

Abstract
Testicular orphan nuclear receptor 4 (TR4) plays essential roles for normal spermatogenesis in male mice. However, its roles in female fertility and ovarian function remain largely unknown. Here we found female mice lacking TR4 (TR4−/−) displayed subfertility and irregular estrous cycles. TR4−/− female mice ovaries were smaller with fewer or no preovulatory follicles and corpora lutea. After superovulation, TR4−/− female mice produced fewer oocytes, preovulatory follicles, and corpora lutea. In addition, more intensive granulosa apoptosis was found in TR4−/− ovaries. Functional analyses suggest that subfertility in TR4−/− female mice can be due to an ovarian defect with impaired folliculogenesis rather than a deficiency in pituitary gonadotropins. Molecular mechanism dissection of defective folliculogenesis found TR4 might induce LH receptor (LHR) gene expression via direct binding to its 5′ promoter. The consequence of reduced LHR expression in TR4−/− female mice might then result in reduced gonadal sex hormones via reduced expression of enzymes involved in steroidogenesis. Together, our results showed TR4 might play essential roles in normal folliculogenesis by influencing LHR signals. Modulation of TR4 expression and/or activation via its upstream signals or unidentified ligand(s) might allow us to develop small molecule(s) to control folliculogenesis.
IN THE UNITED STATES, 15% of women during their reproductive years experience difficulty in conceiving, and infertility affects one in seven couples in the United Kingdom (1). The most common causes of fertility problems are unexplained (30%) or ovulatory failure (27%) (1). Ovarian folliculogenesis is a process of the follicular development starting from the smallest primordial follicles to the largest preovulatory follicles, which ovulate in response to the LH surge. After ovulation, the granulosa cells and theca cells within the preovulatory follicles differentiate into luteal cells to form the corpora lutea (CL) (2). The pituitary gonadotropins FSH and LH, as well as sex hormones, are key hormones that regulate folliculogenesis.
The human and rat testicular orphan nuclear receptor 4 (TR4) cDNAs were initially cloned from human and rat hypothalamus, prostate, and testis libraries with a molecular mass of 67 kDa (3,4). TR4 is widely expressed in both embryonic and adult tissues, suggesting that TR4 affects various developmental and physiological pathways. RT-PCR analysis revealed that TR4 was detectable in the brain, placenta, and ovary (5). Nevertheless, TR4 can regulate transcription of downstream target genes by binding to AGGTCA direct repeat (DR)-like motif located in target genes. TR4 also affects many signaling pathways and modulates the transactivation of other nuclear receptors, such as retinoic acid receptor (RAR), retinoid X receptor (RXR), Vitamin D receptor (VDR), androgen receptor (AR), and estrogen receptor (ER) (6,7,8,9). In addition, TR4 was shown to activate human LH receptor (hLHR) expression in vitro (10). LHR belongs to the large family of G protein-coupled receptors and is responsible for the transduction of the biological actions of LH to their target cells, which is essential for the final stage of follicular maturation and steroidogenesis.
In our previous study, we found sperm production was decreased in TR4−/− male mice, suggesting TR4 plays a critical role in mouse spermatogenesis (11). However, its expression and physiological function in the female reproductive system remain to be explored. Here we found TR4−/− female mice displayed subfertility due to defects in folliculogenesis. Molecular mechanism studies further suggested that TR4 controls folliculogenesis via transcriptional regulation of LHR. Together, our results demonstrate that TR4 might play a critical role in normal folliculogenesis.
RESULTS
Confirmation of Knockout of the TR4 Gene in Female Mice
The deletion of TR4 at both genomic DNA and mRNA levels in TR4−/− female mice was confirmed by PCR analysis of DNA extracted from mouse tail biopsy specimens with two sets of primers that found TR4+/+ female mice have an intact TR4 gene and TR4−/− female mice have DNA deleted between exons 4 and 5 (Fig. 1A). Real-time quantitative PCR (Q-PCR) analyses of RNA extracted from the ovaries of TR4+/+ and TR4−/− female mice also confirmed TR4 mRNA is undetectable in TR4−/− female mice (Fig. 1B).
Figure 1.

Confirmation of Knockout of TR4 Gene in TR4−/− Female Mice
A, PCR analysis of mouse genomic DNA. The wild-type (+/+) and knockout (−/−) give 455- and 760-bp PCR products, respectively. B, Expression of TR4 mRNA in ovarian tissue from TR4+/+ and TR4−/− female mice by Q-PCR (n = 4 mice per group). C–F, Immunostaining of TR4 protein in ovarian sections from TR4+/+ and TR4−/− mice: C and D, TR4−/− ovary shows no TR4 staining in granulosa cell and oocyte; E and F, in TR4+/+ ovary, TR4 staining was found in oocytes and granulosa cells of primary, preantral, and antral follicles and was found in lutea cells of CL.
The loss of TR4 protein in the ovary was further confirmed by immunohistochemical (IHC) staining using an anti-TR4 antibody. As shown in Fig. 1, C and D, no staining can be detected in ovaries of TR4−/− female mice. Because previous results found that TR4 mRNA was expressed in the ovary (5), we investigated which type of cells in ovary express TR4 via IHC analysis. As shown in Fig. 1, E and F, TR4 was detected immunohistochemically in oocytes and granulosa cells, with predominant expression in the nuclei of granulosa cells. The most intensive staining was observed in primary and early antral follicles, and luteal cells with some weak staining also detected in thecal cells and stromal cells (Fig. 1, E and F).
Subfertility of TR4−/− Female Mice
First, we found the number of female TR4 knockout mice was reduced during the knockout mice generation procedures. To check whether TR4 knockout in female mice might cause the subfertility problem, a continuous mating study using sexually mature female mice (n = 5 for each genotype) at 10–12 wk of age mated with fertile male mice was conducted, and the results are shown in Table 1. After 12 wk of mating, female TR4−/− mice consistently exhibited reduced fertility with a significant decrease of litter size (5.25 ± 1.66 pups per litter, P < 0.001) when compared with either TR4+/+ or TR4+/− female mice (8.95 ± 1.39 and 9.15 ± 1.34 pups per litter), respectively (Table 1). In addition, TR4−/− mice also had fewer litters in a 12-wk period when compared with TR4+/+ and TR4+/− (2.4 ± 0.55, 4 ± 0, and 3.8 ± 0.45 litters, respectively, P < 0.05). Together, a prolonged estrous cycle and significantly longer diestrus in TR4−/− mice might contribute to the reduced litter numbers observed in the continuous mating test.
