that oligomeric and dimeric forms of 3HB often constitute a major portion of the products of enzymatic hydrolysis, thus requiring further hydrolysis by 3HB oligomer hydrolase or heating under alkaline conditions to generate 3HB monomers (3, 18, 25, 33).
Bacillus megaterium genes involved in the biosynthesis of nPHB granules have been cloned from strain ATCC 11561 and characterized previously (19, 21, 22). A gene encoding the extracellular PHB depolymerase PhaZ from B. megaterium was recently cloned from strain N-18-25-9 (34). However, little is known about B. megaterium genes involved in the intracellular mobilization of PHB. In this study, we have identified in B. megaterium ATCC 11561 an intracellular PHB depolymerase that could rapidly degrade nPHB granules in vitro without the need for trypsin pretreatment of the nPHB granules. Moreover, almost all the in vitro hydrolytic products released from the degradation of amorphous PHB by this PhaZ were 3HB monomers. This PhaZ could also hydrolyze dPHB with the generation of 3HB monomers. Thus, it appears to be a novel intracellular PHB depolymerase and may have promising potential for biotechnological application in the production of enantiomerically pure (R)-3HB monomers.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. E. coli and B. megaterium cells were grown at 37°C in Luria-Bertani (LB) medium (32), LB medium supplemented with 2% sodium acetate (for the preparation of nPHB granules), or acetate-mineral salts medium (for the measurement of PHB contents, the observation of PHB accumulation by transmission electron microscopy, and the analysis of phaZ1-gfp expression). The acetate-mineral salts medium, which was modified from sucrose-mineral salts medium (16), consisted of 0.1% sodium acetate, 0.4% glucose, 0.1% NaCl, 0.02% MgSO4·7H2O, 1.1% (NH4)2SO4, 0.001% FeSO4·7H2O, 0.001% ZnSO4·7H2O, 0.0007% MnSO4, and 0.0005% CaCl2 in potassium phosphate buffer (0.24% KH2PO4 and 0.23% K2HPO4, pH 7.0). Antibiotics were used at the following concentrations (in micrograms per milliliter): ampicillin, 100; kanamycin, 15; tetracycline, 12 (for E. coli); and chloramphenicol, 3 (for B. megaterium).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Description and/or genotypea | Reference or sourceb |
|---|---|---|
| E. coli strains | ||
| DH5α | F− φ80dlacZΔM15 Δ(lacZYA-argF) recA1 gyrA endA1 relA1 supE44 hsdR17 | Laboratory stock |
| JM109 | recA1 supE44 endA1 hsdR17 gyrA96 relA1 thi Δ(lac-proAB) [F′ traD36 proAB+lacIqZΔM15] | Takara |
| Origami(DE3) | Δ(ara-leu)7697 ΔlacX74 ΔphoA PvuII phoR araD139 galE galK rpsL F′ [lac+ (lacIq) pro] gor522::Tn10 (Tcr) trxB::Kanr (DE3) | Novagen |
| B. megaterium strains | ||
| ATCC 11561 | Wild-type strain | ATCC |
| QM B1551 | Template for gene cloning by PCR | ATCC |
| BM1367 | ATCC 11561 derivative; phaZ1::pGS1874 disruption mutant carrying an in-frame fusion of N-terminal 115 amino acids of PhaZ1 to GFP; Cmr | This study |
| Plasmids | ||
| pET22b | Expression vector for producing His-tagged proteins; Apr | Novagen |
| pQE30 | Expression vector for producing His-tagged proteins; Apr | Qiagen |
| pSG1151 | Vector used for generation of GFP fusion protein; Apr Cmr | 20 |
| pGS1243 | pQE30 carrying the (R)-3HB dehydrogenase gene of B. thuringiensis | 39 |
| pGS1865 | pET22b carrying the wild-type phaZ1 gene | This study |
| pGS1874 | pSG1151 expressing an in-frame fusion of truncated PhaZ1 to GFP; phaZ1-disruptive plasmid | This study |
| pGS1941 | pET22b carrying the mutated phaZ1 gene (S142C substitution) | This study |
Apr, ampicillin resistant; Cmr, chloramphenicol resistant; Kanr, kanamycin resistance; Tcr, tetracycline resistance.
ATCC, American Type Culture Collection.
Construction of plasmids.
To construct plasmids that could overproduce His-tagged PhaZ1 and its S142C variant in E. coli, 0.98-kb DNA fragments carrying the wild-type phaZ1 gene and the mutated phaZ1 gene and flanked by NdeI and XhoI sites were amplified by PCR with primers P1 (5′-GCCAACACATATGTTGGGTTCACTACCT-3′) and P2 (5′-GCCACTCGAGTACTTTTTGACTCATGAAAAA-3′) and cloned between the NdeI and XhoI sites of pET22b (Novagen). This procedure resulted in plasmids pGS1865 and pGS1941, respectively. Site-directed mutagenesis that was used to introduce the S142C mutation was carried out by a two-step PCR method (10). All DNA sequences were confirmed by DNA sequencing.
To construct the phaZ1-disrupting plasmid pGS1874, which could generate an in-frame translational fusion of the N-terminal 115 amino acids of PhaZ1 to green fluorescent protein (GFP), a 0.32-kb DNA fragment carrying the internal sequence of phaZ1 from codon 16 to 115 and flanked by HindIII and EcoRI sites was amplified by PCR and cloned between the HindIII and EcoRI sites of plasmid pSG1151 (20), which carried a modified gfp coding sequence.
Construction of the phaZ1 disruption mutant of B. megaterium.
The phaZ1 disruption mutant BM1367 was created by introducing plasmid pGS1874 into the chromosome of B. megaterium through Campbell-like integration at the phaZ1 locus. Upon the integration of the phaZ1-gfp fusion into the chromosome, expression was driven by the natural promoter of phaZ1 (20), thus producing a fusion protein comprising the N-terminal 115 amino acids of PhaZ1 and GFP. Integrants were selected on LB agar plates for resistance to chloramphenicol. The correctness of phaZ1 disruption was verified by PCR.
Overproduction and purification of His-tagged proteins.
For the overproduction and purification of His-tagged PhaZ1 and its S142C variant from the sonication supernatant, E. coli Origami(DE3) bearing plasmid pGS1865 or pGS1941 was grown in LB medium at 37°C to an absorbance at 600 nm of 0.5 and then treated with 0.3 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 30°C for 16 h. Cells were collected, disrupted by sonication, and centrifuged at 15,000 × g for 10 min. The resulting supernatant was used for the purification of His-tagged proteins by affinity chromatography on an Ni-nitrilotriacetic acid (NTA) agarose column according to the instructions of the matrix manufacturer (Qiagen Inc.). The purification of His-tagged PhaZ of B. thuringiensis from the sonication supernatant was carried out exactly as described previously (39).
