

The use of biophysical assays permitted the identification of a specific human ACC2 carboxyl transferase (CT) domain mutant that binds inhibitors and crystallizes in their presence. This mutant led to determination of the human ACC2 CT domain–CP-640186 complex crystal structure, which revealed differences in the inhibitor conformation from the yeast protein complex that are caused by differing residues in the binding pocket.
Keywords: acetyl-CoA carboxylase, ACC, ACC2, metabolic disorders, fatty-acid metabolism
Abstract
Inhibition of acetyl-CoA carboxylase (ACC) may prevent lipid-induced insulin resistance and type 2 diabetes, making the enzyme an attractive pharmaceutical target. Although the enzyme is highly conserved amongst animals, only the yeast enzyme structure is available for rational drug design. The use of biophysical assays has permitted the identification of a specific C-terminal truncation of the 826-residue human ACC2 carboxyl transferase (CT) domain that is both functionally competent to bind inhibitors and crystallizes in their presence. This C-terminal truncation led to the determination of the human ACC2 CT domain–CP-640186 complex crystal structure, which revealed distinctions from the yeast-enzyme complex. The human ACC2 CT-domain C-terminus is comprised of three intertwined α-helices that extend outwards from the enzyme on the opposite side to the ligand-binding site. Differences in the observed inhibitor conformation between the yeast and human structures are caused by differing residues in the binding pocket.
1. Introduction
Excess accumulation of lipids in both the liver and peripheral tissue is an important contributing factor to the development of insulin resistance and related pathophysiologies including diabetes, hepatic steatosis and the metabolic syndrome (Boden, 1997 ▶; Kelley et al., 1999 ▶; Seppala-Lindroos et al., 2002 ▶). Acetyl-CoA carboxylase (ACC) plays a pivotal role in fat accumulation since its product, malonyl-CoA, is the first pathway-committed precursor for de novo fatty-acid biosynthesis and is also a major negative regulator of fatty-acid oxidation. For this reason, ACC is an attractive candidate for developing drugs that target pathophysiologies related to inappropriate fat accumulation (Harwood et al., 2003 ▶; Harwood, 2005 ▶; Lenhard & Gottschalk, 2002 ▶).
There are two mammalian ACC isozymes, ACC1 and ACC2, which are transcribed by separate genes but have discrete but related physiological roles. ACC1, which is found largely in the cytosol, is the major isoform in lipogenic tissue and supports fatty-acid biosynthesis. ACC2 is the predominant form in skeletal muscle and heart and, at least in rodents, in brown adipose tissue. In muscle and heart it is localized to the outer mitochondrial membrane in proximity to carnitine palmitoyl transferase 1, where it presumably functions as a negative regulator of fatty-acid oxidation (Abu-Elheiga et al., 2000 ▶). Bianchi and coworkers also observed ACC2 in the cytosol in liver (Bianchi et al., 1990 ▶). Both ACC1 and ACC2 are high-molecular-weight proteins (ACC1, 265 kDa; ACC2, 275 kDa) that are 60% identical at the nucleotide level and 75% identical at the amino-acid level (Abu-Elheiga et al., 1997 ▶). They share the same functional domains and key regulatory phosphorylation sites, which are located in homologous regions of the proteins. They differ most significantly by the presence of an N-terminal membrane-targeting sequence on ACC2 (Abu-Elheiga et al., 1997 ▶; Ha et al., 1996 ▶; Harwood et al., 2003 ▶). Both are potentially attractive targets for drug development: ACC1 from the perspective of inhibiting lipid accumulation and ACC2 from the perspective of reducing lipid stores by enhancing fatty-acid oxidation. Because the potential exists for compensatory activation of ACC1 in response to activation of fatty-acid oxidation, targeting both isozymes simultaneously may be beneficial. In actuality, it may be difficult to do otherwise considering how similar the two isozymes are to each other.
The feasibility of targeting ACC has been evaluated using both genetic and chemical means. ACC2-knockout mice exhibited enhanced fatty-acid oxidation and reduced fat storage on a normal laboratory chow diet (Abu-Elheiga et al., 2001 ▶), and resisted the development of obesity, insulin resistance and glucose intolerance when placed on a high-fat diet (Abu-Elheiga et al., 2003 ▶). Together with the fact that ACC1-knockout mice are embryonically lethal (Abu-Elheiga et al., 2005 ▶), these results suggest that ACC2 is the more attractive target for treating lipid-related pathophysiologies. However, ACC2 can support lipogenesis under some circumstances, since ACC2 compensates for ACC1 deficiency in liver-specific ACC1-knockout mice (Harada et al., 2007 ▶). This result is consistent with the observation made by Bianchi and coworkers that in the liver ACC2 is also a cytosolic enzyme and suggests that the product pools of these two isozymes are not strictly segregated and can intermix. Therefore, it is not completely certain that the beneficial effects of knocking out the ACC2 gene derive solely from enhanced fatty-acid oxidation. In another genetic experiment, Savage et al. (2006 ▶) reported that treating high-fat-fed rats with anti-ACC antisense oligonucleotides improved hepatic insulin sensitivity. In this case, suppression of both ACC1 and ACC2 was required for the most significant effect. Finally, using a pharmacological tool, Adler and coworkers reported that treating insulin-resistant high-fat-fed mice with a dual ACC1–ACC2 inhibitor (Harwood et al., 2003 ▶) ameliorated insulin resistance (Adler et al., 2005 ▶; Harwood, 2005 ▶). Considered together, the results of these three approaches argue that a dual ACC1–ACC2 inhibitor could be an efficacious agent for lowering intracellular lipid levels, ameliorating or preventing weight gain and restoring insulin sensitivity.
Based on the available validation studies, we mounted a campaign to identify high-potency ACC1–ACC2 dual inhibitors with appropriate pharmokinetic properties to be useful as therapeutic agents. Compounds were identified using a screen based on inhibition of enzyme catalysis and were optimized by medicinal chemistry guided by structure-based design. One screening hit was a bipiperidine similar to that reported previously (Harwood et al., 2003 ▶). Using enzyme-inhibition studies, Harwood and coworkers reported that CP-610431, a molecule in this structural class, has a high probability of binding to the carboxyl transferase (CT) domain of ACC1 (Harwood et al., 2003 ▶). To guide our optimization efforts, we crystallized the CT domain of human ACC2 bound to compounds in this chemical class and now report the first crystal structure of a metazoan ACC CT domain.