Table 1.
Fertility Test of TR4 Females
| Genotype | n | Litters | Pups | Pups Per Litter | Litters Per Female |
|---|---|---|---|---|---|
| TR4+/+ | 5 | 20 | 179 | 8.95 ± 1.39 | 4 ± 0 |
| TR4+/− | 5 | 19 | 174 | 9.15 ± 1.34 | 3.8 ± 0.45 |
| TR4−/− | 5 | 12 | 63 | 5.25 ± 1.66b | 2.4 ± 0.55a |
Known fertile males were bred with 10- to 11-wk-old female littermates for 12 wk. Results are presented as mean ± sd.
P < 0.05 vs. TR4+/+ and TR4+/−, Student's unpaired t test.
P < 0.001 vs. TR4+/+ and TR4+/−, Student's unpaired t test.
Prolonged and Irregular Estrous Cycles in TR4−/− Female Mice
To examine why the TR4−/− female has reduced reproductive performance, the estrous cycle of TR4+/+ and TR4−/− female mice was checked. TR4+/+ and TR4−/− female mice were examined daily for determination of onset of vaginal opening and were also monitored by vaginal lavage to record estrous cyclicity. TR4−/− females mice exhibited prolonged and irregular cycles characterized by a prolonged period of diestrus (67.4 ± 9.93 vs. 41.2 ± 5.21%) (Fig. 2, A and B). The timing of vaginal opening and first ovulation was also found delayed in TR4−/− (TR4+/+, 37.6 ± 1.81 d; TR4−/−, 51.2 ± 5.8 d) (Fig. 2, C and D).
Figure 2.

The Estrous Cycle (n = 5 Mice Per Group) Measured during a Period of 6 wk
A, Representative estrous cycle in TR4+/+ (upper) and TR4−/− (lower) mice (E, estrus; D, diestrus; P, proestrus); B, percentage of days spent in each estrous cycle in TR4+/+ and TR4−/− mice; C, comparison of vaginal opening in TR4+/+ and TR4−/− female mice at 5+ wk old; D, age of first ovulation in TR4+/+ and TR4−/−mice.
Genital Tract Phenotype of TR4−/− Female Mice
Next, we used comparative histology to examine the reproductive organs from eight age-matched TR4+/+ and TR4−/− female mice at age 12–22 wk. The genital tract weights and appearances during diestrous or estrous stages were also compared. As shown in Fig. 3, A and B, the weights of genital organs (bilateral ovaries, uterus, and vagina) were significantly reduced during the estrous stage (TR4+/+ 203.7 ± 13.1 mg vs. TR4−/− 130.7 ± 16.5 mg) and diestrous stage (TR4+/+ 154.7 ± 26.9 mg vs. TR4−/− 86.7 ± 6.8 mg). More specifically, the ovary sizes were smaller compared with wild-type littermates. Comparative histology of uterine tissues from TR4+/+ and TR4−/− female mice did not reveal any abnormalities in TR4−/− female mice (data not shown). In contrast, histological examination of the ovaries of adult TR4−/− female mice during the diestrous or estrous stage revealed fewer/absent preovulatory follicle and CL (Fig. 3, C–F). All stages of primary, preantral, and antral follicles were present and histologically normal.
Figure 3.

Morphology of the Genital Tracts of Sexually Mature 16-wk-old TR4+/+ and TR4−/− Females at Estrous (A) and Diestrous (B) Stage
The estrous stage of the mice was determined by vaginal smears. TR4−/− mice (left) have smaller genital organ sizes and ovaries compared with TR4+/+ (right). O, Ovary; U, uterus; V, vagina. Ovaries from sexually mature 16-wk-old TR4+/+ females compared with their TR4−/− counterparts at estrous (C and E) and diestrous (D and F) stage. Histological sections revealed that TR4−/− ovaries are smaller with fewer or absent preovulatory follicles (POF) and CL. A, Antral follicle.
Fewer Oocytes in TR4−/− Female Mice after Superovulation
To further test whether the ovarian functional defects in TR4−/− female mice are the result of impaired pre- or postovulatory mechanisms, we induced superovulation with exogenous gonadotropins in immature TR4+/+, TR4+/−, and TR4−/− female mice at 26 d old to determine the oocyte production rate. This treatment can synchronize follicular development and induce ovulation of large numbers of oocytes. The results from 15 mice (Table 2) indicated that TR4−/− female mice produced fewer oocytes compared with TR4+/+ and TR4+/− female mice (TR4−/−, 14.6 ± 4; TR4+/+, 27.2 ± 4.7; TR4+/−, 25.2 ± 3.7, P < 0.05) after superovulation, thus indicating functional defects in TR4−/− ovaries, which might contribute to the reduced fertility.
Table 2.
Superovulation of TR4+/+, TR4+/−, and TR4−/− Genotypes
| Genotype | n | Oocyte Count
|
|
|---|---|---|---|
| Average | Range | ||
| TR4+/+ | 5 | 27.0 ± 4.7 | 20–32 |
| TR4+/− | 5 | 26.2 ± 3.7 | 21–31 |
| TR4−/− | 5 | 14.6 ± 4.0a | 8–19 |
Female mice (n = 5) for each genotype at age 26 d were subjected to superovulation treatment. Oocytes were then surgically extracted from their oviducts, and oocytes were counted after hyaluronidase digestion for 10 min. Results are presented as mean ± sd.
P < 0.05 vs. TR4+/+ and TR4+/−, Student's unpaired t test.
