

Abstract
Azole resistance in Candida albicans can be mediated by the upregulation of the ATP binding cassette transporter genes CDR1 and CDR2. Both genes are regulated by a cis-acting element called the drug-responsive element (DRE), with the consensus sequence 5′-CGGAWATCGGATATTTTTTT-3′, and the transcription factor Tac1p. In order to analyze in detail the DRE sequence necessary for the regulation of CDR1 and CDR2 and properties of TAC1 alleles, a one-hybrid system was designed. This system is based on a P(CDR2)-HIS3 reporter system in which complementation of histidine auxotrophy can be monitored by activation of the reporter system by CDR2-inducing drugs such as estradiol. Our results show that most of the modifications within the DRE, but especially at the level of CGG triplets, strongly reduce CDR2 expression. The CDR2 DRE was replaced by putative DREs deduced from promoters of coregulated genes (CDR1, RTA3, and IFU5). Surprisingly, even if Tac1p was able to bind these putative DREs, as shown by chromatin immunoprecipitation, those from RTA3 and IFU5 did not functionally replace the CDR2 DRE. The one-hybrid system was also used for the identification of gain-of-function (GOF) mutations either in TAC1 alleles from clinical C. albicans isolates or inserted in TAC1 wild-type alleles by random mutagenesis. In all, 17 different GOF mutations were identified at 13 distinct positions. Five of them (G980E, N972D, A736V, T225A, and N977D) have already been described in clinical isolates, and four others (G980W, A736T, N972S, and N972I) occurred at already-described positions, thus suggesting that GOF mutations can occur in a limited number of positions in Tac1p. In conclusion, the one-hybrid system developed here is rapid and powerful and can be used for characterization of cis- and trans-acting elements in C. albicans.
Candida albicans is an opportunistic pathogen causing superficial to deep systemic infection in immunocompromised patients. Due to the increase in the number of people with immunodeficiencies (essentially due to AIDS, transplantation, or chemotherapy), the use of antifungal drugs is now more frequent and has led to development of drug resistance in several fungal species, especially in Candida albicans. Several resistance mechanisms have been described up to now (22). One of the more frequent is the upregulation of multidrug transporters, encoded by CDR1 and CDR2, belonging to the ATP-binding cassette transporter family (1, 2). In azole-resistant isolates, the expression of these pumps is constitutively high. The resulting drug efflux diminishes antifungal efficacy and thus protects the fungus from drug toxic effects. We demonstrated that the expression of pump-encoding genes can also be transiently upregulated by treating azole-susceptible isolates with drugs such as estradiol or fluphenazine, thus mimicking azole-resistant isolates (8). In our laboratory we are particularly interested in the regulation of the expression of multidrug transporter genes.
Previous studies showed that CDR1 and CDR2 promoters contain regulatory sequences crucial for their regulation. The existence of two basal response elements localized between nucleotides −860 and −810 and between −243 and −234 and one negative regulatory element localized within the −289 region was reported (12, 20). Finally, Karnani et al. (14) have identified a steroid response region between −696 and −521 containing two elements (steroid response elements 1 and 2), but none of these elements were shown to be crucial for azole resistance. In contrast, the drug-responsive element (DRE) sequence (5′-CGGAWATCGGATA-3′) in both CDR1 and CDR2 promoters has been characterized and was crucial not only for the upregulation of these genes in azole-resistant strains but also for the transient upregulation of both genes in the presence of different drugs (8).
CDR1 and CDR2 are coregulated with other genes, such as RTA3, IFU5, PDR16, and TAC1, that all contain putative DREs in their promoters (6-8, 24). The expression of these genes was shown to be controlled by the Zn(2)-Cys(6) transcription factor Tac1p (transcriptional activator of CDR genes). Tac1p was shown to be responsible for transient upregulation of CDR genes in azole-susceptible strains in the presence of inducers (6). We previously identified TAC1 hyperactive alleles from clinical azole-resistant strains, which, in contrast to wild-type alleles, conferred constitutive high CDR1 and CDR2 expression on a tac1Δ/tac1Δ mutant (6). Hyperactivity of TAC1 was due to a gain-of-function (GOF) mutation. Seven distinct GOF mutations have been found in 11 analyzed hyperactive alleles (4, 5, 24). In previous studies, Tac1p was shown to bind in vitro and in vivo to the DRE (6, 17). Our in vitro analysis demonstrated the crucial role of the CGG triplets for the binding of Tac1p. Liu et al. (17) performed a genome-wide location analysis of Tac1p in an azole-susceptible strain and demonstrated that 37 promoters including those of CDR1 and CDR2 and of TAC1 itself were bound to Tac1p in an azole-susceptible strain. They deduced a consensus sequence for the Tac1p binding site: 5′-CGGN(4)CGG-3′, which overlapped with the DRE identified by de Micheli et al. (8).
Both elements, the DRE and Tac1p, are crucial for the constitutive and induced upregulation of CDR genes. Further analyses of both elements are, however, needed to better understand the regulation of CDR1 and CDR2. The one-hybrid system provides an interesting tool to study transcriptional regulation of genes. Recently two one-hybrid systems were developed in C. albicans based on a transcription factor coupled with the Staphylococcus aureus lexA DNA binding domain and quantification of the transcriptional activity by β-galactosidase assays (19, 21). Even if these two systems are efficient for measuring the activity of transcription factors, these techniques are time- and material consuming. Moreover, these techniques allow only analysis of trans-acting factors, not cis-acting elements. Therefore an alternative method was needed to rapidly analyze several derivatives of the DRE and several Tac1p variants.
In this study we analyzed more precisely the cis and trans regulators involved in the regulation of CDR2 in C. albicans. A one-hybrid reporter system using a fusion of the CDR2 promoter [P(CDR2)] with HIS3 was designed, enabling both cis and trans elements regulating CDR2 to be tested. We demonstrated that mutations in the DRE, especially at the position of CGG triplets, perturb its function. trans-acting elements regulating CDR2 were tested by reintroducing different TAC1 alleles into the reporter strain lacking TAC1. A library of randomly mutated TAC1 alleles and of TAC1 alleles recovered from clinical isolates was screened in the reporter strain. Seventeen GOF mutations and 5 loss-of-function (LOF) mutations either in the C-terminal part of the protein or between the previously described DNA-binding domain and middle homology region (MHR) were thus identified.
MATERIALS AND METHODS
Strains and media.
The C. albicans strains used in this study are listed in Table 1. Isolates were grown in YEPD (1% Bacto peptone [Difco Laboratories, Basel, Switzerland], 0.5% yeast extract [Difco], and 2% glucose [Fluka]) or in minimal medium (MM):yeast nitrogen base (Difco) and 2% glucose (Fluka, Buchs, Switzerland). When isolates were grown on solid media, 2% agar (Difco) was added. Escherichia coli DH5α was used as a host for plasmid constructions and propagation. DH5α was grown in Luria-Bertani broth (LB) or LB plates supplemented with ampicillin (0.1 mg/ml) (LB-amp) when required.
TABLE 1.
Strains used in this study
| Strain | Parental strain | Genotype | Reference or source |
|---|---|---|---|
| CAI8 | SC5314 | ura3Δ::imm434/ura3Δ::imm434 ade2Δ::hisG/ade2Δ::hisG | 11 |
| DSY2627 | CAI8 | ura3Δ::imm434/ura3Δ::imm434 ade2Δ::hisG/ade2Δ::hisG his3Δ::hisG/his3Δ::hisG | This study |
| DSY2657 | DSY2627 | ade2Δ::pAC3 | This study |
| DSY2773 | DSY2627 | ade2Δ::pAC4 | This study |
| ACY51 | DSY2627 | ade2Δ::pAC126 | This study |
| ACY52 | DSY2627 | ade2Δ::pAC127 | This study |
| ACY53 | DSY2627 | ade2Δ::pAC128 | This study |
| ACY54 | DSY2627 | ade2Δ::pAC129 | This study |
| ACY55 | DSY2627 | ade2Δ::pAC130 | This study |
| ACY56 | DSY2627 | ade2Δ::pAC131 | This study |
| ACY58 | DSY2627 | ade2Δ::pAC133 | This study |
| ACY59 | DSY2627 | ade2Δ::pAC134 | This study |
| ACY60 | DSY2627 | ade2Δ::pAC135 | This study |
| ACY61 | DSY2627 | ade2Δ::pAC136 | This study |
| ACY62 | DSY2627 | ade2Δ::pAC139 | This study |
| ACY63 | DSY2627 | ade2Δ::pAC140 | This study |
| ACY64 | DSY2627 | ade2Δ::pAC137 | This study |
| ACY65 | DSY2627 | ade2Δ::pAC138 | This study |
| ACY101 | DSY2627 | ade2Δ::pAC27 | This study |
| ACY102 | DSY2627 | ade2Δ::pAC28 | This study |
| ACY103 | DSY2627 | ade2Δ::pAC29 | This study |
| ACY104 | DSY2627 | ade2Δ::pAC30 | This study |
| ACY105 | DSY2627 | ade2Δ::pAC31 | This study |
| ACY106 | DSY2627 | ade2Δ::pAC32 | This study |
| ACY107 | DSY2627 | ade2Δ::pAC33 | This study |
| ACY108 | DSY2627 | ade2Δ::pAC34 | This study |
| ACY194 | DSY2627 | ade2Δ::pAC35 | This study |
| ACY195 | DSY2627 | ade2Δ::pAC36 | This study |
| ACY196 | DSY2627 | ade2Δ::pAC37 | This study |
| ACY197 | DSY2627 | ade2Δ::pAC38 | This study |
| ACY198 | DSY2627 | ade2Δ::pAC39 | This study |
| ACY199 | DSY2627 | ade2Δ::pAC40 | This study |
| ACY200 | DSY2627 | ade2Δ::pAC41 | This study |
| ACY201 | DSY2627 | ade2Δ::pAC42 | This study |
| ACY202 | DSY2627 | ade2Δ::pAC43 | This study |
| ACY203 | DSY2627 | ade2Δ::pAC44 | This study |
| ACY204 | DSY2627 | ade2Δ::pAC45 | This study |
| ACY205 | DSY2627 | ade2Δ::pAC46 | This study |
| ACY206 | DSY2627 | ade2Δ::pAC47 | This study |
| ACY207 | DSY2627 | ade2Δ::pAC48 | This study |
| ACY208 | DSY2627 | ade2Δ::pAC49 | This study |
| ACY209 | DSY2627 | ade2Δ::pAC50 | This study |
| ACY210 | DSY2627 | ade2Δ::pAC51 | This study |
| ACY211 | DSY2627 | ade2Δ::pAC52 | This study |
| ACY212 | DSY2627 | ade2Δ::pAC53 | This study |
| ACY213 | DSY2627 | ade2Δ::pAC54 | This study |
| ACY214 | DSY2627 | ade2Δ::pAC55 | This study |
| ACY215 | DSY2627 | ade2Δ::pAC56 | This study |
| ACY216 | DSY2627 | ade2Δ::pAC57 | This study |
| DSY3352 | DSY2627 | ura3Δ::imm434/ura3Δ::imm434 ade2Δ::hisG/ade2Δ::hisG his3Δ::hisG/his3Δ::hisG tac1Δ::hisG/tac1Δ::hisG | This study |
| ACY29 | DSY3352 | ade2Δ::pAC4 | This study |
| ACY31 | DSY2627 | ade2Δ::pAC4 LEU2::pDS1099 | This study |
| ACY36 | DSY2627 | ade2Δ::pAC4 LEU2::pDS1097 | This study |
| ACY39 | DSY2627 | ade2Δ::pAC4 LEU2::pDS178 | This study |
| ACY263 | DSY2627 | ade2Δ::pAC218 | This study |
| ACY264 | DSY3352 | ade2Δ::pAC218 | This study |
| DSY2906 | CAF4-2 | ura3Δ::imm434/ura3Δ::imm434 tac1Δ::hisG/tac1Δ::hisG | 6 |
| ACY1 | DSY2906 | RPS1::pAC9 | This study |
| JCY49 | ACY29 | ade2Δ::pAC4 LEU2::pJC49a | This study |
| JCY60 | ACY29 | ade2Δ::pAC4 LEU2::pJC60 | This study |
| JCY38 | ACY29 | ade2Δ::pAC4 LEU2::pJC38 | This study |
| JCY58 | ACY29 | ade2Δ::pAC4 LEU2::pJC58 | This study |
| JCY53 | ACY29 | ade2Δ::pAC4 LEU2::pJC53 | This study |
| JCY61 | ACY29 | ade2Δ::pAC4 LEU2::pJC61 | This study |
| JCY9 | ACY29 | ade2Δ::pAC4 LEU2::pJC9 | This study |
| JCY16 | ACY29 | ade2Δ::pAC4 LEU2::pJC16 | This study |
| JCY55 | ACY29 | ade2Δ::pAC4 LEU2::pJC55 | This study |
| JCY3 | ACY29 | ade2Δ::pAC4 LEU2::pJC3 | This study |
| JCY59 | ACY29 | ade2Δ::pAC4 LEU2::pJC59 | This study |
| JCY17 | ACY29 | ade2Δ::pAC4 LEU2::pJC17 | This study |
| JCY18 | ACY29 | ade2Δ::pAC4 LEU2::pJC18 | This study |
| JCY19 | ACY29 | ade2Δ::pAC4 LEU2::pJC19 | This study |
| JCY20 | ACY29 | ade2Δ::pAC4 LEU2::pJC20 | This study |
| JCY21 | ACY29 | ade2Δ::pAC4 LEU2::pJC21 | This study |
| JCY22 | ACY29 | ade2Δ::pAC4 LEU2::pJC22 | This study |
| JCY23 | ACY29 | ade2Δ::pAC4 LEU2::pJC23 | This study |
| JCY24 | ACY29 | ade2Δ::pAC4 LEU2::pJC24 | This study |
| JCY25 | ACY29 | ade2Δ::pAC4 LEU2::pJC25 | This study |
| JCY26 | ACY29 | ade2Δ::pAC4 LEU2::pJC26 | This study |
| JCY27 | ACY29 | ade2Δ::pAC4 LEU2::pJC27 | This study |
| JCY28 | ACY29 | ade2Δ::pAC4 LEU2::pJC28 | This study |
Plasmids with the pJC prefix were obtained after rescue from C. albicans strains transformed with random-mutagenized pDS1097.