Prokaryotic ACCs are multi-subunit enzymes with separate proteins for the biotin carboxylase (BC) and biotin carboxyl carrier protein (BCCP) functions and two proteins for the carboxyl transferase (CT) function. Eukaryotes, including yeast, have all these functions condensed within a single polypeptide chain. The structure of the yeast ACC CT protein answered many questions regarding the connection between prokaryotic and eukaryotic CT functionality (Zhang et al., 2003 ▶). Two domains within the yeast CT polypeptide were identified that were sequentially related to the two bacterial protein counterparts. The yeast CT polypeptide dimerizes in a head-to-tail fashion, with an extensive interface between the N-terminal domain of one polypeptide and the C-terminal domain of its partner. An extensive interface between both dimer partners and domains is consistent with the α2β2 organization of the functional prokaryotic CT enzyme. Further studies revealed that the binding sites for ACC inhibitors, including herbicides (Zhang, Tweel & Tong, 2004 ▶) and designed compounds (Zhang, Tweel, Li et al., 2004 ▶), were all at the dimer interface.
Sequentially, the yeast ACC CT domain is 46–52% identical to that of other eukaryotes and starts diverging substantially ∼85 residues prior to the C-terminus. All metazoan ACCs are more homologous throughout this C-terminal region. Although the initial yeast ACC structures were from a construct containing residues 1429–2233, the visible portion of the protein included residues 1480–2196 (Zhang et al., 2003 ▶). The yeast constructs that yielded the inhibitor structures produced visible electron density that included residues 1482–2218 (Zhang, Tweel & Tong, 2004 ▶) and finally 1482–2195 (Zhang, Tweel, Li et al., 2004 ▶). We found that truncating the human ACC2 CT domain to make the equivalent construct, residues 1693–2412, yielded a protein that failed to bind the designed inhibitors. This discovery prompted further examination of the human ACC2 CT domain, with the goal of determining a suitable construct for structural studies that was also competent to bind compounds. Here, we present the results of these studies, which revealed that the C-terminal residues of human ACC2 were necessary for compound binding. These C-terminal residues extend the dimer interface with a set of three intertwined helices.
2. Materials and methods
2.1. Cloning and expression of human ACC2 carboxyl transferase domain cDNA
A total of seven Escherichia coli ACC2 expression constructs with various truncations at both the N- and C-termini were generated by PCR. The names of the constructs and the primers used to amplify the gene are listed in Table 1 ▶.
Table 1. Construct names and primers.
| Construct name | Vector | Primer sequence |
|---|---|---|
| ACC2 (1636–2458) | pET24b | Forward, CATATGATCCGCCAGACCACCACCGGCAGTGCC |
| Reverse, CTCGAGGGTGGAGGCCGGGCTGTCCATGGTAGACAG | ||
| ACC2 (1693–2458) | pET24a | Forward, CATATGCATGGTCACCATCACCACCATCACGAAAACCTGTACTTCCAGGGAAATACTCCGTAC |
| Reverse, CTCGAGCTATCAGGTGGAGGCCGGGCTGTCCATGGTAGACAGCAGGTGAACGACCTGCGCACGCTCAG | ||
| ACC2 (1693–2401) | pET24a | Forward, CATATGCATGGTCACCATCACCACCATCACGAAAACCTGTACTTCCAGGGAAATACTCCGTAC |
| Reverse, CTCGAGCTATCACTCACGGATGGTGGAGCGCGGGCCATCCCCTGCCTG | ||
| ACC2 (1693–2420) | pET24a | Forward, CATATGCATGGTCACCATCACCACCATCACGAAAACCTGTACTTCCAGGGAAATACTCCGTAC |
| Reverse, CTCGAGTTATCATTCAACCAGGCCACGGATGGTCTTGAGGAC | ||
| ACC2 (1693–2441) | pET24a | Forward, CATATGCATGGTCACCATCACCACCATCACGAAAACCTGTACTTCCAGGGAAATACTCCGTAC |
| Reverse, CTCGAGTTATCACTCAGCTGGGCTGATGTGCTGGCTCAGGTAGATCAC | ||
| ACC2 (1693–2451) | pET24a | Forward, CATATGCATGGTCACCATCACCACCATCACGAAAACCTGTACTTCCAGGGAAATACTCCGTAC |
| Reverse, CTCGAGTTATCACTCAGACAGCAGGTGAACGACCTGCGCACGCTC | ||
| ACC2 (1693–2458) | pET24a-MBP | Forward, GGATCCAATACTCCGTACGTCACCAAGGATCTGCTCCAGGCCAAG |
| Reverse, CTCGAGCTATCAGGTGGAGGCCGGGCTGTCCATGGTAGACAGCAGGTGAACGACCTGCGCACGCTCAG |
PCR was performed with each corresponding primer pair using an in-house human ACC2 (accession No. AAR37018) construct that had been codon-optimized for E. coli expression. Each reaction contained 2 µl (∼1 ng) ACC2 DNA template, 20 mM Tris–HCl pH 8.4, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM each of the four deoxyribonucleotide triphosphates, 0.5 µM each of primers and 0.5 unit high-fidelity Taq DNA polymerase (Roche) in a total volume of 50 µl. PCR amplification conditions were 2 min of denaturation at 367 K followed by 30 cycles of 367 K for 30 s, 333 K for 30 s and 345 K for 2 min and a final extension step of 345 K for 15 min. DNA bands were purified using a Qiagen kit, digested with NdeI/XhoI (for cloning into pET24) or BamHI/XhoI (for cloning into pET24-MBP) and ligated to a restricted pET24 or pET24-MBP vector. The resulting constructs in pET24 [ACC2 (1693–2401), ACC2 (1693–2420), ACC2 (1693–2441) and ACC2 (1693–2451)] contained an N-terminal 6×His tag with a TEV-cleavable linker that leaves a glycine-residue artifact after proteolysis. The resulting construct in pET24-MBP [ACC2 (1693–2458)] contained an N-terminal 6×His-MBP tag with a thrombin-cleavable linker that leaves a Gly-Ser artifact after proteolysis. Each construct was then confirmed by double-stranded DNA sequencing.
The plasmids were transformed into BL21 (DE3) for protein expression. A single colony was picked and inoculated in 40 ml 2×YT medium in the presence of 50 µg ml−1 kanamycin and incubated at 310 K until the OD600 reached 0.6; IPTG was then added to a final concentration of 0.5 mM to induce protein expression at 293 K overnight. Soluble protein expression was evaluated by SDS–PAGE.