Defective Folliculogenesis after Superovulation in TR4−/− Female Ovaries
To determine whether the reduced fertility is due to impaired follicle development or ovulation dysfunction in TR4−/− female mice, the morphology and histology of ovaries from 4-wk-old prepubertal TR4+/+ and TR4−/− mice with or without superovulation were compared. The genital organs and ovaries were smaller in TR4−/− female mice, and histology showed no significant difference in the number of growing follicles (Fig. 4, A and D). We injected pregnant mare serum gonadotropin (PMSG) into TR4+/+ and TR4−/− female mice for 48 h (n = 4 for each genotype), compared the morphology and follicular compartments in TR4+/+ and TR4−/− female mice and found that both TR4+/+ and TR4−/− female mice responded appropriately with swollen uterine horns. However, TR4−/− ovaries were still smaller than TR4+/+ ovaries. Histology showed fewer preovulatory follicles and more atretic follicles in the TR4−/− females ovaries (Fig. 4, B and E). The statistical analyses indicate a significant difference in the number of preovulatory follicles in TR4−/− (0.13 ± 0.03/mm2) and TR4+/+ (0.36 ± 0.07/mm2) mice (P < 0.05, Fig. 4G). Furthermore, we tested TR4+/+ and TR4−/− female mice after stimulation with PMSG for 48 h and then followed with 72 h human chorionic gonadotropin (hCG) treatment to induce CL formation. We found that TR4−/− female mice exhibited significantly fewer and smaller CL compared with their TR4+/+ counterparts (Fig. 4, C and F). The statistical analyses indicate a significant difference in the number of CL in TR4−/− (1.22 ± 0.38/mm2) and TR4+/+ (2 ± 0.35/mm2) female mice (P < 0.05, Fig. 4H).
Figure 4.

Morphology of the Genital Tracts in Immature TR4+/+ and TR4−/− Females with or without Superovulation
A and D, Genital organs of 4-wk-old TR4−/− mice are a smaller size compared with TR4+/+. The ovaries were histologically similar. B and E, PMSG for 48 h. Both TR4+/+ and TR4−/− respond to exogenous gonadotropins properly with fewer preovulatory follicles in the TR4−/− ovaries. C and F, PMSG for 48 h followed by hCG for an additional 72 h. Many CL were induced in the TR4+/+ ovaries, compared with fewer and smaller CL in the TR4−/− ovaries. G, Statistical analysis of the number of the follicular compartments in TR4+/+ and TR4−/− ovaries after PMSG for 48 h (n = 4 mice per group). H, Statistical analysis of the number and size of CL in both genotypes (n = 4 mice per group). Statistical significance was determined by using Student's unpaired and two-tailed t test. *, P < 0.05; **, P < 0.01. Representative sections are shown. A, Antral follicle; AF, atretic follicle (includes primordial, primary, preantral, and antral follicles); EA, early antral follicle; O, ovary; P, primary follicle; PF, preantral follicles; POF, preovulatory follicle; U, uterus; V, vagina. Bar, 5 mm (A–C) and 200 μm (D–F).
Superovulation experiments demonstrated that defective folliculogenesis occurred in TR4−/− female mice. We also found both serum LH and FSH concentrations were not significantly different in TR4−/− female mice as compared with TR4+/+ female mice (data not shown). These results strongly suggested that the subfertility in TR4−/− female mice is due to an ovarian defect rather than a deficiency in the pituitary gonadotropin axis. Our data identified that the defective folliculogenesis is the primary cause of subfertility and suggested that follicular development is unaffected until the late antral stage. A major decrease in preovulatory follicle numbers causes a consequent decrease in ovulation rate and number of CL formed in TR4−/− females.
Higher Granulosa Cell Apoptosis in TR4−/− Ovaries
Because treatment with exogenous PMSG induces granulosa cell proliferation and differentiation and increases the numbers of developing follicles by rescuing them from atresia, we then injected mice with PMSG for 48 h to compare apoptosis in different follicle stages between TR4+/+ and TR4−/− female ovaries. Using terminal transferase dUTP nick end labeling (TUNEL) assays, we found granulosa cell apoptosis in many preantral and early antral follicles in the TR4−/− ovaries, whereas wild type littermates have less apoptosis (Fig. 5A). The percentages of atretic follicles that contain five or more apoptotic granulosa cells were significantly greater in TR4−/− (59.17 ± 11.9% of preantral, 60 ± 13% of early antral) than TR4+/+ (30 ± 9.9% of preantral, 14.7 ± 4.13% of early antral) ovaries (P < 0.05, Fig. 5B). Similar phenomena were also observed in the ovaries of 16-wk-old TR4−/− female mice that received PMSG for 48 h and hCG for an additional 10 h (data not shown).
Figure 5.
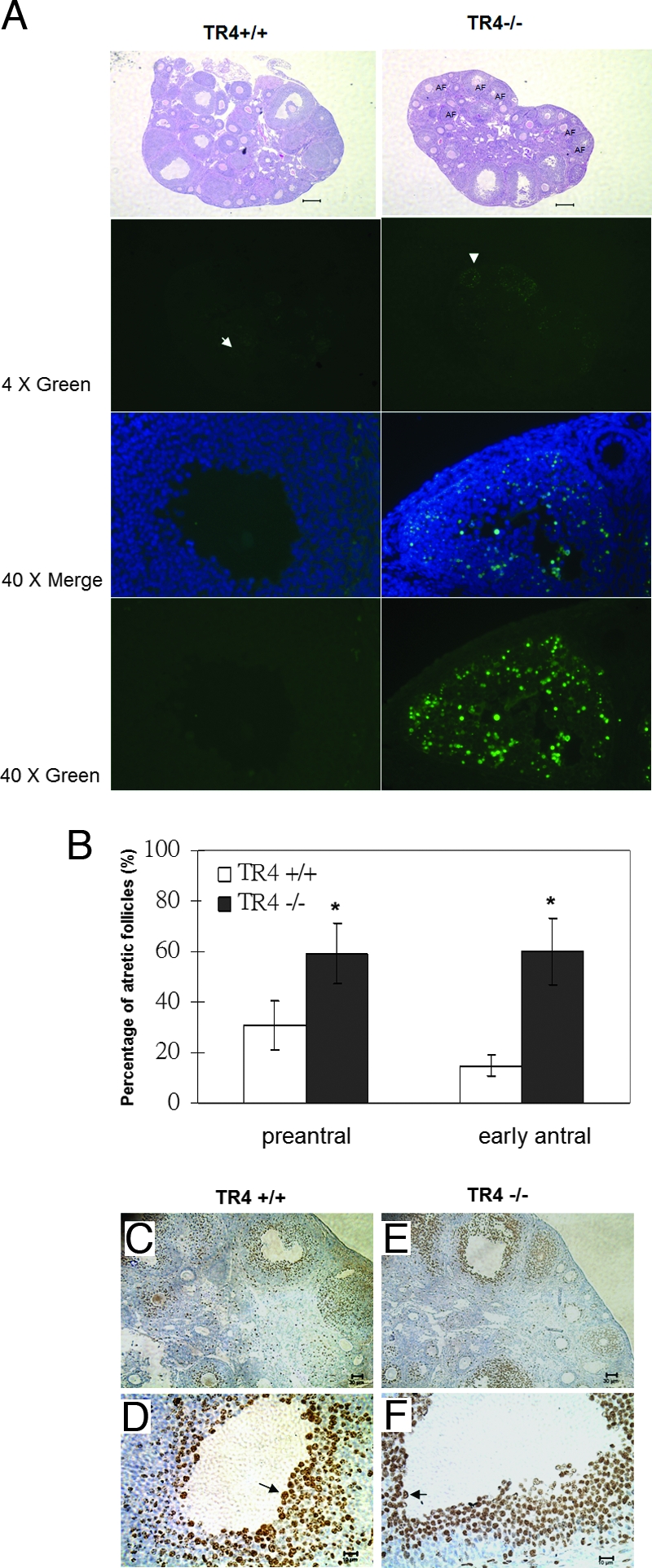
Detection of Apoptosis and Proliferation in Follicles of 4+-wk-old TR4+/+ and TR4−/− Females Treated with PMSG for 48 h (n = 3 for Each Genotype) by Using TUNEL Assay and BrdU
A, Superovulated ovaries from TR4+/+ and TR4−/− females were compared. Little TUNEL-positive staining was found in granulosa cells of preantral and early antral follicles in TR4+/+ ovaries, whereas intensive granulosa apoptosis was seen in many preantral and early antral follicles in the TR4−/− ovaries. The sections were also subjected to hematoxylin and eosin staining (lower panels). B, Statistical analysis of the percentage of the apoptotic follicles in both genotypes. Statistical significance determined by using Student's unpaired and two-tailed t test is indicated: *, P < 0.05. Data were obtained by scoring of at least four sections of each ovary stained for DAPI (DNA) and TUNEL (apoptosis). C–F, Immunostaining of granulosa cells in TR4+/+ and TR4−/− females showing incorporation of BrdU. The arrows indicate BrdU-positive cells. Bar, 200 μm (A and B), 30 μm (D and F), and 10 μm (C and E).