Yeast transformation.
C. albicans cells from a 0.2-ml stationary-phase culture were resuspended in 0.1 ml of a solution containing 200 mM lithium acetate (pH 7.5), 40% (wt/vol) polyethylene glycol 8000, 15 mg/ml dithiothreitol, and 250 μg/ml denatured salmon sperm DNA. Transforming DNA (1 to 5 μg) was added to the yeast suspension, which was incubated for 60 min at 44°C. Transformation mixtures were plated directly onto selective plates.
Immunoblots.
C. albicans cell extracts for immunoblotting were prepared by an alkaline extraction procedure from cells grown to mid-log phase. Briefly, cells (optical density at 540 nm, 5) were resuspended in an Eppendorf tube with 500 μl water and 150 μl of a solution containing 1.85 M NaOH and 7.5% β-mercaptoethanol. This mixture was incubated on ice for 10 min. Proteins were then precipitated with 150 μl of a 50% trichloroacetic acid solution, and the suspension was left on ice for another 10 min. Precipitated proteins were centrifuged at maximal speed in a microcentrifuge for 15 min. The sediment was resuspended in 50 μl of loading buffer (40 mM Tris-HCl [pH 6.8], 8 M urea, 5% sodium dodecyl sulfate [SDS], 0.1 M EDTA, 1% β-mercaptoethanol, and 0.1 mg/ml bromophenol blue) and incubated at 37°C for 10 min. Nonsolubilized material was eliminated by a centrifugation step for 10 min. Ten microliters of solubilized yeast proteins was separated by 10% SDS-polyacrylamide gel electrophoresis and transferred by Western blotting onto a nitrocellulose membrane. Immunodetections of Cdr1p and Cdr2p were performed with rabbit polyclonal anti-Cdr1p and anti-Cdr2p antibodies as described previously (8).
Construction of gene disruption cassettes.
For the disruption of HIS3, a region containing the entire open reading frame (ORF) was amplified from genomic DNA using the cloning primers HIS3-XHO and HIS3-XBA (Table 2). PCR fragments were cloned into pBluescript KS(+) to yield pDS970. Deletions within cloned regions were carried out by PCR with deletion primers CAHIS-PST and CAHIS-BGL and with cloning constructs as templates. The 3.7-kb PstI-BglII fragment comprising the URA3-blaster cassette from pMB7 was cloned into the PCR fragment previously digested with PstI and BglII to obtain deletion construct pAC1. For transformation in C. albicans, linear fragments were obtained by digestion of deletion constructs with ApaI and SacI. The deletion of TAC1 in DSY2627 was performed as previously described, yielding strain ACY29 (6).
TABLE 2.
Primers used in this study
| Primer | Sequence |
|---|---|
| Amplification of ADE2 | |
| ADE-xho | GCGCAAACTCGAGAATGATTAATCAATGATCACCATAAACTTG |
| ADE2-pst | GCGCAAACTGCAGAAGAAAAAGACACTTAAGTTTTAATTATAAG |
| HIS3 inactivation | |
| HIS3-XHO | GCGCAAACTCGAGCCAAGGTTTTAATTCAATTTTGGGTTGAGG |
| HIS3-XBA | GCGCAAATCTAGACAGAGGTTCAATTATTCGAAATCCAGCAAT |
| CAHIS-PST | GCGCAAACTGCAGGTAGAGAATTTATTTATTTATTTATTTATT |
| CAHIS- BGL | ACACAGAAAGATCTAATAGTAAAAAAACGCCTGCTTAC |
| Amplification TAC1 | |
| Zinc2-5-BamB | GCAAGGATCCAAGAAGAAGTGGATAATTTTGATTAC |
| Zinc2-3-Xho | GCAACTCGAGAGTATATTCTGTTGGGAAAGGGGTGAG |
| Construction of pAC9, pAC3 and pAC44 | |
| SPHI-ZINC2C | GCGCAAAGCATGCAAATCCCCAAATTATTGTCAAAGAAAAA |
| SALI-ZINC2C | GCGCAAAGTCGACATAATGGACACTTCACTGTCACTGGGA |
| HIS3-KPN | GCGCAAAGGTACCAAGGTTTTAATTCAATTTTGGGTTGAGG |
| HIS3-ATG | TTCACTAATTAACACATACAATAAAAACATATGTCACGAGAAGCTTTAATCAATAGAATA |
| CDR2-ANTI | ATGTTTTTATTGTATGTGTTAATTAGTGAA |
| CDR2prom Nsi- | GCGCAAACTCGAGGTATGTGCAAAAACTGATAATATACCTCTG |
| CDR2-DEL3 | AGTATTCATAATAGAGGCTTTGAAAACAA |
| CDR2-DEL5 | GTATTAATTTTTACGTATTTTCTTTGTGTTATTCAATTCTTGTTTTCAAAGCCT |
| Construction of pAC218 | |
| IFU5promF1000xho | GCGCAAACTCGAGCCAGTATTATGAGAGCAAAGATCATGCGCG |
| IFU5promR-his | CTAGAAGTCTCCGCCGCCAAAGTCACC |
| IFU5-HIS | GGTGACTTTGGCGGCGGAGACTTCTAGATGTCACGAGAAGCTTTAATCAATAGAATA |
| CDR2 promoter mutations | |
| CDR2 DREmini | GCGCAAACTCGAGTTCACGGAAATCGGATATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegpolyT | GCGCAAACTCGAGTTCACGGAAATCGGATAGTTTTCAAAGCCTCTAT |
| DREIIdegw/oTc | GCGCAAACTCGAGTTCACGGAAACGGATATTTTTTTTTGTTTTCAAAGCC |
| DREdeg-CGG1 | GCGCAAACTCGAGTTCAATTAAATCGGATATTTTTTTTTGTTTTCAAAGCC |
| DREdeg-AAA | GCGCAAACTCGAGTTCACGGCCCTCGGATATTTTTTTTTGTTTTCAAAGCC |
| DREdeg-CGG2 | GCGCAAACTCGAGTTCACGGAAATATTATATTTTTTTTTGTTTTCAAAGCC |
| DREdeg-ATA | GCGCAAACTCGAGTTCACGGAAATCGGCGCTTTTTTTTTGTTTTCAAAGCC |
| DREdeg-part1 | GCGCAAACTCGAGTTCAATTCCCTCGGATATTTTTTTTTGTTTTCAAAGCC |
| DREdeg-part2 | GCGCAAACTCGAGTTCACGGAAATATTCGCTTTTTTTTTGTTTTCAAAGCC |
| DREdeg delete | GCGCAAACTCGAGTTCACGGAAATTTTTTTTTGTTTTCAAAGCC |
| DREdeg1T | CGCAAACTCGAGAATTCACGGAAATCGGATATGTTTTCAAAGCCTCTAT |
| DREdeg2T | CGCAAACTCGAGAATTCACGGAAATCGGATATTGTTTTCAAAGCCTCTAT |
| DREdeg3T | CGCAAACTCGAGAATTCACGGAAATCGGATATTTGTTTTCAAAGCCTCTAT |
| DREdeg4T | CGCAAACTCGAGAATTCACGGAAATCGGATATTTTGTTTTCAAAGCCTCTAT |
| DREdeg5T | CGCAAACTCGAGAATTCACGGAAATCGGATATTTTTGTTTTCAAAGCCTCTAT |
| DREdeg6T | CGCAAACTCGAGAATTCACGGAAATCGGATATTTTTTGTTTTCAAAGCCTCTAT |
| DREdeg7T | CGCAAACTCGAGAATTCACGGAAATCGGATATTTTTTTGTTTTCAAAGCCTCTAT |
| DREdeg8T | CGCAAACTCGAGAATTCACGGAAATCGGATATTTTTTTTGTTTTCAAAGCCTCTAT |
| DRE-degV1 | GCGCAAACTCGAGTTCACGGAAATCGGATAGGGGGTTTTGTTTTCAAAGCC |
| DRE-degV2 | GCGCAAACTCGAGTTCACGGAAATCGGATATTTTTGGGGGTTTTCAAAGCC |
| DRE-degTT | GCGCAAACTCGAGTTCACGGAAATCGGATAGGGGGGGGGGTTTTCAAAGCC |
| DREdegPoly T1 | GCGCAAACTCGAGTTCACGGAAATCGGATATCCCCTTTTGTTTTCAAAGCC |
| DREdegPoly T2 | GCGCAAACTCGAGTTCACGGAAATCGGATATTCCCTTTTGTTTTCAAAGCC |
| DREdegPoly T3 | GCGCAAACTCGAGTTCACGGAAATCGGATATTTCCTTTTGTTTTCAAAGCC |
| DREdegPoly T4 | GCGCAAACTCGAGTTCACGGAAATCGGATATTTTCTTTTGTTTTCAAAGCC |
| DREIIdegC1 | GCGCAAACTCGAGTTCANGGAAATCGGATATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegG1 | GCGCAAACTCGAGTTCACNGAAATCGGATATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegG2 | GCGCAAACTCGAGTTCACGNAAATCGGATATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegA1 | GCGCAAACTCGAGTTCACGGNAATCGGATATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegA2 | GCGCAAACTCGAGTTCACGGANATCGGATATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegA3 | GCGCAAACTCGAGTTCACGGAANTCGGATATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegTc | GCGCAAACTCGAGTTCACGGAAANCGGATATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegC2 | GCGCAAACTCGAGTTCACGGAAATNGGATATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegG3 | GCGCAAACTCGAGTTCACGGAAATCNGATATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegG4 | GCGCAAACTCGAGTTCACGGAAATCGNATATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegA4 | GCGCAAACTCGAGTTCACGGAAATCGGNTATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegT1 | GCGCAAACTCGAGTTCACGGAAATCGGANATTTTTTTTTGTTTTCAAAGCC |
| DREIIdegA5 | GCGCAAACTCGAGTTCACGGAAATCGGATNTTTTTTTTTGTTTTCAAAGCC |
| DRE-IFU5 | CGCAAACTCGAGTTGTATATATCCGATTTCCGATTTCCCTGTTTTCAAAGCCTCTATTATGAATAC |
| DRE-RTA3 | CGCAAACTCGAGTCCACACGGAACTCGGAAATTATGCGTTTTCAAAGCCTCTATTATGAATAC |
| DRE-CDR1 | CGCAAACTCGAGTTGAGACGGATATCGGATATTTTTTTTGTTTTCAAAGCCTCTATTATGAATAC |
| DRE-TAC1-457 | CGCAAACTCGAGTGGAAATAGTGGCGAAGGCAAATTGAAAATTCCGGATAGTTTTCAAAGCCTCTATTATGAATAC |
| DRE-TAC1-501 | CGCAAACTCGAGCTTTTTATTTTCCGTTGCTTCTTCCGTGCTCCGCGTTTTCAAAGCCTCTATTTGAATAC |
| DRE-PDR16-ZCB-1 | CGCAAACTCGAGCCTAAAATCGGATTCGGAATTAACAAATGGTTTTCAAAGCCTCTATTATGAATAC |
| qPCR | |
| CDR1-CHIP2-F | TTTCAACATATTAGAATCGAATCATTACG |
| CDR1-CHIP2-R | GCGGCTGTGTGTTTGTGTG |
| CDR2-CHIP2-F | AATTCAAACACAAACAATAAGGCTGT |
| CDR2-CHIP2-R | GCAATCATTGTGGTATACATCGGA |
| RTA3-CHIP2-F | AGATCCACACGGAACTCGGA |
| RTA3-CHIP2-R | TGATAACACACATGGGTTGTAAGATATTT |
| IFU5-CHIP-R | AATACATGCCAGTATTATGAGA |
| IFU5-CHIP-F | TGCTTACATTAGCAAATATGAG |
| ACT1-RTPCR-F | GTTCCCAGGTATTGCTGAAC |