2.2. Purification of human ACC2 carboxyl transferase domain
The cell paste was resuspended in 25 mM HEPES, 100 mM NaCl, 20 mM imidazole pH 7.5. Cells were lysed at 277 K by passage through a homogenizer two times at 69 MPa. The lysate was clarified by centrifugation at 20 rev min−1 in a TZ-28 rotor (Sorvall) for 1 h followed by filtering through a 0.22 µm filter (Pall). The lysate was loaded onto a 20 ml Nickel Sepharose FF column (GE Healthcare Biosciences) equilibrated in 25 mM HEPES, 100 mM NaCl, 20 mM imidazole pH 7.5. Human ACC2 CT-domain protein was eluted using a gradient of imidazole to 300 mM. Pooled fractions were diluted threefold with 25 mM HEPES pH 7.5 and applied onto a 22 ml Q Sepharose column (GE) equilibrated in 25 mM HEPES pH 7.5. The protein was eluted using a gradient of NaCl to 500 mM. Tag removal was carried out on the pooled fractions by adding protease at a 1:100 ratio and 4 mM EDTA and digesting overnight at 277 K. TEV protease cleavage left a glycine residue at the N-terminus, while thrombin cleavage left a Gly-Ser artifact at the N-terminus. The sample was desalted on a 50 ml HiPrep Desalting column (GE) and passed back over a 5 ml HisTrap Ni Sepharose FF column. The flowthrough was collected and applied onto a 70 ml hydroxyapatite column (BioRad, Ceramic, 20 µm) equilibrated in 20 mM sodium phosphate pH 7.5. The protein was eluted using a gradient of sodium phosphate to 200 mM. Pooled fractions were concentrated using a JumboSep 30 kDa molecular-weight cutoff (Pall) and applied onto a 320 ml Superdex 200 column (GE) equilibrated in 25 mM HEPES, 200 mM NaCl pH 7.5. Pooled fractions were concentrated to 5–7 mg ml−1 using a CentriPrep YM-10 (Millipore) and delivered fresh at 277 K. Optimal results were not obtained using frozen samples.
2.3. Circular-dichroism spectroscopy
An Aviv 62DS/202.01 circular-dichroism spectrometer was used to measure the ellipticity at 222 nm as a function of temperature using a five-position Peltier-controlled sample holder. Measurements were made in 0.1 cm path-length quartz cuvettes with a sample volume of 0.3 ml. The ACC2 concentration was 4–7 µM in PBS. Compounds were dissolved in ethanol prior to their addition to the protein samples and equivalent volumes of ethanol were added to the samples containing protein only. Thermal transitions were analyzed by nonlinear least-squares curve fitting to the Boltzmann equation
where Θt 222nm is the ellipticity at 222 nm at temperature t, Θmin 222nm and Θmax 222nm are the minimal and maximal ellipticities at 222 nm, respectively, T m is the midpoint of the thermal transition and and dt is the steepness of the transition.
2.4. SYPRO Orange dye binding
A PerkinElmer ABI Prism 7700 Sequence Detector was utilized to monitor the fluorescence emission intensity of SYPRO Orange dye as a function of temperature. Standard 96-well PCR plates with optical plastic covers were used in these experiments. Each well contained a final volume of 40 µl; the final concentration of purified ACC2 was 1–2 µM, the SYPRO Orange dye (5000× stock in DMSO) was diluted 50-fold in DMSO and 0.4 µl of this working dilution was used in each well. The dye was added to the protein stock solution prior to filling the plate, into which the compounds to be tested had been previously dispensed. Compounds were added in DMSO, either to result in one test concentration, usually 10 µM, or as a serial dilution in DMSO. Compounds were dispensed at 100× in DMSO, resulting in another 0.4 µl of DMSO per well, which brought the final DMSO concentration to 2%(v/v). A small effect of DMSO on the thermal transition temperature of ACC2 was observed over the range 0–6%(v/v) DMSO (an increase of 0.4 K at 2%); therefore, all measurements, with or without compound, were made with the same final DMSO concentration of 2%(v/v). Thermal transitions were fitted to a modified Boltzman equation,
where Flt is the fluorescence intensity at temperature t, Flt pre and Flt post are the minimal and maximal fluorescence intensities at temperature t, respectively, T m is the midpoint of the thermal transition and dt is the steepness of the transition. Flt pre and Flt post were treated as linear functions of temperature, where Flt pre = m 1 × t + b 1 and Flt post = m 2 × t + b 2, where m 1, b 1 and m 2, b 2 are the slope and y intercept of the pre-transition and post-transition baselines at temperature t, respectively. This modification accounts for nonzero slopes in the baselines of the fluorescence data.
2.5. Crystallization of human ACC2 carboxyl transferase domain
All human ACC2 CT-domain crystallization trials were performed with protein in the buffer from the final purification step (25 mM HEPES pH 7.5, 200 mM NaCl). All successful crystallization trials also required the addition of a coenzyme A analogue to the protein solution. For example, acetyl coenzyme A was added to the human ACC2 CT domain [ACC2 (1693–2451)] solution to a final concentration of 2 mM (from a 100 mM stock in 10 mM HEPES pH 7.5) and the complex was concentrated to 15 mg ml−1 using a CentriPrep YM-3 (Millipore) to produce the protein solution used for the crystal structures reported here. In separate trials, 2 mM acetyl coenzyme A, acetoacetyl coenzyme A, butyryl coenzyme A or malonyl coenzyme A (Sigma) were added to the human ACC2 CT-domain [ACC2 (1693–2451)] solution in attempts to optimize crystal diffraction. Although clear electron density for cofactors was never observed, optimization of crystals containing proprietary compounds suggested that better diffraction data were consistently obtained when 2 mM malonyl coenzyme A was substituted in place of 2 mM acetyl coenzyme A.
Initial crystals of the ACC2–acetyl coenzyme A complex were obtained in Index Screen (Hampton Research) condition No. 88 [20%(w/v) PEG 3350, 0.2 M triammonium citrate pH 7.0] by hanging-drop vapor-diffusion equilibration in 96-well Neuro Probe trays (Neuro Probe Inc.) using 0.3 µl protein solution and 0.3 µl well solution. Optimized crystals of the ACC2–acetyl coenzyme A complex or the ACC2–malonyl coenzyme A complex were grown by mixing 2 µl protein solution with 2 µl well solution and equilibrating using hanging-drop vapor diffusion at 295 K in 24-well trays containing 1 ml crystallization solution in the well. All trays employed a reagent-concentration grid around the observed crystallization conditions containing 17–22%(w/v) PEG 3350, 0.1–0.25 M triammonium citrate pH 7.0 and 5%(v/v) glycerol. Initial crystal optimization produced clusters of multiple thin plates that were manually crushed into microcrystal seed stock and stored in 30 µl 20%(w/v) PEG 3350, 0.2 M triammonium citrate pH 7.0 and 5%(v/v) glycerol. Another round of optimization used the same protocol as described above, but with microcrystals streak-seeded into the crystallization drops using a horse hair. Microseeding produced large single plates (∼0.5 × 0.35 × 0.1 mm) that took three weeks to grow. These crystals were extremely fragile and required vapor-diffusion fixation with glutaraldehyde (Lusty, 1999 ▶) for 15 min prior to further handling. Apo crystals were quickly dunked into well solution plus 25%(v/v) ethylene glycol and plunge-frozen in liquid nitrogen.