To determine the proliferation rate in ovary, bromodeoxyuridine (BrdU) incorporation was performed in vivo. There were many proliferating granulosa cells in a wide range of follicles from both TR4−/− and TR4+/+ mice. The positive brown staining is predominantly located within the granulosa cell nuclei of different follicle stages after PMSG treatment (Fig. 5, C–F). BrdU staining also appeared to be more intense in the granulosa cell layers closest to the oocytes.
Protein and mRNA Expression of LHR Are Reduced in TR4−/− Female Mice
Using IHC, we found immunoreactive staining of LHR on the cell surface and cytoplasmic regions of stromal, thecal, and luteal cells. LHR were located predominantly in the CL with moderate staining found in theca and stromal cells around follicles and negative staining in primary, preantral, early antral, or antral follicles (Fig. 6, A–D). We also found the reduced LHR expression in luteal cells of TR4−/− female mice as compared with their wild-type littermates (Fig. 6, A and C). We also applied Q-PCR to quantitate the LHR mRNA expression in ovaries. As shown in Fig. 6E, the LHR mRNA expression is lower in TR4−/− ovaries after 48 h PMSG treatment without addition of hCG (data not shown) or the additional 22 h of hCG (Fig. 6E).
Figure 6.

The Change of Expression Pattern of LHR Signal Pathway in TR4 Knockout Mice
A–D, Immunostaining of LHR protein in ovarian sections from TR4+/+ and TR4−/− mice: A and C, LHR was moderately expressed on both centrally and peripherally located luteinizing cells in TR4−/− CL; B and D, intensely expressed LHR was found in both centrally and peripherally located luteinizing cells in TR4+/+ CL. E, Quantitative real-time RT-PCR was performed to determine gene expression of LHR, prolactin receptor (PRLR), ERα, ERβ, FSHR, StAR, P450scc, and P450-arom using RNA from 4+-wk-old TR4+/+ and TR4−/− ovaries treated with PMSG for 48 h and then hCG for 22 h (n = 4 mice per group). F, Serum estradiol and progesterone level in 4+-wk-old mice after PMSG for 48 h and then hCG for 22 h decreased in TR4−/− females (n = 4 mice per group). Statistical significance was determined by using Student's unpaired and two-tailed t test: *, P < 0.05.
Reduced LHR Expression Results in Reduced Serum Estradiol and Progesterone via Reduced Expression of Key Enzymes Involved in Steroidogenesis
Because LHR signals play essential roles in ovarian sex hormone production via steroidogenesis, we were interested to see whether sex hormones were altered in TR4−/− female mice. We found both serum estradiol and progesterone concentrations were significantly decreased in TR4−/− female mice as compared with TR4+/+ female mice after PMSG stimulation for 48 h with an additional 22 h of hCG (Fig. 6F). We further assayed the gene expression of several key enzymes involved in steroidogenesis and found steroidogenic acute regulatory protein (StAR) and P450 side-chain cleavage (P450scc) were also reduced in TR4−/− female mice. FSH receptor (FSHR), ERα, ERβ, and P450-arom play important roles in folliculogenesis, and their expression levels are both similar in TR4−/− and TR4+/+ female mice (Fig. 6E). Together, our data from Fig. 6, A–F, suggested that TR4 might modulate LHR gene expression to control estradiol and progesterone production and play important roles in the normal ovarian function.
TR4 Induces LHR Gene Expression via Binding Directly to TR4 Response Element Located in LHR 5′ Promoter
To further dissect the molecular mechanism by which TR4 regulates LHR gene expression, we performed a promoter gene assay using a luciferase reporter plasmid linked to various lengths of 5′ promoter of the mouse LHR (mLHR) gene (12) in CV-1 cells. As expected, addition of TR4 can induce LHR expression at the transcriptional level via two different LHR 5′ promoter constructs (Fig. 7A).
Figure 7.