The purification of His-tagged PhaZ1 from inclusion bodies was conducted according to a previously described method (28) with slight modification. E. coli Origami(DE3) bearing plasmid pGS1865 was first grown in LB medium at 37°C to an absorbance at 600 nm of 0.5 and then treated with 0.3 mM IPTG for 2 h. Cells were harvested by centrifugation, resuspended in 50 mM phosphate buffer (pH 7.4), and then disrupted by a French press. The sediment obtained from centrifugation at 15,000 × g for 10 min was resuspended in 50 mM phosphate buffer (pH 8.0) containing 0.15 M NaCl and 1 mM EDTA. After incubation on ice for 20 min, the solution was centrifuged at 15,000 × g for 10 min. The insoluble fraction was washed with phosphate buffer (pH 8.0) containing 2 M urea and 0.5% Triton X-100 and then dissolved in phosphate buffer (pH 8.0) containing 6 M guanidine hydrochloride with gentle stirring at 4°C for 1 h. After centrifugation at 15,000 × g for 10 min, the supernatant containing denatured PhaZ1 was incubated at 4°C for 1 h with Ni-NTA agarose preequilibrated with 6 M guanidine hydrochloride. The mixture was then applied to an empty column and washed with 50 mM phosphate buffer (pH 8.0) containing 8 M urea, 5 mM imidazole, and 300 mM NaCl. PhaZ1 was refolded on-column by applying a decreasing gradient of urea (8 to 0 M) and then eluted by using a stepwise gradient of imidazole (100, 200, 300, and 400 mM) in the elution buffer containing 50 mM potassium phosphate (pH 6.0), 300 mM NaCl, and 10% glycerol. The purification of His-tagged B. thuringiensis PhaZ from inclusion bodies was conducted in the same way as described for PhaZ1 from B. megaterium.
Preparation of nPHB granules, aPHB granules, and dPHB.
nPHB granules were isolated from French press-disrupted B. megaterium cells that were grown at 37°C in LB medium supplemented with 2% sodium acetate for 6 h by the method of sucrose density gradient centrifugation as described previously (21). Trypsin-pretreated nPHB granules were obtained by preincubation of 0.7 mg of nPHB granules with 4 μg of trypsin in the reaction solution at 37°C for 10 min. aPHB granules were prepared from dPHB according to the details in a previous report (1) by using sodium deoxycholate as a surfactant. dPHB was isolated from B. megaterium cells by a procedure involving sodium hypochlorite digestion and subsequent solvent extraction with acetone-ether (2:1, vol/vol) as described previously (5).
Turbidimetric determination of PHB depolymerase activity.
The decrease in the turbidity of a suspension made from nPHB granules, aPHB granules, or dPHB due to the hydrolysis of PHB by PHB depolymerase was monitored as described previously (9). The turbidities of reaction mixtures were assayed spectrophotometrically at 650 nm by using a SpectraMax 190 microplate reader (Molecular Devices Corp.). Unless otherwise specified, each reaction mixture (200 μl) contained 100 mM Tris-HCl (pH 8.0); 0.7 mg of nPHB granules, aPHB granules, or dPHB; and 3 μg of purified PHB depolymerase. Trypsin-pretreated nPHB granules were preincubated with 4 μg of trypsin at 37°C for 10 min prior to the addition of purified PHB depolymerase. One unit of activity is defined as a decrease of 1 U in the optical density at 650 nm in 1 min.
To investigate the effects of various chemical reagents on the aPHB-hydrolyzing activity of PhaZ1, chemical reagents were preincubated with PhaZ1 in the reaction mixture for 10 min prior to the addition of the substrate aPHB.
Assay for esterase and lipase activities.
Spectrophotometric measurement of yellow p-nitrophenol generated from the hydrolysis of p-nitrophenyl esters by PhaZ1 was performed exactly as described previously (39). The lipase activity toward triolein or tributyrin was also measured exactly as described previously (39). A lipase from “Chromobacterium viscosum” (Sigma) was used as a positive control.
Quantitation of PHB and 3HB monomers.
The analysis of PHB contents in B. megaterium cells or the measurement of the amounts of PHB in nPHB granules and dPHB was carried out by gas chromatography as described previously (39), as well as by the method involving spectrophotometric evaluation of crotonic acid generated from the digestion of PHB with concentrated sulfuric acid (17).
The amount of 3HB monomers produced from the hydrolysis of dPHB or trypsin-pretreated nPHB granules by PHB depolymerase was quantified by the enzymatic method using NAD+-dependent (R)-3HB dehydrogenase according to the description in a previous report (44) and as reported previously by us (39).
In vitro nPHB granule-binding assay.
The assay mixture contained 10 mM Tris-HCl (pH 7.4), the purified His-tagged S142C variant (22 ng/μl) of B. megaterium PhaZ1 or His-tagged PhaZ (20 ng/μl) from B. thuringiensis, and nPHB granules (50 μg/μl) isolated from B. megaterium as described above. After incubation at 30°C for 15 min, the assay mixture was separated into the supernatant and the granule-containing pellet by centrifugation. The supernatant was subjected to sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis (SDS-10% PAGE) and analyzed by Western blotting with anti-His tag antibody (Santa Cruz Biotechnology, Inc.) as the probe. The pellet was washed twice with 100 μl of 10 mM Tris-HCl (pH 7.4) at room temperature. The washed pellet was resuspended in the sample buffer for SDS-PAGE, heated to 100°C for 5 min, and centrifuged again. Its supernatant was analyzed by the same Western blotting procedure.
Transmission electron microscopy.
B. megaterium cells were grown in acetate-mineral salts medium. Cell pellets were washed and incubated at room temperature with fresh fixative solution containing 2% glutaraldehyde, 3% paraformaldehyde, 5% sucrose, and 0.1 M sodium phosphate buffer (pH 7.4). After 1 h of primary fixation, cell pellets were washed twice with 0.1 M sodium phosphate buffer (pH 7.4) and subjected to secondary fixation with 1% osmium tetroxide solution at room temperature for 1 h. Cell pellets were then washed twice with distilled water and stained with 1% uranyl acetate at 4°C overnight. Cells were finally embedded in Spurr's epoxy resin according to a procedure described previously (36). Thin sections were examined on a JEM 2000EXII transmission electron microscope (JEDL Inc.).
Western blot analysis.
Immunoblots were prepared by transferring proteins from SDS-10% polyacrylamide gels onto nitrocellulose membranes (Bio-Rad). After being blocked with a blocking solution containing phosphate-buffered saline (pH 7.4), 5% skim milk, and 0.1% Tween 20, membranes were incubated with rabbit polyclonal anti-GFP antibody (Santa Cruz Biotechnology, Inc.), anti-His tag antibody, or anti-GroEL antibody (Sigma) in the blocking solution. Immunoreactive bands were detected with horseradish peroxidase-coupled anti-rabbit immunoglobulin G (Santa Cruz Biotechnology, Inc.). The signals were visualized with the Western Lightning Plus chemiluminescence reagent kit according to the instructions of the manufacturer (PerkinElmer).
Protein sequence analysis.
The B. megaterium genome BLAST server at Northern Illinois University was used for database searches. Overall amino acid identities and similarities between proteins were calculated with the CLUSTAL W software (35). The signal peptide sequence was analyzed by the SignalP software (24).
Other methods.
Genomic DNA of B. megaterium was isolated as described previously (26). The transformation of B. megaterium cells by the protoplast method was carried out as described previously (2). Protein concentrations were determined by the bicinchoninic acid protein assay method according to the instructions of the assay kit manufacturer (Pierce Biotechnology, Inc.) with bovine serum albumin as the standard.
Nucleotide sequence accession number.