Compounds, including CP-640186, were soaked into cross-linked crystals using the following protocol. Cross-linked crystals were placed into a 4.5 µl drop of harvest solution [20%(w/v) PEG 3350, 0.2 M triammonium citrate pH 7.0 and 5%(v/v) glycerol] and 0.5 µl 20 mM compound in DMSO was added to the drop to give a final compound concentration of 2 mM. The harvest-solution drop containing the crystals and compound was suspended over 1 ml harvest solution for 5 h using a single well from a hanging-drop tray. Crystals were removed from the harvest drop, quickly dunked into harvest solution plus 25%(v/v) ethylene glycol and plunge-frozen in liquid nitrogen.
2.6. Crystal structure determination and refinement
An initial 3.6 Å data set for apo human ACC2 CT domain was collected using a Rigaku 007HF rotating-anode X-ray generator (Rigaku USA) with a MAR345 DTB imaging-plate detector (MAR USA). This data set provided the initial crystal parameters; an orthorhombic space group with unit-cell parameters a = 82, b = 171, c = 295 Å. Using a molecular weight of 86 037 Da, the Matthews coefficient suggested the presence of four molecules per asymmetric unit. Molecular replacement was carried out using the program MOLREP (Vagin & Teplyakov, 1997 ▶) and confirmed that the space group was P212121. The search model was composed of a monomer of the yeast ACC CT domain (PDB code 1uyr; Zhang, Tweel & Tong, 2004 ▶) truncated to span residues 1482–2133. This procedure correctly located three of the four subunits and confirmed that the human ACC2 CT domain formed a dimer with a similar interface as observed for the yeast protein. A fourth monomer was placed manually to act as the dimer partner for the third monomer, which further reduced the R factor and accounted for the extensive positive difference density. No building or refinement was performed using this data set.
The large unit-cell edge of the crystal form suggested that the lower mosaicity from a smaller beam obtained at a synchrotron might improve diffraction quality. Diffraction data collected at the Southeast Regional Collaborative Access Team (SER-CAT) and the General Medicine/Cancer (GM/CA) CAT at the Advanced Photon Source (APS) confirmed this suspicion, with the mosaicity dropping by 0.5°. A 3.4 Å data set from crystals of apo human ACC2 CT domain was collected on SER-CAT 22-BM at the APS and was used for building and refinement. A starting human ACC2 model was built from a monomer of the truncated yeast protein (Zhang, Tweel & Tong, 2004 ▶) using Swiss-PDB Viewer (Guex & Peitsch, 1997 ▶) to incorporate the human numbering and sequence. Monomers were positioned by molecular replacement using the program MOLREP (Vagin & Teplyakov, 1997 ▶) followed by rigid-body refinement and fourfold electron-density map averaging using noncrystallographic symmetry. The human protein model was refined against this data set using CNX2002 (Brünger et al., 1998 ▶) and built using QUANTA05 (Morris, 2004 ▶). Strict fourfold noncrystallographic symmetry was enforced for the first round of refinement and strict twofold noncrystallographic symmetry was enforced for the next two rounds, followed by noncrystallographic restraints. The apo protein was refined to R = 23.1, R free = 28.4 with r.m.s.d.s from ideality of 0.009 Å and 1.4° for bond lengths and angles, respectively. The final model contained four copies of the human ACC2 CT domain spanning residues 1695–2449 and no water molecules.
The 3.19 Å resolution ACC2 CT domain–CP-640186 complex data were collected on GM/CA CAT 23-IDD at the APS and the structure was solved by molecular replacement using the human apo structure. The model was further refined to R = 21.8, R free = 26.8 using REFMAC5 (Murshudov et al., 1997 ▶) and built using Coot (Emsley & Cowtan, 2004 ▶). The final model contained four protein molecules, four molecules of CP-640186 and 76 water molecules. The r.m.s.d.s from ideal geometry were 0.007 Å and 1.4° for bond lengths and angles, respectively. Fourfold noncrystallographic symmetry constraints were not imposed on this structure. All four copies of the CP-640186 molecule were independently placed into 3σ difference density using the ligand-fitting feature in Coot and refined independently. Data-collection and processing statistics are summarized in Table 2 ▶.
Table 2. X-ray data-collection and refinement statistics for human ACC2 CT-domain structures.
Values in parentheses are for the highest resolution shell.
| Apo protein | CP-640186 complex | |
|---|---|---|
| Data collection | ||
| X-ray source | APS 22-BM | APS 23-IDD |
| Wavelength (Å) | 1.0 | 1.0 |
| Space group | P212121 | P212121 |
| Unit-cell parameters (Å) | a = 82.4, b = 170.8, c = 296.6 | a = 82.3, b = 170.7, c = 295.4 |
| Resolution range (Å) | 20.0–3.4 (3.55–3.4) | 20.0–3.19 (3.27–3.19) |
| Observations | 194635 | 2507021 |
| Unique reflections | 54871 | 64397 |
| Redundancy | 7.0 | 7.5 (7.5) |
| Completeness (%) | 94.1 (95.1) | 99.9 (99.9) |
| 〈I/σ(I)〉 | 10.6 (4.0) | 13.1 (4.8) |
| Rmerge† (%) | 12.3 (44.6) | 11.0 (45.7) |
| Refinement statistics | ||
| R factor‡ (%) | 23.1 (33.0) | 21.8 (29.4) |
| Rfree‡ (%) | 28.4 (35.0) | 26.8 (37.8) |
| No. of protein/water atoms | 22847/0 | 22930/76 |
| R.m.s.d. bond distances (Å) | 0.010 | 0.007 |
| R.m.s.d. bond angles (°) | 1.42 | 0.954 |
| Wilson B value (Å2) | 55.0 | 46.4 |
| Average protein B value (Å2) | 57.8 | 40.3 |
| Ramachandran plot | ||
| Most favored ϕ and ψ angles (%) | 87.0 | 94.9 |
| Additional allowed ϕ and ψ angles (%) | 12.8 | 4.8 |
R
merge = 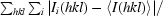
 .
.
R factor and R
free = 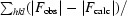
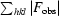 ; R
free was calculated for a randomly chosen 7% of reflections in both structures.
; R
free was calculated for a randomly chosen 7% of reflections in both structures.
