

Abstract
Bacterial cytokinesis is coupled to the localized synthesis of new peptidoglycan (PG) at the division site. This newly generated septal PG is initially shared by the daughter cells. In Escherichia coli and other gram-negative bacteria, it is split shortly after it is made to promote daughter cell separation and allow outer membrane constriction to closely follow that of the inner membrane. We have discovered that the LytM (lysostaphin)-domain containing factors of E. coli (EnvC, NlpD, YgeR, and YebA) are absolutely required for septal PG splitting and daughter cell separation. Mutants lacking all LytM factors form long cell chains with septa containing a layer of unsplit PG. Consistent with these factors playing a direct role in septal PG splitting, both EnvC-mCherry and NlpD-mCherry fusions were found to be specifically recruited to the division site. We also uncovered a role for the LytM-domain factors in the process of β-lactam-induced cell lysis. Compared to wild-type cells, mutants lacking LytM-domain factors were delayed in the onset of cell lysis after treatment with ampicillin. Moreover, rather than lysing from midcell lesions like wild-type cells, LytM− cells appeared to lyse through a gradual loss of cell shape and integrity. Overall, the phenotypes of mutants lacking LytM-domain factors bear a striking resemblance to those of mutants defective for the N-acetylmuramyl-l-alanine amidases: AmiA, AmiB, and AmiC. E. coli thus appears to rely on two distinct sets of putative PG hydrolases to promote proper cell division.
Cytokinesis in Escherichia coli and other gram-negative bacteria proceeds via the coordinated constriction of their envelope layers (outer membrane, inner membrane, and peptidoglycan [PG]) (12, 13, 34, 89). This coordination is achieved by a multi-protein division machine referred to as the septal ring or divisome (20). Assembly of the septal ring begins with the polymerization of the bacterial tubulin protein, FtsZ, into a ring structure just underneath the inner membrane at the prospective site of cell division (8). Once formed, this so-called Z-ring facilitates the recruitment of a number of essential and nonessential division proteins to the division site for the assembly of the trans-envelope divisome organelle (20).
A major function of the cytokinetic machinery is to promote the synthesis of the PG layer that will eventually fortify the new poles of the developing daughter cells. PG is a polysaccharide polymer composed of repeating units of N-acetyl-glucosamine (GlcNAc) and N-acetyl-muramic acid (MurNAc) linked by a β-1,4-glycosidic bond (46). Attached to the MurNAc sugar is a short peptide that is used to form cross-links between adjacent polysaccharide strands (46). Such cross-links allow for the construction of a cell-shaped PG meshwork that surrounds the cell membrane and protects it from osmotic rupture.
A new wave of zonal PG synthesis is initiated at the division site during each cell cycle (23, 25, 72, 77, 91). Several of the major PG synthases called penicillin-binding proteins are components of the divisome organelle and play important roles in the synthesis of PG during division (7, 21, 62, 67, 73, 74, 80, 81, 88, 90). The septal PG layer produced by these and perhaps other components of the divisome is thought to be initially shared by the daughter cells (46). In gram-positive bacteria, this septal PG layer is typically split some time after the daughter cells have been compartmentalized by membrane fusion (11). In gram-negative bacteria, however, the septal PG layer is split shortly after it is formed to allow constriction of the outer membrane to closely follow that of the inner (cytoplasmic) membrane (12, 13, 34, 89). This gives rise to the characteristic constricted appearance of predivisional cells of E. coli and its relatives.
PG hydrolysis is required to promote septal PG splitting and eventual daughter cell separation (87). E. coli, like many bacteria, encodes a vast array of factors with known or predicted PG hydrolase activity (at least 30 genes and 11 different protein families) (29, 31, 87). In most cases, the loss of individual PG hydrolase factors has little effect on growth and division, suggesting that there is significant functional overlap between the various hydrolases (87). This dearth of phenotypic information has consequently made it difficult to understand the physiological roles of PG hydrolases and identify the subset of these factors needed for septal PG splitting. An approach that has helped overcome this limitation in E. coli, however, has been the systematic deletion of all members of a particular PG hydrolase family from the genome (22, 44, 45, 63). Thus far, of all the families of PG hydrolases encoded by E. coli, the factors that play the predominant role in cell separation appear to be the LytC-type N-acetylmuramyl-l-alanine amidases: AmiA, AmiB, and AmiC (44, 45, 69). Loss of all three of these amidases results in a severe defect in cell separation and the formation of extremely long cell chains. This chaining phenotype can be exacerbated by the loss of members of other classes of PG hydrolases like the lytic transglycosylases or d,d-endopeptidases (44, 68). However, relative to strains defective for the amidases, mutants lacking multiple lytic transglycosylases or d,d-endopeptidases alone do not display significant chaining phenotypes in E. coli. These PG hydrolases therefore appear to be playing more of an ancillary role in cell separation.
The LytM (lysostaphin/peptidase M23)-domain containing factors (referred to as LytM factors for convenience) are a widely distributed class of putative PG hydrolases that have been poorly characterized with regard to their role in PG biogenesis in E. coli and other bacteria (31). The most well-studied members of this family of factors, LytM and lysostaphin, are metallo-endopeptidases that cleave the pentaglycine cross-bridges found in staphylococcal PG (9, 30, 64). Based on this activity, other LytM factors are also likely to be PG hydrolases but with altered cleavage specificity because pentaglycine cross-bridges are only found among the staphylococci (75). Indeed, the LytM protein, gp13, from the Bacillus subtilis phage Φ29 was recently shown to be a d,d-endopeptidase that cleaves the meso-diaminopimelic acid-d-Ala cross-links of B. subtilis PG (17).
E. coli encodes four factors with identifiable LytM-domains: EnvC, NlpD, YgeR, and YebA (29) (Fig. 1). Of the four, only EnvC has been studied in appreciable detail. EnvC− mutants have a mild cell separation (chaining) defect when grown in medium containing salt and a severe division defect when grown at high temperatures in medium lacking salt (5, 42, 48, 71). In addition, purified EnvC protein was found to possess PG hydrolase activity using a gel-based zymogram assay, and an EnvC-green fluorescent protein (GFP) fusion exported to the periplasm via the Tat system was shown to be recruited to the division site (5). In all, these results support a model in which EnvC is targeted to the division site to participate directly in septal PG splitting and daughter cell separation.
FIG. 1.

Predicted domain structure of the E. coli LytM factors. Shown is a diagram depicting the predicted domain architecture of the four E. coli factors with identifiable LytM domains. Abbreviations: LytM, LytM domain; LysM, LysM PG-binding domain (29); CC, coiled coil; T, transmembrane domain; SS, signal sequence; SS*, lipoprotein signal sequence. The UniProtKB/Swiss-Prot accession numbers are as follows: EnvC (P37690), NlpD (P0ADA3), YebA (P0AFS9), and YgeR (Q46798).
In the present study, we investigated the physiological role(s) of the entire set of E. coli LytM factors by generating mutant strains lacking all possible combinations of them. We found that, like the amidases, LytM factors play a critical role in daughter cell separation. Furthermore, studies of their subcellular localization revealed that NlpD is recruited to the division site along with EnvC, indicating that both of these LytM factors are likely to be participating directly in the septal PG splitting process. We also discovered that mutants lacking multiple LytM factors lyse more slowly and display an altered morphological response relative to wild-type (WT) cells when they are treated with ampicillin. This finding suggests that in addition to cell separation, LytM proteins play a role in the lytic mechanism of β-lactam antibiotics.
MATERIALS AND METHODS
Media and bacterial strains.
Cells were grown in LB (1% tryptone, 0.5% yeast extract, 0.5% NaCl) or minimal M9 medium (61) supplemented with 0.2% Casamino Acids and 0.2% sugar (glucose, maltose, or arabinose as indicated). Unless otherwise indicated, antibiotics were used at 10 (chloramphenicol), 15 (ampicillin), or 20 (kanamycin [Kan]) μg/ml.
The bacterial strains used in the present study are listed in Table 1. Mutants lacking multiple LytM factors or multiple amidases were constructed by successive rounds of P1 transduction (61) and the curing of the Kan-resistance (Kanr) cassettes flanked by frt sites using plasmid pCP20 as described previously (18) (see Table 1 for construction details). The desired chromosomal modification was confirmed by using diagnostic PCR every time a Kanr cassette was transduced or cured. When a strain contained multiple lesions, diagnostic PCR was used to confirm the status of each lesion. No difference in phenotype was observed when strains containing the Kanr cassette were compared to the corresponding strains in which the cassette was removed by using FLP recombinase.
TABLE 1.
Bacterial strains used in this study
| Strain | Genotypea | Source, reference, or constructionb |
|---|---|---|
| BW25113 | Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) rph-1 Δ(rhaD-rhaB)568 hsdR514 | 1 |
| JW5646 | BW25113 ΔenvC725::Kanr | 1 |
| JW2712 | BW25113 ΔnlpD747::Kanr | 1 |
| JW2833 | BW25113 ΔygeR787::Kanr | 1 |
| JW2428 | BW25113 ΔamiA764::Kanr | 1 |
| JW5449 | BW25113 ΔamiC742::Kanr | 1 |
| TB10 | rph1 ilvG rfb-50 λΔcro-bio nad::Tn10 | 51 |
| TD8 | TB10 ΔyebA::Kanr | This study |
| HSC078 | araD139 Δ(argF-lac)U169 rpsL150 relA1 flbB5301 deoC1 pstF25 rbsR ara+ ΔamiB::Kanr | Dan Kahne |
| TB28 | rph1 ilvG rfb-50 ΔlacIZYA::frt | 5 |
| TB134 | TB28 ΔenvC725::Kanr | P1(JW5646) × TB28 |
| TB140 | TB28 ΔenvC725::frt | TB134/pCP20 |
| TB139 | TB28 ΔnlpD747::Kanr | P1(JW2712) × TB28 |
| TB145 | TB28 ΔnlpD747::frt | TB139/pCP20 |
| TB156 | TB28 ΔenvC725::frt ΔnlpD747::Kanr | P1(JW2712) × TB140 |
| TB135 | TB28 ΔamiA764::Kanr | P1(JW2428) × TB28 |
| TB141 | TB28 ΔamiA764::frt | TB135/pCP20 |
| TB137 | TB28 ΔamiC742::Kanr | P1(JW5449) × TB28 |
| TB147 | TB28 ΔamiA764::frt ΔamiC742::Kanr | P1(JW5449) × TB141 |
| TB150 | TB28 ΔamiA764::frt ΔamiC742::frt | TB147/pCP20 |
| TB171 | TB28 ΔamiA764::frt ΔamiC742::frt ΔamiB::Kanr | P1(HSC078) × TB150 |
| TD4 | TB28 ΔygeR787::Kanr | P1(JW2833) × TB28 |
| TD5 | TB28 ΔygeR787::frt | TD4/pCP20 |
| TD9 | TB28 ΔyebA::Kanr | P1(TD8) × TB28 |
| TD10 | TB28 ΔyebA::frt | TD9/pCP20 |
| TD6 | TB28 ΔnlpD747::frt ΔygeR787::Kanr | P1(JW2833) × TB145 |
| TD7 | TB28 ΔnlpD747::frt ΔygeR787::frt | TD6/pCP20 |
| TD11 | TB28 ΔnlpD747::frt ΔyebA::Kanr | P1(TD8) × TB145 |
| TD12 | TB28 ΔnlpD747::frt ΔyebA::frt | TD11/pCP20 |
| TD17 | TB28 ΔygeR787::frt ΔenvC725::Kanr | P1(JW5646) × TD5 |
| TD18 | TB28 ΔyebA::frt ΔenvC725::Kanr | P1(JW5646) × TD10 |
| TD13 | TB28 ΔygeR787::frt ΔyebA::Kanr | P1(TD8) × TD5 |
| TD14 | TB28 ΔygeR787::frt ΔyebA::frt | TD13/pCP20 |
| TD15 | TB28 ΔnlpD747::frt ΔygeR787::frt ΔyebA::Kanr | P1(TD8) × TD7 |
| TD16 | TB28 ΔnlpD747::frt ΔygeR787::frt ΔyebA::frt | TD15/pCP20 |
| TD19 | TB28 ΔnlpD747::frt ΔyebA::frt ΔenvC725::Kanr | P1(JW5646) × TD12 |
| TD20 | TB28 ΔnlpD747::frt ΔygeR787::frt ΔenvC725::Kanr | P1(JW5646) × TD7 |
| TD21 | TB28 ΔygeR787::frt ΔyebA::frt ΔenvC725::Kanr | P1(JW5646) × TD14 |
| TD22 | TB28 ΔnlpD747::frt ΔygeR787::frt ΔyebA::frt ΔenvC725::Kanr | P1(JW5646) × TD16 |
The Kanr cassette is flanked by frt sites for removal by FLP recombinase. An frt scar remains after removal of the cassette using FLP expressed from pCP20.