| ACT1-RTPCR-R | CAATGGATGGACCAGATTCG |
| ControlChIP-Ch3F | AGGAGCTGGACATAGTTG |
| ControlChIP-Ch3R | CCAGCAGTGAATGATACG |
| TAC1 sequencing | |
| Zinc2-604 | ATAAGAGTGGCATGTGATA |
| Zinc2-1123 | GATGCCAACGAATTATTGA |
| Zinc2-1708 | CAGAATTCGTTGGAGAATA |
| Zinc2-2242 | GCCTTGTTACAATCAAGAA |
| Zinc2-2683 | GCAGCATATCTTGTATTAG |
| Zinc2-3224 | ATGCTCAGTCACCAAGTTA |
| Zinc2-3087c | GGTGTTCCTGCTACCACAA |
| Zinc2-3078c | GGTGTTCCTGCTACCACAA |
| Zinc2-1798c | ACATCAACAATGCTTCTAC |
| Zinc2-1247c | TCTTCACCGTATGAACCTA |
| Zinc2-778c | CGTTGCTATTGGCGGTTGA |
| Zinc2-1169 | TGTTGGTACTCATTCAATT |
| Zinc2-1722 | TTGGAGAATAGTGCCATTT |
| Zinc2-1510 | GCTACCAAGCGAAGGAGAT |
| Zinc2-2465c | TCTCTCGCCTAATTGACGT |
| TAC1 truncated allele | |
| JC-TAC | GCGCAAACTCGAGTTATGGGGTCAAATATTCTTC |
Construction of the one-hybrid P(CDR2)-HIS3 system and its modification.
Fusion of CDR2 promoter region with HIS3 was performed by two consecutive PCRs. First, the CDR2 promoter and HIS3 were amplified separately from genomic DNA of strain SC5314 using the primers CDR2prom-Nsi with CDR2-anti and HIS3-ATG with HIS3-KPN (Table 2). The HIS3-ATG primer allowed introduction of the last 30 bp of the 3′-end sequence of the CDR2 promoter before the HIS3 ATG start codon. Then, a second PCR was performed using the two previous PCR products as templates, which can hybridize via the 3′ end of the CDR2 promoter sequence present in both PCR products. This second PCR was performed using the external primers CDR2prom-Nsi and HIS3-KPN.
This PCR product consisting of the fusion of the CDR2 promoter with the HIS3 ORF was inserted between the XhoI and KpnI restriction sites of pDS961, which is pBluescript KS(+) carrying ADE2 between the XhoI and PstI sites. This ligation resulted in the plasmid pAC3.
Other constructs were obtained by amplifying the CDR2 promoter-HIS3 fusion from pAC3 using different CDR2 promoter-modifying primers listed in Table 2. The PCR products were then cloned in pDS961 using XhoI and KpnI restriction sites. These modifications were made within a CDR2 minimal promoter [−224 relative to the start codon; P(CDR2mini)] that was adapted from pAC3 to yield pAC4.
In the case of the CDR2 promoter lacking the DRE, a fusion between the 5′ and 3′ regions of the P(CDR2)-HIS3 fusion flanking the DRE was performed. For this purpose, two PCRs using the primers CDR2prom-Nsi with CDR2-DEL3 and CDR2-DEL5 with HIS3-KPN were first performed. A second PCR using the two previous PCR products as templates was performed. This second PCR was performed using the external primers CDR2prom-Nsi and HIS3-KPN.
Construction of one-hybrid reporter strains.
Reporter strains were obtained by transformation of C. albicans DSY2627 with the different CDR2- or CDR2-modified promoter-HIS3 constructs. These plasmids were linearized by NsiI and transformed into C. albicans, thus allowing integration into the genomic ADE2 locus.
Strain ACY29 was transformed with pDS178-derived plasmids (8) containing different TAC1 alleles cloned between XhoI and BamHI restriction sites. These plasmids were linearized by SalI and transformed into C. albicans, thus allowing integration into the genomic LEU2 locus.
One-hybrid screening.
C. albicans strains analyzed for recovery of histidine auxotrophy were grown overnight in MM-yeast nitrogen base containing uridine and adenine with or without histidine under constant agitation at 30°C and diluted from a starting culture containing 1.5 ×107 cells/ml with serial 10-fold dilutions. Five microliters of each dilution was spotted onto MM and in some cases onto MM with CDR2 inducers such as estradiol, terbinafine, benomyl, diamide (10 μg/ml), and fluphenazine (100 μg/ml). Cells were also spotted onto MM containing histidine as a growth control. Plates were incubated for 48 h at 34°C.
Chromatin immunoprecipitations (ChIP).
TAC1 was cloned by using SalI and SphI into CIP-ACT-C-ZZ (3), which introduced protein A at the C terminus of Tac1p. Our construct (pAC9) was introduced into the RPS1 locus of a strain lacking TAC1 (DSY2906). To verify the functionality of the fusion protein, the expression of CDR2 and CDR1 was controlled in the absence and presence of fluphenazine by immunoblotting as described above.
For ChIP assays, 200 ml of YEPD was inoculated with 2 ml of an overnight culture of strains to be tested and strains were grown at 30°C under agitation until the culture reach a density of 1.5 × 107 cells/ml. This culture was then treated for 15 min with 1% formaldehyde with occasional agitation. The reaction was stopped by addition of glycine at 125 mM and incubation for 5 min at room temperature. The culture was washed twice in cold TBS (20 mM Tris-HCl [pH 7.6], 150 mM NaCl). The pellet was resuspended in 500 μl ice-cold lysis buffer (50 mM HEPES-KOH [pH 7.5], 1% Triton X-100, 140 mM NaCl, 0.1% Na deoxycholate, 1 mM EDTA, and Roche protease inhibitor mixture). The solution was transferred in a 2-ml tube, and glass beads were added. The mixture was agitated at 4°C and 1,400 rpm in an Eppendorf Thermomixer block for 45 min. Lysates were recovered by puncturing the bottoms of tubes with a 23-gauge needle and by centrifugation at 1,000 rpm for 2 min. The recovered lysates were next sonicated four times (10 s) at full power with a sonicator (Soniprep 150; MSE Ltd., London, United Kingdom). The sonicated lysates were centrifuged for 20 min at 4°C and 10,000 × g. The supernatants were recovered, and 25 μl of this crude extract was conserved at −80°C as the input control of the experiment. In parallel, 40 μl of protein G-Dynabeads (corresponding to the quantity needed for one sample) was incubated for 2 h at room temperature with 5 ml of rabbit anti-protein A antibody (Sigma), washed twice with phosphate-buffered saline-bovine serum albumin (5 mg/ml) using a magnetic device, and resuspended in 40 μl of phosphate-buffered saline-bovine serum albumin. Beads were added to 250 μl of the crude extracts, and the solution was incubated overnight with tilt rotation at 4°C. Beads were next washed twice for 5 min with 600 μl lysis buffer on a rotator, with wash buffer (10 mM Tris-HCl [pH 8.0], 0.5% Na deoxycholate, 0.25 M LiCl, 1 mM EDTA, 0.5% NP-40), and for 5 min in 600 μl Tris-EDTA (TE). Beads were resuspended in 60 μl TE-1% SDS and incubated for 10 min at 65°C with shaking. Supernatants were recovered using a magnetic device, and beads were retreated for 10 min at 65°C with TE-1% SDS. Both supernatants were pooled and supplemented with 130 μl of TE-1% SDS. Only 100 μl of TE-1% SDS was added to 25 μl of the crude input. Immunoprecipitated samples and input samples were incubated overnight at 65°C. After addition of 240 μl TE, 40 μg of glycogen, and 200 μg of proteinase K, tubes were incubated for 2 h at 37°C. After addition of 50 ml LiCl (5 M), samples were treated twice with phenol-chloroform. Recovered DNA was then precipitated with Na acetate and absolute ethanol. Pellets were then washed in 70% ethanol and resuspended in 60 μl TE (30 μl for input samples).
Quantitative real-time PCR and normalization.
Real-time PCRs were performed using the QuantiTect SYBR green PCR kit (Qiagen) in a final volume of 25 μl according to the manufacturer's recommendations. PCRs were performed in duplicate for the standard curve and in triplicate for the samples to be quantified. DNA of the input samples was diluted 100-fold, and immunoprecipitated samples were diluted 10-fold to be in the range of the standard curve. Quantitative real-time PCRs were performed on an ABI Prism 7000 sequence detection system with the following program: 2 min at 55°C, 10 min at 95°C, and 45 cycles of 15 s at 95°C and 1 min at 60°C.