3. Results
3.1. Yeast sequence similarity to animal and human ACCs
Our major objective was to use the human ACC2 CT domain in the rational design of potent selective inhibitors. Prior to this study, only the yeast ACC had yielded a publicly available eukaryotic ACC structure, and the possibility existed that differences in compound binding result from sequence differences between yeast and human. Overall, the yeast ACC has ∼45% pairwise-sequence identity to animal ACCs, with 46–52% pairwise sequence identity in the CT domain. However, the C-terminal 85 residues of yeast ACC are ∼13% identical to those of animal ACCs. Human ACC2 is more typical of animal ACCs. Overall, human ACC2 shows 58–88% pairwise sequence identity to animal ACCs, with similar (60–87%) pairwise sequence identity within the CT domain. Within the last 85 residues, human ACC2 has 30% pairwise sequence identity to arthropod ACCs and 52–75% identity to other mammalian ACCs (Fig. 1 ▶). Thus, the yeast ACC is similar to the animal enzyme overall, but there are regions, notably the C-terminus, that contain significant divergence. It was necessary for us to determine whether these differences produced significant systematic changes to the compound-binding pocket that might influence drug-discovery efforts.
Figure 1.

Sequence comparison between animal and yeast ACC CT-domain C-termini. Residues identical to the human protein are shaded green. Residues involved in the dimer interface are shown in solid black boxes. Residues making visible contact in only one dimer are shown in dashed black boxes. Proline residues facilitating the kinking of the terminal helices are surrounded by a blue box. The sequences compared are Homo sapiens ACC2 (hACC2; accession No. AAR37018), Mus musculus ACC2 (mACC2; accession No. NP_598665), Rattus norvegicus ACC2 (rACC2; accession No. NP_071529), Gallus gallus ACC (cACC; accession No. AAA48701), H. sapiens ACC1 (hACC1; accession No. NP_942131), M. musculus ACC1 (mACC1; accession No. AAS13685), Anopheles gambiae ACC (agACC; accession No. XP_314071), Drosophila melanogaster ACC (dmACC; accession No. NP_610342) and Saccharomyces cerevisiae ACC (yACC; accession No. NP_014413).
Initial attempts at crystallizing the human ACC2 CT domain used the full domain, residues 1693–2458, and produced no crystals. The yeast structures were then used to provide guidance for designing further constructs. The fragment of yeast ACC CT domain bound to the synthetic inhibitor CP-640186 included residues 1476–2233, but the structure only included residues 1482–2195 (Zhang, Tweel, Li et al., 2004 ▶), equivalent to human residues 1693–2412. When the human ACC2 was truncated to include the conserved amino acids, residues 1693–2401, or the visible residues, 1693–2420, no crystals were obtained. At this point, a more careful examination of the protein’s functional properties was initiated, with the results of these experiments guiding further construct selection.
3.2. Functional selection of human ACC2 carboxyl transferase domain
The carboxyl transferase domain of human ACC2 was a target for structural determination, specifically in complex with inhibitors, in order to enable and facilitate structure-guided chemistry efforts. Unfortunately, this isolated domain lacks much of the enzymatic activity that was measured for full-length ACC2, so measurement of direct binding of inhibitors was required in order to select an appropriate construct for structural determination.
Progressively truncated constructs of the carboxyl transferase domain of human ACC2 were engineered, expressed and purified as described above. Several approaches were used to assess the ligand-binding competence of these constructs. The objective was to identify the smallest domain construct which retains inhibitor binding and to use that construct for crystallography.
3.2.1. Fluorescence polarization assay for compound binding
A known inhibitor of the catalytic activity of full-length human ACC2, CP-640188, has intrinsic fluorescent properties, making it suitable for use as a direct ligand-binding reagent, as detected by changes in its fluorescence polarization when bound to ACC2.
CP-640188 (Fig. 2 ▶ a) has an excitation maximum at 365 nm and an emission maximum at 413 nm. Using these wavelengths, the polarization of the free ligand was measured and this polarization increased as human ACC2 carboxyl terminal domain (1693–2458) was titrated (Fig. 2 ▶ b). Fitting this binding curve to a standard isotherm resulted in an apparent K d of ≤1.2 µM for ACC2 binding to CP-640188. Other inhibitors, such as GSK723A, were shown to displace the fluorescent CP-640188 (Fig. 2 ▶ c). Fitting these displacement curves resulted in an apparent K d of 5.6 µM for GSK723A competition of CP-640188 from ACC2. Similarly, a truncated form of human ACC2 carboxyl transferase domain (1693–2451) could be used to titrate the ligand polarization and other inhibitors, GSK723A and GSK193A, were also shown to displace the fluorescent CP-640188 (Fig. 2 ▶ d).
Figure 2.

Fluorescence polarization and anisotropy measurements of ligand binding to the human ACC2 CT domain. (a) The structures of CP-640188 and CP-640186, which were used to establish the fluorescence and thermal melting assays for ligand binding and as a complex for crystal structure determination, respectively. (b) Saturable fluorescence polarization signal (excitation at 365 nm, emission at 413 nm) obtained by titrating 1 µM CP-640188 with human ACC2 (1693–2458). (c) 1 µM ACC2 (1693–2458)/1.5 µM CP-640188 polarization (triangles) and anisotropy (circles) signals decrease when competed with nonfluorescent GSK723A. (d) Fluorescence polarization (black) and anisotropy (white) for the active ACC2 truncation (1693–2451) at 1 µM; 1 µM CP-640188 was present for all measurements except ‘ACC2/malCoA only’, which shows the background fluorescence polarization signal. ‘PBS’ represents the fluorescence polarization signal for 1 µM CP-640188 in the absence of ACC2 protein. ACC2L is ACC2 (1693–2458). ‘ACC2/malCoA’ is ACC2 (1693–2451) complexed with malonyl-coenzyme A and where indicated GSK723A and GSK193A are competing ligands present at 10 µM.
3.2.2. Thermal stability assay for compound binding to human ACC2 CT domain
Thermodynamically, the binding of a small-molecule ligand to a protein results in a net decrease in the free energy of the system. This is reflected in a stabilization of the protein–ligand complex relative to the protein alone; that is, more energy (i.e. a higher temperature) is required to unfold the protein–ligand complex than for the protein alone.
Two methodologies were used to assess the ligand-dependent thermal stability of human ACC2 CT domain in vitro: circular-dichroism spectroscopy (CD) and fluorescent-dye binding. In this application, both of these techniques measure optical changes as a function of increasing temperature and both of those signals are sensitive to the unfolding of protein in response to heating. CD directly monitors a protein’s secondary-structural composition, most notably the α-helical content. By observing the CD signal at a characteristic wavelength, 222 nm, with increasing temperature, the loss of α-helical structure can be measured. This is representative of the overall folding and thus the stability of the protein. The other technique depends upon the fluorescent properties of a dye, SYPRO Orange, which preferentially binds to exposed hydrophobic regions of proteins. Exposure of this dye to a hydrophobic environment results in an increase of its relative fluorescence emission. When a protein unfolds, i.e. in response to heating, its hydrophobic core becomes exposed to solvent and to SYPRO Orange dye binding if present. Similarly to that described above for CD, observation of the fluorescence signal at a specific emission wavelength with increasing temperature can be used to monitor the unfolding and thus the stability of the protein.