Strain constructions by P1 transduction are described using the shorthand: P1(donor) × recipient. In all cases, transductants were selected on LB Kan plates. Strains resulting from the removal of a Kanr cassette using pCP20 are indicated as: parental strain/pCP20.
All deletion alleles were constructed to be in-frame deletions. All except those of amiB and yebA were constructed as part of the Keio collection (1). The ΔamiB::Kanr allele was a generous gift of the Kahne lab. It was constructed as described previously (18) by using the primers 5′-TATCGCATCAGAAATTGGTTGGTAGCGACGCTGCTGCTGCTGGTGTAGGCTGGAGCTGCTT C-3′ and 5′-TTAGTTTGGCAGCGTGCGATCTGGCGTCGTCACCGTACTGGCCATATGAATATCCTCCTTAGTTCC-3′ and the Kanr cassette from pKD4 (18). The ΔyebA::Kanr deletion was constructed to be identical to the corresponding deletion in the Keio collection. The Kanr cassette was amplified from pKD13 by using the primers 5′-GAGCTGCCTGAAAGGAGATTAATGAGGAAGTGATTACGTGATTCCGGGGATCCGTCGACC-3′ and 5′-GGCTGCGAATGGATGTTAATTAATCAAACCGTAGCTGCGGTGTAGGCTGGAGCTGCTTCG-3′. The resulting product was electroporated into strain TB10 to generate the chromosomal deletion as described previously (51).
Plasmids.
Plasmids used in the present study are listed in Table 2. Vectors with R6K origins are all derivatives of the CRIM plasmids developed by Haldimann and Wanner (41). The vectors were either maintained in the cloning strain DH5α(λpir), where they replicate as plasmids, or they were integrated into phage attachment sites (HK022 or λ) by using the helper vectors pTB102 or pInt-ts, respectively, as described previously (41). Single-copy integrants were identified by using diagnostic PCR (41). Integrated vectors were transferred between strains by P1-mediated transduction. Plasmids pCP20 (18), pCAH63 (41), pKD3 (18), pKD13 (18), pBAD33 (40), pMLB1113 (19), pTB26 (5), pTB102 (6), pTB183 (3), pTB222 (3), and pFB250 (3) were described previously. The construction of plasmids used in the present study is described below. In all cases, PCR was performed using KOD polymerase (Novagen) according to the manufacturer's instructions. Restriction sites for use in plasmid constructions are underlined in the primer sequences given below. Plasmid DNA or PCR fragments were purified by using a Qiaprep spin miniprep kit (Qiagen) or a Qiaquick PCR purification kit (Qiagen), respectively. Site-directed mutagenesis was performed by using the QuikChange method (Stratagene).
TABLE 2.
Plasmids used in this study
| Plasmid | Genotype | Origin | Source or reference |
|---|---|---|---|
| pCP20 | bla cat cI857 repA(Ts) PR::flp | pSC101 | 18 |
| pTB102 | cat cI857 repA(Ts) PR::intHK022 | pSC101 | 6 |
| pInt-ts | bla cI857 repA(Ts) PR::intλ | pSC101 | 41 |
| pTB314 | attHK022 bla lacIq Plac::nlpD-mCherry | R6K | This study |
| pTB316 | attHK022 bla lacIq Plac::envC-mCherry | R6K | This study |
| pTD38 | attHK022 bla lacIq Plac::ygeR-mCherry | R6K | This study |
| pTD39 | attHK022 bla lacIq Plac::yebA-mCherry | R6K | This study |
| pTD25 | attλ cat Para::envC | R6K | This study |
The mCherry fusion constructs used in the present study are all under the control of the WT lac promoter (Plac) and a linked copy of lacIq. They encode the full-length reading frames of envC, nlpD, yebA, and ygeR or the signal sequence of dsbA (dsbAss) fused to mCherry. The constructs all produce a C-terminal fusion protein with the amino acid linker LEGPAGL between the LytM factor or signal sequence and mCherry (see Table 2). The construction of these and other plasmids created for the present study are described in detail below.
pTD25 [attλ cat Para::envC] was constructed in a number of steps starting with the parent vector pTB183 [attHK022 bla Plac::gfp-zapA] (3). The backbone of pTB183, including bla, the R6K ori, and attHK022, was amplified by using the primers 5′-GTCAAGATCTCCGCGGTTGAAGACGAAAGGGCCTCG-3′ and 5′-ATGAGTCGACAAGCTTATCGATCTCAC-3′. The lacIq region of pMLB1113 (19) was amplified by using the primers 5′-GTCAGTCGACCCGGATCCGGTCTAGAGGCTCGAGGGGAATTCCACCATCGAATGGTGCAAAACCTTTCGCG-3′ and 5′-GTCAAGATCTGCCGGAAGCATAAAGTGTAAAGCC-3′. The two PCR fragments were each digested with BglII and SalI and ligated together to generate plasmid pTD16. The arabinose promoter region of pBAD33 (40) was amplified by using the primers 5′-GTCAAGATCTGAATTCCGCCATTCAGAGAAGAAACCAATTG-3′ and 5′-GTCATCTAGACCCAAAAAAACGGGTATGGAGA-3′. The promoter fragment was digested with BglII and XbaI and ligated with appropriately digested pTD16 to yield pTB265 (attHK022 bla Para). The NcoI-NheI attHK022 region of pTB265 was replaced by the corresponding attλ region of pCAH63 (41) to generate pTB274 (attλ bla Para). The BglII-NotI bla containing fragment of pTB274 was replaced with a BglII-NotI-digested PCR fragment containing the cat gene to generate pTB277 (attλ cat Para). The cat PCR fragment was generated by PCR from plasmid pKD3 (18) using the primers 5′-GTCAAGATCTCTACCTGTGACGGAAGATCACTTCG-3′ and 5′-TGACGCGGCCGCCTTCATTTAAATGGCGCGCCTTAC-3′. The envC gene was amplified from TB28 DNA using the primers 5′-GCTATCTAGACCATGAGGGGAAAGGCGATTAATAC-3′ and 5′-GTCAAAGCTTGGAGAGCGCCAACAGAGCGGC-3′. The resulting PCR fragment was digested with XbaI and HindIII and ligated with appropriately digested pTB183 (3) to generate pTD11 (attHK022 bla Plac::envC). Finally, the XbaI-HindIII fragment from pTD11 containing envC was inserted into the corresponding sites of pTB277 to generate pTD25 (attλ cat Para::envC).
The constructs used to produce mCherry fusions to the LytM factors (pTB314, pTB316, pTD38, and pTD39) were assembled in numerous steps. Before we describe the details of their construction, we must first describe the construction of several intermediate plasmids. One of the important intermediates is pTB263 (attHK022 bla Plac::dsbAss-gfpsf). It was constructed in several steps from pTB225 (attHK022 bla Plac::zipA-gfpsf). To construct pTB225, the gfp containing XhoI-HindIII fragment of pTB222 (attHK022 bla Plac::zipA-gfp) (3) was replaced with a corresponding fragment containing gfpsf encoding a superfolding variant of GFP (66). The gfpsf gene with codons optimized for E. coli and the desired restriction sites was synthesized and cloned for us by Epoch Biolabs (Sugar Land, TX). To generate pMM2 (attHK022 bla Plac::zipA-gfpsf), the XhoI-HindIII fragment of pTB225 containing gfpsf was replaced by a modified gfpsf PCR fragment with additional restriction sites for generating gene fusions. This fragment was generated by using pTB225 as a template and the primers 5′-GTCACTCGAGGGTCCGGCTGGTCTGTCTAAAGGTGAAG-3′ and 5′-GTACAAGCTTGTCGACTTATTAGGATCCGCCAGCACCTTTGTAGAGCTCATCCATGCC GTG-3′. Synthetic oligonucleotides encoding the dsbA signal sequences 5′-TATGAAAAAGATTTGGCTGGCGCTGGCTGGTTTAGTTTTAGCGTTTAG CGCATCGGCGGCGCAGTATGAAGATC-3′ and 5′-TCGAGATCTTCATACTGCGCCGCCGATGCGCTAAACGCTAAAACTAAACCAGCCAGCGCC AGCCAAATCTTTTTCA-3′ were annealed and ligated with NdeI-XhoI-digested pMM2, thus replacing the zipA gene with dsbAss to generate pTB263 (attHK022 bla Plac::dsbAss-gfpsf).
Another important intermediate is pTU136 (attHK022 bla Plac::dsbAss-mCherry). Multiple steps were required for its assembly. pTU134 (attHK022 bla Plac::dsbAss-mCherry) was first constructed by replacing the gfpsf containing XbaI-SalI fragment of pTB263 with the corresponding mCherry containing fragment from pTB275 [bla Plac::malF(TM)-mCherry]. pTB275 is identical to pFB250 (3) except that an internal Met codon in mCherry was changed to a Leu codon using site-directed mutagenesis to prevent internal translational starts. The primers used for mutagenesis were 5′-GGCGAGGAGGATAACCTGGCTATCATTAAAGAGT-3′ and its reverse complement. The mCherry gene in pTU134 contains an internal HindIII site. This was removed by site-directed mutagenesis to generate pTU135. The primers used for this mutagenesis were 5′-GTACGGTTCTAAAGCGTATGTTAAACACCCAG-3′ and its reverse complement. The dsbAss-mCherry gene fusion of pTU135 was subcloned into the corresponding XbaI-HindIII sites of pTU127 to generate pTU136 (attHK022 bla Plac::dsbAss-mCherry). pTU127 is a pTB263 derivative containing envC. A portion of the envC gene (codons 34 to 419) was amplified from TB28 DNA using the primers 5′-GTCAGGATCCGGTGATGAGCGTGACCAACTCAAATCTATTC-3′ and 5′-GTCAAAGCTTGGAGAGCGCCAACAGAGCGGC-3′. The resulting fragment was digested with BamHI and HindIII and inserted into appropriately digested pTB263 to generate pTU127 [attHK022 bla Plac::dsbAss-gfpsf-envC(34-419)].
pTB314 (attHK022 bla Plac::nlpD-mCherry), pTB316 (attHK022 bla Plac::envC-mCherry), and pTD38 (attHK022 bla Plac::ygeR-mCherry) were constructed in a similar fashion. The nlpD gene was amplified from TB28 DNA by using the primers 5′-GTACCATATGAGCGCGGGAAGCCCA-3′ and 5′-GTCACTCGAGTCGCTGCGGCAAATAACGC-3′. The ygeR gene was amplified from TB28 DNA by using the primers 5′-GTACCATATGAGTGCGGGACGCCTGAATA-3′ and 5′-CTAGCTCGAGTGATTTGCTACCCGAACAGCCC-3′. The resulting nlpD and ygeR fragments were digested with NdeI and HindIII and used to replace the zipA containing NdeI-HindIII fragment of pTB225 to generate pTD9 (attHK022 bla Plac::nlpD-gfpsf) and pTD10 (attHK022 bla Plac::nlpD-gfpsf), respectively. The envC containing Xba-XhoI fragment from pTB26 (5) was used to replace the corresponding zipA fragment from pTB225 to generate pTB226 (attHK022 bla Plac::envC-gfpsf). The gfpsf containing XhoI-HindIII fragments of pTD9, pTD10, and pTB226 were replaced with the mCherry containing XhoI-HindIII fragment of pTU136 to generate pTB314, pTD38, and pTB316, respectively. The construction of pTD39 (attHK022 bla Plac::yebA-mCherry) was slightly different from the other mCherry constructs. The yebA gene was amplified from TB28 DNA by using the primers 5′-GTACCATATGCAACAGATAGCCCGCTCTGTC-3′ and 5′-GTACGGATCCATCAAACCGTAGCTGCGGCAC-3′. The resulting fragment was digested with NdeI and BamHI and used to replace the corresponding zipA fragment of pTB225 to generate pTD8 (attHK022 bla Plac::yebA-gfpsf). The gfpsf containing BamHI-SalI fragment of pTD8 was replaced with the mCherry containing BamHI-SalI fragment of pTB275 to generate pTD39.
Fluorescence microscopy.