Draft sequence data on phaZ1 from B. megaterium QM B1551 were obtained from Northern Illinois University and the Institute of Genome Science at the University of Maryland School of Medicine through the website at http://www.bios.niu.edu/b_megaterium/index.html. A DNA fragment encompassing the phaZ1 gene of B. megaterium ATCC 11561 was amplified by PCR with primers designed according to the draft sequence of phaZ1 of B. megaterium QM B1551 and was subjected to DNA sequencing by Mission Biotech Co., Ltd. (Taipei, Taiwan). The obtained nucleotide sequence was deposited in GenBank under accession number FJ175152.
RESULTS
Identification of a putative intracellular PHB depolymerase from B. megaterium by a BLAST search.
Since the draft genome sequence of B. megaterium QM B1551 just became available upon request (43), we attempted to use amino acid sequences of known intracellular PHB depolymerases as probes to identify putative intracellular PHB depolymerases of B. megaterium. When R. eutropha PhaZd1 (GenBank accession number BAE44206) (1) was used as the probe in a BLAST search, we identified a putative PHB depolymerase tentatively designated PhaZ1. PhaZ1 from B. megaterium QM B1551 shows 25.2% overall amino acid identity and 39.8% similarity to PhaZd1 from R. eutropha.
B. megaterium genes involved in the biosynthesis of nPHB granules were cloned previously from strain ATCC 11561 (21). Because we would like to study genes involved in the intracellular mobilization of PHB within strain ATCC 11561, we designed primers according to the flanking sequence of the phaZ1 gene from strain QM B1551 and used chromosomal DNA from strain ATCC 11561 as a template to amplify a DNA fragment containing the phaZ1gene of strain ATCC 11561 by PCR. DNA sequencing analysis revealed that the phaZ1 gene of strain ATCC 11561 encodes a putative protein of 323 amino acids containing a pentapeptide sequence (G-L-S142-A-G) that is similar to the lipase box motif (G-X-S-X-G) (12). The catalytic triad of R. eutropha PhaZd1 was previously predicted to be Ser190, Asp266, and His330. The putative oxyanion hole was predicted to be His108 (1). Analysis of the amino acid sequence alignment of B. megaterium PhaZ1 and R. eutropha PhaZd1 suggests that, in B. megaterium PhaZ1, Ser142, Asp218, and His291 may constitute the putative catalytic triad and His59 may be the putative oxyanion hole (Fig. 1). There are only two cysteine residues (Cys61 and Cys99) in the amino acid sequence of PhaZ1. The positions of these two cysteines in B. megaterium PhaZ1 roughly accord with those of the cysteines in PhaZd1 from R. eutropha (Cys110 and Cys148) (1) (Fig. 1). No potential signal peptide sequence at the N terminus of PhaZ1 was predicted by the SignalP software (24). PhaZ1 shows no significant amino acid similarity to other currently known intracellular or extracellular amorphous PHB-degrading PhaZ enzymes (Table 2), including B. thuringiensis PhaZ (39), P. denitrificans PhaZ (6), P. lemoignei PhaZ7 (7), R. eutropha PhaZa1 (31), and Rhodospirillum rubrum PhaZ1 (a periplasmic PhaZ) (8). However, a large region within PhaZ1, which spans amino acids 27 to 320, shows 50.2% identity and 72.7% similarity to most of the catalytic domain of the extracellular PHB depolymerase PhaZ from B. megaterium N-18-25-9 (34). The putative catalytic triads plus oxyanion holes and two conserved cysteine residues of these two proteins are present at corresponding positions (Fig. 1). The extracellular B. megaterium PhaZ was previously demonstrated to be a dPHB-degrading depolymerase (34).
FIG. 1.

Alignment of amino acid sequences of PhaZ1 and other PhaZs. PhaZ1, PhaZ1 from B. megaterium ATCC 11561; PhaZBm, the catalytic domain (amino acids 25 to 331) of the extracellular PHB depolymerase PhaZ from B. megaterium N-18-25-9; PhaZd1, PhaZd1 from R. eutropha H16. Identical residues are marked below with asterisks. The lipase box-like sequences are boxed and underlined. Residues of the proposed catalytic triad and oxyanion hole are boxed. Conserved cysteine residues are shown in white on black. GenBank accession numbers are as follows: PhaZ1, FJ175152; PhaZBm, BAF35850; and PhaZd1, BAE44206.
TABLE 2.
Percentages of identities and similarities between PhaZ1 from B. megaterium ATCC 11561 and other bacterial PHB depolymerases
| PHB depolymerase | Protein length (amino acids) | % Identity/% similarity to B. megaterium PhaZ1 |
|---|---|---|
| B. megaterium PhaZ1 | 323 | |
| B. thuringiensis PhaZ | 300 | 6.9/11.6 |
| P. denitrificans PhaZ | 447 | 13.1/25.0 |
| P. lemoignei PhaZ7 | 380 | 18.0/29.2 |
| R. eutropha PhaZa1 | 419 | 8.2/13.3 |
| R. eutropha PhaZd1 | 362 | 25.4/40.1 |
| R. rubrum PhaZ1 | 362 | 6.0/9.5 |
Overproduction and purification of His-tagged PhaZ1.
To examine the PHB-hydrolyzing activity of PhaZ1, we first constructed plasmid pGS1865, which could overproduce His-tagged PhaZ1 in E. coli. The sonication supernatant was prepared from E. coli harboring pGS1865. The His-tagged PhaZ1 was then purified by affinity chromatography on an Ni-NTA agarose column. The purified His-tagged PhaZ1 migrated as a major protein band with an apparent molecular mass of approximately 38 kDa on an SDS-10% polyacrylamide gel (Fig. 2). This estimated value is close to that calculated from its amino acid composition (35,735 Da) plus the mass of a His tag. A minor band present just below the major band of PhaZ1, as shown in Fig. 2, is probably a degradation product of PhaZ1 because it could be detected together with the major band by Western blot analysis using the anti-His tag antibody (data not shown). Since we could not exclude the possibility that a trypsin-like protease might happen to be present as a very minor contaminating protein along with purified PhaZ1, we also purified PhaZ1 by another method involving the solubilization and refolding of inclusion body proteins of E. coli harboring pGS1865 as described in Materials and Methods. It was found that the purity of His-tagged PhaZ1 purified by this method was increased (Fig. 2).
FIG. 2.

SDS-PAGE analysis of purified His-tagged PhaZ1 and the S142C variant. Whole-cell extracts of E. coli transformants carrying the PhaZ1-producing plasmid pGS1865 (A) or the S142C variant-producing plasmid pGS1941 (B) and purified His-tagged proteins were subjected to SDS-10% PAGE and stained with Coomassie blue. (A) Lanes: 1 and 2, whole-cell extracts of E. coli carrying pGS1865 without and with IPTG induction, respectively; 3, His-tagged PhaZ1 purified from the sonication supernatant of E. coli; and 4, refolded His-tagged PhaZ1 purified from inclusion bodies of E. coli. (B) Lanes: 1 and 2, whole-cell extracts of E. coli carrying pGS1941 without and with IPTG induction, respectively; and 3, His-tagged S142C variant purified from the sonication supernatant of E. coli. The arrows to the right indicate the positions of His-tagged PhaZ1 and the His-tagged S142C variant.
PHB depolymerase activity and other enzymatic activities of B. megaterium PhaZ1.