To normalize data, quantitative PCR (qPCR) signals derived from the ChIP samples were divided by the qPCR signals derived from the input samples. Background signals were next removed. For this purpose the ratio obtained previously for CAF2-1, which did not carry the TAC1-protein A fusion, was subtracted from the ratio obtained for ACY1 containing the TAC1-protein A construct. Finally the enrichment of the gene of interest was compared to that of control genes by dividing the normalized values obtained for our sequences of interest (DREs of CDR1, CDR2, RTA3, and IFU5) by the normalized value obtained for the control sequence. The control sequence is located in the promoter of orf19.5970, situated on chromosome 3 between CDR1 and CDR2. This gene was chosen because of its location between two Tac1p target genes (CDR1 and CDR2) and because it was not regulated by Tac1p either in the presence of fluphenazine or in azole-resistant strains in which CDR genes were upregulated, as shown by microarray experiments (6).
Analysis of TAC1 alleles from clinical isolates.
TAC1 was amplified from the genomic DNA of the clinical strains and cloned into pDS178 (Table 3). In order to distinguish the two putative TAC1 alleles present in a strain, at least three plasmids were analyzed by restriction fragment length polymorphism (RFLP) using the restriction enzyme EcoRI or AciI or both (see Table 4). For strains heterozygous for the mating type-like (MTL) locus, from 3 to 20 plasmids could be analyzed. For each distinct TAC1 allele, at least two different TAC1-containing plasmids carrying the same RFLP profile were sequenced and introduced at the LEU2 locus of the one-hybrid reporter strain after SalI digestion. When isolates were heterozygous for the MTL but had no distinct TAC1 RFLP profiles, from 3 to 20 distinct TAC1-containing plasmids from a given strain were sequenced in order to discriminate distinct sequences.
TABLE 3.
Plasmids used in this study
| Vector | Backbone | Description | Source or reference |
|---|---|---|---|
| pBluescript KS(+) | Cloning vector | Stratagene, La Jolla, CA | |
| pDS178 | pRC2312 | pRC2312-derived plasmid containing URA3 and LEU2 | 8 |
| pDS1097 | pDS178 | Insertion of TAC1-1 between XhoI and BamHI sites | 6 |
| pDS1099 | PDS178 | Insertion of TAC1-5 between XhoI and BamHI sites | 6 |
| pDS961 | pBS-KS(+) | Insertion of ADE2 | This study |
| pAC3 | pDS961 | Insertion of P(CDR2) (−720-1)-HIS3 fusion between XhoI and PstI sites | This study |
| pAC4 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DRE-CDR2-mini and HIS3-KPN | This study |
| pAC27 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegpolyT and HIS3-KPN | This study |
| pAC28 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegw/oTc and HIS3-KPN | This study |
| pAC29 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg-CGG1 and HIS3-KPN | This study |
| pAC30 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg-AAA and HIS3-KPN | This study |
| pAC31 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg-CGG2 and HIS3-KPN | This study |
| pAC32 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg-ATA and HIS3-KPN | This study |
| pAC33 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg-part1and HIS3-KPN | This study |
| pAC34 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg-part2 and HIS3-KPN | This study |
| pAC35 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg delete and HIS3-KPN | This study |
| pAC37 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DRE-degV1 and HIS3-KPN | This study |
| pAC38 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DRE-degV2 and HIS3-KPN | This study |
| pAC39 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DRE-degTT and HIS3-KPN | This study |
| pAC40 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdegPoly T1 and HIS3-KPN | This study |
| pAC41 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdegPoly T2 and HIS3-KPN | This study |
| pAC42 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdegPoly T3 and HIS3-KPN | This study |
| pAC43 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdegPoly T4 and HIS3-KPN | This study |
| pAC44 | pDS961 | Insertion of P(CDR2)w/oDRE-HIS3 amplified from pAC3 by fusion of the PCR products from CDR2 nsi- with CDR2 DEL3 and HIS-KPN with CDR2 DEL5 | This study |
| pAC45A,G,Ta | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegC1 and HIS3-KPN | This study |
| pAC46C,T,Aa | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegG1 and HIS3-KPN | This study |
| pAC47C,T,Aa | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegG2 and HIS3-KPN | This study |
| pAC48G,C,Ta | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegA1 and HIS3-KPN | This study |
| pAC49G,C,Ta | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegA2 and HIS3-KPN | This study |
| pAC50G,C,Ta | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegA3 and HIS3-KPN | This study |
| pAC51G,C,Aa | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegTC and HIS3-KPN | This study |
| pAC52A,G,Ta | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegC2 and HIS3-KPN | This study |
| pAC53C,T,Aa | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegG3 and HIS3-KPN | This study |
| pAC54C,T,Aa | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegG4 and HIS3-KPN | This study |
| pAC55G,C,Ta | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegA4 and HIS3-KPN | This study |
| pAC56G,C,Aa | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegT1 and HIS3-KPN | This study |
| pAC57G,C,Ta | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREIIdegA5 and HIS3-KPN | This study |
| pAC126 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DRE-PDR16-zcb-1 and HIS3-KPN | This study |
| pAC127 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DRE-TAC1-501 and HIS3-KPN | This study |
| pAC128 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DRE-TAC1-457 and HIS3-KPN | This study |
| pAC129 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DRE-CDR1 and HIS3-KPN | This study |
| pAC130 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DRE-RTA3 and HIS3-KPN | This study |
| pAC131 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DRE-IFU5 and HIS3-KPN | This study |
| pAC133 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg1T and HIS3-KPN | This study |
| pAC134 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg2T and HIS3-KPN | This study |
| pAC135 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg3T and HIS3-KPN | This study |
| pAC136 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg4T and HIS3-KPN | This study |
| pAC137 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg5T and HIS3-KPN | This study |
| pAC138 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg6T and HIS3-KPN | This study |
| pAC139 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg7T and HIS3-KPN | This study |
| pAC140 | pDS961 | Insertion of P(CDR2)-HIS3 amplified from pAC3 with DREdeg8T and HIS3-KPN | This study |
| CIpACT-C-ZZ | Clp10 | 3 | |
| pAC9 | CIpACT-C-ZZ | Insertion of TAC1 ORF amplified with SPHI-ZINC2C and SALI-ZINC2C | This study |
| pAC218 | pDS961 | Insertion of P(IFU5)(−999-1)-HIS3 between XhoI and KpnI sites | This study |
These plasmids have three versions, each containing one of the bases indicated.
TABLE 4.
Summary of the analysis of TAC1 alleles cloned from clinical isolates and screened by the P(CDR2)-HIS3 reporter system
| Patient | Origin | Strain | Fluconazole MIC (μg/ml) | MTL | Plasmid name | EcoRI profile | AciI profile | Sequence profilec | One-hybrid phenotypee | TAC1 GOF mutation(s) |
|---|---|---|---|---|---|---|---|---|---|---|
| A | CHUVd | DSY281 | 1 | a/α | 1607-1 | A | WT | |||
| 1607-2 | B | WT | ||||||||
| DSY284 | 32 | a/α | 1608-8 | A | Allele A | HYP | H741Ya | |||
| 1608-99 | A | Allele B | HYP | G980E + H741Y | ||||||
| B | DSY2014 | 0.125 | a/α | 1609-13 | A | WT | ||||
| 1609-12 | B | WT | ||||||||
| DSY2015 | 16 | a/α | 1621-71b | A | HYP | R673Q | ||||
| C | DSY2019 | 4 | α/α | 1610-19 | A | WT | ||||
| 1610-16 | B | HYP | N972D | |||||||
| DSY2020 | 32 | α/α | 1622-76 | B | HYP | N972D | ||||
| D | DSY2250 | 1 | a/α | 1611-23 | A | WT | ||||
| 1611-24 | B | WT | ||||||||
| DSY2251 | 16 | a/α | 1477-A | A | WT | |||||
| 1477-B | B | HYP | N972D | |||||||
| E | CHUV | DSY544 | 0.5 | a/α | 1612-30 | A | WT | |||
| 1612-26 | B | WT | ||||||||
| DSY775 | 128 | a/a | 1618-56 | A | HYP | G980W | ||||
| F | CHUV | DSY1280 | 1 | a/α | 1635-4 | A | HYP | E461K | ||
| 1617-121 | B | HYP | E461K | |||||||
| DSY1292 | 16 | a/α | 1490-A | A | A | HYP | N972S | |||
| 1490-C | A | B | HYP | N972S | ||||||
| G | CHUV | DSY2242 | 8 | a/a | 1619-61 | B | HYP | G980E | ||
| DSY2243 | 1 | a/a | 1637-3 | B | WT | |||||
| H | CHUV | DSY555 | 8 | a/α | 1620-68 | A | WT | |||
| 1620-92 | B | HYP | R693K | |||||||
| DSY556 | 128 | α/α | 1488-3 | A | HYP | R693K | ||||
| DSY557 | 128 | α/α | 1493-12 | B | HYP | R693K | ||||
| I | CHUV | DSY757 | 2 | a/α | 1623-83 | A | WT | |||
| 1623-82 | B | WT | ||||||||
| DSY758 | 16 | a/α | 1624-86 | B | Allele A | HYP | A736V | |||
| 1624-88 | B | Allele B | HYP | A736V | ||||||
| J | CHUV | DSY2260 | 8 | a/α | 1638-1 | A | HYP | E841Ga | ||
| 1638-3 | B | HYP | E841G | |||||||
| DSY2262 | 64 | a/α | 1613-31 | A | A | HYP | E841G | |||
| 1639-1 | A | B | HYP | E841G | ||||||
| K | CHUV | DSY482 | 8 | a/α | 1614-36 | B | WT | |||
| 1614-138 | A | WT | ||||||||
| DSY488 | 16 | α/α | 1487-3 | A | HYP | A736T | ||||
| L | CHUV | DSY520 | 32 | a/α | 1615-44 | A | WT | |||
| 1615-41 | B | HYP | N972D | |||||||
| DSY522 | 128 | a/α | 1616-46 | B | HYP | N972D | ||||
| 1474-B | A | HYP | N972D | |||||||
| M | CHUV | DSY2305 | 2 | a/α | 1633-3 | A | A | Allele A | WT | |
| 1633-4 | A | A | Allele B | WT | ||||||
| DSY2306 | 8 | a/a | 1634-1 | A | From allele A | HYP | E461K | |||
| N | CHUV | DSY2023 | 16 | α/α | 1528-10 | A | HYP | N972D | ||
| DSY2025 | 16 | α/α | 1529-2 | A | HYP | N972D |
This putative GOF mutation was not verified by site-directed mutagenesis.
Only one TAC1 allele was cloned even if the sequence of the genomic DNA indicated a polymorphism at the MTL.
TAC1 sequence analysis was performed after failure of TAC1 allele discrimination by RFLP in MTL heterozygous strains.
CHUV, University Hospital Lausanne and University Hospital Center.
WT, wild type; HYP, hyperactive.
Next, two distinct transformants for each allele were tested on the different media in order to determine the property of the introduced TAC1 allele.
Random mutagenesis.