TR4 Mediates the Expression of LHR
A, Regulation of LHR promoter activity in cultured cells. TR4 activates the LHR promoter luciferase reporter genes pLHR(−173)-Luc and pLHR (−2040)-Luc, which contain the DR1 motif. CV-1 cells were cotransfected with LHR promoter luciferase reporter and PCMX or TR4 (PCMX-TR4). *, P < 0.05 vs. control. B, ChIP of mouse TM3 cells using TR4-specific antisera. PCR amplification was performed of mLHR promoter (−5 to −301) that includes the region containing the DR1 motif. Lane 1, marker; lane 2, input control; lane 3, control IP without normal mouse IgG; lane 4, control IP with normal mouse IgG; lane 5, PCR product obtained from immunoprecipitates using TR4 antisera no. 15, which is specific for TR4. C, cAMP assay. TM3 cells were transfected with pRetro-shiTR4 and control vector. After 24 h, TM3-shiTR4 and TM3 control cells were incubated in serum-free medium for 1 h and then further cultured in the presence of 100 ng/ml LH with or without 10 μm forskolin for 2 h. The cells were harvested, and TR4 expression level was determined by Western blotting. The intracellular cAMP levels were measured as described in Materials and Methods. The experiment was repeated twice. A Student's t test was used to compare the mean of cAMP levels between the vector control group and TR4siRNA groups treated with LH. *, P < 0.05.
Through sequence analysis of the LHR gene (13), we identified one direct repeat (DR1) fragment as a putative TR4 binding site, located between −153 and −164 bp (AGGTCAAAGGAGA). Using the chromatin immunoprecipitation (ChIP) assay, we further confirmed in vivo binding between TR4 protein and the LHR gene in mouse TM3 cells. After formaldehyde cross-linking and shearing of chromatin by sonication, protein/DNA complexes were immunoprecipitated with antibodies against normal IgG or TR4 antibody. PCR was performed with site-specific primers that cover the putative TR4 binding site. As shown in Fig. 7B, lane 5, TR4 was recruited to the DR1 region of the mLHR promoter, and no PCR product was observed in mouse IgG control pull-down, (Fig. 7B, lanes 3 and 4). Together, both 5′ promoter assay and ChIP assays demonstrated that TR4 could induce LHR gene expression via direct binding to its TR4 response element located on its 5′ promoter.
cAMP Assay
To confirm the effect of TR4 in LH/LHR signaling pathway, TR4 small interference RNA (siRNA) was used to knock down TR4 protein in TM3 cells. The influence of LH stimulation signal was measured by the secondary message of intracellular cAMP amount. As shown in Fig. 7C, the LH-stimulated intracellular cAMP amount was reduced 50% after transfecting TR4-siRNA into TM3 cells compared with the control. However, forskolin treatment in TR4 knockdown cells could compensate for the decreased effect of intracellular cAMP after LH stimulation. This result indicates that TR4 may play a role in LH/LHR signal.
DISCUSSION
A previous result indicated TR4 expression was detectable in ovary (5), and we also showed positive signals in oocytes and granulosa cells. TR4−/− mice are produced at a lower rate than the Mendelian ratios, with a significantly lower proportion of TR4−/− female mice produced (14). This study identifies the primary cause of subfertility in TR4−/− female mice as defective folliculogenesis rather than a deficiency in the pituitary gonadotropin axis. However, follicular development is unaffected until the final phase of follicular maturation. Here, we revealed novel functions of the orphan receptor, TR4, in female reproduction.
The LH-LHR signal pathway plays essential roles in many ovarian functions including the final phase of follicular maturation, ovulation, and in maintaining the CL (15,16). The lack of LHR in female mice [LHR knockout (LuRKO)] caused smaller ovaries, delayed vaginal opening, and decreased estradiol and progesterone production. Furthermore, LuRKO ovaries revealed the presence of antral follicles but absence of preovulatory follicles or CL, suggesting that normal expression of LHR is essential for ovulation as well as follicular maturation from the antral to preovulatory stage (17,18).
TR4−/− female mice also showed smaller ovaries and significantly fewer preovulatory follicles and CL compared with their TR4+/+ counterparts. The LHR, two LH response genes (P450scc and StAR), and the luteal marker PRLR (17,18), had reduced expression levels in TR4−/− female mice (Fig. 6E). Also, we found that TR4 regulates LHR by binding directly to the TR4 response element (Fig. 7A). However, the number of CLs and LHR expression are both reduced in TR4−/− female mice, and the reduced CL number may be due to reduced expression of LHR or vice versa. Therefore, it is difficult to conclude cause and effect from the results in our study, and more experiments to elucidate the relationship between cause and effect are needed.
We also found some gene expression levels that are similar in TR4+/+ and TR4−/− female mice that have been documented well to play important roles in early folliculogenesis. Follicular development is unaffected until late antral stage in TR4−/− female mice, which apparently is due to normal FSH action. This finding is also supported by the fact that FSHR, ERα, ERβ, and P450-arom mRNA expression levels are similar in TR4−/− and TR4+/+ female mice (Fig. 6 E). Interestingly, we found a high expression of growth differentiation factor 9 (GDF-9) and bone morphogenetic protein 15 (BMP-15) (19), both of which play essential roles in early stages of follicular development (data not shown). This may be due to an elevated proportion of oocytes with fewer numbers of preovulatory follicles in TR4−/− female mice.
Human females with defects of LHR expression also have defective folliculogenesis, anovulation, and delayed feminization at puberty, amenorrhea, and infertility (20,21,22,23). Our findings showing defective folliculogenesis in TR4−/− female mice with defective LHR expression match very well the phenotypes shown in LuRKO mice and human females with defective LHR expression, which strengthens our conclusion that TR4 might play important roles via induction of LHR expression at the transcriptional level.
The increase of preantral follicular atresia in TR4−/− female mice may have several factors, such as multiple paracrine and autocrine signals, that determine the follicle apoptosis. Ovarian steroid hormone plays an important role in ovarian cell death, and estradiol acts as a survival factor in granulosa cells (24). Progesterone also maintains granulosa cell viability indirectly by stimulating granulosa cells to synthesize basic fibroblast growth factor (25). Cell contact is also found to be important in the resistance of granulosa cells to apoptosis and in which gap junction protein is known to be elevated by estradiol (26). We found both estradiol and progesterone level decreases in TR4−/− female mice, which might be the cause of the increased follicular atresia. After some follicles reach the stage of the preovulatory phase, the rest of the follicles are gradually eliminated by the apoptotic process. In TR4−/− female mice, fewer follicles can reach the preovulatory stage, and the rest of the follicles are gradually eliminated by the apoptotic process. So this is another possibility of increased apoptosis in TR4-null mice. Our previous study showed that the TR4 can modulate apoptosis via regulation of Bcl-2 gene expression. Because Bcl-2 acts as a survival signal in the ovary, increased apoptosis in TR4 might be caused by down-regulation of Bcl-2 expression in TR4−/− female mice (27).