All fluorescence micrographs in the present study were captured by using a Nikon TE2000 inverted microscope outfitted with a Nikon Intensilight illuminator, a Coolsnap HQ2 charge-coupled device camera from Photometrics, a Nikon CFI Plan Apo VC ×100 objective lens (1.4 NA) for differential interference contrast (DIC) imaging or a CFI Plan Apo DM ×100 objective lens (1.4 NA) for phase contrast imaging. Filter cubes for fluorescence image acquisition were from Chroma. For FM1-43 images we used the ET-GFP filter set (Chroma 49002). The DAPI (4′,6′-diamidino-2-phenylindole) images were taken by using the ET-DAPI filter set (Chroma 49000), and the mCherry and Texas Red images were taken by using the ET-mCherry filter set (Chroma 49008). Images were captured by using Nikon Elements software, exported to Adobe Photoshop CS3, and cropped for figure preparation in Adobe Illustrator CS3.
To assess the division phenotypes of the various mutants under study (Fig. 2 and 3 and Table 3), overnight cultures of each strain were grown in LB at 37°C. Cells were then diluted 1:100 into fresh LB and grown at 37°C to an optical density at 600 nm (OD600) between 0.4 and 0.7. A portion of the culture (0.5 ml) was then stained with 5 μg of FM1-43FX (Invitrogen)/ml for 10 min at room temperature. The culture was then adjusted to 33 mM sodium phosphate (pH 7.4), and the cells were fixed by the addition of formaldehyde and glutaraldehyde to 2.4 and 0.04%, respectively. Fixation was carried out for 30 min at room temperature, after which the cells were washed several times with phosphate-buffered saline (PBS; 137 mM NaCl, 10 mM phosphate, 2.7 mM KCl [pH 7.4]) and resuspended in PBS to a final OD600 of ∼2.5. The cells were then stained with 250 ng of DAPI/ml, immobilized on agarose pads, and visualized by using DIC and fluorescence optics as described above (see the legend to Fig. 2 for further details). Images of many fields of cells from each strain were captured, and the measurements in Table 3 were made using these images and the ObjectJ analysis plugin (N. A. Vischer and S. Nastase [http://simon.bio.uva.nl/objectj/]) for ImageJ (W. A. Rasband [http://rsb.info.nih.gov/ij/]).
FIG. 2.
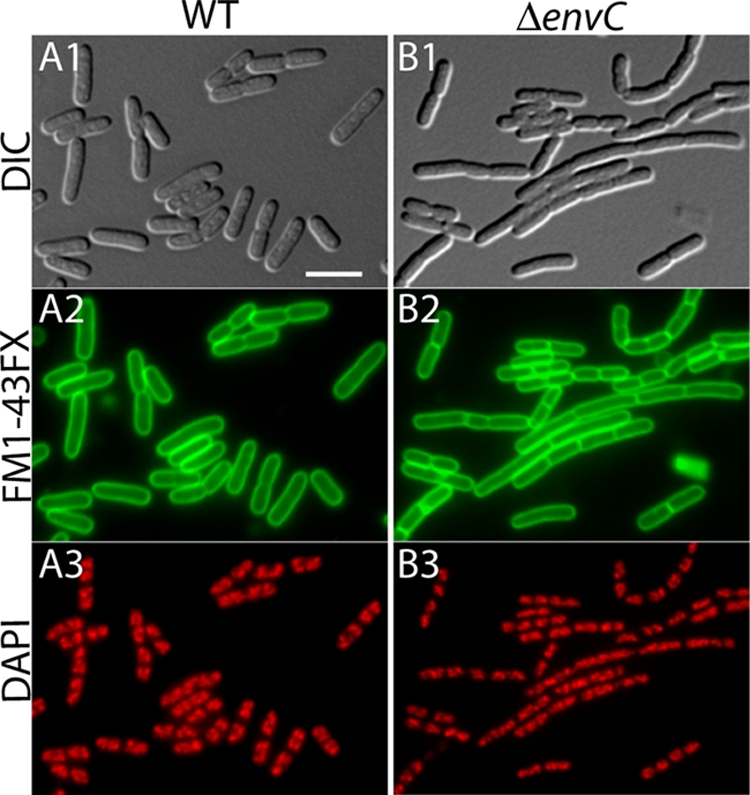
EnvC− mutants have a mild cell separation phenotype. Shown are representative micrographs of fixed cells of TB28 (WT) (A) and TB134 (ΔenvC) (B). Cells in exponential growth (OD600 = 0.5 to 0.7) in LB at 37°C were stained with the membrane dye FM1-43-FX, fixed, and stained with DAPI. They were visualized with DIC (panels 1) and fluorescence optics (panels 2 and 3). The FM1-43-FX channel was pseudo-colored green (panels 2), and the DAPI channel was pseudo-colored red (panels 3). Bar, 4 μm.
FIG. 3.

Severe chaining defect of cells lacking multiple LytM factors. Shown are representative micrographs of fixed cells of TB156 (ΔenvC ΔnlpD) (A), TD22 (Δlyt4 = ΔenvC ΔnlpD ΔygeR ΔyebA) (B), and TB171 (Δami3 = ΔamiA ΔamiB ΔamiC) (C). Cells were grown, processed, and imaged as described in the legend to Fig. 2. Bar, 4 μm.
TABLE 3.
Division phenotypes of LytM− mutants
| Strain | Relevant genotype | No. of cellsa | Total length (μm)b | Avg length (μm)c | Total no. of septad | Length/septum (μm)e | Length/segment (μm)f | No. of septa/cell |
|---|---|---|---|---|---|---|---|---|
| TB28 | WT | 203 | 806 | 4.0 | 101 | 8.0 | 2.7 | 0.5 |
| TB140 | ΔenvC | 124 | 823 | 6.6 | 244 | 3.4 | 2.2 | 2.0 |
| TB145 | ΔnlpD | 338 | 848 | 2.5 | 101 | 8.4 | 1.9 | 0.3 |
| TD10 | ΔyebA | 280 | 831 | 3.0 | 91 | 9.1 | 2.2 | 0.3 |
| TD5 | ΔygeR | 244 | 826 | 3.4 | 115 | 7.2 | 2.3 | 0.5 |
| TB156 | ΔenvC ΔnlpD | 27 | 846 | 31.3g | 316 | 2.7 | 2.5 | 11.7 |
| TD18 | ΔenvC ΔyebA | 92 | 806 | 8.8 | 298 | 2.7 | 2.1 | 3.2 |
| TD17 | ΔenvC ΔygeR | 94 | 804 | 8.6 | 290 | 2.8 | 2.6 | 3.1 |
| TD12 | ΔnlpD ΔyebA | 302 | 821 | 2.7 | 79 | 10.4 | 2.2 | 0.3 |
| TD7 | ΔnlpD ΔygeR | 303 | 838 | 2.8 | 139 | 6.0 | 1.9 | 0.5 |
| TD14 | ΔyebA ΔygeR | 277 | 816 | 3.0 | 103 | 7.9 | 2.1 | 0.4 |
| TD19 | ΔenvC ΔnlpD ΔyebA | 20 | 1078 | 53.8 | 479 | 2.3 | 2.2 | 24.0 |
| TD20 | ΔenvC ΔnlpD ΔygeR | 20 | 1256 | 62.8 | 576 | 2.2 | 2.1 | 28.8 |
| TD21 | ΔenvC ΔyebA ΔygeR | 77 | 837 | 10.9 | 350 | 2.4 | 2.0 | 4.6 |
| TD16 | ΔnlpD ΔyebA ΔygeR | 289 | 804 | 2.8 | 118 | 6.8 | 2.0 | 0.4 |
| TD22 | ΔenvC ΔnlpD ΔyebA ΔygeR | 21 | 1784 | 84.9 | 760 | 2.4 | 2.3 | 36.2 |
| TB171 | ΔamiA ΔamiB ΔamiC | 25 | 1605 | 64.2 | 815 | 2.0 | 1.9 | 32.6 |
Cell chains were considered single cells regardless of the number of segments they contained.
Total length refers to the cumulative length of all cells measured.
That is, the total length/number of cells.
Visible membrane constrictions were counted as septa in addition to completed membrane septa visible in cell chains.
Length/septum refers to the total length/total number of septa. This is a measure of the frequency at which septa are observed. The number is much lower in chaining cells because septa persist for an abnormally long time.
The number of cell segments indicates the number of cells plus the number of septa. The “length/segment” is the total length/total number of segments. In normal (nonchaining) cells, this measurement is similar to the average cell length (pole-pole distance), but the value is smaller because predivisional cells contain two segments and are counted as two cells instead of one (i.e., some pole-to-septa measurements are taken into account, as well as pole-pole measurements). In chaining cells, the length/segment measurement reports mainly on the distance between adjacent septa, since septa greatly outnumber poles.
Many of the cell chains in these mutants extended outside the field of view due to their extreme length. The average length and septa/cell measurements are therefore minimum values (indicated in boldface). Despite this, the measurements still reflect the relative magnitude of the cell separation defects for the strains in question.
To determine the subcellular localization of the LytM factors, cells expressing the fusions were grown by diluting overnight cultures 1:100 in M9-maltose medium supplemented with IPTG (isopropyl-β-d-thiogalactopyranoside) as indicated in the legend to Fig. 7. Cells were then grown at 30°C to an OD600 of 0.5 to 0.7, transferred to an agarose pad on a microscope slide, and immediately visualized using DIC and fluorescence optics (see the legend to Fig. 7 for details).
FIG. 7.

Altered mode of ampicillin-induced lysis in cells lacking LytM factors. (A) Growth curves of TB28(attλTD25) [WT (Para::envC)] and TD22(attλTD25) [Δlyt4 (Para::envC)] with (open symbols) or without (closed symbols) ampicillin treatment (5 μg/ml). Cells were removed from the cultures in panel A at various time points for fixation. Fixed cells were then visualized by phase-contrast microscopy to monitor cell morphology. TB28(attλTD25) cells growing without ampicillin are shown in panels B (41 min) and D (50 min). TB28(attλTD25) cells treated with ampicillin are shown in panels C (41 min), E (50 min), and J (80 min). TD22(attλTD25) cells growing without ampicillin are shown in panels F (41 min) and H (50 min). TD22(attλTD25) cells treated with ampicillin are shown in panels G (41 min), I (50 min), K (80 min), L (110 min), and M (130 min). Arrows in panels C and E point to lysed or lysing cells displaying septal lesions. Double arrows in panels L and M point to cells losing their shape as they are lysing. Bar, 8 μm.
Electron microscopy (EM).
For the analysis of thin sections, cells from colonies of various strains were grown overnight on LB plates at 30°C and were resuspended in PBS containing 5% bovine serum albumin (BSA; Sigma). Samples were rapidly frozen under high pressure using a Leica EM PACT2 freezer. Cells were then freeze substituted with a solution of 1% osmium tetroxide and 0.1% uranyl acetate in acetone over 3 days at −90°C. The samples were then brought to room temperature, washed with acetone, and infiltrated and embedded in TAAB Epon. Thin sections were cut by using a Reichert Ultracut-S microtome, mounted on copper grids, and poststained with lead citrate. Thin sections were visualized by using a JOEL 1200EX microscope outfitted with a charge-coupled device camera. Images were imported to Adobe Photoshop and Illustrator CS3 for figure preparation.
Ampicillin lysis.
To monitor ampicillin-induced lysis of the Lyt4− and Ami3− mutants, cells were grown overnight at 37°C in LB without arabinose. The resulting cultures were diluted 1:200 into fresh LB lacking arabinose and grown at 37°C to an OD600 of ∼0.6. Cells were then further diluted 1:3 into LB medium with or without 5 μg of ampicillin/ml. Growth was then continued at 37°C, and the OD600 of each culture was measured every 10 min. In addition, 0.5-ml aliquots of culture were removed at various times for cell fixation as described above (see Results and the legend to Fig. 7 for further details).
Purification of sacculi.
Sacculi were prepared as described by Priyadarshini et al. (69) with modifications. Cells grown in 1 liter of LB were harvested at an OD600 of 0.5 by centrifugation (4,000 × g) for 15 min at 4°C, and the pellets were resuspended in 20 ml of PBS. The resulting cell suspension was added to a flask containing 80 ml of boiling 5% sodium dodecyl sulfate (SDS) with vigorous stirring. The samples were boiled for 30 min and incubated overnight at room temperature. The following morning, sacculi were sedimented by ultracentrifugation at 100,000 × g for 1 h at 25°C. The pellets were washed three times with water, resuspended in 1 ml of PBS, treated with α-chymotrypsin (300 μg/ml, final concentration), and incubated overnight at 37°C. The next morning the sacculi were treated again with 300 μg of α-chymotrypsin/ml for at least 3 h at 37°C, and then 0.25 ml of 5% SDS was added (1% final concentration), followed by incubation at 95°C for 2 h. Sacculi were pelleted by centrifugation at 21,000 × g for 30 min at room temperature and washed with 1 ml of water repeatedly until the SDS concentration was <0.01% as determined using the method of Hayashi (43). The final pellet was dissolved in 1 ml of water containing 0.02% sodium azide and stored at 4°C.