We then prepared nPHB granules from B. megaterium cells grown in LB medium supplemented with 2% sodium acetate for 6 h, since at this growth stage no endogenous PHB depolymerase activity was present in the prepared nPHB granules. We also prepared amorphous aPHB granules as well as dPHB as described in Materials and Methods. A turbidimetric method that monitored the decrease in turbidity of a suspension made from nPHB granules, aPHB granules, or dPHB was used to examine the degradation of PHB by PHB depolymerase. The result showed that, in a control experiment, purified His-tagged PhaZ from B. thuringiensis needed trypsin pretreatment of nPHB granules for PHB degradation (Fig. 3A). This result is consistent with the data in our previous report (39). Interestingly, B. megaterium His-tagged PhaZ1 purified from the sonication supernatant of E. coli cells harboring plasmid pGS1865 could rapidly hydrolyze nPHB granules without the need for trypsin pretreatment of the granules (Fig. 3A). The specific PHB-degrading activity of PhaZ1 against nPHB granules is approximately 101 U/mg of protein.
FIG. 3.

Turbidimetric determination of PHB-degrading activities of PHB depolymerases. A decrease in the turbidity of a reaction mixture (200 μl) containing 0.7 mg of nPHB granules, aPHB granules, or dPHB was detected spectrophotometrically at 650 nm. (A to D) Three micrograms of purified His-tagged PhaZ1 from B. megaterium or His-tagged PhaZ from B. thuringiensis was used in each assay. nPHB granules were isolated from B. megaterium cells that were grown in LB medium supplemented with 2% sodium acetate for 6 h. Symbols: (A) solid circles, nPHB granules plus PhaZ1; solid triangles, nPHB granules plus PhaZ; hollow triangles, trypsin-pretreated nPHB granules plus PhaZ; hollow circles, nPHB granules alone; (B) solid circles, nPHB granules plus refolded PhaZ1; solid triangles, nPHB granules plus refolded PhaZ; hollow circles, nPHB granules alone; (C) solid circles, trypsin-pretreated nPHB granules plus refolded PhaZ1; solid triangles, trypsin-pretreated nPHB granules plus refolded PhaZ; hollow circles, trypsin-pretreated nPHB granules alone; (D) solid circles, aPHB granules plus PhaZ1; solid triangles, aPHB granules plus refolded PhaZ1; hollow circles, aPHB granules alone. (E and F) Different amounts of purified His-tagged PhaZ1 from B. megaterium or His-tagged PhaZ from B. thuringiensis were used in the assays. Symbols: (E) solid circles, dPHB plus 3 μg of PhaZ1; solid triangles, dPHB plus 3 μg of PhaZ; hollow circles, dPHB alone; (F) solid circles, dPHB plus 15 μg of PhaZ1; solid triangles, dPHB plus 15 μg of PhaZ; hollow circles, dPHB alone. OD650, optical density at 650 nm.
Likewise, B. megaterium PhaZ1 purified by the method involving solubilization and refolding of inclusion body proteins of E. coli cells harboring pGS1865 could also efficiently hydrolyze nPHB granules without the need for trypsin pretreatment of the granules (Fig. 3B). As a negative control, B. thuringiensis PhaZ purified by the same method still could not hydrolyze nPHB granules unless the nPHB granules were pretreated with trypsin (Fig. 3B and C). It seemed that the efficiency of refolding of B. thuringiensis PhaZ was not as high as that of B. megaterium PhaZ1. When aPHB granules were used as the substrate, B. megaterium PhaZ1 purified either from the sonication supernatant or from inclusion bodies could also efficiently degrade aPHB granules (Fig. 3D). Surprisingly, B. megaterium PhaZ1 showed the capability of degrading dPHB in a concentration-dependent manner, although the efficiency of hydrolysis was much lower than that of the hydrolysis of nPHB granules (Fig. 3E and F). In a negative control experiment, B. thuringiensis PhaZ was still incapable of degrading dPHB (Fig. 3E and F). The result of the control experiment is consistent with the data in our previous report (39).
Analyses of products of enzymatic hydrolysis of nPHB granules or dPHB by using NAD+-dependent (R)-3HB dehydrogenase as described in Materials and Methods revealed time-dependent generation of 3HB monomers from PHB degradation by PhaZ1 (Fig. 4). The amounts of 3HB monomers produced from PHB degradation by B. megaterium PhaZ1 and B. thuringiensis PhaZ after 2 h of reaction time corresponded to approximately 92% and 42% of total 3HB equivalents present in nPHB granules, respectively (Fig. 4). In other words, B. megaterium PhaZ1 produced about 2.2-fold more 3HB monomers than B. thuringiensis PhaZ, and almost all the hydrolytic products of PHB degradation by B. megaterium PhaZ1 were monomeric 3HB. On the other hand, the amount of 3HB monomers generated from dPHB degradation by B. megaterium PhaZ1 was dependent on the concentration of PhaZ1 under the assay conditions. B. thuringiensis PhaZ still could not degrade dPHB under similar conditions (Fig. 4).
FIG. 4.
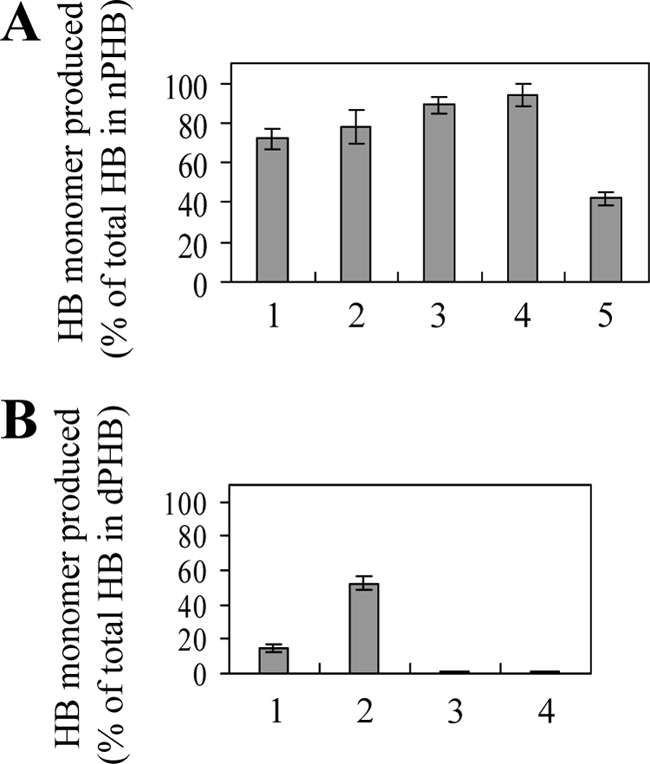
Production of 3HB monomers from in vitro degradation of nPHB granules or dPHB by PHB depolymerases. The reaction mixture (200 μl) contained 0.7 mg of nPHB granules or dPHB. (A) Amounts of 3HB monomers produced from the degradation of nPHB granules divided by the total 3HB equivalent in PHB. Lanes: 1 to 4, amounts produced from the degradation of nPHB granules in the absence of trypsin by 3 μg of B. megaterium PhaZ1 for 5, 10, 30, and 120 min, respectively, and 5, amounts produced from the degradation of trypsin-pretreated nPHB granules by 3 μg of B. thuringiensis PhaZ for 120 min. (B) Amounts of 3HB monomers generated from the degradation of dPHB for 180 min divided by the total 3HB equivalent in PHB. Lanes: 1 and 2, amounts produced from the degradation by 3 and 15 μg of B. megaterium PhaZ1, respectively, and 3 and 4, amounts produced from the degradation by 3 and 15 μg of B. thuringiensis PhaZ, respectively. Each value is the mean of three determinations. Each error bar indicates the standard error of the mean.