Random mutagenesis of the TAC1-1 allele cloned in pDS178, yielding pDS1097 (Table 3), was performed using E. coli XL1-Red (Stratagene, La Jolla, CA) by following the instructions of the manufacturer. Briefly, 100-μl aliquots of competent XL1-Red were transformed with 50 ng of pDS1097 carrying TAC1-1. Three independent transformations were performed, and products were plated onto LB-amp plates for 16 h. Plates were divided in quarters, and colonies of each quarter were recovered and incubated in 5 ml LB-amp and agitated overnight at 37°C. Minipreparations of DNA were produced, and 1 μl of extracted DNA was retransformed in DH5α. All DH5α colonies were recovered and pooled into 10 ml LB-amp. Seven milliliters was used to inoculate 500 ml LB-amp. Cultures were grown for 4 to 5 h at 37°C, and plasmid DNA was extracted. Sixty independent C. albicans transformations were performed using 10 μg of randomly mutagenized plasmid DNA linearized by SalI.
Rescue of mutated TAC1 alleles.
Mutated TAC1 alleles were amplified from genomic DNA of the C. albicans strains transformed with a randomly mutagenized plasmid library using the M13-forward and M13-reverse primers. DNA amplicons were digested by XhoI and BamHI, ligated into pDS178, and transformed in DH5α. Rescued plasmids were then sequenced and introduced into ACY29 to verify phenotypes initially observed in C. albicans.
Sequencing of the TAC1 alleles.
The rescued TAC1 alleles were sequenced in a 3130XL genetic analyzer (Applied Biosystem). Sequences analysis was performed using ContigExpress and VectorNTI software (Infomax; Invitrogen Life Sciences, Basel, Switzerland).
TAC1 alleles from clinical strains of our collections were cloned in pDS178 and sequenced with a 96-capillary 3730XL DNA analyzer (Applied Biosystems). The resulting data were analyzed using the Mutation Surveyor software (SoftGenetics).
RESULTS
Construction of the C. albicans one-hybrid system.
In order to study the activity of the CDR2 promoter and the importance of the DRE for its activity, we designed a one-hybrid system based on the restoration of histidine prototrophy in a C. albicans reporter strain. Briefly, HIS3 was introduced under the control of the CDR2 promoter (−506 to −1) in a strain auxotrophic for histidine. Since CDR2 expression is almost absent in normal growth conditions, we expected that a P(CDR2)-HIS3 chimeric fusion would not be transcribed. In contrast, in the presence of CDR2 inducers like estradiol, we expected that HIS3 would be transcribed (Fig. 1A). The P(CDR2)-HIS3 reporter activities can thus be determined in a strain lacking HIS3 by monitoring the restoration of growth in the absence of histidine. To enable the monitoring of P(CDR2)-HIS3 reporter activities, strain CAI8, which is already auxotrophic for uridine and adenine, was made auxotrophic for histidine by targeted deletion of HIS3, leading to strain DSY2627. In parallel, the P(CDR2)-HIS3 chimeric construct was introduced into a plasmid carrying ADE2. This construct was introduced into DSY2627 at the ADE2 locus, yielding DSY2657.
FIG. 1.

Schematic view of the promoter-reporter system. (A) Testing of cis-acting elements of the CDR2 promoter. The promoter-reporter system is based on a reporter construct consisting of the CDR2 promoter regulating HIS3 introduced at the ADE2 locus in a strain lacking HIS3. The reporter strain is unable to grow in the absence of histidine unless the CDR2 promoter is activated by inducers such as fluphenazine or estradiol. (B) Testing of trans-acting elements of the CDR2 promoter. Properties of the TAC1 alleles can be assayed in the reporter strain lacking TAC1, encoding the CDR2 regulator. The parental strain is unable to grow in the absence of histidine even in the presence of a CDR2 inducer, since TAC1 is absent. Introduction of a wild-type TAC1 allele should restore histidine prototrophy, but only in the presence of an inducer. A hyperactive TAC1 allele can confer constitutive activation on the CDR2 promoter and thus growth in the absence of histidine. In contrast, a nonfunctional TAC1 allele cannot confer the ability to grow in the absence of histidine even in the presence of a CDR2 inducer.
The functionality of this system was verified by the following experiments. CAI8, DSY2627, and DSY2657 were spotted onto MM supplemented with uridine and adenine in the presence or absence of histidine. All strains were able to grow in the presence of histidine. However, only CAI8 could grow as expected in the absence of this amino acid (Fig. 2). To verify that histidine auxotrophy could be complemented by treatment with CDR2-inducing agents in DSY2657 that carries the one-hybrid system, strains were also spotted onto MM containing either estradiol, fluphenazine, or terbinafine. As shown in Fig. 2, DSY2657 was able to grow in the absence of histidine but only in the presence of CDR2 promoter-inducing drugs, in contrast to DSY2627, which did not contain the reporter system. To verify the inducer specificity of the system, the strains were also spotted onto MM containing drugs unable to induce CDR2 such as diamide or benomyl. Neither DSY2657 nor DSY2627 was able to grow on these drug-supplemented media, thus confirming the specificity of the one-hybrid system (Fig. 2). The dosage of estradiol permitting growth of DSY2627 ranged between 10 and 20 μg/ml (see Fig. S1 in the supplemental material). These experiments demonstrated the functionality and specificity of the one-hybrid screening system based on CDR2 promoter activity controlling HIS3. The system was therefore used to probe cis and trans CDR2 promoter regulatory elements in C. albicans.
FIG. 2.

Functionality of the P(CDR2)-HIS3 reporter system. Serial dilutions of overnight cultures were plated onto MM supplemented with histidine or containing different drugs as indicated. Plates were incubated for 48 h at 34°C.
Identification of the minimal DRE sequence required for drug responsiveness of the CDR2 promoter.
The DRE was previously demonstrated to be crucial for the regulation of CDR1 and CDR2 in azole-susceptible strains treated with CDR-inducing drugs or in azole-resistant strains in which CDR genes are upregulated (8). Nevertheless, the minimal requirements of this sequence are still not precisely described, and it is difficult to determine putative DREs in the promoters of other genes. The DRE consensus derived from previous studies performed with the CDR1 and CDR2 promoters is a 20-bp sequence (5′-CGGAWATCGGATATTTTTTT-3′) (8). The DRE was therefore systematically mutated in the context of the P(CDR2)-HIS3 reporter system in order to identify the minimal sequence requirement of the CDR2 promoter. We used a minimal CDR2 promoter [−224 from start codon; P(CDR2mini)] as the starting promoter context. This minimal promoter contained the full DRE and was inducible by estradiol (see Fig. S1 in the supplemental material). Growth of reporter strains containing each DRE variant in histidine-free medium in the presence of CDR2 inducers was monitored (Fig. 3B). Two domains could be distinguished in the CDR2 DRE: (i) a 13-bp region (CGGAAATCGGATA) forming two hexamers (CGGAA/TA) separated by a central thymidine (the “core” domain) and (ii) a poly(T) sequence of nine thymidines downstream of the core domain in the CDR2 DRE but of seven thymidines in the CDR1 DRE. The two hexamers of the core could be themselves divided into two triplets, one being a CGG triplet and the other an AWA triplet. All these different DRE segments were transversed or deleted, and the resulting constructs were inserted into DSY2773, which contained a portion of the CDR2 promoter (−224 to −1).
FIG. 3.

Activity of the CDR2 promoter carrying a degenerate DRE. (A) Summary of reporter activities of all tested constructs. Growth of the strains carrying the P(CDR2)-HIS3 constructs was evaluated on MM in the absence or presence of 10 μg/ml estradiol. −, the reporter system is nonfunctional, meaning that the strain containing it is unable to grow in the presence of estradiol or shows constitutive growth even in the absence of estradiol; +, the reporter system is functional, meaning that the growth of the associated strain is identical to that of the positive control carrying wild-type DRE (DSY2673); +/−, reduced growth compared to the positive control. Underlined nucleotides indicate the localization of the DRE core or the putative DRE sequences. Letters in lowercase correspond to the CDR2 promoter sequence flanking the degenerate DRE or the putative DREs of CDR1 and CDR2 coregulated genes; letters in uppercase correspond to the putative DRE sequence; and letters in boldface correspond to transversed nucleotides. (B) Growth of reporter strains carrying degenerate DREs in the P(CDR2)-HIS3 reporter system. Serial dilutions of overnight cultures were plated onto MM supplemented with histidine (his) or with estradiol. (Top) Numbers 9 to 16 indicate the types of constructs (detailed in panel A) carrying degenerate DREs. (Bottom) P(CDR2)-HIS3 constructs contained DREs from other gene promoters as indicated in panel A. Plates were incubated for 48 h at 34°C.
A total of 24 strains containing individual P(CDR2)-HIS3 mutated constructs were spotted onto selective media (data not shown). In contrast to what was found for DSY2773, which carried the CDR2 DRE wild-type sequence, the majority of the modifications performed in the DRE abolished the activity of the promoter, as the strains were unable to grow on MM even in the presence of estradiol (Fig. 3A). Constructs 10 and 11, containing a DRE with a poly(T) stretch of six and seven thymidines, still allowed the induction of CDR2 by estradiol, but to a reduced extent compared to DSY2773 (Fig. 3). This result is consistent with the fact that the poly(T) of the CDR1 DRE contains only seven thymidines and is still fully functional (Fig. 3).
Since nucleotide changes in the DRE were performed within the 13-bp core of the DRE (5′-CGGAAATCGGATA-3′) (Fig. 3), we decided to replace each base of the core DRE with the three other possible bases. Results are presented in Fig. 4B and summarized in Fig. 4A. We observed that the DRE sequence cannot tolerate base substitutions, especially at the position of the first CGG triplet. We observed that the AATC bases from positions 5 to 8 and the A at position 11 with respect to the core sequence accept base substitutions since reporter strains carrying the P(CDR2)-HIS3 reporter constructs were still able to grow on selective medium in the presence of a CDR2 inducer (Fig. 4). Permissive mutations to T or G at position 5 and to C at position 7 were also found in the DRE of CDR1 and in the putative DRE of RTA3, which is a gene coregulated with CDR1 and CDR2. Unexpectedly, changes to C or T at position 4 and A or T at position 10 resulted in a constitutive activation of the P(CDR2)-HIS3 reporter system (Fig. 4).
FIG. 4.

Nucleotide replacements in the CDR2 DRE. (A) Possibility of base replacement in the DRE core sequence. For each position, the growth conferred in MM supplemented with estradiol (10 μg/ml) by the base(s) replacing the wild-type nucleotide is indicated. −, the reporter system is nonfunctional, meaning that the strain containing it is unable to grow in the presence of estradiol or shows constitutive growth even in the absence of estradiol; +, the reporter system is functional, meaning that the growth of the associated strain is identical to that of the positive control carrying wild-type DRE (DSY2673); +/−, reduced growth compared to the positive control. (B) Growth of reporter strains carrying DREs with systematic nucleotide replacement in the P(CDR2)-HIS3 reporter system. Serial dilutions of overnight cultures were plated onto MM supplemented with histidine (his) or with estradiol. Nucleotide positions in the DRE and replacements are indicated. Plates were incubated for 48 h at 34°C. Each strain spotted is representative of three transformants. WT, wild type.
Sequence analysis of the promoters of CDR1 and CDR2 coregulated genes identified the presence of a DRE-like sequence (5′-CGGWWWTCGGWWW-3′) with the possibility of one mismatch (Fig. 3A). In order to test the functionality of these DRE-like sequences, they were substituted into the CDR2 DRE in the one-hybrid system designed here. Attempts to use full-length promoters (for example, the IFU5 promoter) in our reporter system failed due to intrinsic TAC1-independent HIS3 reporter activity (see Fig. S2 in the supplemental material). Except for the CDR1 DRE, none of these putative regulatory sequences could restore growth in the absence of histidine in the context of the CDR2 promoter (Fig. 3). In conclusion, the DRE sequence of CDR2 accepts only a few modifications such as base substitution from positions 5 to 8 (AATC) and required a poly(T) stretch of at least four Ts at the 3′ end.