Labeling sacculi with Texas Red and Alexa 488-WGA.
The peptide moieties of sacculi were stained with Texas Red as described by Priyadarshini et al. (69) with modifications. A 10-μl portion of a sacculi suspension was mixed with 35 μl of 0.1 M NaHCO3 buffer (pH 8.6), followed by the addition of 5 μl of 5 mg of Texas Red-succinimidyl ester (SE)/ml dissolved in dimethyl sulfoxide (Invitrogen/Molecular Probes) with vigorous vortexing. The suspension was incubated in the dark for 15 min, and the reaction was terminated with the addition of 5 μl of 5 mg of lysine/ml. The sample was washed once with 0.1 M NaHCO3 buffer and two times with water by centrifugation at 21,000 × g for 20 min at room temperature and resuspension in the appropriate solution. The final pellet was resuspended in 50 μl of water, and 5 μl was coated onto a glass microscope slide and air dried. Next, 3 μl of water was spotted onto the dried sacculi, and a coverslip was placed on the water droplet. Sacculi were then imaged by using fluorescence microscopy as described above. Glycan strands of sacculi were labeled with Alexa 488-conjugated wheat germ agglutinin (Alexa 488-WGA; Invitrogen-Molecular Probes). A suspension of sacculi (5 μl) was spotted onto a microscope slide and air dried. Then, 5 μl of 100 μg of Alexa 488-WGA/ml in PBS containing BSA (1 mg/ml) was then spotted onto the dried sacculi, and a coverslip was placed over the droplet. Labeled sacculi were then imaged by using fluorescence microscopy as described above.
Muropeptide analysis.
Muropeptides were prepared as described by Glauner et al. (38) with modifications. Briefly, sacculi (equivalent to cells in 200 ml of culture) isolated as described above were treated with 10 U of mutanolysin (Sigma, St. Louis, MO) in 50 mM sodium phosphate buffer (pH 6.0) at 37°C overnight. Further digestion was performed the next morning by addition of another 5 U of mutanolysin and incubation at 37°C for at least 3 h. The digests were centrifuged (21,000 × g, 10 min, room temperature), and the supernatants were reduced with ∼1 mg of sodium borohydride in 50 mM sodium borate (pH 9.0) at room temperature for 30 min, followed by the addition of 20% phosphoric acid to adjust pH to <4. The reduced samples were lyophilized and resuspended in 100 μl of water for analysis by high-performance liquid chromatography (HPLC).
HPLC was performed with a Dionex Ultimate 3000 system equipped with a Jupiter Proteo C12 column (250 by 4.6 mm, 4 μm, 90 Å) from Phenomenex. The samples were injected onto the column at 50°C and eluted at a flow rate of 0.5 ml/min with 1% acetonitrile in water containing 0.1% (vol/vol) trifluoroacetic acid for 10 min. A convex gradient of 1 to 10% acetonitrile (Dionex gradient programmer, profile 3) over 60 min in water containing 0.1% trifluoroacetic acid was then initiated. Muropeptides were detected by determining the absorbance at 206 nm. To identify each peak, fractions were collected, lyophilized, resuspended in water, and analyzed with a Voyager-DE MALDI-TOF mass spectrometer (Perseptive Biosystems). Based on their molecular masses, all major peaks were identified as known muropeptides. Some minor peaks still require identification, however. Based on identified peak area, the relative amounts of the muropeptides were calculated as described by Glauner et al. (38). Minor unidentified peaks did not affect the calculations.
RESULTS
Mutants lacking multiple LytM factors have a severe cell separation defect.
We set out to determine whether, in addition to EnvC, the other LytM factors encoded by E. coli are also playing a role in daughter cell separation. To do so, we generated a set of mutant strains lacking all possible combinations of the four E. coli LytM factors—EnvC, NlpD, YgeR, and YebA—and studied their growth and division phenotypes. We used strains from the Keio deletion collection (1) as the source of our deletion alleles except for ΔyebA. Deletions were combined in our WT background TB28 (MG1655 [ΔlacIZYA]) by alternating rounds of P1 transduction and removal of the Kanr cassette using FLP recombinase from the curable plasmid pCP20 (18) (see Materials and Methods for details). Cells of each mutant were visualized microscopically to study their morphology and division pattern. For the microscopic analysis, exponentially growing cultures of each mutant were stained with the fixable membrane dye FM1-43FX (Invitrogen). The cells were then fixed and stained with DAPI after fixation. Images of the cells were captured by using both DIC and fluorescence optics. To quantify the magnitude of any observed division defects, measurements of cell length along with the number of septa present in each cell or cell chain were performed by using ObjectJ software (Table 3). Representative micrographs for WT and select mutant cells are shown in Fig. 2 and 3.
Of all the single LytM− mutants, only TB140 (ΔenvC) displayed an obvious division phenotype (Table 3 and Fig. 2). As reported previously for other EnvC− mutants, a high percentage of TB140 cells formed short chains of about three to four cell units per chain (5, 42, 48, 71). Similar to the single mutants, only double LytM− mutants harboring an envC deletion had an observable division phenotype (Table 3). When combined with ΔenvC, the loss of YebA or YgeR only slightly exacerbated the division phenotype resulting from an EnvC− defect alone (Table 3). Strikingly, however, the combination of envC and nlpD deletions in strain TB156 led to a significant block in cell separation (Table 3). EnvC− NlpD− cells formed very long cell chains with fairly regularly spaced septa visible both with the membrane stain and as cell constrictions in the DIC images (Fig. 3A). The depth of the constrictions in these chains observable by DIC indicate that a significant degree of outer membrane invagination is still possible in these mutants. In addition to the cell separation defect, TB156 cells also displayed a growth defect on solid media and formed colonies that were smaller and more opaque than those of the WT (Fig. 4A). Both the chaining and growth defects of TB156 were corrected upon the expression of nlpD from an integrated chromosomal expression construct (see Fig. 6G and data not shown). Thus, the exacerbated chaining phenotype of TB156 (ΔenvC ΔnlpD) relative to TB140 (ΔenvC) is truly due to the loss of NlpD and not polar effects of the deletion and insertion mutation on nearby genes.
FIG. 4.
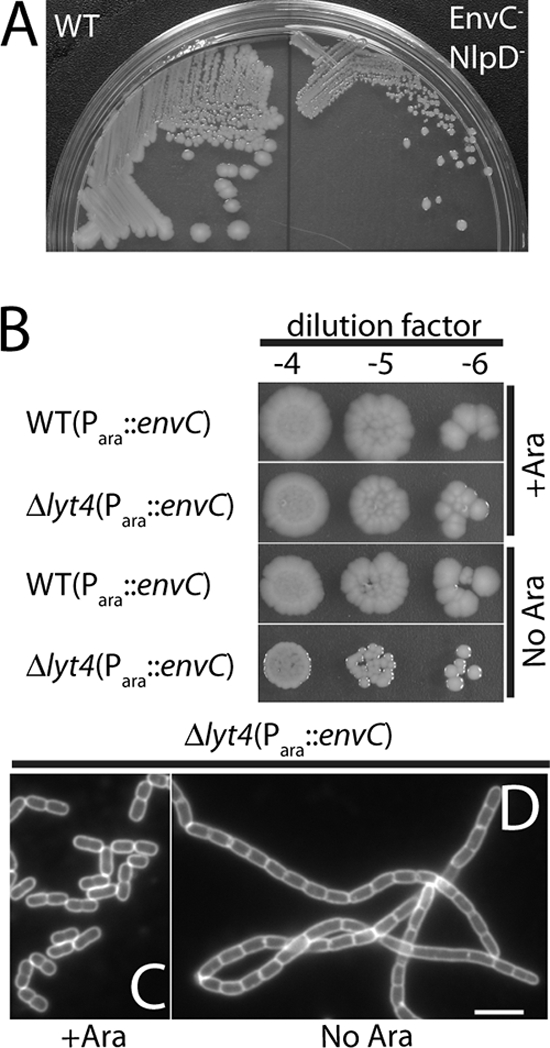
Growth of mutants lacking multiple LytM factors. (A) TB28 (WT) (left) and TB156 (ΔenvC ΔnlpD) (right) cells were plated on LB agar and incubated overnight at 37°C. Notice the significant difference in the size and appearance of the TB156 colonies relative to those of TB28. (B) Overnight cultures of TB28(attλTD25) [WT(Para::envC)] and TD22(attλTD25) [Δlyt4(Para::envC)] were grown in M9 arabinose at 37°C. The cells were harvested by centrifugation, washed three times in LB, and resuspended to their original culture volume in LB. The suspensions were adjusted to the same OD600 and serially diluted, and 5 μl of each dilution was spotted onto LB agar with or without arabinose supplementation. Plates were incubated overnight at 37°C. The photos in panels A and B were taken with a Nikon D40 camera outfitted with a 60-mm f/2.8D AF Micro-Nikkor lens and mounted on a copy stand. (C and D) FM1-43FX-stained TD22(attλTD25) cells grown in LB at 37°C with or without arabinose, respectively. An overnight culture grown in LB supplemented with 0.2% arabinose at 37°C was diluted 1:200 into LB with or without 0.2% arabinose. Cultures were grown to an OD600 of 0.6 and processed as described in the legend for Fig. 2. Bar, 4 μm.
FIG. 6.

Subcellular localization of the LytM factors. Shown are representative micrographs of TB140(attHKTB316) [ΔenvC (Plac::envC-mCherry)] (A), TB145(attHKTB314) [ΔnlpD (Plac::nlpD-mCherry)] (B), TD5(attHKTD38) [ΔygeR (Plac::ygeR-mCherry)] (C), and TD9(attHKTD39) [ΔyebA (Plac::yebA-mCherry)] (D). Cells were grown to an OD600 between 0.4 and 0.7 in M9-maltose supplemented with 0, 50, 100, or 250 μM IPTG for panels A, B, C, and D, respectively, and imaged live on agarose pads with DIC (panels 1) or fluorescence (panels 2) optics. Arrows highlight prominent septal localization patterns in panels A and B. (E to G) Representative micrographs of TB156 (ΔenvC ΔnlpD) cells containing attHKTU136 (Plac::dsbAss-mCherry) (E), attHKTB316 (Plac::envC-mCherry) (F), or attHKTB314 (Plac::nlpD-mCherry) (G). Cells were grown to and OD600 of ∼0.5 in LB supplemented with 0 (E and F) or 100 (G) μM IPTG. They were then stained with FM1-43FX, fixed, and visualized by fluorescence microscopy. Bar, 4 μm.
The same basic trends observed for the single and double LytM− mutants were also uncovered in our analysis of triple and quadruple LytM− mutants. Again, only mutants lacking envC displayed a division defect and, relative to a single ΔenvC mutant, the severity of this defect was only enhanced significantly in mutants combining ΔenvC and ΔnlpD. The contributions of YebA and YgeR to cell separation became more apparent when they were inactivated in the context of an EnvC− NlpD− background. The loss of YebA and/or YgeR in combination with the loss of both EnvC and NlpD led to an increasingly severe cell separation defect with the chaining phenotype displayed by the respective triple mutants (TD19 and TD20) being more severe than the double EnvC− NlpD− mutant (TB156). The most severe chaining phenotype of all was displayed by the quadruple LytM− mutant (Δlyt4; Lyt4−) (Table 3 and Fig. 3B). Like the double EnvC− NlpD− mutant, the Δlyt4 strain showed fairly regularly spaced membrane septa visualized using the membrane dye. The cell constrictions visible by DIC, on the other hand, appeared more shallow in the Δlyt4 strain than in the double EnvC− NlpD− mutant, suggesting that the loss of all LytM factors leads to an early block in the outer membrane constriction process. The Δlyt4 mutant had a cell separation defect comparable to that of a mutant lacking all three amidases (AmiA, AmiB, AmiC; Δami3; Ami3−) (Fig. 3B and C and Table 3). In both cases the mutants appeared to be completely defective for normal cell separation such that they only seemed to separate upon physical shearing or the lysis of a cell within the chain. We therefore conclude that the LytM factors, like the amidases, are absolutely required to promote the normal cell separation process. Moreover, our analysis suggests that among the LytM factors, EnvC plays the dominant role in cell separation followed closely in relative importance by NlpD. YebA and YgeR, on the other hand, only appear to be playing minor roles in the division process under the conditions tested.