The hydrolyzing activities of PhaZ1 toward p-nitrophenyl esters and triglycerides were also examined. PhaZ1 shows only very weak activity toward p-nitrophenylbutyrate (ca. 0.083 μmol/min/mg of protein) but is inactive against p-nitrophenyldecanoate, p-nitrophenyllaurate, and p-nitrophenylpalmitate. No lipase activity of PhaZ1 toward triolein or tributyrin was detected, whereas the C. viscosum lipase used as a positive control could degrade these two substrates (data not shown).
The effects of various chemical reagents on the activity of PhaZ1 were also investigated. As was the case for PhaZd1 of R. eutropha (1) and PhaZ of P. denitrificans (6), the in vitro aPHB-degrading activity of PhaZ1 was not inhibited by 1 mM or 10 mM freshly prepared phenylmethylsulfonyl fluoride (PMSF) (data not shown). It is well known that PMSF, which is often used as a serine protease inhibitor, does not inhibit all serine proteases and serine lipases. Dithiothreitol (DTT) at 2 mM, iodoacetamide at 1 mM or 10 mM, N-ethylmaleimide at 1 mM, and H2O2 at 2 or 40 mM also did not inhibit the in vitro aPHB-degrading activity of PhaZ1 (data not shown). PhaZ1 showed maximum activity toward aPHB granules at pH 8.0 to 8.5 and at temperatures of 40 to 45°C.
In vitro nPHB granule-binding assay.
In order to explore the possible mechanism for trypsin-independent degradation of nPHB granules by PhaZ1, we first examined the in vitro nPHB granule-binding ability of PhaZ1. nPHB granules were isolated from B. megaterium cells grown in LB medium supplemented with 2% sodium acetate for 6 h as mentioned above. Because the incubation of purified water-soluble His-tagged PhaZ1 with isolated nPHB granules in the nPHB granule-binding assay resulted in the rapid degradation of nPHB granules by PhaZ1, we used instead the purified water-soluble His-tagged S142C variant of PhaZ1 (Fig. 2), in which the putative nucleophile (Ser142 within the lipase box) was replaced with a cysteine residue. Purified water-soluble His-tagged PhaZ from B. thuringiensis was used separately in the nPHB granule-binding assay for comparison, because it would not degrade nPHB granules without trypsin pretreatment of the granules. After the incubation of nPHB granules with either the His-tagged S142C variant or His-tagged PhaZ, the reaction mixture was separated into the supernatant and the granule-containing pellet by centrifugation. Proteins in each fraction were run on an SDS-polyacrylamide gel and analyzed by Western blotting with anti-His tag antibody as the probe. The results showed that the water-soluble His-tagged S142C variant of PhaZ1 could bind to nPHB granules whereas the water-soluble His-tagged PhaZ from B. thuringiensis displayed no affinity for nPHB granules under similar assay conditions (Fig. 5). These results are consistent with the notion that the ability of a PHB depolymerase to bind to nPHB granules correlates with its ability to degrade nPHB granules in a trypsin-independent manner. It should be mentioned that, during the incubation of PhaZ1 with nPHB granules, PhaZ1 with a His tag at the N terminus was somehow susceptible to degradation at the C terminus since the degradation product of PhaZ1 could still be detected by the anti-His tag antibody (Fig. 5).
FIG. 5.

Results of an in vitro nPHB granule-binding assay. nPHB granules were isolated from B. megaterium cells grown at 37°C in LB medium supplemented with 2% sodium acetate for 6 h. The purified His-tagged S142C variant (22 ng/μl) of B. megaterium PhaZ1 or His-tagged PhaZ (20 ng/μl) from B. thuringiensis was incubated with nPHB granules (50 μg/μl) at 30°C for 15 min. The incubation mixture was then separated into the supernatant and the granule-containing pellet by centrifugation. Proteins in the supernatant and pellet were subjected to SDS-10% PAGE and analyzed by Western blotting with anti-His tag antibody as the probe. In, input His-tagged protein; T, total proteins before separation into fractions by centrifugation; S, supernatant; P, pellet.
Effects of phaZ1 disruption on PHB accumulation in vivo.
To investigate the role of the B. megaterium phaZ1 gene in PHB degradation in vivo, we constructed the phaZ1 disruption mutant BM1367 as described in Materials and Methods. B. megaterium cells were grown in acetate-mineral salts medium, which contained 0.1% sodium acetate, 0.4% glucose, and various mineral salts. Phase-contrast microscopy revealed that no free spores were produced after 23, 25, and 27 h of cultivation in acetate-mineral salts medium (data not shown). As shown in Fig. 6A, the PHB contents of the phaZ1 mutant after 23, 25, and 27 h of cultivation were substantially higher than those of wild-type B. megaterium cells grown for the corresponding periods of time. These results were also confirmed by transmission electron microscopy, which showed comparable accumulation of nPHB granules in the wild type and the phaZ1 mutant after 10 h of growth (during the PHB-synthesizing stage) in acetate-mineral salts medium (Fig. 6B, panels a and c) but significantly higher accumulation of nPHB granules in the phaZ1 mutant than in the wild type after 23 h of growth (during the PHB-degrading stage) in acetate-mineral salts medium (Fig. 6B, panels b and d). Taken together, these results support the notion that PhaZ1 is functional in PHB mobilization in vivo.
FIG. 6.

Effects of phaZ1 disruption on PHB accumulation in vivo and on the hydrolysis of nPHB granules in vitro. Wild-type B. megaterium and the phaZ1 mutant BM1367 were grown in acetate-mineral salts medium at 37°C for the indicated periods of time. (A) Growth curves and PHB contents. Circles, wild type; triangles, phaZ1 mutant. Each value representing PHB contents is the mean of at least three determinations. Each error bar indicates the standard error of the mean. OD600, optical density at 600 nm. (B) Electron micrographs of thin sections of wild-type B. megaterium and the phaZ1 mutant showing the accumulation of nPHB granules. The scale bars represent 2 μm. (C) Turbidimetric analysis of the in vitro hydrolysis of nPHB granules by sonication supernatants from wild-type B. megaterium and the phaZ1 mutant grown for 23 h. Equal amounts (350 μg) of total proteins in sonication supernatants prepared from wild-type B. megaterium and the phaZ1 mutant were used in the assays. Solid circles, sonication supernatant from wild-type B. megaterium plus nPHB granules; solid triangles, sonication supernatant from the phaZ1 mutant plus nPHB granules; hollow circles, nPHB granules alone.