Determination of binding of Tac1p on the DRE by ChIP assays.
It was previously shown that the DNA-binding motif of Tac1p was able to bind in vitro to the DREs of the CDR1 and CDR2 promoters (6). We then asked if this binding could occur in vivo. For this purpose, we tagged a wild-type form of Tac1p in the C terminus with protein A. After verification that the chimeric protein was functional (data not shown), we performed ChIP experiments. We confirmed that Tac1p was able to bind to regions of 123 and 150 bp flanking the DREs of CDR1 and CDR2 (Fig. 5). The enrichments were 439-fold ± 44-fold and 173-fold ± 32-fold, respectively, compared to that for a control sequence located in the orf19.5970 promoter situated on chromosome 3 between the CDR1 and CDR2 loci. Surprisingly, even if the putative DREs of RTA3 and IFU5 were not functional in our one-hybrid reporter system, Tac1p could bind their promoters in a region covering the DRE-like sequence with enrichments of 162-fold ± 16-fold and 147-fold ± 31-fold compared to that for the control sequence, respectively (Fig. 5). This binding did not require treatments with fluphenazine or estradiol, and these drugs did not modify the enrichment of DRE sequences (data not shown). Moreover, parallel ChIP experiments performed with a TAC1 hyperactive allele fused to protein A gave results identical to those with a wild-type TAC1 allele (data not shown) and thus confirmed the constitutive binding of Tac1p to its targets independently of any activation.
FIG. 5.
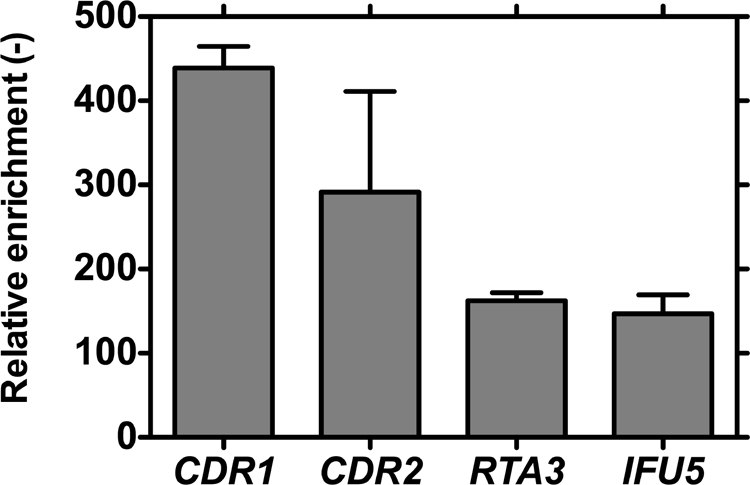
Tac1p binds to DRE-containing promoters. After cross-linking to the DNA, the Tac1p-protein A fusion was immunoprecipitated using anti-protein A antibody. Specific sequences flanking the DREs of CDR1 and CDR2 or putative DREs of RTA3 and IFU5 were amplified by real-time PCR on the precipitated DNA. A sequence located on chromosome 3 in the promoter of orf19.5970, which is situated between CDR1 and CDR2, was also amplified and used as an unspecific sequence. Results were expressed as increases in enrichment relative to that for unspecific sequence amplification and are representative of three independent experiments.
Screening of different TAC1 alleles by the one-hybrid P(CDR2)-HIS3 system.
The CDR2 one-hybrid system was designed to study elements regulating the activity of the CDR2 promoter either in cis or in trans. In this part of the study, we focused on the CDR2 promoter activity driven by different TAC1 alleles. For this purpose, the two TAC1 alleles were deleted in DSY2627, yielding DSY3352. The P(CDR2)-HIS3 reporter system carried by pAC3 was next introduced into the latter strain, giving ACY29, which was used as the recipient strain for different TAC1 alleles from azole-susceptible or azole-resistant clinical strains. We expected to be able to rapidly discriminate between wild-type, hyperactive, and nonfunctional TAC1 alleles, as represented in Fig. 1B. For example, a wild-type TAC1 allele should confer on ACY29 the ability to grow on a medium lacking histidine only in the presence of CDR2 inducers such as estradiol and fluphenazine. In contrast, TAC1 hyperactive alleles should confer on the strain the ability to grow in the absence of histidine or of CDR2 inducers. Finally, a strain lacking TAC1 or carrying a nonfunctional allele of TAC1 should not be able to grow in the absence of histidine even if a CDR2 inducer is added, since the activity of the CDR2 promoter was shown to be Tac1p dependent (6).
To verify the functionality of the system, TAC1-1 and TAC1-5, which are wild-type and hyperactive alleles, respectively, were introduced into ACY29, giving ACY36 and ACY31. ACY31 grew in the absence of histidine regardless of the presence of inducers. ACY36 grew in the absence of histidine only if an inducer was present (Fig. 6A, left). A control strain (ACY29) lacking TAC1 was unable to grow in the absence of histidine, even if an inducer was added. These experiments therefore validated the one-hybrid system for discriminating between wild-type, hyperactive, and absent or nonfunctional TAC1 alleles.
FIG. 6.

Verification of growth phenotypes obtained in the promoter-reporter strains receiving randomly mutated TAC1 alleles selected for their potential hyperactivity. (A) Each strain was plated onto MM in the presence or absence of histidine. A hyperactive TAC1 allele should confer growth in the absence of histidine. (B) Immunodetection of Cdr1p and Cdr2p in C. albicans strains carrying randomly mutated TAC1 alleles. C. albicans strains were grown in YEPD to mid-log phase and exposed (+) or not (−) to fluphenazine (10 μg/ml) for 20 min. ACY29, tac1Δ/tac1Δ reporter strain; CTRhyp, hyperactive control strain (ACY29 carrying TAC1-5 [hyperactive]); CTRwt, control wild-type strain (ACY29 carrying wild-type TAC1-1); CTRnf, nonfunctional control strain (ACY29 carrying an empty plasmid). For each tested strain, except for JCY3, the GOF mutation responsible for Tac1p hyperactivity is indicated.
Detection of TAC1 hyperactive and TAC1 wild-type alleles from a collection of clinical isolates.
We previously showed that GOF mutations such as single nucleotide substitutions or codon deletions led to the conversion of a TAC1 wild-type allele into a hyperactive allele. Up to now, seven distinct GOF mutations have been identified among 26 analyzed TAC1 alleles (4, 5); however, this number of GOF mutations is probably not exhaustive. In this part of the study, we addressed the occurrence of GOF mutations using the one-hybrid reported system designed here. We selected 29 clinical strains, grouped in 13 pairs, and one triplet of related strains from 14 different patients. Each group of isolates originating from the same patient contained at least one azole-susceptible and one azole-resistant strain (Table 4). The azole MICs for each strain were determined as well as their mating status (Table 4). Among the 29 strains, 18 were MTL a/α, 4 were MTL a/a, and 7 were MTL α/α. Since TAC1 is located close to the MTL, heterozygosity of the MTL often correlates with TAC1 heterozygosity (4, 5). We therefore, attempted to discriminate two distinct TAC1 alleles in these strains.
Among 12 MTL heterozygous strains out of a total of 18, two distinct TAC1 alleles could be discriminated. A summary of the results is presented in Table 4. We can observe that, except for DSY2015 and DSY2019, we were able to clone two distinct TAC1 alleles in strains heterozygous for the MTL and only one TAC1 allele in strains homozygous for the MTL. In isolate DSY2015 (a/α), only one TAC1 allele was cloned even after the analysis by RFLP and by sequencing at least 30 distinct plasmids. In contrast to DSY2019, which is homozygous for the MTL, two distinct TAC1 alleles were cloned.
In all the 29 remaining isolates we identified 47 distinct TAC1 alleles. The 47 plasmids containing distinct TAC1 alleles were introduced into the P(CDR2)-HIS3 reporter system. Among these 47 TAC1 alleles we could distinguish 19 wild-type and 28 hyperactive TAC1 alleles. Alignments of the obtained TAC1 sequences allowed us to deduce putative GOF mutations in the hyperactive TAC1 alleles. Out of 11 deduced distinct GOF mutations (G980E, G980W, N972D, N972S, R693K, R673Q, A736V, A736T, E461K, E841G, H741Y), 3 were already described (G980E, N972D, and A736V) (4, 24) and 8 were new GOF mutations (N972S, A736T, R693K, R673Q, G980W, E461K, E841G, and H741Y), including three occurring at already-described positions (N972S, A736T, and G980W) (4, 24). Introduction of six out of eight newly identified GOF mutations by site-directed mutagenesis converted the TAC1-1 wild-type allele into a hyperactive allele using the one-hybrid screening system (data not shown).
Detection of GOF and LOF mutations in TAC1 by random mutagenesis.
To increase the number of potential GOF mutations that can be obtained in TAC1, random mutagenesis was performed on a TAC1 wild-type allele. Importantly, random mutagenesis could also result in LOF alleles. A collection of TAC1 mutant alleles obtained in XL1-Red E. coli (approximately 2,200 plasmids) was screened using the one-hybrid reporter system in order to detect hyperactive or nonfunctional TAC1 alleles. From this library, approximately 3,000 C. albicans strains were generated in the ACY29 background for screening onto MM as shown in Fig. 1B.
From the 3,000 C. albicans transformants, pools of 70 strains were spotted onto a medium lacking histidine in order to select His+ strains potentially carrying the TAC1 hyperactive alleles. This first selection allowed the recovery of 42 clones on the His medium. The TAC1 alleles of 28 isolates out of 42 His+ colonies were successfully recovered and sequenced. Detected polymorphisms compared to the TAC1-1 initial allele are presented in Table 5 and Fig. 6A. In 19 out of the 28 recovered alleles, seven single nonsilent point mutations (N972D, N972I, I255stop [introduction of a stop codon in place of codon 255], W239L, I794V, A736V, and T225A) were detected and designated GOF mutations. Five other strains (JCY52, JCY54, JCY55, and JCY57) carry TAC1 alleles with several mutations, but in each strain one of them was already defined as a GOF mutation (A736V for JCY52 and JCY54, N977D for JCY55, T225A for JCY57, and E641K for JCY59). TAC1 alleles in JCY1 and JCY58 contained several mutations but shared one of them (A736T). We therefore considered that this mutation was responsible for the hyperactivity of TAC1 in these two strains. Finally, the TAC1 allele recovered from the last strain (JCY3) carried several TAC1 mutations, but none of them have already been described. We were thus unable to deduce those responsible for TAC1 hyperactivity. To verify that the phenotypes so far observed were due to the identified mutations, each rescued TAC1 hyperactive allele was reintroduced into the reporter strain ACY29. All strains except JCY3 were able to grow in the absence of histidine (Fig. 6A). This phenotype correlated with a high expression of CDR1 and CDR2 in all strains except the one carrying TAC1 from JCY3, as shown by Western blot analysis (Fig. 6B).
TABLE 5.