As described above for TB156 (ΔenvC ΔnlpD), mutants with severe chaining phenotypes generally displayed a significant growth defect on solid media and formed smaller colonies than did the WT or other nonchaining mutants (Fig. 4A). Colonies from these strains also typically became mucoid when their growth was continued at room temperature for an additional day or two after an initial overnight incubation at 30 or 37°C. It is important to note that growth defects for the chaining mutants were only observed on solid media. When mutants with severe chaining phenotypes were grown in liquid media, their growth rates during exponential phase were very similar to those of WT cells (see Fig. 7A). The reason for the differing growth characteristics of the chaining mutants on solid versus liquid media is not known. Nevertheless, to avoid the potential selection of mutations that suppress the growth defect associated with a strong cell chaining phenotype when cells are propagated on agar, we reconstructed several of the multiple LytM− mutants by introducing a ΔenvC::Kanr allele into the desired background containing an additional, arabinose-inducible copy of envC (Para::envC). This envC expression construct (attλTD25) was present on a plasmid integrated at the lambda attachment site. Transductants were selected on medium containing arabinose and tested for growth and division on media with or without added arabinose. In all cases, the mutants grew as well as the WT and showed normal division patterns when grown with arabinose. In the absence of arabinose, the growth and chaining phenotypes observed for particular mutant combinations were essentially equivalent to those observed in the deletions constructed without attλTD25. For example, TD22(attλTD25) [Δlyt4 (Para::envC)] grew well and separated normally when grown with arabinose, while it grew poorly and formed long chains when grown without arabinose (Fig. 4C and D). Importantly, TD22(attλTD25) had essentially the same plating efficiency when grown with or without arabinose, indicating that suppressor mutations are not required for survival in the absence of the full set of LytM factors (Fig. 4B).
Other than the growth and cell separation phenotypes observed for mutants lacking multiple LytM factors, we noticed that cells lacking NlpD, YgeR, or YebA were shorter on average than WT cells (Table 3). This was true for single ΔnlpD, ΔygeR, and ΔyebA mutants or mutants where these alleles were combined with other deletions. The short cells did not appear to be appreciably wider than WT cells (data not shown) such that the defect appears primarily to affect cell length control. The reason for this defect is not known, but it suggests that several of the LytM factors may be involved in promoting cell elongation or in the regulation of division timing. Importantly, the short cell phenotype of the ΔnlpD mutant could be corrected by the expression of nlpD in trans, indicating that the phenotype was not due to polar effects of the deletion on the expression of the downstream rpoS gene (data not shown). Further work is required to determine the exact cause of the “short-cell” phenotype in ΔnlpD, ΔygeR, and ΔyebA mutants.
Mutants lacking LytM factors are defective in septal PG splitting.
Given that the LytM factors are thought to be PG hydrolase enzymes and that the chaining phenotype of the Δlyt4 mutant closely resembles that of the Δami3 mutant, the most likely explanation for the separation defect displayed by cells lacking multiple LytM factors is a failure in the process of septal PG splitting. To test this, we used EM to visualize septa from both normal cells and chain forming mutants. Cells from colonies of TB28 (WT), TB156 (ΔenvC ΔnlpD), and TD22 (Δlyt4) were suspended in buffer containing 5% BSA to induce mild plasmolysis and serve as a cryoprotectant. Cells were then rapidly frozen under high pressure and processed for the analysis of thin sections by transmission EM (see Materials and Methods for details). As predicted from the bright-field and fluorescence images in Fig. 3, many of the septa visible in thin sections of the chaining mutants showed a defect in outer membrane constriction. In the examples shown in Fig. 5A to C, inner membrane fusion looks to be complete, and the cytoplasmic contents of the adjacent cell segments appear to be separated. Constriction of the outer membrane, on the other hand, seems to be obstructed by what is likely to be a layer of unsplit PG located between the adjacent units of the chain (Fig. 5A to C). In stark contrast to the septa of chaining cells, septa visualized from thin sections of dividing WT cells showed the expected tight coordination between inner and outer membrane constriction without an observable unsplit septal PG intermediate (Fig. 5D and E).
FIG. 5.
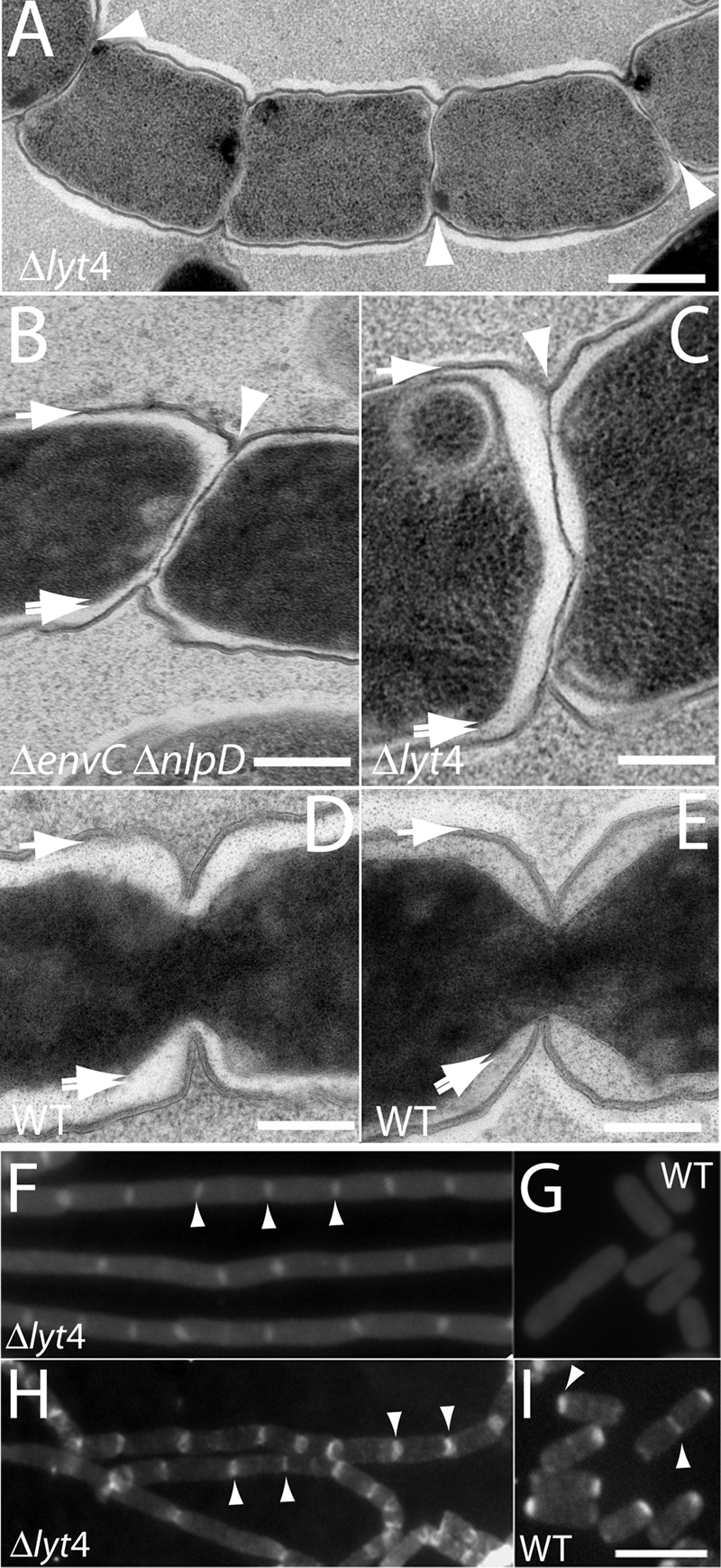
Septal PG splitting defect in mutants lacking LytM factors. TD22 (Δlyt4) (A and C), TB156 (ΔenvC ΔnlpD) (B), or TB28 (WT) (D and E) cells were mildly plasmolyzed and processed for EM (see the text for details). Shown are representative thin sections highlighting the septal PG splitting defect of TD22 and TB156 cells (A to C). The TB28 thin sections are provided for comparison to show that the constriction of the outer membrane and PG layer is normally tightly coordinated with the constriction of the inner membrane (D and E). The single arrows point to the outer membrane and PG layer surrounding the cell body. These two layers were not resolved at the cell periphery. The double arrows point to the inner membrane surrounding the darkly stained cytoplasm, and the arrowheads point to what appears to be an unsplit layer of septal PG in the chaining mutants. (F and G) Fluorescence micrographs of purified PG sacculi from TD22 (Δlyt4) (F) and TB28 (WT) (G) stained with amino-reactive Texas Red SE. (H and I) Fluorescence micrographs of purified PG sacculi from TD22 (Δlyt4) (H) and TB28 (WT) (I) stained with an Alexa 488-labeled lectin (WGA). Examples of intensely staining septa or poles are highlighted with arrow heads. Bars: 500 nm (A), 200 nm (B to E), 4 μm (F to I).
As an independent method to examine the status of septal PG in the mutants, we purified PG sacculi from WT and Δlyt4 cells and stained them with two different fluorescent reagents: Texas Red SE to label the free amino groups of peptides and Alexa 488-conjugated WGA, a lectin that binds the polysaccharide strands. Labeled sacculi from the Δlyt4 mutant retained the chainlike appearance of the cells they were isolated from (Fig. 5F and H). In addition, using both reagents, bands or rings of intense staining were observed in the mutant sacculi at fairly regular intervals, a finding consistent with idea that these regions correspond to depositions of septal PG (Fig. 5F and H). Similar results were obtained previously when sacculi from a Δami3 mutant were stained with an amino reactive dye (69). In contrast to our results with the mutant sacculi, the staining patterns observed for WT sacculi differed depending on the labeling reagent used (Fig. 5G and I). WT sacculi were uniformly labeled with Texas Red SE, whereas Alexa 488-WGA appeared to preferentially label septal and polar regions (Fig. 5G and I). Strikingly, only one pole was typically enriched for Alexa 488-WGA, suggesting some asymmetry in the composition or structure of PG between the two poles (Fig. 5I). We do not currently know whether it is the old pole or the new pole that is labeled, nor do we know why the septal and polar regions of WT cells are preferentially stained with Alexa 488-WGA relative to Texas Red SE. A potential source of the disparity is the dramatic difference in the molecular weights of the reagents. WGA protein is likely to have greater problems accessing its binding sites within the cross-linked PG meshwork than the small molecule Texas Red SE. However, this disparity alone is not likely to generate such a distinct labeling pattern for Alexa 488-WGA. Further work is required to determine the reasons for the preferential labeling of septal and polar regions of sacculi with Alexa 488-WGA and why one pole is preferentially labeled over the other.
Based on the combination of our EM analysis and PG labeling experiments, we conclude that the cell separation phenotype observed for Δlyt4 cells is most likely the result of a severe defect in septal PG splitting. What remains to be resolved is what the architecture of this unsplit PG layer is and whether or not its construction can be completed to form a solid “disk” of unsplit PG connecting adjacent cell segments. Arguing against disk formation, regularly spaced, darkly stained rings of PG material (SP-rings) were observed previously by Heidrich et al. (45) and Priyadarshini et al. (69) when Ami3− sacculi were analyzed by EM after negative staining with uranyl acetate. These rings have been interpreted to represent intermediates in septal PG assembly where the formation of a complete PG septum has been delayed or stalled (69). According to this view, PG splitting by the amidases is required to sustain septal PG synthesis. Since the staining properties of uranyl acetate are not always predictable, we do not believe that it is possible to tell whether the SP-rings observed in the previous reports are indeed “open” in the middle. The darkly stained septal PG rings observed in Ami3− sacculi could just as readily be interpreted to mark the outer edge of a solid disk of PG that for some reason stains more robustly with uranyl acetate than the disk's interior. In addition, if PG septa in chaining mutants were often largely incomplete rings as suggested by Priyadarshini et al. (69), the probability of slicing through a hole in a PG-ring during thin sectioning would be much higher than that of slicing through the edge. We would therefore expect thin sections such as those shown in Fig. 5A to C to be rare relative to those displaying little or no (PG) material between adjacent cytoplasmic compartments. Although they themselves were rare, when thin sections cutting through the long axis of a cell chain were observed, they more often than not looked like the examples shown in Fig. 5A to C. These results therefore argue for a completed or very nearly completed disk of unsplit PG being present at most of the septa in the LytM− cell chains. This argument is indirect, however, as are those favoring the idea of an incomplete ring of septal PG being present at the septa of cell chains. Accordingly, more direct visualization methods are needed to definitively address the architecture of the PG at the septa of chaining mutants.