B. megaterium PhaZ1 is an intracellular PHB depolymerase.
In contrast to the N terminus of the extracellular PHB depolymerase PhaZ from B. megaterium N-18-25-9, which contains a typical signal peptide sequence (34), the N terminus of B. megaterium PhaZ1 does not contain a potential signal peptide sequence as predicted by the SignalP program (24). However, since the region of PhaZ1 spanning amino acids 27 to 320 shows 50.2% identity to the catalytic domain of the extracellular PHB depolymerase PhaZ from B. megaterium N-18-25-9 and PhaZ1 can also hydrolyze dPHB, one is tempted to speculate that PhaZ1 is an extracellular PHB depolymerase. As described above, the disruption of the phaZ1 gene significantly affected intracellular PHB mobilization as revealed by transmission electron microscopy and the measurement of PHB contents. In contrast, the disruption of the gene encoding the extracellular PHB depolymerase of B. megaterium ATCC 11561 did not affect intracellular PHB mobilization (data not shown). These results support the notion that PhaZ1 functions as an intracellular PHB depolymerase. We further used sonication supernatants prepared from wild-type B. megaterium and the phaZ1 mutant BM1367 grown in acetate-mineral salts medium for 23 h to carry out turbidimetric analyses. As shown in Fig. 6C, the sonication supernatant of the wild type displayed PHB depolymerase activity whereas the sonication supernatant of the phaZ1 mutant lost this activity. Overall, this evidence indicates that PhaZ1 is indeed an intracellular PHB depolymerase.
Western blot analysis of the expression of a translational fusion of phaZ1-gfp.
We further examined the time course of expression of phaZ1 in B. megaterium. At first, we constructed a B. megaterium strain carrying a chromosomal in-frame translational fusion of full-length phaZ1 to gfp in a manner similar to the construction of the phaZ1 disruption mutant BM1367. However, after 6 to 25 h of growth in acetate-mineral salts medium, no production of the PhaZ1-GFP fusion protein could be detected by Western blotting using a polyclonal anti-GFP antibody as the probe (data not shown). We then constructed a B. megaterium strain carrying a chromosomal in-frame translational fusion of phaZ1 to gfp with the deletion of the C-terminal 7 amino acids of PhaZ1, but still no PhaZ1-GFP fusion protein could be detected under similar conditions. Although the exact reasons are unknown, one possible explanation is that some portion of PhaZ1 in the fusion protein may interact with GFP, leading to a local conformational change in GFP. This conformational change in GFP may make it more susceptible to degradation by proteases.
Therefore, we resorted to using the phaZ1 disruption mutant BM1367 to examine the expression of the phaZ1-gfp fusion in B. megaterium. This mutant carried a chromosomal in-frame translational fusion of phaZ1 to gfp and could produce a fusion protein comprising the N-terminal 115 amino acids of PhaZ1 and GFP. The expression of the chromosomal phaZ1-gfp fusion was driven by the natural promoter of phaZ1 as described in the paper on the construction of the original vector pSG1151 (20). Whole-cell extracts were prepared from B. megaterium cells that were grown in acetate-mineral salts medium for various periods of time. Western blot analysis using a polyclonal anti-GFP antibody showed time-dependent production of the PhaZ1-GFP fusion protein in strain BM1367 (Fig. 7). The bands representing the PhaZ1-GFP fusion protein were absent in the extract from wild-type B. megaterium because it did not carry the phaZ1-gfp fusion. The detection of the presence of GroEL protein, whose level in cells remained constant under the assay conditions, was employed as an internal loading control. There seemed to be a time lag between the appearance of the PhaZ1-GFP fusion protein and the onset of a difference in PHB content between the wild type and the phaZ1 mutant (Fig. 6A and Fig. 7).
FIG. 7.

Western blot analysis of the expression of a translational fusion of phaZ1 to gfp. Wild-type B. megaterium and its derivative BM1367 were grown in acetate-mineral salts medium at 37°C for the indicated periods of time. BM1367 carried a chromosomal in-frame translational fusion of the phaZ1 region encoding the N-terminal 115 amino acids of PhaZ1 to gfp, and the expression of the fusion gene was driven by the natural promoter of phaZ1. Equal numbers of cells were collected and equal amounts of proteins from whole-cell extracts were subjected to SDS-10% PAGE and Western blotting. The PhaZ1-GFP fusion protein was detected with polyclonal anti-GFP antibody. The detection of the presence of GroEL protein on the same gel with the polyclonal antibody against GroEL was used as an internal loading control.
DISCUSSION
In this study, we have found that the intracellular PHB depolymerase PhaZ1 of B. megaterium has at least three unique properties that were not seen in other known intracellular PHB depolymerases: (i) PhaZ1 can rapidly hydrolyze nPHB granules in vitro without the need for trypsin pretreatment of the granules; (ii) almost all the in vitro hydrolytic products produced from the degradation of amorphous PHB by PhaZ1 are 3HB monomers; and (iii) PhaZ1 can degrade, albeit with lower efficiency than that of nPHB granule degradation, semicrystalline dPHB with the generation of 3HB monomers. It was previously reported that an intracellular medium-chain-length PHA depolymerase from Pseudomonas putida can also degrade medium-chain-length PHA in native PHA granules, regardless of the presence of granule-associated proteins (4). It is not clear why these two intracellular PhaZs can degrade native granules in vitro without the need for trypsin pretreatment of the granules. PhaZ7 of P. lemoignei is the only currently known extracellular PHB depolymerase specific for the degradation of nPHB granules in a trypsin-independent manner (7). In the in vitro nPHB granule-binding experiment carried out previously with the water-soluble PhaZ7, nPHB granules were completely hydrolyzed to form water-soluble products. This result led the authors of the study to suggest that PhaZ7 can efficiently bind to nPHB granules (7). A similar situation occurred with PhaZ1 from B. megaterium, so the S142C variant of PhaZ1 was used instead in our nPHB granule-binding assay. Results obtained from our nPHB granule-binding experiments support the notion that the ability of a PhaZ protein to bind to native granules correlates with its ability to degrade native granules in a trypsin-independent manner.
Bacterial extracellular dPHB-degrading PhaZs are known to have a dPHB-binding domain at the C terminus, and two types of dPHB-binding domains have been identified (13). The dPHB-binding ability is lost in the extracellular PhaZs that lack the C-terminal dPHB-binding domain, and accordingly, these truncated PhaZs can no longer hydrolyze dPHB. There is no similarity between these dPHB-binding domains and PhaZ1 from B. megaterium or PhaZ7 from P. lemoignei. In the future, it will be interesting to identify in these proteins the motif(s) or domain(s) required for nPHB granule binding.
PhaZ1 of B. megaterium ATCC 11561 shows 25.4% overall amino acid identity and 40.1% similarity to PhaZd1 of R. eutropha. Both proteins contain only two cysteine residues, roughly conserved at positions 110 and 148 in PhaZd1 and positions 61 and 99 in PhaZ1. It was previously found that the in vitro enzymatic activity of purified PhaZd1 can be completely inhibited by DTT (DTT at 0.1 and 1 mM results in 86 and 100% inhibition, respectively). This discovery led to the suggestion that the in vivo catalytic activity of PhaZd1 might be regulated by intracellular oxidation-reduction states (1). However, in our case, DTT at 2 mM could not significantly inhibit the aPHB-degrading activity of PhaZ1. Another difference is that, when R. eutropha cells were grown in minimum salt medium, the PhaZd1 protein appeared predominantly at the PHB-synthesizing stage and disappeared rapidly when cells entered the PHB-degrading stage (1). In our case, the PhaZ1 protein of B. megaterium cells grown in the acetate-mineral salts medium appeared predominantly at the PHB-degrading stage. Based on the fact that most of the in vitro hydrolytic products of PHB degradation by PhaZd1 are 3HB oligomers, it has been suggested previously that PhaZd1 probably acts as an endotype depolymerase (1). Since almost all the final hydrolytic products produced from in vitro PHB degradation by B. megaterium PhaZ1 are 3HB monomers, PhaZ1 probably works, at least in part, as an exotype depolymerase.