In conclusion, our data provide the in vivo evidence showing TR4 can influence folliculogenesis via alteration of LH-LHR signals. In humans, over 30% of infertility and subfertility are unexplained, and some patients have a poor response to exogenous gonadotropin treatment (28). It will be of great interest to determine whether reduced, but not total absence of, LH-LHR signal transduction would cause ovarian dysfunction. In addition, identification of small compounds that function as either ligands or modulators for signaling pathways to regulate TR4 activity would be important to control TR4-mediated LHR-mediated folliculogenesis in the clinical application. This might allow us to apply the small compound(s) to improve fertility via control of this newly identified pathway from TR4 to LHR.
MATERIALS AND METHODS
Generation of TR4−/− Mice
The strategy of generation of TR4−/− mice was described previously (14). All mice were housed in the Vivarium facility of the University of Rochester Medical Center and maintained on a 12-h light, 12-h dark cycle. The animals were provided a standard diet with constant access to food and water.
Genotyping of TR4−/− Mice
Mice used in these studies were genotyped from tail biopsy specimen DNA via PCR analyses. The primers used were the same as previously described (11).
Vaginal Smear Analysis and Fertility of Female Mice
Female TR4+/+and TR4−/− mice were individually housed, and vaginal smears were taken daily over a period of at least 6 consecutive weeks. For fertility testing, 10- to 12-wk-old female TR4+/+, TR4+/−, and TR4−/− mice (n = 5) for each genotype were subjected to a continuous mating study for 12 wk. One female mouse was housed with one 10- to 12-wk-old known fertile male mouse, and male mice were rotated biweekly for 12 wk, after which the female was separated to monitor daily for an additional 23-d period. The number of pups and litters was recorded.
Superovulation, Serum Hormone Levels, and Oocyte Quantitation
For all experiments, prepubescent 26- to 28-d-old TR4+/+ and TR4−/− littermate female mice of each genotype (n = 12 mice per genotype) were injected ip with 5 IU PMSG (Sigma Chemical Co., St. Louis, MO). A portion of the mice were killed 48 h after PMSG injection (n =4 mice per genotype), whereas the remaining mice received a single ip injection of 5 IU hCG (Sigma) to induce ovulation. Mice receiving hCG were then killed 22 h (n =4 per genotype) or 72 h after hCG administration (n =4 per genotype). At the time of killing, blood was obtained by cardiac puncture and was allowed to clot. After 15 min of centrifugation at 3000 × g, the serum was collected. The total serum estradiol and progesterone levels were measured using ELISA kits (Diagnostic System Laboratories, Inc., Webster, TX). The genital tracts were excised, and genital organ (ovary, uterus, and vagina) weights were recorded. One ovary from each animal was fixed in 4% paraformaldehyde for histological examination, and the other ovary was frozen at −70 C for RNA extraction. In the 22- and 72-h post-hCG groups, the oocyte/cumulus masses were surgically harvested from the oviducts and cultured in DMEM (Cellgro, Manassas, VA) supplemented with 10% fetal calf serum and antibiotics. Oocytes were counted after 0.3% hyaluronidase (Sigma) disassociation from surrounding cumulus cells.
Histology
Paraffin-embedded ovaries were serially sectioned at 5 μm. Every tenth section was stained with hematoxylin and eosin. The number of follicles for each ovary was counted and was normalized by the total ovarian area in the section (29). The ovary area was measured with Photoshop version 6.0 (Adobe Systems, San Jose, CA). The follicle types, including primary, preantral, early antral, antral, atretic, preovulatory follicles, and CL, were classified as described previously (29). Data are presented as mean ± sd.
IHC, BrdU Incorporation, and Apoptosis Assay
For IHC, mouse ovaries were fixed overnight in 4% paraformaldehyde. Five-micrometer-thick ovarian sections were deparaffinized and rehydrated using an alcohol gradient. Tissue sections were subjected to a microwave antigen retrieval technique Sections were incubated with mouse anti-TR4 or rabbit anti-LHR overnight at 4 C. In negative controls, normal serum was substituted for primary antibody. Avidin-biotin complex kits were obtained from Vector Laboratories (Burlingame, CA). The proliferative activity of granulosa cells was detected by BrdU incorporation, using a BrdU labeling kit from Zymed Laboratories (South San Francisco, CA). To label cells in the S phase, mice were injected ip with 100 μg BrdU/g body weight. For cell apoptosis detection, the Fluorescence FragEL DNA fragmentation kit (EMD Biosciences, San Diego, CA) was used on the tissue sections.
Q-PCR of Gene Expression
For Q-PCR analysis, total RNA was isolated from the ovaries of TR4+/+and TR4−/− mice, using the total RNA isolation system (Promega, Madison, WI). First-strand cDNA synthesis was achieved using the Superscript II RNase H reverse transcriptase kit (Invitrogen, Carlsbad, CA). The relative abundance of target mRNA was quantified relative to the control β-actin gene expression from the same reaction. Q-PCR amplifications of reverse-transcribed first-strand DNA samples were performed using the iCycler iQ PCR cycle and detection system (Bio-Rad Laboratories, Inc., Hercules, CA).
Cell Culture, Transfections, and Luciferase Assays
CV-1 cells were maintained in DMEM supplemented with 10% fetal bovine serum (10). TM3 cells were maintained in a 1:1 mixture of Ham's F-12 medium and DMEM supplemented with 4 mm glutamine and adjusted to contain 1.5 g/liter sodium bicarbonate, 4.5 g/liter glucose, 1 mm sodium pyruvate, 100 U/ml penicillin, 100 mg/ml streptomycin, 5% horse serum, and 2.5% fetal bovine serum (30). For luciferase assays, CV-1 cells were cultured in 12-well plates (Corning, Corning, NY) at a concentration of 5 × 104 cells per well and transfected with 1 μg DNA using SuperFect (QiAGEN, Valencia, CA) according to the manufacturer's protocol. The internal control plasmid pRL-TK (Promega) was cotransfected in all transfection experiments. After 48 h transfection, the cells were harvested, and luciferase assays were performed using the dual-luciferase kit (Promega). Luciferase activity was measured using a luminometer.