Muropeptide analysis of mutant sacculi.
To better understand the potential PG modifications brought about by the LytM factors, we analyzed the chemical composition of sacculi isolated from the Δlyt4 mutant and compared it to the composition of WT sacculi (Table 4) . For the analysis, purified sacculi were completely digested with the muramidase mutanolysin and the resulting fragments were reduced with sodium borohydride and separated by reversed-phase HPLC. Three independent sacculi preparations were analyzed for each strain, and the composition of the mutant PG was not found to be significantly different from that of WT (Table 4). Similarly, the muropeptide composition of sacculi from a Δami3 strain was also reported to be comparable to that of WT sacculi (45). Thus, even though Ami3− and Lyt4− mutants are defective for cell separation and have sacculi that are presumably greatly enriched for unsplit septal PG, only minor (if any) differences in the overall PG composition relative to the WT are detectable.
TABLE 4.
Muropeptide analysis of PG from Δlyt4 cellsa
| Muropeptide or characteristic | % Total muropeptides
|
|
|---|---|---|
| TB28 (WT) | TD22 (Δlyt4) | |
| Monomeric (total) | 51.7 ± 3.0 | 50.4 ± 0.9 |
| Monomeric (tetrapeptide) | 31.5 ± 0.0 | 36.0 ± 0.0 |
| Monomeric (tripeptide) | 5.5 ± 0.0 | 6.7 ± 0.0 |
| Dimeric (total) | 43.3 ± 1.7 | 43.9 ± 0.8 |
| Trimeric (total) | 5.1 ± 1.5 | 5.7 ± 0.2 |
| Cross-linkaged | 24.5 ± 1.7 | 24.9 ± 0.5 |
| Anhydro-MurNAc | 1.8 ± 0.3 | 2.2 ± 0.2 |
| Lys-Arg peptidesc | 11.1 ± 1.6 | 11.5 ± 0.6 |
| Glycan strand lengthb | 55.6 ± 9.3 | 46.4 ± 3.5 |
Results are the averages ± standard deviations from three independent PG preparations for each strain.
Calculated from the anhydro-MurNAc levels. The values are numbers of disaccharides.
Lys-Arg peptides are monomeric muropeptides that were attached to Lpp. Lpp was removed with chymotrypsin treatment, leaving Lys-Arg attached.
Percentage of muropeptide in cross-links.
EnvC and NlpD are recruited to the cell division site.
The discovery that all of the LytM factors are playing a role in septal PG splitting and daughter cell separation prompted us to investigate their subcellular localization to determine whether they are recruited to the division site to participate in the division process. We previously reported the recruitment of EnvC to the division site using an EnvC-GFP fusion artificially targeted to the periplasm through the Tat export system (5). This artificial targeting was necessary because EnvC contains a canonical Sec signal sequence at its N terminus (42), and GFP is not fluorescent when it is secreted to the periplasm in an unfolded conformation via the Sec system (28). None of the other LytM factors are predicted to contain a simple Sec signal sequence. YebA is predicted to be a bitopic membrane protein with N-in/C-out topology, and NlpD and YgeR are known or predicted to contain an N-terminal lipoprotein signal peptide, respectively (47) (Fig. 1). We thought it unlikely that the Tat-targeting approach would work for transmembrane proteins or lipoproteins. We therefore used fusions to the monomeric red fluorescent protein, mCherry (79), to study the subcellular localization of the LytM factors since mRFP, and its derivatives have recently been shown to be functional and fluorescent after Sec export (15, 57).
Consistent with our previous results with Tat-targeted EnvC-GFP, EnvC-mCherry was specifically recruited to the division site (Fig. 6A). In addition, NlpD-mCherry was also greatly enriched at division sites (Fig. 6B). However, unlike EnvC-mCherry, which showed a midcell localization pattern in both short unconstricted cells and longer cells with constrictions, NlpD-mCherry only appeared to be enriched at division sites in cells with a clear constriction. This suggests that EnvC is likely to be recruited to the division machinery at an earlier stage in its assembly than NlpD. Both the EnvC-mCherry and the NlpD-mCherry fusions were functional and could correct the division phenotypes of EnvC− NlpD− cells (Fig. 6E to G). In the latter case, production of NlpD-mCherry in TB156 (ΔenvC ΔnlpD) dramatically reduced the size of the chains formed, reducing their length such that the phenotype was comparable to an EnvC− defect alone.
Neither YgeR-mCherry nor YebA-mCherry displayed a strong septal localization pattern (Fig. 6C to D). YgeR-mCherry displayed a patchy peripheral localization with a mild enrichment at the division sites of cells with a deep constriction (Fig. 6C). YebA-mCherry, on the other hand, displayed a fairly uniform peripheral localization with an apparent enrichment at the poles in many cells (Fig. 6D). Since the phenotypes of YgeR− and YebA− cells were subtle, even in the absence of other LytM-domain factors, it was difficult to assess the functionality of the YgeR-mCherry and YebA-mCherry fusions. However, as a whole, the localization patterns of the mCherry fusions to the LytM factors correlate nicely with their respective importance in daughter cell separation. The factors that play the most significant role in cell separation, EnvC and NlpD, display a clear septal recruitment signal when fused to mCherry, whereas mCherry fusions to those that only appear to play minor roles in separation, YgeR and YebA, do not display a strong septal localization pattern.
LytM− mutants lyse aberrantly after ampicillin treatment.
Despite years of study, the exact mechanism by which penicillin induces cell lysis remains a mystery (2, 70). The most penicillin-sensitive step in the cell cycle, however, appears to be the division process since E. coli cells undergoing penicillin-induced lysis, or lysis induced by other broad spectrum β-lactams such as ampicillin, often display membrane protrusions emanating from midcell before they rupture (26, 55, 56, 76, 83). A simple hypothesis to explain this is that by inhibiting PG cross-linking, penicillin causes a catastrophic defect in the splitting of septal PG during division. This, in turn, is likely the result of misregulated or runaway PG hydrolase activity in the absence of productive synthesis. Consistent with this idea, blocking cell division has been shown to have protective effects when E. coli cells are treated with some β-lactams (24, 36, 60), and in several bacterial systems, the inactivation of PG hydrolase enzymes has been shown to slow or even prevent lysis by penicillin and its derivatives (39, 44, 85, 86).
It was previously shown that E. coli mutants lacking the amidases along with several lytic transglycosylases display a delayed lytic response to ampicillin treatment in addition to their severe cell separation defect (44). The discovery that LytM-domain factors are critical for proper cell separation and their relatedness to known PG hydrolases prompted us to test whether mutants lacking multiple LytM-domain factors are also delayed in their lytic response to β-lactams and whether or not the mechanism of the lytic event is altered in the absence of these factors. For this, we grew TB28(attλTD25) [WT (Para::envC)] and TD22(attλTD25) [Δlyt4 (Para::envC)] cells overnight in LB without arabinose supplementation, diluted them 1:200 into fresh LB medium lacking arabinose, and grew them to mid-log phase at 37°C. At this point, TD22(attλTD25) cells displayed a cell separation phenotype consistent with the depletion of EnvC. Once EnvC depletion was confirmed, a portion of each culture was diluted 1:3 into fresh LB broth with or without 5 μg of ampicillin/ml (t = 0 at the time of this final dilution). The OD600 of each culture was then monitored to follow their growth and lysis (Fig. 7A), and aliquots of cells were removed and fixed at several time points so that their morphology could be analyzed when the growth measurements were completed. As shown in Fig. 7A, the Lyt4− mutant grew at about the same rate as the WT strain in the absence of ampicillin. Conversely, in the presence of ampicillin the onset of lysis in the Lyt4− mutant was delayed relative to WT and lytic process itself appeared to proceed at a reduced rate (Fig. 7A).
For WT cells, ampicillin-induced lysis was evident from the OD600 measurements at about 40 min after ampicillin treatment (Fig. 7A). Cells at this time point were elongated relative to untreated control cells and 20 to 30% of them appeared to be lysing through midcell lesions (blebs) characteristic of β-lactam induced lysis of E. coli (Fig. 7C) (26, 55, 56, 76, 83). A higher proportion of lysed (phase-light) or lysing cells were observed at the 50-min time point consistent with the drop in the OD600 of the culture. Finally, by 80 min postdilution, 70 to 80% of the WT cells in the ampicillin-treated culture appeared as lysed cell “ghosts” (Fig. 7E). Morphological analysis of ampicillin-treated Lyt4− cells revealed that they not only lyse more slowly than WT cells, they also appear to lyse via a completely different pathway. Unlike the WT, very few lysed Lyt4− cells were observed at the 41-, 50-, or 80-min time points after ampicillin treatment (Fig. 7G, I, and K). However, starting at about 50 min posttreatment, cell segments in the ampicillin treated chains appeared to grow wider that those of the untreated controls (Fig. 7G, I, K, L, and M). This widening continued at later time points until the segments in the treated samples lost much of their rod-like character and began to lyse. Thus, rather than lysing rapidly through an apparent septal catastrophe like WT cells, ampicillin-treated Lyt4− chains appear to lyse via a gradual loss of cell shape and integrity. Ampicillin-induced lysis of Ami3− chains was also studied, and these cells displayed a similarly aberrant progression to lysis (data not shown). Although other explanations cannot be ruled out, our results suggest that the activities of the LytM factors and the amidases are required to promote the septal lesions formed upon β-lactam treatment of normally dividing E. coli cells. Furthermore, given their subcellular localization and potential PG hydrolase activity (Fig. 6A and B) (4, 5, 45), it is quite possible that these factors are directly responsible for generating the PG lesions observed at the septa of β-lactam treated cells.
DISCUSSION
The importance of PG hydrolases in bacterial cell separation was first appreciated some time ago. Cell chains formed during growth under specialized conditions could be separated by the addition of lysozyme or partially purified lytic enzymes (27, 58). Also, mutants with reduced autolytic activity were found to grow as long chains and, in some cases, such chains were found to be connected by a layer of unsplit PG (32, 49). Although these early results highlighted the general importance of PG hydrolases in cell division, only relatively recently have the specific PG hydrolase factors responsible for promoting daughter cell separation begun to be uncovered. Much of the initial success in this regard has come from studies of gram-positive bacteria where several classes of known or predicted PG hydrolases have been implicated in division. These include: amidases, d,l,-endopeptidases (NlpC/P60 family, and CHAP-domain family), N-acetyglucosaminidases, muramidases, and lytic transglycosylases (10, 14, 33, 37, 50, 52-54, 59, 65, 78, 82, 84, 92, 94).
Only in the last several years have enzymes responsible for septal PG splitting in gram-negative bacteria such as E. coli and Neisseria gonorrhoeae been identified. In both organisms, factors with amidase activity have been implicated (4, 35, 44, 45, 69), and in Neisseria a lytic transglycosylase with a significant role in cell separation has also been identified (16). Other than these enzymes, the only additional factor known to be important for septal PG splitting and cell separation in gram-negative bacteria is the E. coli LytM factor, EnvC (5, 42, 48, 71). It is one of the few LytM factors for which a biological function has been described, and the only one implicated in cell division (31). To further understand the function of LytM proteins, we set out to determine whether or not the other LytM factors encoded in the E. coli genome are also involved in cell separation and, if so, what the relative importance of each factor is. Of the collection of mutants we generated lacking individual LytM factors and all possible combinations of them, only mutants with an EnvC− defect failed to separate normally. This definitively established that EnvC is the most critical LytM factor for proper cell separation in E. coli. A role for the other LytM factors in septal PG splitting was uncovered when we inactivated them in the context of an EnvC− mutant. From this analysis, NlpD was found to be the next most important of the LytM factors. The combined loss of EnvC and NlpD led to a severe cell chaining defect, indicating that it is primarily NlpD function that mitigates the loss of EnvC (Table 3 and Fig. 2 and 3). In contrast to NlpD, YebA, and YgeR only appear to play minor, yet observable, roles in cell separation (Table 3). Their loss only mildly exacerbates the division defects of mutants already displaying a strong phenotype. Thus, the genetic results suggest that EnvC and NlpD are specialized division factors, while YebA and YgeR are likely to participate in other aspects of PG biogenesis and only have weak cell separation activity. Consistent with this idea, both EnvC and NlpD are specifically recruited to the division site, whereas YebA and YgeR show a more dispersed peripheral localization pattern.