When the intracellular PhaZ from B. thuringiensis was used as the probe in a BLAST search, we identified a homolog (298 amino acids) in B. megaterium QM B1551, tentatively designated PhaZ2. Meanwhile, a BLAST search using R. eutropha PhaZd1 as the probe also identified a third putative PHB depolymerase (322 amino acids) in B. megaterium QM B1551, which was tentatively designated PhaZ3. Our preliminary data showed that both PhaZ2 and PhaZ3 could hydrolyze nPHB granules in vitro but that the pretreatment of nPHB granules with trypsin was absolutely required (H.-J. Chen, S.-C. Pan, and G.-C. Shaw, unpublished results). These findings suggest that the B. megaterium genome encodes at least three intracellular PHB depolymerases. These PHB depolymerases may act together or separately for the in vivo hydrolysis of PHB during the PHB-degrading stage, and some of them may be partially redundant in function. It is not unprecedented that multiple intracellular PHB depolymerases are present in one bacterial species. R. eutropha H16, a paradigmatic PHB-producing bacterium, is known to encode distinct intracellular PHB depolymerases (1, 42).
Recently, it was found that, in the presence of free coenzyme A (CoA), purified PHB synthase from R. eutropha can catalyze in vitro PHB degradation with 3HB-CoA as the reaction product, although the observed thiolytic activity is rather low (40). Furthermore, nPHB granules isolated from R. eutropha were also found to be able to catalyze the thiolysis of PHB in the presence of free CoA, with the generation of 3HB-CoA (41). The thiolytic activity of isolated nPHB granules is likely to be attributed to the nPHB granule-associated PhaZa1, which uses the sulfhydryl group of a cysteine residue at the catalytic center as the nucleophile (41). Whether similar thiolysis is involved in PHB mobilization in B. megaterium remains to be clarified, but this is an intriguing possibility because PHB contents in the phaZ1 mutant still decreased with time during the PHB-degrading stage.
It is well known that the monomeric units of PHB naturally synthesized in bacteria as a carbon reserve are optically active and exist in R configuration. The hydrolysis of bacterial PHB can produce enantiomerically pure (R)-3HB, which can be used as a chiral starting material for many industrial and medical applications (23, 32). A common problem encountered in using PhaZa1 from R. eutropha or PhaZ7 from P. lemoignei in the fermentative production of (R)-3HB is that oligomeric and dimeric forms of 3HB often constitute the main hydrolytic products (3, 18, 25, 33). Due to the feature that almost all the hydrolytic products produced from the degradation of nPHB granules by PhaZ1 are monomeric 3HB, PhaZ1 has promising potential for biotechnological application in (R)-3HB production by fermentation.
Acknowledgments
We thank Patricia S. Vary and her group for allowing access to the draft genome sequence of B. megaterium QM B1551. Sequencing of the genome of B. megaterium QM B1511 was accomplished with support from the National Science Foundation of the United States. We also thank Wan-Jr Syu for continuing interest and support.
This research was supported by grant NSC 94-2311-B-010-013 from the National Science Council and a grant, Aim for the Top University Plan, from the Ministry of Education of the Republic of China.
Footnotes
Published ahead of print on 26 June 2009.
REFERENCES
- 1.Abe, T., T. Kobayashi, and T. Saito. 2005. Properties of a novel intracellular poly(3-hydroxybutyrate) depolymerase with high specific activity (PhaZd) in Wautersia eutropha H16. J. Bacteriol. 187:6982-6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang, S., and S. N. Cohen. 1979. High frequency transformation of Bacillus subtilis protoplasts by plasmid DNA. Mol. Gen. Genet. 168:111-115. [DOI] [PubMed] [Google Scholar]
- 3.Chen, G. Q., and Q. Wu. 2005. Microbial production and applications of chiral hydroxyalkanoates. Appl. Microbiol. Biotechnol. 67:592-599. [DOI] [PubMed] [Google Scholar]
- 4.de Eugenio, L. I., P. Garcia, J. M. Luengo, J. M. Sanz, J. S. Roman, J. L. Garcia, and M. A. Prieto. 2007. Biochemical evidence that phaZ gene encodes a specific intracellular medium chain length polyhydroxyalkanoate depolymerase in Pseudomonas putida KT2442: characterization of a paradigmatic enzyme. J. Biol. Chem. 282:4951-4962. [DOI] [PubMed] [Google Scholar]
- 5.Delafield, F. P., M. Doudoroff, N. J. Palleroni, C. J. Lusty, and R. Contopoulos. 1965. Decomposition of poly-β-hydroxybutyrate by pseudomonads. J. Bacteriol. 90:1455-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao, D., A. Maehara, T. Yamane, and S. Ueda. 2001. Identification of the intracellular polyhydroxyalkanoate depolymerase gene of Paracoccus denitrificans and some properties of the gene product. FEMS Microbiol. Lett. 196:159-164. [DOI] [PubMed] [Google Scholar]
- 7.Handrick, R., S. Reinhardt, M. L. Focarete, M. Scandola, G. Adamus, M. Kowalczuk, and D. Jendrossek. 2001. A new type of thermoalkalophilic hydrolase of Paucimonas lemoignei with high specificity for amorphous polyesters of short chain-length hydroxyalkanoic acids. J. Biol. Chem. 276:36215-36224. [DOI] [PubMed] [Google Scholar]
- 8.Handrick, R., S. Reinhardt, P. Kimmig, and D. Jendrossek. 2004. The “intracellular” poly(3-hydroxybutyrate) (PHB) depolymerase of Rhodospirillum rubrum is a periplasm-located protein with specificity for native PHB and with structural similarity to extracellular PHB depolymerases. J. Bacteriol. 186:7243-7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Handrick, R., S. Reinhardt, D. Schultheiss, T. Reichart, D. Schuler, V. Jendrossek, and D. Jendrossek. 2004. Unraveling the function of the Rhodospirillum rubrum activator of polyhydroxybutyrate (PHB) degradation: the activator is a PHB-granule-bound protein (phasin). J. Bacteriol. 186:2466-2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higuchi, R., B. Krummel, and R. K. Saiki. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16:7351-7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horowitz, D. M., and J. K. M. Sanders. 1994. Amorphous, biomimetic granules of polyhydroxybutyrate: preparation, characterization, and biological implications. J. Am. Chem. Soc. 116:2695-2702. [Google Scholar]
- 12.Jaeger, K. E., S. Ransac, B. W. Dijkstra, C. Colson, M. van Heuvel, and O. Misset. 1994. Bacterial lipases. FEMS Microbiol. Rev. 15:29-63. [DOI] [PubMed] [Google Scholar]
- 13.Jendrossek, D., and R. Handrick. 2002. Microbial degradation of polyhydroxyalkanoates. Annu. Rev. Microbiol. 56:403-432. [DOI] [PubMed] [Google Scholar]