ChIP
Formaldehyde was added to TM3 cells to achieve a final concentration of 1% formaldehyde. Fixation was allowed to proceed at room temperature for 10 min and then was stopped by addition of glycine to a final concentration of 0.125 m. The cells were washed with PBS and collected by centrifugation. The cells were incubated with cell lysis buffer [5 mm Pipes (pH 8.0), 85 mm KCl, and 0.5% Nonidet P-40 with Roche protease inhibitor mixture] on ice for 10 min. The nuclei were collected by centrifugation, resuspended in nuclear lysis buffer [50 mm Tris (pH 8.1), 10 mm EDTA, and 1% SDS with Roche protease inhibitor mixture], and incubated on ice for 10 min. The samples were put on ice and sonicated with a Branson Sonifier 250 at a setting of 5 for eight 20-sec pulses to an average length of 600 bp (confirmed by electrophoresis) and microcentrifuged. The chromatin solution was precleared with protein A-Sepharose (Pierce, Rockford, IL) for 30 min at 4 C. IPs were performed overnight at 4 C with 1 μg anti-TR4 antibody. After immunoprecipitation, 45 μl protein A-Sepharose and 2 μg of salmon sperm DNA were added and incubated for 1 h at 4 C with rotation. Precipitates were washed sequentially for 3–5 min each in low-salt buffer [0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl (pH 8.1), 150 mm NaCl], high-salt buffer [0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl (pH 8.1), 500 mm NaCl], LiCl wash buffer [0.25 m LiCl, 1% Nonidet P-40, 1% deoxycholate, 1 mm EDTA, 10 mm Tris-HCl (pH 8.1)], and twice in 1× Tris-EDTA buffer. Then we extracted precipitates three times with 1% SDS and 0.1 m NaHCO3. Eluates were pooled and incubated in a 65 C water bath for 4 h to reverse the formaldehyde cross-linking, and we purified DNA fragments using QIAquick Spin Kit (QIAGEN). Total input chromatin samples were resuspended in 30 μl Tris-EDTA and diluted to 1:100. Each 50 μl of PCR mixture contained 5 μl IP sample, 1.5 mm MgCl2, 50 ng of each primer, 20 μm dNTP, 1× PCR buffer (Promega), and 1.25 U Taq DNA polymerase (Promega). With 45 cycles of amplification, 10 μl of each of the PCR products was subjected to electrophoresis on a 2% agarose gel, and the DNA was stained with ethidium bromide and visualized under UV light. The sequences of primers used for PCR follow: mLHR promoter DR1 (sense), 5′-GGCAGAGCAGAGTTCAAAGC-3′, and mLHR promoter DR1 (antisense), 5′-CGCCAGCCTGAGTGTGAG-3′.
cAMP Assay
TM3 Leydig cells were plated in 24-well culture dishes at 2 to 4 × l04 cells/ml in cultured medium (DMEM/Ham's F12 1:1, 5% horse serum, 2.5% fetal bovine serum, penicillin G at 100 U/ml, streptomycin at 100 μg/ml) and allowed to grow for 24 h. The cells were cotransfected with 0.35 μg pRetro-shNS control sequence (GTTCTCCGAACGTGTCACG; Cellogenetics, Inc., Ijamsville, MD) or pRetro-shiTR4 and 1/20 β-galactosidase plasmid DNA by Lipofectamine according to the protocol of the manufacturer (Life Technologies, Grand Island, NY). Cells were cultured for 48 h after transfection before this experiment. To control the transfection efficiency, cells were harvested, and the activity of β-galactosidase in the cell lysate was assayed as described previously (31). Cells were washed twice and preincubated for 15 min in serum-free medium supplemented with 0.5 mm isobutylmethylxanthine (Sigma). Triplicate wells were then incubated in the presence of 100 ng/ml LH, with or without 10 μm forskolin for 2 h. The cells were harvested, and intracellular cAMP was extracted by incubation with 0.1 m HCl for 10 min. Cell extracts were clarified by centrifugation and cAMP determined using a commercial EIA kit (Cayman Chemical Co., Ann Arbor, MI) according to the manufacturer's instructions.
Acknowledgments
We thank Karen Wolf for help with manuscript preparation.
Footnotes
This work was supported by the George H. Whipple Professorship Endowment and National Institutes of Health Grant CCA122285).
Disclosure Statement: The authors have nothing to declare.
First Published Online January 3, 2008
Abbreviations: BrdU, Bromodeoxyuridine; ChIP, chromatin immunoprecipitation; CL, corpus lutea; DR, direct repeat; ER, estrogen receptor; FSHR, FSH receptor; hCG, human chorionic gonadotropin; hLHR, human LH receptor; LuRKO, LHR knockout; mLHR, mouse LHR; PMSG, pregnant mare serum gonadotropin; P450scc, P450 side-chain cleavage; Q-PCR, quantitative PCR; siRNA, small interfering RNA; StAR, steroidogenic acute regulatory protein; TR4, testicular orphan nuclear receptor 4; TUNEL, terminal transferase dUTP nick end labeling.