The phenotypes displayed by mutants lacking multiple LytM factors are strikingly similar to those observed previously for mutants defective for multiple PG amidases (4, 44, 45, 69). This suggests that LytM factors and the amidases are likely working closely with one another to promote septal PG splitting and daughter cell separation. Determining the mechanism by which these factors are cooperating during cytokinesis remains a major challenge. Thus far, very little is known about the biochemical activity of EnvC and the other E. coli LytM factors (31). Based on their similarity to lysostaphin and LytM, they are predicted to be metallo-endopeptidases capable of hydrolyzing PG. Consistent with this idea, EnvC was shown to form zones of clearing in a gel-based zymogram assay (5). Conversely, several attempts at detecting PG hydrolysis by purified EnvC in solution as opposed to the in-gel assay have been unsuccessful (T. Uehara, unpublished results). Also, the chemical composition of PG sacculi isolated from the Δlyt4 mutant failed to provide us with clues as to the potential cleavage specificity of the LytM factors. Thus, for the time being, the precise biochemical activity of EnvC and the other E. coli LytM factors remains a mystery. Biochemical studies of the E. coli amidases are also in their infancy. N-acetylmuramyl-l-alanine amidase activity has only been detected for AmiA and AmiC, but this was done using a crude extract from cells overproducing these factors rather than assaying activity from purified enzymes (45). Moreover, the activity of AmiB has not yet been definitively established (45). Future studies of the biochemistry of LytM factors and the amidases alike are therefore clearly needed if we are to understand how these enzymes promote proper septal PG splitting.
Another important unresolved question relates to the regulation of the LytM factors and the amidases. In general, surprisingly little is known about the regulation of PG hydrolases, especially considering the potential for disrupting this regulation as a strategy for the development of new classes of lytic antibiotics. Only very recently has the first posttranslational regulator of PG hydrolase activity been identified, the IseA protein from B. subtilis that apparently serves as a PG hydrolase inhibitor to reduce their activity as cells enter the stationary phase (93). Given the importance of protecting the PG layer from unwanted degradation, many other regulatory strategies are likely to be actively used. A potential window into these regulatory mechanisms may come from studies of β-lactam-induced lysis. As described above, cell division sites in E. coli are especially sensitive to β-lactams since membrane lesions are often observed to emanate from midcell just prior to lysis (Fig. 7C and 7E) (26, 55, 56, 76, 83). Lysis can be delayed or prevented in cells temporarily blocked for cell division (24, 36, 60). We and others have also found that β-lactam-induced lysis in E. coli is delayed in mutants lacking the amidases or the LytM factors (Fig. 7A) (44). In addition, we have shown here that the mode of cell lysis is dramatically altered in cell separation mutants. Instead of lysing from midcell lesions, chaining mutants lyse from a gradual loss of cell shape (Fig. 7). Taken together, these results, as well as similar studies from many other bacterial systems, support the commonly held notion that β-lactams promote rapid lysis by subverting the regulation of PG hydrolysis at the division site during cytokinesis. Consistent with this idea, EnvC, NlpD, and AmiC all appear to be recruited to the division site during cell division (4, 5). The subcellular localization of AmiB has not been previously reported, but our preliminary studies suggest that it is also greatly enriched at the division site (T. Dinh, unpublished data). Thus, most of the factors implicated in septal PG splitting are perfectly positioned to promote the improper PG degradation that ultimately results in cell lysis after treatment with β-lactams. Continued study of the septal PG splitting process in E. coli is therefore likely to teach us as much about PG hydrolase regulation and the mechanism of β-lactam-induced lysis as it will about the division process itself.
Acknowledgments
We thank Steve Sandler for the synthetic mCherry gene, the Kahne lab for the ΔamiB::Kanr strain, and the E. coli Genetic Stock Center for supplying mutants from the Keio Collection. We also thank Maria Ericsson, Elizabeth Benecchi, and Louise Trakimas at the HMS EM facility for their help preparing and visualizing the thin sections of the LytM− mutants. Special thanks go to John Collier and Adam Barker for help with mass spectrometry.
This study was supported by funds from the Armenise-Harvard Foundation, the Massachusetts Life Science Center, and the Burroughs Wellcome Fund. T.G.B. holds a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund.
Footnotes
Published ahead of print on 12 June 2009.
REFERENCES
- 1.Baba, T., T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner, and H. Mori. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bayles, K. W. 2000. The bactericidal action of penicillin: new clues to an unsolved mystery. Trends Microbiol. 8:274-278. [DOI] [PubMed] [Google Scholar]
- 3.Bendezú, F. O., and P. A. de Boer. 2008. Conditional lethality, division defects, membrane involution, and endocytosis in mre and mrd shape mutants of Escherichia coli. J. Bacteriol. 190:1792-1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernhardt, T. G., and P. A. de Boer. 2003. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol. Microbiol. 48:1171-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernhardt, T. G., and P. A. de Boer. 2004. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol. Microbiol. 52:1255-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernhardt, T. G., and P. A. de Boer. 2005. SlmA, a nucleoid-associated, FtsZ binding protein required for blocking septal ring assembly over chromosomes in Escherichia coli. Mol. Cell 18:555-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertsche, U., T. Kast, B. Wolf, C. Fraipont, M. E. Aarsman, K. Kannenberg, M. von Rechenberg, M. Nguyen-Distèche, T. den Blaauwen, J. V. Höltje, and W. Vollmer. 2006. Interaction between two murein (peptidoglycan) synthases, PBP3 and PBP1B, in Escherichia coli. Mol. Microbiol. 61:675-690. [DOI] [PubMed] [Google Scholar]
- 8.Bi, E. F., and J. Lutkenhaus. 1991. FtsZ ring structure associated with division in Escherichia coli. Nature 354:161-164. [DOI] [PubMed] [Google Scholar]
- 9.Browder, H., W. Zygmunt, J. Young, and P. Tavormina. 1965. Lysostaphin: enzymatic mode of action. Biochem. Biophys. Res. Commun. 19:383-389. [DOI] [PubMed] [Google Scholar]
- 10.Buist, G., J. Kok, K. J. Leenhouts, M. Dabrowska, G. Venema, and A. J. Haandrikman. 1995. Molecular cloning and nucleotide sequence of the gene encoding the major peptidoglycan hydrolase of Lactococcus lactis, a muramidase needed for cell separation. J. Bacteriol. 177:1554-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burdett, I. D. 1979. Electron microscope study of the rod-to-coccus shape change in a temperature-sensitive rod mutant of Bacillus subtilis. J. Bacteriol. 137:1395-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burdett, I. D., and R. G. Murray. 1974. Electron microscope study of septum formation in Escherichia coli strains B and B-r during synchronous growth. J. Bacteriol. 119:1039-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burdett, I. D., and R. G. Murray. 1974. Septum formation in Escherichia coli: characterization of septal structure and the effects of antibiotics on cell division. J. Bacteriol. 119:303-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carroll, S. A., T. Hain, U. Technow, A. Darji, P. Pashalidis, S. W. Joseph, and T. Chakraborty. 2003. Identification and characterization of a peptidoglycan hydrolase, MurA, of Listeria monocytogenes, a muramidase needed for cell separation. J. Bacteriol. 185:6801-6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen, J. C., P. H. Viollier, and L. Shapiro. 2005. A membrane metalloprotease participates in the sequential degradation of a Caulobacter polarity determinant. Mol. Microbiol. 55:1085-1103. [DOI] [PubMed] [Google Scholar]
- 16.Cloud, K. A., and J. P. Dillard. 2004. Mutation of a single lytic transglycosylase causes aberrant septation and inhibits cell separation of Neisseria gonorrhoeae. J. Bacteriol. 186:7811-7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen, D. N., Y. Y. Sham, G. D. Haugstad, Y. Xiang, M. G. Rossmann, D. L. Anderson, and D. L. Popham. 2009. Shared catalysis in virus entry and bacterial cell wall depolymerization. J. Mol. Biol. 387:607-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Boer, P. A., R. E. Crossley, and L. I. Rothfield. 1989. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in Escherichia coli. Cell 56:641-649. [DOI] [PubMed] [Google Scholar]
- 20.den Blaauwen, T., M. A. de Pedro, M. Nguyen-Distèche, and J. A. Ayala. 2008. Morphogenesis of rod-shaped sacculi. FEMS Microbiol. Rev. 32:321-344. [DOI] [PubMed] [Google Scholar]
- 21.den Blaauwen, T., M. E. Aarsman, N. O. Vischer, and N. Nanninga. 2003. Penicillin-binding protein PBP2 of Escherichia coli localizes preferentially in the lateral wall and at mid-cell in comparison with the old cell pole. Mol. Microbiol. 47:539-547. [DOI] [PubMed] [Google Scholar]
- 22.Denome, S. A., P. K. Elf, T. A. Henderson, D. E. Nelson, and K. D. Young. 1999. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J. Bacteriol. 181:3981-3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Pedro, M. A., J. C. Quintela, J. V. Höltje, and H. Schwarz. 1997. Murein segregation in Escherichia coli. J. Bacteriol. 179:2823-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Pedro, M. A., J. V. Höltje, and H. Schwarz. 2002. Fast lysis of Escherichia coli filament cells requires differentiation of potential division sites. Microbiology 148:79-86. [DOI] [PubMed] [Google Scholar]
- 25.de Pedro, M. A., W. D. Donachie, J. V. Höltje, and H. Schwarz. 2001. Constitutive septal murein synthesis in Escherichia coli with impaired activity of the morphogenetic proteins RodA and penicillin-binding protein 2. J. Bacteriol. 183:4115-4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donachie, W. D., and K. J. Begg. 1970. Growth of the bacterial cell. Nature 227:1220-1224. [DOI] [PubMed] [Google Scholar]
- 27.Fan, D. P. 1970. Autolysin(s) of Bacillus subtilis as dechaining enzyme. J. Bacteriol. 103:494-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feilmeier, B. J., G. Iseminger, D. Schroeder, H. Webber, and G. J. Phillips. 2000. Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J. Bacteriol. 182:4068-4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finn, R. D., J. Tate, J. Mistry, P. C. Coggill, S. J. Sammut, H. R. Hotz, G. Ceric, K. Forslund, S. R. Eddy, E. L. Sonnhammer, and A. Bateman. 2008. The Pfam protein families database. Nucleic Acids Res. 36:D281-D288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Firczuk, M., A. Mucha, and M. Bochtler. 2005. Crystal structures of active LytM. J. Mol. Biol. 354:578-590. [DOI] [PubMed] [Google Scholar]
- 31.Firczuk, M., and M. Bochtler. 2007. Folds and activities of peptidoglycan amidases. FEMS Microbiol. Rev. 31:676-691. [DOI] [PubMed] [Google Scholar]
- 32.Forsberg, C., and H. J. Rogers. 1971. Autolytic enzymes in growth of bacteria. Nature 229:272-273. [DOI] [PubMed] [Google Scholar]
- 33.Fukushima, T., A. Afkham, S. Kurosawa, T. Tanabe, H. Yamamoto, and J. Sekiguchi. 2006. A new d,l-endopeptidase gene product, YojL (renamed CwlS), plays a role in cell separation with LytE and LytF in Bacillus subtilis. J. Bacteriol. 188:5541-5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fung, J., T. J. MacAlister, and L. I. Rothfield. 1978. Role of murein lipoprotein in morphogenesis of the bacterial division septum: phenotypic similarity of lkyD and lpo mutants. J. Bacteriol. 133:1467-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia, D. L., and J. P. Dillard. 2006. AmiC functions as an N-acetylmuramyl-l-alanine amidase necessary for cell separation and can promote autolysis in Neisseria gonorrhoeae. J. Bacteriol. 188:7211-7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.García del Portillo, F., M. A. de Pedro, D. Joseleau-Petit, and R. D'Ari. 1989. Lytic response of Escherichia coli cells to inhibitors of penicillin-binding proteins 1a and 1b as a timed event related to cell division. J. Bacteriol. 171:4217-4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.García, P., M. P. González, E. García, R. López, and J. L. García. 1999. LytB, a novel pneumococcal murein hydrolase essential for cell separation. Mol. Microbiol. 31:1275-1281. [DOI] [PubMed] [Google Scholar]