CP-640188 was observed to shift the thermal transition of the ACC2 carboxyl transferase domain construct (1693–2451) by 10–14 K at saturation (Fig. 3 ▶ a). The ability of this ligand to stabilize ACC2 was lost as the constructs were progressively truncated, indicating that the shorter constructs were not able to bind this inhibitor (Fig. 3 ▶ b). The smallest construct made which retained measurable CP-640188 binding was (1693–2451), therefore making it most suitable for crystallography.
Figure 3.

Thermal denaturation is influenced by ligand binding to human ACC2 CT domain. (a) A reproducible and saturable increase in the melting temperature of ACC2 (1693–2451) (circles) is observed when 13 µM (gray diamonds) and 50 µM (triangles) CP-640188 is added to 4.4 µM human ACC2. (b) The compound-induced shift (triangles) in the midpoint of the CD thermal transition of ACC2 (circles) is retained when seven residues are truncated but lost when 17 residues are truncated from the C-terminus. ACC2 was at 4–7 µM and where indicated CP-640188 was present at 33–50 µM. Lines are drawn only to indicate trends. (c) Multiple compounds induce a titratable melting-temperature shift in ACC2 (1693–2451). ACC2 at 1–2 µM was titrated with 0–10 µM CP-640188 (circles), GSK723A (squares) and GSK193A (triangles) and subjected to thermal denaturation in the presence of (1×) SYPRO Orange fluorescent dye. The resulting T m values from fitted curves are plotted against compound concentration.
Human ACC2 (1653–2451) carboxyl transferase domain was titrated with three known ACC2 inhibitors: CP-640188, GSK723A and GSK193A (Fig. 3 ▶ c). All three of these ligands increased the thermal transition of this ACC2 carboxyl transferase domain by 7–10 K at saturation, indicating that this construct was competent to bind various inhibitors. Fitting of these binding curves to a standard isotherm resulted in apparent K d values of ≤1.1 µM for CP-640188, ≤0.8 µM for GSK723A and ≤2.2 µM for GSK193A. These values are presented as ‘less than or equal to’ (≤) because the experimental conditions impose a tight binding limit owing to the relatively high protein concentration required for these measurements. Having the ability to bind different chemical entities offered increased confidence in the selection of this construct for the pursuit of crystallography of ACC2 complexes.
3.3. The crystal structure of human ACC2 CT domain shows a novel C-terminus
The determination that hACC2 (1653–2451) was functionally competent to bind compounds paralleled its successful application in crystallization trials. This ACC2 CT construct was crystallized as a complex with CP-640186 (CP186; Fig. 2 ▶ a) in 25%(w/v) PEG 3350, 5%(v/v) glycerol and 0.2 M triammonium citrate pH 7.0. Crystal formation required the presence of a coenzyme A derivative (e.g. acetyl or malonyl) in the crystallization solution, but good electron density for this cofactor was never observed. However, interpretable electron density was observed for both the ligand and the C-terminus of the protein.
The model used to determine the structure of the human ACC2 CT domain was built from the yeast structure and only included the sequentially conserved residues 1482–2133 (human 1693–2337). Nevertheless, the electron density generated from the original molecular-replacement phases indicated that the protein extended substantially past the model terminus (Fig. 4 ▶ a). Using the observed noncrystallographic symmetry to average this electron density (Fig. 4 ▶ b) revealed a series of three intertwined helices that extended to residue 2449 (Fig. 4 ▶ c). These three helices extend outward from the bulk of the protein complex (Figs. 5a and 5b ▶) and are flexible. Despite their flexibility, elimination of any part of this structure resulted in a protein that was functionally unable to bind compound.
Figure 4.

Electron density is observed beyond the C-terminus of the molecular-replacement search model. (a) Electron density for the C-terminal region is observed immediately following molecular replacement with yeast ACC CT domain (PDB code 1uyr) (red). A ribbon showing the final human structure is shown for comparison (yellow). (b) The C-terminal electron density was improved by fourfold noncrystallographic averaging based on the positions of the conserved protein residues. (c) The electron density surrounding one of the C-terminal regions in the final refined model. All electron-density maps are contoured at 1σ.
Figure 5.

Comparison between the human and yeast ACC CT domains. (a) This cartoon diagram shows the human ACC2 CT-domain dimer (light and dark red protein, orange ligand) superimposed on the structure of the yeast ACC CT domain (light and dark blue protein, blue ligand). The C-terminal region and antiparallel helical extensions are indicated. (b) A detailed view of one copy of the flexible C-terminus is included. Sequence limits for the three helices are given and prolines at the helical kinks are shown with thick bonds. (c) The residues responsible for the folding of the antiparallel helical extensions are indicated.
The contacts between the C-terminal helices (Fig. 5 ▶ b) are maintained by the amphipathic nature of the sequence and the kinking is facilitated by two prolines: Pro2423 and Pro2439. The flexibility of the helices led to a loss of the strict twofold symmetry of the protein complex at the C-terminus, as well as during the overlay between noncrystallographically related molecules. This lack of symmetry provides the caveat that the conformational details may be influenced by crystal packing. Fortunately, there was sufficient mass superimposition to yield improvement during the initial fourfold electron-density averaging. These additional helices only increased the size of the dimer interface by 293 Å2 per molecule, but many of the side chains were not visible in the crystal structure. The final helix started at residue 2439, just prior to the longest nonbinding construct. Although we know that these C-terminal helices are necessary for compound binding, we have not directly measured their effect on domain dimerization.
The human ACC2 CT-domain structure also contains residues 2252–2284 (yeast 2048–2080) which were not present in the search model (Figs. 5a and 5c ▶). As was the case for the C-terminal residues, the electron density was visible in the initial molecular-replacement map (Fig. 6 ▶ a), the helical secondary structure was defined after fourfold noncrystallographic averaging (Fig. 6 ▶ b) and the final map positioned the residues and allowed the turn to be built (Fig. 6 ▶ c). The final average refined B factor for this loop is 48.3 Å2, which is comparable to that of the entire protein. This stretch continues from the previously observed α-helix initiated at residue 2241 (yeast 2037) until Pro2253 generates a right-angle turn outward from the protein dimer. The next stretch of residues (2254–2284) forms a pair of antiparallel amphipathic α-helices separated by a three-residue turn following Pro2265. The two helices are pinned together at the proximal ends by a hydrogen bond between Tyr2255 and Glu2283 and a salt bridge between Asp2252 and Arg2282 (Fig. 5 ▶ c).