Mutations encoded by hyperactive TAC1 alleles after random mutagenesis of TAC1-1
| Strain | TAC1-encoded mutation(s)a |
|---|---|
| JCY1 | L131S, S199N, N396S, A736T, D776N, E829Q |
| JCY2 | NR |
| JCY3 | F14Y, K215E, L563S, I571V, Y720H |
| JCY4 | NR |
| JCY5 | NR |
| JCY6 | NR |
| JCY7 | NR |
| JCY8 | NR |
| JCY9 | N972D |
| JCY10 | N972D |
| JCY11 | N972D |
| JCY12 | NR |
| JCY13 | N972I |
| JCY14 | N972I |
| JCY15 | NR |
| JCY16 | N972I |
| JCY37 | NR |
| JCY38 | I255stop* |
| JCY39 | NR |
| JCY40 | I255stop* |
| JCY41 | I255stop* |
| JCY42 | NR |
| JCY43 | I255stop* |
| JCY44 | NR |
| JCY45 | T225A |
| JCY46 | T225A |
| JCY47 | T225A |
| JCY48 | NR |
| JCY49 | T225A |
| JCY50 | T225A |
| JCY51 | NR |
| JCY52 | S651P, L706S, A736V |
| JCY53 | A736V |
| JCY54 | S264L, W442R, Y674H, A736V |
| JCY55 | L17S, M170V, S199N, R206H, V207A, A377V, N396S, N772K, D776N, R869Q, N977D |
| JCY56 | As for JCY55 |
| JCY57 | T225A, K640R |
| JCY58 | E154R, G500D, V510A, A736T |
| JCY59 | N93Y, L237F, E461K, S518L, E681V |
| JCY60 | W239L |
| JCY61 | I794V |
| JCY62 | I794V |
These analyses allowed us to detect 10 GOF mutations located at eight distinct positions. Four of them (N972I, I255stop, W239L, and I794V) were new GOF mutations, including one new substitution (N972I) but at a position (N972) already described in clinical isolates with another substitution. Six of them (N972D, A736V, N977D, T225A, E461K, and A736T) have already been described in clinical isolates.
TAC1 random mutagenesis coupled with one-hybrid system analysis could also detect LOF mutations in TAC1. For this purpose, all of the 3,000 previously obtained C. albicans transformants of the library were replica plated onto complete medium and MM supplemented with estradiol. Nonfunctional alleles should grow on complete medium but not onto MM supplemented with estradiol. Forty-six strains had this phenotype. As previously performed for hyperactive alleles, the TAC1 alleles of 12 strains out of the 46 were successfully recovered and sequenced. Results are presented in Table 6. Four strains carried TAC1 alleles with single point mutations (K215stop, F368S, N871D, and L392F). They were considered LOF mutations. Two strains (JCY18 and JCY23) each carried two mutations but with one in common (K763stop). This mutation was probably responsible for the nonfunctionality of the allele. For the last four alleles recovered from JCY17, JCY22, JCY26, and JCY27, TAC1 alleles carried too many mutations to deduce those responsible for nonfunctionality. Surprisingly, two strains (JCY24 and JCY25) carried TAC1 alleles with no mutations either in the ORF or in the 500-bp promoter compared to TAC1-1. The phenotypes were verified as previously explained by reintroducing each rescued TAC1 allele into the reporter strain. Transformants were plated onto complete medium and onto MM in the presence or absence of estradiol. All the strains except JCY25 (no mutation) and JCY27 (T158A, N218S, L949S) (indication of growth for JCY25 and JCY27 was considered a false-positive result) were unable to grow in the absence of histidine and when estradiol was added, in contrast to ACY36, carrying TAC1-1 (Fig. 7 [CTRwt]). This phenotype correlated with the inability of strains, except for JCY25 and JCY27, to overexpress transiently CDR1 and CDR2 in the presence of fluphenazine as the inducer (Fig. 7B). This analysis therefore allowed us to determine five LOF mutations, among which two introduced early stop codons (TAA). The nonfunctionality of the TAC1 allele from the JCY24 strain cannot be attributed to any mutations either in the ORF or in a 500-bp promoter.
TABLE 6.
Mutations encoded by nonfunctional TAC1 alleles after random mutagenesis of TAC1-1
| Strain | TAC1-encoded mutation(s)a |
|---|---|
| JCY17 | L244S, D304V, L732F |
| JCY18 | S382G, K763stop* |
| JCY19 | K215stop* |
| JCY20 | F368S |
| JCY21 | N871D |
| JCY22 | E132G, Y395N |
| JCY23 | F641Y, K763stop* |
| JCY24 | — |
| JCY25 | — |
| JCY26 | N441D, N591D |
| JCY27 | T158A, N218S, L949S |
| JCY28 | L392F |
—, no mutation found in the TAC1 ORF and in the TAC1 1,000-bp promoter; *, mutation resulting from introduction of a stop codon corresponding to an Ochre mutation (TAA) into TAC1.
FIG. 7.

Verification of phenotypes obtained in the promoter-reporter strain receiving randomly mutated TAC1 alleles selected for their potential nonfunctionality. Each strain was plated onto MM in the presence or absence of histidine or with 10 μg/ml estradiol as a TAC1 inducer. Strains carrying nonfunctional TAC1 alleles should not grow in the presence of an inducer, in contrast to strains carrying wild-type alleles. ACY29, tac1Δ/tac1Δ reporter strain; CTRhyp, hyperactive control strain (ACY29 carrying TAC1-5 [hyperactive]); CTRwt control wild-type strain (ACY29 carrying wild-type TAC1-1); CTRnf, nonfunctional control strain (ACY29 carrying pDS178). For each tested strain, except for JCY17, JCY22, JCY26, and JCY27, which carried more than one TAC1 mutation, and JCY24 and JCY25, which carried no TAC1 mutations, the GOF mutation responsible for Tac1p hyperactivity is indicated. *, JCY18 carrying K763stop and S382G GOF mutations in TAC1; **, JCY23 carrying K763stop and F641Y GOF mutations in TAC.
Results reported in Fig. 8 showed that the GOF and LOF mutations in Tac1p identified so far were essentially located in the C-terminal part of the protein and in a region located between the DNA-binding domain and the MHR of the protein. Although these mutations do not converge to small protein domains, they probably delimit functional domains of the protein.
FIG. 8.

Map of Tac1p with GOF (black bars) and LOF (gray bars) mutations obtained by random mutagenesis and found in clinical isolates. The Mrr1p and CgPdr1p maps are based on data from Dunkel et al. (9) and from Ferrari et al. (10), respectively.
DISCUSSION
In this work, we developed a screening system allowing the analysis of cis- and trans-acting factors of a given gene by growing reporter strains on different media. This system is based on the properties of CDR2 and absence of expression of this gene in normal growth conditions. When the CDR2 promoter is coupled with a reporter gene, for example HIS3, the absence of CDR2 promoter activity can be observed as absence of growth on a selective MM. Any modification in the transcriptional activity of CDR2 can therefore be monitored by restoration of growth on the same MM. For example, since several drugs are known to induce CDR2, their presence in the MM agar restored growth of the P(CDR2)-HIS3 reporter strains. The P(CDR2)-HIS3 reporter system was used here to determine the minimal sequence requirement of the major CDR2 cis-acting regulatory element, the DRE, and next to screen a collection of TAC1 alleles encoding the major CDR2 trans-acting regulatory factor. The P(CDR2)-HIS3 reporter system allowed the identification of several mutations in Tac1p conferring GOF that is associated with increased CDR1 and CDR2 expression or resulting in LOF. The P(CDR2)-HIS3 one-hybrid system is convenient since it does not necessitate further processing as with a lacZ-dependent system (19, 21) or any special fluorimetric equipment, as for green fluorescent protein reporter systems (13). However, the P(CDR2)-HIS3 system still remains qualitative. However, semiquantitative results may be obtained for Saccharomyces cerevisiae by dose-dependent inhibition of HIS3 activity by 3-aminotriazole (3-AT), which can be added to MM. We attempted to inhibit HIS3 activity with 3-AT. When the hyperactive allele TAC1-5 was expressed, growth could be gradually abolished at 3-AT concentrations from 1 to 10 mM (see Fig. S3 in the supplemental material). On the other hand, a concentration of 3-AT as low as 0.1 mM was sufficient to turn off estradiol induction in the presence of a TAC1 wild-type allele (see Fig. S3 in the supplemental material). In some cases, it was difficult to visualize estradiol inducibility even if 3-AT was added. This was mainly due to high intrinsic reporter activity, as observed for the P(IFU5)-HIS3 fusion (see Fig. S2 in the supplemental material).
Dissection of the CDR2 DRE indicated that only a few nucleotides can be replaced in the DRE deduced from the CDR1 and CDR2 promoters without loss of promoter activity. This result is consistent with our previous reports showing that transversion in the CGG triplets of the DRE sequence abolished its binding to Tac1p (6). Results of this study show that the 4-bp spacing between the two CGG triplets in the DRE is crucial for its function, since deletion in the central part of the DRE (T in position 7) abolished CDR2 promoter activity. This result is consistent with the ChIP analysis performed on Tac1p promoter targets. Liu et al. (17) determined that the consensus Tac1p DNA binding sequence is CGG(N4)CGG, and this suggests that the spacing between the two CGG triplets could be 4 nucleotides. This region, although restrictive for its length, appeared to be in our analysis the most permissive region in the DRE for nucleotide substitutions (Fig. 4). Moreover, we note that replacement of the first A of this region resulted in constitutive activation of the system. We might claim that we introduced a novel trans-activator binding site, leading to constitutive recruitment of the transcriptional machinery. Nevertheless the constitutive CDR2 promoter activity that we observed with these constructs was always TAC1 dependent, since the reporter activities obtained by hyperactive constructs were absent in a strain lacking TAC1 (data not shown).
Liu et al. (17) did not include in their proposed Tac1p binding site a group of 7 to 10 nucleotides downstream of the second CGG triplet of the DRE. In our analysis, this sequence group in the CDR2 DRE could not be replaced or shortened by more than 4 nucleotides without eliminating CDR2 promoter activity. Consistent with our observations is that other CDR1 and CDR2 coregulated genes such as RTA3, IFU5, and PDR16 have putative DREs in their promoters and each DRE has a poly(T/A) sequence of at least 7 nucleotides (Fig. 3). These observations lead us to distinguish the DRE consensus sequence 5′-CGG(N4)CGGWWWWWWW-3′, necessary for transcriptional machinery recruitment, from a Tac1p-binding domain. This DRE consensus overlaps, but is still different from, that of the Tac1p-binding domain described by Liu et al. (17). This highlights that the promoter environment may be important for the function of the DRE present in TAC1-dependent genes.
Replacements of the complete CDR2 DRE by putative DREs of coregulated genes failed to complement CDR2 DRE activity. Unless detection levels for the reporter activities of the chimeric constructs were too low to be detected in the phenotypic plate assay, these results suggest that the promoter environment may play a role in the recognition of the DRE by trans-acting factors (Fig. 3). The putative DREs of PDR16 and TAC1 analyzed in this study (Fig. 3) do not fit with the proposed alternative consensus DRE sequence, and this could explain why they cannot replace the CDR2 DRE. Electrophoretic mobility shift assays with these putative DREs or with deletions of these DREs in the native promoters could help to better define their role in gene regulation and especially their ability to bind Tac1p. It is also possible that Tac1p may recognize other regulatory sequences in distinct promoter contexts due to formation of heterodimers with other transcription factors, as demonstrated for Pdr1p ad Pdr3p in S. cerevisiae (18). Moreover, Pdr1p and Pdr3p necessitate several binding regions for normal target gene expression (15). These effects could be operating in the promoters of RTA3, IFU5, and PDR16.