- 14.Jendrossek, D. 2002. Extracellular polyhydroxyalkanoate depolymerases: the key enzymes of PHA degradation, p. 41-83. In Y. Doi and A. Steinbüchel (ed.), Biopolymers, vol. 3b. Polyesters II. Wiley-VCH, Weinheim, Germany. [Google Scholar]
- 15.Jendrossek, D. 2007. Peculiarities of PHA granules preparation and PHA depolymerase activity determination. Appl. Microbiol. Biotechnol. 74:1186-1196. [DOI] [PubMed] [Google Scholar]
- 16.Kolodziej, B. J., and R. A. Slepecky. 1964. Trace metal requirements for sporulation of Bacillus megaterium. J. Bacteriol. 88:821-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Law, J. H., and R. A. Slepecky. 1961. Assay of poly-β-hydroxybutyric acid. J. Bacteriol. 82:33-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee, S. Y., and Y. Lee. 2003. Metabolic engineering of Escherichia coli for production of enantiomerically pure (R)-(−)-hydroxycarboxylic acids. Appl. Environ. Microbiol. 69:3421-3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee, T. R., J. S. Lin, S. S. Wang, and G. C. Shaw. 2004. PhaQ, a new class of poly-β-hydroxybutyrate (PHB)-responsive repressor, regulates phaQ and phaP (phasin) expression in Bacillus megaterium through interaction with PHB. J. Bacteriol. 186:3015-3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis, P. J., and A. L. Marston. 1999. GFP vectors for controlled expression and dual labelling of protein fusions in Bacillus subtilis. Gene 227:101-110. [DOI] [PubMed] [Google Scholar]
- 21.McCool, G. J., and M. C. Cannon. 1999. Polyhydroxyalkanoate inclusion body-associated proteins and coding region in Bacillus megaterium. J. Bacteriol. 181:585-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCool, G. J., and M. C. Cannon. 2001. PhaC and PhaR are required for polyhydroxyalkanoic acid synthase activity in Bacillus megaterium. J. Bacteriol. 183:4235-4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merrick, J. M., R. Steger, and D. Dombroski. 1999. Hydrolysis of native poly(hydroxybutyrate) granules (PHB), crystalline PHB, and artificial amorphous PHB granules by intracellular and extracellular depolymerases. Int. J. Biol. Macromol. 25:129-134. [DOI] [PubMed] [Google Scholar]
- 24.Nielsen, H., J. Engelbrecht, S. Brunak, and G. von Heijne. 1997. A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int. J. Neural Syst. 8:581-599. [DOI] [PubMed] [Google Scholar]
- 25.Park, S. J., S. Y. Lee, and Y. Lee. 2004. Biosynthesis of R-3-hydroxyalkanoic acids by metabolically engineered Escherichia coli. Appl. Biochem. Biotechnol. 113-116:373-379. [PubMed] [Google Scholar]
- 26.Pospiech, A., and B. Neumann. 1995. A versatile quick-prep of genomic DNA from gram-positive bacteria. Trends Genet. 11:217-218. [DOI] [PubMed] [Google Scholar]
- 27.Potter, M., and A. Steinbuchel. 2005. Poly(3-hydroxybutyrate) granule-associated proteins: impacts on poly(3-hydroxybutyrate) synthesis and degradation. Biomacromolecules 6:552-560. [DOI] [PubMed] [Google Scholar]
- 28.Rehm, B. H., Q. Qi, B. B. Beermann, H. J. Hinz, and A. Steinbuchel. 2001. Matrix-assisted in vitro refolding of Pseudomonas aeruginosa class II polyhydroxyalkanoate synthase from inclusion bodies produced in recombinant Escherichia coli. Biochem. J. 358:263-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rehm, B. H. 2003. Polyester synthases: natural catalysts for plastics. Biochem. J. 376:15-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ren, Q., A. Grubelnik, M. Hoerler, K. Ruth, R. Hartmann, H. Felber, and M. Zinn. 2005. Bacterial poly(hydroxyalkanoates) as a source of chiral hydroxyalkanoic acids. Biomacromolecules 6:2290-2298. [DOI] [PubMed] [Google Scholar]
- 31.Saegusa, H., M. Shiraki, C. Kanai, and T. Saito. 2001. Cloning of an intracellular poly[d(−)-3-hydroxybutyrate] depolymerase gene from Ralstonia eutropha H16 and characterization of the gene product. J. Bacteriol. 183:94-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sambrook, J., and D. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 33.Shiraki, M., T. Endo, and T. Saito. 2006. Fermentative production of (R)-(−)-3-hydroxybutyrate using 3-hydroxybutyrate dehydrogenase null mutant of Ralstonia eutropha and recombinant Escherichia coli. J. Biosci. Bioeng. 102:529-534. [DOI] [PubMed] [Google Scholar]
- 34.Takaku, H., A. Kimoto, S. Kodaira, M. Nashimoto, and M. Takagi. 2006. Isolation of a Gram-positive poly(3-hydroxybutyrate) (PHB)-degrading bacterium from compost, and cloning and characterization of a gene encoding PHB depolymerase of Bacillus megaterium N-18-25-9. FEMS Microbiol. Lett. 264:152-159. [DOI] [PubMed] [Google Scholar]
- 35.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian, J., A. J. Sinskey, and J. Stubbe. 2005. Kinetic studies of polyhydroxybutyrate granule formation in Wautersia eutropha H16 by transmission electron microscopy. J. Bacteriol. 187:3814-3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tokiwa, Y., and B. P. Calabia. 2004. Degradation of microbial polyesters. Biotechnol. Lett. 26:1181-1189. [DOI] [PubMed] [Google Scholar]
- 38.Tokiwa, Y., and C. U. Ugwu. 2007. Biotechnological production of (R)-3-hydroxybutyric acid monomer. J. Biotechnol. 132:264-272. [DOI] [PubMed] [Google Scholar]
- 39.Tseng, C. L., H. J. Chen, and G. C. Shaw. 2006. Identification and characterization of the Bacillus thuringiensis phaZ gene, encoding new intracellular poly-3-hydroxybutyrate depolymerase. J. Bacteriol. 188:7592-7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uchino, K., and T. Saito. 2006. Thiolysis of poly(3-hydroxybutyrate) with polyhydroxyalkanoate synthase from Ralstonia eutropha. J. Biochem. 139:615-621. [DOI] [PubMed] [Google Scholar]
- 41.Uchino, K., T. Saito, B. Gebauer, and D. Jendrossek. 2007. Isolated poly(3-hydroxybutyrate) (PHB) granules are complex bacterial organelles catalyzing formation of PHB from acetyl coenzyme A (CoA) and degradation of PHB to acetyl-CoA. J. Bacteriol. 189:8250-8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uchino, K., T. Saito, and D. Jendrossek. 2008. Poly(3-hydroxybutyrate) (PHB) depolymerase PhaZa1 is involved in mobilization of accumulated PHB in Ralstonia eutropha H16. Appl. Environ. Microbiol. 74:1058-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vary, P. S., R. Biedendieck, T. Fuerch, F. Meinhardt, M. Rohde, W. D. Deckwer, and D. Jahn. 2007. Bacillus megaterium—from simple soil bacterium to industrial protein production host. Appl. Microbiol. Biotechnol. 76:957-967. [DOI] [PubMed] [Google Scholar]
- 44.Williamson, D. H., J. Mellanby, and H. A. Krebs. 1962. Enzymic determination of D(−)-β-hydroxybutyric acid and acetoacetic acid in blood. Biochem. J. 82:90-96. [DOI] [PMC free article] [PubMed] [Google Scholar]