- 38.Glauner, B., J. V. Höltje, and U. Schwarz. 1988. The composition of the murein of Escherichia coli. J. Biol. Chem. 263:10088-10095. [PubMed] [Google Scholar]
- 39.Goodell, E. W., R. Lopez, and A. Tomasz. 1976. Suppression of lytic effect of beta lactams on Escherichia coli and other bacteria. Proc. Natl. Acad. Sci. USA 73:3293-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guzman, L. M., J. J. Barondess, and J. Beckwith. 1992. FtsL, an essential cytoplasmic membrane protein involved in cell division in Escherichia coli. J. Bacteriol. 174:7716-7728. [PMC free article] [PubMed] [Google Scholar]
- 41.Haldimann, A., and B. L. Wanner. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J. Bacteriol. 183:6384-6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hara, H., S. Narita, D. Karibian, J. T. Park, Y. Yamamoto, and Y. Nishimura. 2002. Identification and characterization of the Escherichia coli envC gene encoding a periplasmic coiled-coil protein with putative peptidase activity. FEMS Microbiol. Lett. 212:229-236. [DOI] [PubMed] [Google Scholar]
- 43.Hayashi, K. 1975. A rapid determination of sodium dodecyl sulfate with methylene blue. Anal. Biochem. 67:503-506. [DOI] [PubMed] [Google Scholar]
- 44.Heidrich, C., A. Ursinus, J. Berger, H. Schwarz, and J. V. Höltje. 2002. Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli. J. Bacteriol. 184:6093-6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heidrich, C., M. F. Templin, A. Ursinus, M. Merdanovic, J. Berger, H. Schwarz, M. A. de Pedro, and J. V. Höltje. 2001. Involvement of N-acetylmuramyl-l-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol. Microbiol. 41:167-178. [DOI] [PubMed] [Google Scholar]
- 46.Höltje, J. V. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62:181-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ichikawa, J. K., C. Li, J. Fu, and S. Clarke. 1994. A gene at 59 min on the Escherichia coli chromosome encodes a lipoprotein with unusual amino acid repeat sequences. J. Bacteriol. 176:1630-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ichimura, T., M. Yamazoe, M. Maeda, C. Wada, and S. Hiraga. 2002. Proteolytic activity of YibP protein in Escherichia coli. J. Bacteriol. 184:2595-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ingram, L. O., and H. C. Aldrich. 1974. Cell separation in blue-green bacteria. J. Bacteriol. 118:708-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ishikawa, S., Y. Hara, R. Ohnishi, and J. Sekiguchi. 1998. Regulation of a new cell wall hydrolase gene, cwlF, which affects cell separation in Bacillus subtilis. J. Bacteriol. 180:2549-2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson, J. E., L. L. Lackner, C. A. Hale, and P. A. de Boer. 2004. ZipA is required for targeting of DMinC/DicB, but not DMinC/MinD, complexes to septal ring assemblies in Escherichia coli. J. Bacteriol. 186:2418-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kajimura, J., T. Fujiwara, S. Yamada, Y. Suzawa, T. Nishida, Y. Oyamada, I. Hayashi, J. Yamagishi, H. Komatsuzawa, and M. Sugai. 2005. Identification and molecular characterization of an N-acetylmuramyl-l-alanine amidase Sle1 involved in cell separation of Staphylococcus aureus. Mol. Microbiol. 58:1087-1101. [DOI] [PubMed] [Google Scholar]
- 53.Layec, S., B. Decaris, and N. Leblond-Bourget. 2008. Diversity of Firmicutes peptidoglycan hydrolases and specificities of those involved in daughter cell separation. Res. Microbiol. 159:507-515. [DOI] [PubMed] [Google Scholar]
- 54.Layec, S., J. Gérard, V. Legué, M. P. Chapot-Chartier, P. Courtin, F. Borges, B. Decaris, and N. Leblond-Bourget. 2009. The CHAP domain of Cse functions as an endopeptidase that acts at mature septa to promote Streptococcus thermophilus cell separation. Mol. Microbiol. 71:1205-1217. [DOI] [PubMed] [Google Scholar]
- 55.Lederberg, J. 1956. Bacterial protoplasts induced by penicillin. Proc. Natl. Acad. Sci. USA 42:574-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lederberg, J. 1957. Mechanism of action of penicillin. J. Bacteriol. 73:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewenza, S., D. Vidal-Ingigliardi, and A. P. Pugsley. 2006. Direct visualization of red fluorescent lipoproteins indicates conservation of the membrane sorting rules in the family Enterobacteriaceae. J. Bacteriol. 188:3516-3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lominski, I., J. Cameron, and G. Wyllie. 1958. Chaining and unchaining Streptococcus faecalis: a hypothesis of the mechanism of bacterial cell separation. Nature 181:1477. [DOI] [PubMed] [Google Scholar]
- 59.Mesnage, S., F. Chau, L. Dubost, and M. Arthur. 2008. Role of N-acetylglucosaminidase and N-acetylmuramidase activities in Enterococcus faecalis peptidoglycan metabolism. J. Biol. Chem. 283:19845-19853. [DOI] [PubMed] [Google Scholar]
- 60.Miller, C., L. E. Thomsen, C. Gaggero, R. Mosseri, H. Ingmer, and S. N. Cohen. 2004. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science 305:1629-1631. [DOI] [PubMed] [Google Scholar]
- 61.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 62.Morlot, C., A. Zapun, O. Dideberg, and T. Vernet. 2003. Growth and division of Streptococcus pneumoniae: localization of the high-molecular-weight penicillin-binding proteins during the cell cycle. Mol. Microbiol. 50:845-855. [DOI] [PubMed] [Google Scholar]
- 63.Nelson, D. E., and K. D. Young. 2000. Penicillin binding protein 5 affects cell diameter, contour, and morphology of Escherichia coli. J. Bacteriol. 182:1714-1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Odintsov, S. G., I. Sabala, M. Marcyjaniak, and M. Bochtler. 2004. Latent LytM at 1.3Å resolution. J. Mol. Biol. 335:775-785. [DOI] [PubMed] [Google Scholar]
- 65.Ohnishi, R., S. Ishikawa, and J. Sekiguchi. 1999. Peptidoglycan hydrolase LytF plays a role in cell separation with CwlF during vegetative growth of Bacillus subtilis. J. Bacteriol. 181:3178-3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pédelacq, J. D., S. Cabantous, T. Tran, T. C. Terwilliger, and G. S. Waldo. 2006. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24:79-88. [DOI] [PubMed] [Google Scholar]
- 67.Pogliano, J., K. Pogliano, D. S. Weiss, R. Losick, and J. Beckwith. 1997. Inactivation of FtsI inhibits constriction of the FtsZ cytokinetic ring and delays the assembly of FtsZ rings at potential division sites. Proc. Natl. Acad. Sci. USA 94:559-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Priyadarshini, R., D. L. Popham, and K. D. Young. 2006. Daughter cell separation by penicillin-binding proteins and peptidoglycan amidases in Escherichia coli. J. Bacteriol. 188:5345-5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Priyadarshini, R., M. A. de Pedro, and K. D. Young. 2007. Role of peptidoglycan amidases in the development and morphology of the division septum in Escherichia coli. J. Bacteriol. 189:5334-5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rice, K. C., and K. W. Bayles. 2008. Molecular control of bacterial death and lysis. Microbiol. Mol. Biol. Rev. 72:85-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rodolakis, A., P. Thomas, and J. Starka. 1973. Morphological mutants of Escherichia coli: isolation and ultrastructure of a chain-forming envC mutant. J. Gen. Microbiol. 75:409-416. [DOI] [PubMed] [Google Scholar]
- 72.Ryter, A., Y. Hirota, and U. Schwarz. 1973. Process of cellular division in Escherichia coli growth pattern of E. coli murein. J. Mol. Biol. 78:185-195. [DOI] [PubMed] [Google Scholar]
- 73.Scheffers, D. J., and J. Errington. 2004. PBP1 is a component of the Bacillus subtilis cell division machinery. J. Bacteriol. 186:5153-5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scheffers, D. J., L. J. Jones, and J. Errington. 2004. Several distinct localization patterns for penicillin-binding proteins in Bacillus subtilis. Mol. Microbiol. 51:749-764. [DOI] [PubMed] [Google Scholar]
- 75.Schleifer, K. H., and O. Kandler. 1972. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36:407-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schwarz, U., A. Asmus, and H. Frank. 1969. Autolytic enzymes and cell division of Escherichia coli. J. Mol. Biol. 41:419-429. [DOI] [PubMed] [Google Scholar]
- 77.Schwarz, U., A. Ryter, A. Rambach, R. Hellio, and Y. Hirota. 1975. Process of cellular division in Escherichia coli: differentiation of growth zones in the sacculus. J. Mol. Biol. 98:749-759. [DOI] [PubMed] [Google Scholar]
- 78.Sham, L. T., W. L. Ng, H. C. T. Tsui, R. J. Arnold, and M. E. Winkler. 2009. Influences of capsule on cell shape and chain formation of wild-type and pcsB mutants of serotype 2 Streptococcus pneumoniae. J. Bacteriol. 191:3024-3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shaner, N. C., R. E. Campbell, P. A. Steinbach, B. N. Giepmans, A. E. Palmer, and R. Y. Tsien. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22:1567-1572. [DOI] [PubMed] [Google Scholar]
- 80.Spratt, B. G. 1975. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc. Natl. Acad. Sci. USA 72:2999-3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spratt, B. G. 1977. Temperature-sensitive cell division mutants of Escherichia coli with thermolabile penicillin-binding proteins. J. Bacteriol. 131:293-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stapleton, M. R., M. J. Horsburgh, E. J. Hayhurst, L. Wright, I. M. Jonsson, A. Tarkowski, J. F. Kokai-Kun, J. J. Mond, and S. J. Foster. 2007. Characterization of IsaA and SceD, two putative lytic transglycosylases of Staphylococcus aureus. J. Bacteriol. 189:7316-7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Staugaard, P., F. M. van den Berg, C. L. Woldringh, and N. Nanninga. 1976. Localization of ampicillin-sensitive sites in Escherichia coli by electron microscopy. J. Bacteriol. 127:1376-1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sugai, M., H. Komatsuzawa, T. Akiyama, Y. M. Hong, T. Oshida, Y. Miyake, T. Yamaguchi, and H. Suginaka. 1995. Identification of endo-β-N-acetylglucosaminidase and N-acetylmuramyl-l-alanine amidase as cluster-dispersing enzymes in Staphylococcus aureus. J. Bacteriol. 177:1491-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tomasz, A. 1979. The mechanism of the irreversible antimicrobial effects of penicillins: how the beta-lactam antibiotics kill and lyse bacteria. Annu. Rev. Microbiol. 33:113-137. [DOI] [PubMed] [Google Scholar]
- 86.Tomasz, A., A. Albino, and E. Zanati. 1970. Multiple antibiotic resistance in a bacterium with suppressed autolytic system. Nature 227:138-140. [DOI] [PubMed] [Google Scholar]
- 87.Vollmer, W., B. Joris, P. Charlier, and S. Foster. 2008. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 32:259-286. [DOI] [PubMed] [Google Scholar]
- 88.Wang, L., M. K. Khattar, W. D. Donachie, and J. Lutkenhaus. 1998. FtsI and FtsW are localized to the septum in Escherichia coli. J. Bacteriol. 180:2810-2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weigand, R. A., K. D. Vinci, and L. I. Rothfield. 1976. Morphogenesis of the bacterial division septum: a new class of septation-defective mutants. Proc. Natl. Acad. Sci. USA 73:1882-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weiss, D. S., K. Pogliano, M. Carson, L. M. Guzman, C. Fraipont, M. Nguyen-Distèche, R. Losick, and J. Beckwith. 1997. Localization of the Escherichia coli cell division protein Ftsl (PBP3) to the division site and cell pole. Mol. Microbiol. 25:671-681. [DOI] [PubMed] [Google Scholar]
- 91.Wientjes, F. B., and N. Nanninga. 1989. Rate and topography of peptidoglycan synthesis during cell division in Escherichia coli: concept of a leading edge. J. Bacteriol. 171:3412-3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamada, S., M. Sugai, H. Komatsuzawa, S. Nakashima, T. Oshida, A. Matsumoto, and H. Suginaka. 1996. An autolysin ring associated with cell separation of Staphylococcus aureus. J. Bacteriol. 178:1565-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamamoto, H., M. Hashimoto, Y. Higashitsuji, H. Harada, N. Hariyama, L. Takahashi, T. Iwashita, S. Ooiwa, and J. Sekiguchi. 2008. Post-translational control of vegetative cell separation enzymes through a direct interaction with specific inhibitor IseA in Bacillus subtilis. Mol. Microbiol. 70:168-182. [DOI] [PubMed] [Google Scholar]
- 94.Yamamoto, H., S. Kurosawa, and J. Sekiguchi. 2003. Localization of the vegetative cell wall hydrolases LytC, LytE, and LytF on the Bacillus subtilis cell surface and stability of these enzymes to cell wall-bound or extracellular proteases. J. Bacteriol. 185:6666-6677. [DOI] [PMC free article] [PubMed] [Google Scholar]