Figure 6.

Electron density is observed for a loop spanning residues 2252–2284 of the human ACC2 CT domain that was not present in the molecular-replacement search model. A stick representation showing the final human structure is shown for comparison (yellow). (a) Electron density for the loop is observed immediately following molecular replacement with yeast ACC CT domain (PDB code 1uyr). (b) The electron density is improved by fourfold noncrystallographic averaging based on the positions of the yeast protein residues. (c) The electron density surrounding a representative loop in the final refined model. All electron-density maps are contoured at 1σ.
3.4. Compounds bind to the human ACC2 CT domain at the dimer interface
The binding mode and pocket surrounding CP186 in the human ACC2 CT domain closely resembled those of the yeast ACC CT domain (Fig. 7 ▶ a). As observed in the yeast ACC structure, CP186 binds at the dimer interface and interacts with residues from both copies of the ACC2 protein. The large anthracene moiety is not surrounded by hydrophobic residues, but by flexible polar residues that adopt rotamers pointing the polar groups away from the ligand. Although there are residues with polar side chains surrounding the ligand, the only two polar interactions are with the protein main chain: a hydrogen bond between the carbonyl adjacent to the anthracene and the main-chain amide N atom of Glu2230 (yeast Glu2026) and a weak hydrogen bond between the carbonyl adjacent to the morpholine and the main-chain amide N atom of Gly2162 (yeast Gly1958). Otherwise, the C-shaped molecule simply fits nicely into a complementary pocket shape formed by the surrounding residues.
Figure 7.

Stereo figure showing the comparison of CP-640186 binding to human (pink) and yeast (cyan) ACC CT domains. Residues that differ sequentially between yeast and human and that contact the ligand are indicated with thick bonds. Hydrogen bonds are indicated by magenta dashed lines.
Of the 22 residues surrounding the ligand at a distance of less than 5 Å, six differ between human and yeast, including the substitution of Leu1965 for Ile1762, Val1968 ▶ for Met1765, Ala2125 for Pro192, Phe2130 for His1925, Glu2232 for Gln2028 and Glu2236 for Gly2032 ▶ (Fig. 7 ▶). Although those changes did not substantially change the binding mode for this compound, the conformations of the piperidine and the terminal morpholine differed from the yeast structure in all four copies of CP186 observed in human ACC2. This is a significant result because all four molecular copies of CP186 were individually placed into 3σ difference density and the complex was refined without noncrystallographic constraints. Thus, the observed difference in ligand conformation appears to be a direct consequence of the residue substitution on both sides of the morpholine. The substitution of Ala2125 for yeast Pro1920 creates space on one side of the morpholine ring, while the substitution of Leu1965 for yeast Ile1762 reduces space on the other. This suggests that subtle changes in the pocket shape and surface polarity may lead to differences in compound binding to yeast and human ACCs.
4. Summary and implications for drug discovery
Acetyl-coenzyme A carboxylase (ACC) is responsible for the pivotal step in fatty-acid anabolism: the synthesis of malonyl-CoA from acetyl-CoA. The diseases associated with fatty-acid accumulation, such as insulin resistance, diabetes and metabolic syndrome, suggest that ACC is an attractive target for regulating this pathway by small molecules. Previous chemistry efforts (Harwood et al., 2003 ▶) demonstrated that ACC could be inhibited by small molecules and structural efforts (Zhang, Tweel, Li et al., 2004 ▶) identified a unique binding site in the CT domain. Although the residues surrounding the compound are similar in the yeast and human ACCs, human ACC2 did not bind compound when truncated to the equivalent size protein in the yeast structure. We suspected that the C-terminal sequence divergence between yeast and metazoan ACC may play a role in this functional distinction and mutagenesis combined with compound-binding assays bore out this notion.
Two methods were identified for measuring compound binding to the human ACC2 CT domain, a direct binding method utilizing the intrinsic fluorescence of the anthracene moiety in CP-640188 and a thermal melting assay that measured the stability imparted to the protein by compound binding. Both methods indicated that a seven-residue C-terminal truncation was tolerated for compound binding but a 17-residue C-terminal truncation produced protein unable to bind compound. Thus, the window for crystallization success was within a ten-residue segment of an 826-residue protein, representing fairly tight constraints on construct design. Performing the functional assays led to the identification of useful constructs before protein scale-up, purification and crystallization trials. Thus, up-front small-scale construct functional assaying represents a successful strategy for medium-throughput triaging prior to committing to a lengthy crystallization campaign.
As expected, the overall fold of the human ACC2 CT domain was similar to that of yeast with the exception of the C-terminus, where animal ACC sequences differ significantly from that of yeast. Human ACC2 terminated in a series of three intertwined α-helices extending out from the bulk of the ACC dimer. The unique conformation of this C-terminal structure is maintained by the amphipathic nature of the α-helices and a pair of prolines in the terminal sequence that facilitates the directional changes. These helices were very mobile in the crystal structure, with average B factors that were 60 Å2 higher than that of the entire protein. However, they were clearly visible in the unbiased electron density obtained while solving the structure and are present in all refined structures. The high pairwise sequence identity in the terminal residues suggests that this structure is maintained amongst the other animal ACCs. In fact, the two prolines that permit the intertwining are conserved in all animal ACCs but do not exist in the yeast protein (Fig. 1 ▶).
Functional studies demonstrated that the ability of human ACC2 to bind compound depended on the correct folding of the C-terminal helices, but we do not know whether this functional dependence is maintained throughout the animal kingdom. The observed functional dependence is somewhat surprising given that the C-terminus is located on the opposite side of the complex from the ligand-binding site. One possible explanation lies in the fact that the ligand binds at the dimer interface, which may be modified or stabilized by the C-terminal structure. If true, the effect must indeed be subtle as it had no clear impact on the protein’s melting temperature. By comparison, ligand binding raised the melting temperature by 16 K. The propensity to bind ligand is clearly related to crystallization, as the shorter functionally dead constructs did not yield crystals. Unfortunately, without the nonfunctional structures for comparison, the mechanism by which the C-terminus of the animal ACCs imparts functionality from the opposite side of the molecule is unresolved.