References
- Schmidt L, Munster K, Helm P 1995 Infertility and the seeking of infertility treatment in a representative population. Br J Obstet Gynaecol 102:978–984 [DOI] [PubMed] [Google Scholar]
- Murphy BD 2000 Models of luteinization. Biol Reprod 63:2–11 [DOI] [PubMed] [Google Scholar]
- Chang C, Da Silva SL, Ideta R, Lee Y, Yeh S, Burbach JP 1994 Human and rat TR4 orphan receptors specify a subclass of the steroid receptor superfamily. Proc Natl Acad Sci USA 91:6040–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose T, Fujimoto W, Tamaai T, Kim KH, Matsuura H, Jetten AM 1994 TAK1: molecular cloning and characterization of a new member of the nuclear receptor superfamily. Mol Endocrinol 8:1667–1680 [DOI] [PubMed] [Google Scholar]
- Lee YF, Lee HJ, Chang C 2002 Recent advances in the TR2 and TR4 orphan receptors of the nuclear receptor superfamily. J Steroid Biochem Mol Biol 81:291–308 [DOI] [PubMed] [Google Scholar]
- Lee YF, Shyr CR, Thin TH, Lin WJ, Chang C 1999 Convergence of two repressors through heterodimer formation of androgen receptor and testicular orphan receptor-4: a unique signaling pathway in the steroid receptor superfamily. Proc Natl Acad Sci USA 96:14724–14729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YF, Young WJ, Burbach JP, Chang C 1998 Negative feedback control of the retinoid-retinoic acid/retinoid X receptor pathway by the human TR4 orphan receptor, a member of the steroid receptor superfamily. J Biol Chem 273:13437–13443 [DOI] [PubMed] [Google Scholar]
- Lee YF, Young WJ, Lin WJ, Shyr CR, Chang C 1999 Differential regulation of direct repeat 3 vitamin D3 and direct repeat 4 thyroid hormone signaling pathways by the human TR4 orphan receptor. J Biol Chem 274:16198–16205 [DOI] [PubMed] [Google Scholar]
- Shyr CR, Hu YC, Kim E, Chang C 2002 Modulation of estrogen receptor-mediated transactivation by orphan receptor TR4 in MCF-7 cells. J Biol Chem 277:14622–14628 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Dufau ML 2000 Nuclear orphan receptors regulate transcription of the gene for the human luteinizing hormone receptor. J Biol Chem 275:2763–2770 [DOI] [PubMed] [Google Scholar]
- Mu X, Lee YF, Liu NC, Chen YT, Kim E, Shyr CR, Chang C 2004 Targeted inactivation of testicular nuclear orphan receptor 4 delays and disrupts late meiotic prophase and subsequent meiotic divisions of spermatogenesis. Mol Cell Biol 24:5887–5899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hefnawy T, Krawczyk Z, Nikula H, Vihera I, Huhtaniemi I 1996 Regulation of function of the murine luteinizing hormone receptor promoter by cis- and trans-acting elements in mouse Leydig tumor cells. Mol Cell Endocrinol 119:207–217 [DOI] [PubMed] [Google Scholar]
- Huhtaniemi IT, Eskola V, Pakarinen P, Matikainen T, Sprengel R 1992 The murine luteinizing hormone and follicle-stimulating hormone receptor genes: transcription initiation sites, putative promoter sequences and promoter activity. Mol Cell Endocrinol 88:55–66 [DOI] [PubMed] [Google Scholar]
- Collins LL, Lee YF, Heinlein CA, Liu NC, Chen YT, Shyr CR, Meshul CK, Uno H, Platt KA, Chang C 2004 Growth retardation and abnormal maternal behavior in mice lacking testicular orphan nuclear receptor 4. Proc Natl Acad Sci USA 101:15058–15063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indrapichate K, Meehan D, Lane TA, Chu SY, Rao CV, Johnson D, Chen TT, Wimalasena J 1992 Biological actions of monoclonal luteinizing hormone/human chorionic gonadotropin receptor antibodies. Biol Reprod 46:265–278 [DOI] [PubMed] [Google Scholar]
- Ascoli M, Fanelli F, Segaloff DL 2002 The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev 23:141–174 [DOI] [PubMed] [Google Scholar]
- Zhang FP, Poutanen M, Wilbertz J, Huhtaniemi I 2001 Normal prenatal but arrested postnatal sexual development of luteinizing hormone receptor knockout (LuRKO) mice. Mol Endocrinol 15:172–183 [DOI] [PubMed] [Google Scholar]
- Pakarainen T, Zhang FP, Nurmi L, Poutanen M, Huhtaniemi I 2005 Knockout of luteinizing hormone receptor abolishes the effects of follicle-stimulating hormone on preovulatory maturation and ovulation of mouse Graafian follicles. Mol Endocrinol 19:2591–2602 [DOI] [PubMed] [Google Scholar]
- Moore RK, Erickson GF, Shimasaki S 2004 Are BMP-15 and GDF-9 primary determinants of ovulation quota in mammals? Trends Endocrinol Metab 15:356–361 [DOI] [PubMed] [Google Scholar]
- Latronico AC, Anasti J, Arnhold IJ, Rapaport R, Mendonca BB, Bloise W, Castro M, Tsigos C, Chrousos GP 1996 Brief report: testicular and ovarian resistance to luteinizing hormone caused by inactivating mutations of the luteinizing hormone-receptor gene. N Engl J Med 334:507–512 [DOI] [PubMed] [Google Scholar]
- Huhtaniemi IT, Themmen AP 2005 Mutations in human gonadotropin and gonadotropin-receptor genes. Endocrine 26:207–217 [DOI] [PubMed] [Google Scholar]
- Latronico AC, Chai Y, Arnhold IJ, Liu X, Mendonca BB, Segaloff DL 1998 A homozygous microdeletion in helix 7 of the luteinizing hormone receptor associated with familial testicular and ovarian resistance is due to both decreased cell surface expression and impaired effector activation by the cell surface receptor. Mol Endocrinol 12:442–450 [DOI] [PubMed] [Google Scholar]
- Wu SM, Leschek EW, Rennert OM, Chan WY 2000 Luteinizing hormone receptor mutations in disorders of sexual development and cancer. Front Biosci 5:D343–D352 [DOI] [PubMed] [Google Scholar]
- Billig H, Furuta I, Hsueh AJ 1993 Estrogens inhibit and androgens enhance ovarian granulosa cell apoptosis. Endocrinology 133:2204–2212 [DOI] [PubMed] [Google Scholar]
- Peluso JJ, Pappalardo A 1999 Progesterone maintains large rat granulosa cell viability indirectly by stimulating small granulosa cells to synthesize basic fibroblast growth factor. Biol Reprod 60:290–296 [DOI] [PubMed] [Google Scholar]
- Blaschuk OW, Farookhi R 1989 Estradiol stimulates cadherin expression in rat granulosa cells. Dev Biol 136:564–567 [DOI] [PubMed] [Google Scholar]
- Kim E, Ma WL, Lin DL, Inui S, Chen YL, Chang C 2007 TR4 orphan nuclear receptor functions as an apoptosis modulator via regulation of Bcl-2 gene expression. Biochem Biophys Res Commun 361:323–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraretti AP, Gianaroli L, Magli MC, Bafaro G, Colacurci N 2000 Female poor responders. Mol Cell Endocrinol 161:59–66 [DOI] [PubMed] [Google Scholar]
- Cheng G, Weihua Z, Makinen S, Makela S, Saji S, Warner M, Gustafsson JA, Hovatta O 2002 A role for the androgen receptor in follicular atresia of estrogen receptor β knockout mouse ovary. Biol Reprod 66:77–84 [DOI] [PubMed] [Google Scholar]
- Mather JP 1980 Establishment and characterization of two distinct mouse testicular epithelial cell lines. Biol Reprod 23:243–252 [DOI] [PubMed] [Google Scholar]
- Heinlein CA, Ting HJ, Yeh S, Chang C 1999 Identification of ARA70 as a ligand-enhanced coactivator for the peroxisome proliferator-activated receptor γ. J Biol Chem 274:16147–16152 [DOI] [PubMed] [Google Scholar]