In this study, a collection of TAC1 alleles isolated from clinical samples was screened in our reporter system in order to determine their transcriptional properties. From the 29 strains investigated, 47 alleles were distinguished by RFLP and sequencing. Among them, 28 were assigned as hyperactive while the remaining 19 were categorized as wild type. Alignment of these 47 new sequences with already described TAC1 alleles (4, 5, 24) allowed deduction of the GOF mutations responsible for their hyperactivity. Eleven GOF mutations were thus identified, and among them 3 have already been described (4, 5, 24).
Up to now, among the 39 TAC1 hyperactive alleles out of a total of 75 available alleles, only 19 GOF mutations could be identified at 15 positions. Moreover, some positions were more frequently encountered than others among the known GOF substitutions. For example, substitutions at position 736 or 980 were found in four unrelated azole-resistant strains isolated from distinct patients and having distinct multilocus sequence typing profiles (data not shown) containing six and four distinct hyperactive alleles, respectively. However, substitutions at position 972 (N972D, N972I, and N972S) were the most frequent, since they occurred in six unrelated azole-resistant strains, corresponding to 11 distinct hyperactive TAC1 alleles. In addition, mutations at positions 736 and 972, obtained by random mutagenesis of a TAC1 wild-type allele, were also frequently found since they were present in 28 TAC1 mutated rescued alleles. Six strains carried mutations at position 972 (21%) and five at position 736 (18%) (Table 5).
Since a limited number of possible GOF positions were detected, our results suggest that some positions may be more strongly selected than others. It is possible that such mutations are responsible for strong TAC1 activity. On the other hand, the selection of TAC1 hyperactive alleles by P(CDR2)-HIS3 promoter activities could favor the phenotypic selection of strong TAC1 hyperactive mutants on MM, since the HIS3 gene is strongly expressed in these strains. This hypothesis has still to be demonstrated by quantification of the CDR2 promoter activities with a quantitative reporter system or quantitative RT-PCR.
The results obtained with TAC1 random mutagenesis did not increase efficiently the collection of TAC1 GOF mutations from clinical isolates since only three additional GOF mutations (W239L, I255stop, and I794V) were recovered at new positions, including those in a truncated protein. This may indicate either that the number of possible GOF mutations in TAC1 is limited or that the number of selected mutants was not sufficient to saturate the mutagenesis. In general, it is recommended that at least three plasmids per nucleotide of the target sequence be obtained in order to assemble a library with every single nucleotide mutated. Both hypotheses could be true since, first, the 2,200 different plasmids recovered from mutagenic E. coli barely covered TAC1 (2,943 bp) and, second, only 11 GOF mutations were found at eight distinct positions in 28 TAC1 alleles of clinical strains.
In contrast, the analysis of 122 Candida glabrata strains allowed discrimination of 70 distinct CgPDR1 alleles, including 12 wild-type and 58 hyperactive alleles (10). Among these, 58 hyperactive CgPDR1 alleles carried a distinct GOF mutation (10) (Fig. 8). For MRR1 from C. albicans, 14 azole-resistant strains have been analyzed up to now, delivering 19 wild-type MRR1 alleles and 15 distinct hyperactive alleles (9). Almost all hyperactive alleles contained a different GOF mutation, except for two alleles, each encoding the P683H mutation (9). This situation is different from that for TAC1, in which 39 hyperactive alleles carried only 19 distinct GOF mutations.
In Tac1p, the seven GOF mutations located at four positions in the C-terminal extremity of the protein between position 962 and the C-terminal end (Fig. 8) may delimit the minimal transcriptional activation domain of the protein. A GOF mutation located between positions 673 and 841 in the middle of the protein may be situated in an unknown regulatory region. Finally, three GOF and three LOF mutations were located between the DNA binding domain and the MHR, which may correspond to an inhibitory domain as described for Pdr1p in S. cerevisiae (16). Finally only one GOF mutation was found in the MHR at position 461. With the exception of this position, no GOF mutation has been found up to now in the MHR. Mrr1p GOF mutations are grouped in two distinct regions, which is similar to our observation on Tac1p. One region was located between the DNA binding domain and the MHR, and a large one was located in the middle of the protein. No mutations have been found up to now in the C-terminal extremity of the protein. In the case of CgPDR1, the GOF mutations were located in three main regions including the putative inhibitory domain, the MHR, and the C-terminal transcriptional activation domain. Tac1p chimeric proteins could be constructed and tested in the P(CDR2)-HIS3 one-hybrid system to test their functionality and thus determine the role of each Tac1p domain. Other Tac1p variants, especially those lacking the DNA binding domain, could also be used in the lexA one-hybrid system, in which only transcriptional activation domains can be tested. Recently, a study performed using S. cerevisiae suggested that Pdr1p is able to directly bind azoles and thus acts as a nuclear receptor (23). It could be relevant to test this hypothesis in Tac1p using estradiol or fluphenazine and to determine the ligand domains of the protein.
Finally our P(CDR2)-HIS3 promoter reporter system could be adapted to analyze DRE-like regions located in promoters of CDR genes of other species such as Candida dubliniensis and Candida tropicalis and to screen their TAC1 alleles in a species-adapted promoter reporter system. More generally, such a system can be useful to rapidly investigate regulators of other genes by a simple phenotypic assay on selective media.
Supplementary Material
Acknowledgments
This work was supported by a grant of the Swiss National Research Foundation, no. 3200B0-100747/1, and partly by an EC grant to the EURESFFUN consortium (contract LSHM-CT-2005-518199).
We thank Sélène Ferrari for her technical advice on ChIP assays and her advice on the manuscript and Françoise Ischer for her technical support.
Footnotes
Published ahead of print on 26 June 2009.
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1.Akins, R. A. 2005. An update on antifungal targets and mechanisms of resistance in Candida albicans. Med. Mycol. 43285-318. [DOI] [PubMed] [Google Scholar]
- 2.Barker, K. S., and P. D. Rogers. 2006. Recent insights into the mechanisms of antifungal resistance. Curr. Infect. Dis. Rep. 8449-456. [DOI] [PubMed] [Google Scholar]
- 3.Blackwell, C., C. L. Russell, S. Argimon, A. J. Brown, and J. D. Brown. 2003. Protein A-tagging for purification of native macromolecular complexes from Candida albicans. Yeast 201235-1241. [DOI] [PubMed] [Google Scholar]
- 4.Coste, A., A. Selmecki, A. Forche, D. Diogo, M. E. Bougnoux, C. d'Enfert, J. Berman, and D. Sanglard. 2007. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot. Cell 61889-1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coste, A., V. Turner, F. Ischer, J. Morschhauser, A. Forche, A. Selmecki, J. Berman, J. Bille, and D. Sanglard. 2006. A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics 1722139-2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coste, A. T., M. Karababa, F. Ischer, J. Bille, and D. Sanglard. 2004. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot. Cell 31639-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Deken, X., and M. Raymond. 2004. Constitutive activation of the PDR16 promoter in a Candida albicans azole-resistant clinical isolate overexpressing CDR1 and CDR2. Antimicrob. Agents Chemother. 482700-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Micheli, M., J. Bille, C. Schueller, and D. Sanglard. 2002. A common drug-responsive element mediates the upregulation of the Candida albicans ABC transporters CDR1 and CDR2, two genes involved in antifungal drug resistance. Mol. Microbiol. 431197-1214. [DOI] [PubMed] [Google Scholar]
- 9.Dunkel, N., J. Blass, P. Rogers, and J. Morschhäuser. 2008. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol. Microbiol. 4827-840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrari, S., F. Ischer, D. Calabrese, B. Posteraro, M. Sanguinetti, G. Fadda, B. Rohde, C. Bauser, O. Bader, and D. Sanglard. 2009. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 1e1000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fonzi, W. A., and M. Y. Irwin. 1993. Isogenic strain construction and gene mapping in Candida albicans. Genetics 134717-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaur, N. A., N. Puri, N. Karnani, G. Mukhopadhyay, S. K. Goswami, and R. Prasad. 2004. Identification of a negative regulatory element which regulates basal transcription of a multidrug resistance gene CDR1 of Candida albicans. FEMS Yeast Res. 4389-399. [DOI] [PubMed] [Google Scholar]
- 13.Hiller, D., S. Stahl, and J. Morschhäuser. 2006. Multiple cis-acting sequences mediate upregulation of the MDR1 efflux pump in a fluconazole-resistant clinical Candida albicans isolate. Antimicrob. Agents Chemother. 502300-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karnani, N., N. A. Gaur, S. Jha, N. Puri, S. Krishnamurthy, S. K. Goswami, G. Mukhopadhyay, and R. Prasad. 2004. SRE1 and SRE2 are two specific steroid-responsive modules of Candida drug resistance gene 1 (CDR1) promoter. Yeast 21219-239. [DOI] [PubMed] [Google Scholar]
- 15.Katzmann, D. J., T. C. Hallstrom, Y. Mahe, and W. S. Moye-Rowley. 1996. Multiple Pdr1p/Pdr3p binding sites are essential for normal expression of the ATP binding cassette transporter protein-encoding gene PDR5. J. Biol. Chem. 27123049-23054. [DOI] [PubMed] [Google Scholar]
- 16.Kolaczkowska, A., M. Kolaczkowski, A. Delahodde, and A. Goffeau. 2002. Functional dissection of Pdr1p, a regulator of multidrug resistance in Saccharomyces cerevisiae. Mol. Genet. Genomics 26796-106. [DOI] [PubMed] [Google Scholar]
- 17.Liu, T., S. Znaidi, K. Barker, L. Xu, R. Homayouni, S. Saidane, J. Morschhäuser, A. Nantel, M. Raymond, and P. Rogers. 2007. Genome-wide expression and location analyses of the Candida albicans Tac1p regulon. Eukaryot. Cell 62122-2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mamnun, Y. M., R. Pandjaitan, Y. Mahe, A. Delahodde, and K. Kuchler. 2002. The yeast zinc finger regulators Pdr1p and Pdr3p control pleiotropic drug resistance (PDR) as homo- and heterodimers in vivo. Mol. Microbiol. 461429-1440. [DOI] [PubMed] [Google Scholar]
- 19.Martchenko, M., A. Levitin, and M. Whiteway. 2007. Transcriptional activation domains of the Candida albicans Gcn4p and Gal4p homologs. Eukaryot. Cell 6291-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puri, N., S. Krishnamurthy, S. Habib, S. E. Hasnain, S. K. Goswami, and R. Prasad. 1999. CDR1, a multidrug resistance gene from Candida albicans, contains multiple regulatory domains in its promoter and the distal AP-1 element mediates its induction by miconazole. FEMS Microbiol. Lett. 180213-219. [DOI] [PubMed] [Google Scholar]
- 21.Russell, C. L., and A. J. Brown. 2005. Expression of one-hybrid fusions with Staphylococcus aureus lexA in Candida albicans confirms that Nrg1 is a transcriptional repressor and that Gcn4 is a transcriptional activator. Fungal Genet. Biol. 42676-683. [DOI] [PubMed] [Google Scholar]
- 22.Sanglard, D., and F. C. Odds. 2002. Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect. Dis. 273-85. [DOI] [PubMed] [Google Scholar]
- 23.Thakur, J., H. Arthanari, F. Yang, S. Pan, X. Fan, J. Breger, D. Frueh, K. Gulshan, D. Li, E. Mylonakis, K. Struhl, W. Moye-Rowley, B. Cormack, G. Wagner, and A. Näär. 2008. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 452604-609. [DOI] [PubMed] [Google Scholar]
- 24.Znaidi, S., X. De Deken, S. Weber, T. Rigby, A. Nantel, and M. Raymond. 2007. The zinc cluster transcription factor Tac1p regulates PDR16 expression in Candida albicans. Mol. Microbiol. 2440-452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.