Another unresolved effect was the requirement of a CoA derivative for crystallization. No crystals were obtained in the absence of any CoA and the use of different CoA derivatives (e.g. acetyl, malonyl etc.) during crystallization had an impact on diffraction quality. Despite numerous cocrystallization and crystal-soaking attempts with millimolar concentrations of CoA derivatives, the phases resulting from the refined crystal structures only produced patchy electron density in the pocket adjacent to CP186, where acetyl-CoA was built into the yeast structure (Fig. 8 ▶; Zhang et al., 2003 ▶). Superimposition of this structure with the human ACC2–CP186 complex suggests that the CoA cofactors are almost in direct contact with the inhibitor and might share several contact residues, such as Thr1960, Leu2229 and Ile2237.
Figure 8.

The acetyl-coenzyme A moiety binds in a pocket immediately adjacent to the ligand CP186. The yeast structure with acetyl-coenzyme A (PDB code 1od2; Zhang et al., 2003 ▶) was superimposed on the human ACC2–CP186 structure (pink and magenta ribbon and C atoms) to show the relative positions of the inhibitor (orange C atoms) and cofactor (blue C atoms).
Successful elucidation of the human ACC2 structure confirmed the binding site for CP186 observed in yeast ACC and also illustrated some potential problems modeling compound binding because of amino-acid differences between human and yeast. Adjacent to the CP186 anthracene moiety the yeast ACC has a short flexible Gly2032 and a neutral Gln2028. In contrast, the human ACC2 has two negatively charged glutamic acids (2232 and 2236) in the same positions. For this ligand, glutamic acids 2232 and 2236 adopt rotamers that point their charged groups away from the anthracene and towards His2291 and Arg2240, respectively. At the other end of the ligand pocket, three hydrophobic residue substitutions subtly change the pocket shape: Leu1965 for yeast Ile1762, Val1968 for yeast Met1765 and Ala2125 for yeast Pro1920. CP186 is anchored by the large anthracene at one end and two main-chain hydrogen bonds throughout the molecule. Diverse compounds with various anchor points may interact differently with pocket residues and these differences could be magnified or lead to distinct binding modes when comparing yeast and human structures.
Supplementary Material
PDB reference: human ACC2 CT domain, 3ff6, r3ff6sf
Acknowledgments
Thanks to the staff of the General Medicine and Cancer Institute (GM/CA) and Southeast Regional (SER) Collaborative Access Teams (CATs) for the guidance provided during the challenging data collection. GM/CA CAT has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). SER-CAT is a collaboratively funded facility at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science under contract No. DE-AC02-06CH11357.
References
- Abu-Elheiga, L., Almarza-Ortega, D. B., Baldini, A. & Wakil, S. J. (1997). J. Biol. Chem.272, 10669–10677. [DOI] [PubMed]
- Abu-Elheiga, L., Brinkley, W. R., Zhong, L., Chirala, S. S., Woldegiorgis, G. & Wakil, S. J. (2000). Proc. Natl Acad. Sci. USA, 97, 1444–1449. [DOI] [PMC free article] [PubMed]
- Abu-Elheiga, L., Matzuk, M. M., Abo-Hashema, K. A. H. & Wakil, S. J. (2001). Science, 291, 2613–2616. [DOI] [PubMed]
- Abu-Elheiga, L., Matzuk, M. M., Kordari, P., Oh, W., Shaikenov, T., Gu, Z. & Wakil, S. J. (2005). Proc. Natl Acad. Sci. USA, 102, 12011–12016. [DOI] [PMC free article] [PubMed]
- Abu-Elheiga, L., Oh, W., Kordari, P. & Wakil, S. J. (2003). Proc. Natl Acad. Sci. USA, 100, 10207–10212. [DOI] [PMC free article] [PubMed]
- Adler, A. L., Perham, M. A., McNally, T., Gu, Y.-G., Weitzberg, M., Voorbach, M. J., Beutel, B., Sham, H., Jacobson, P., Zinker, B. & Camp, H. C. (2005). Diabetes, 54, A354.
- Bianchi, A., Evans, J. L., Iverson, A. J., Nordlund, A. C., Watts, T. D. & Witters, L. A. (1990). J. Biol. Chem.265, 1502–1509. [PubMed]
- Boden, G. (1997). Diabetes, 46, 3–10. [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Guex, N. & Peitsch, M. C. (1997). Electrophoresis, 18, 2714–2723. [DOI] [PubMed]
- Ha, J., Lee, J. K., Kim, K. S., Witters, L. A. & Kim, K. H. (1996). Proc. Natl Acad. Sci. USA, 93, 11466–11470.
- Harada, N., Oda, Z., Hara, Y., Fujinami, K., Okawa, M., Ohbuchi, K., Yonemoto, M., Ikeda, Y., Ohwaki, K., Aragane, K., Tamai, Y. & Kusunoki, J. (2007). Mol. Cell. Biol.27, 1881–1888. [DOI] [PMC free article] [PubMed]
- Harwood, H. J. Jr (2005). Expert Opin. Ther. Targets, 9, 267–281. [DOI] [PubMed]
- Harwood, H. J. Jr et al. (2003). J. Biol. Chem.278, 37099–37111. [DOI] [PubMed]
- Kelley, D. E., Goodpaster, B., Wing, R. R. & Simoneau, J. A. (1999). Am. J. Physiol.277, E1130–E1141. [DOI] [PubMed]
- Lenhard, J. M. & Gottschalk, W. K. (2002). Adv. Drug Deliv. Rev.54, 1199–1212. [DOI] [PubMed]
- Lusty, C. J. (1999). J. Appl. Cryst.32, 106–112.
- Morris, R. J. (2004). Acta Cryst. D60, 2133–2143. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Savage, D. B., Choi, C. S., Samuel, V. T., Liu, Z. X., Zhang, D., Wang, A., Zhang, X. M., Cline, G. W., Yu, X. X., Geisler, J. G., Bhanot, S., Monia, B. P. & Shulman, G. I. (2006). J. Clin. Invest.116, 817–824. [DOI] [PMC free article] [PubMed]
- Seppala-Lindroos, A., Vehkavaara, S., Hakkinen, A. M., Goto, T., Westerbacka, J., Sovijarvi, A., Halavaara, J. & Yki-Jarvinen, H. (2002). J. Clin. Endocrinol. Metab.87, 3023–3028. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Zhang, H., Tweel, B., Li, J. & Tong, L. (2004). Structure, 12, 1683–1691. [DOI] [PubMed]
- Zhang, H., Tweel, B. & Tong, L. (2004). Proc. Natl Acad. Sci. USA, 101, 5910–5915. [DOI] [PMC free article] [PubMed]
- Zhang, H., Yang, Z., Shen, Y. & Tong, L. (2003). Science, 299, 2064–2067. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human ACC2 CT domain, 3ff6, r3ff6sf
PDB reference: human ACC2 CT domain, 3ff6, r3ff6sf