

Abstract
Objective:
To examine caloric intake, dietary composition, and body mass index (BMI) in participants in the Prospective Huntington At Risk Observational Study (PHAROS).
Methods:
Caloric intake and macronutrient composition were measured using the National Cancer Institute Food Frequency Questionnaire (FFQ) in 652 participants at risk for Huntington disease (HD) who did not meet clinical criteria for HD. Logistic regression was used to examine the relationship between macronutrients, BMI, caloric intake, and genetic status (CAG <37 vs CAG ≥37), adjusting for age, gender, and education. Linear regression was used to determine the relationship between caloric intake, BMI, and CAG repeat length.
Results:
A total of 435 participants with CAG <37 and 217 with CAG ≥37 completed the FFQ. Individuals in the CAG ≥37 group had a twofold odds of being represented in the second, third, or fourth quartile of caloric intake compared to the lowest quartile adjusted for age, gender, education, and BMI. This relationship was attenuated in the highest quartile when additionally adjusted for total motor score. In subjects with CAG ≥37, higher caloric intake, but not BMI, was associated with both higher CAG repeat length (adjusted regression coefficient = 0.26, p = 0.032) and 5-year probability of onset of HD (adjusted regression coefficient = 0.024; p = 0.013). Adjusted analyses showed no differences in macronutrient composition between groups.
Conclusions:
Increased caloric intake may be necessary to maintain body mass index in clinically unaffected individuals with CAG repeat length ≥37. This may be related to increased energy expenditure due to subtle motor impairment or a hypermetabolic state.
GLOSSARY
- BEE
= basal energy expenditure;
- BMI
= body mass index;
- FFQ
= Food Frequency Questionnaire;
- HD
= Huntington disease;
- OR
= odds ratio;
- PD
= Parkinson disease;
- PHAROS
= Prospective Huntington At Risk Observational Study;
- TEE
= total energy expenditure;
- UHDRS
= Unified Huntington’s Disease Rating Scale.
Individuals who have Huntington disease (HD) have lower body mass index (BMI) than age-matched controls,1–5 and these differences increase as HD advances.1,3 Weight loss of ≥1 kg/year was associated with more severe motor impairment and increasingly severe chorea in 927 HD cases followed for a mean of 3.4 years, while weight gain was more common in those with shorter disease duration (0–2 years).2 Weight loss in HD may be multifactorial and related to decreased caloric intake from dysphagia, increased energy expenditure due to physical activity (e.g., chorea and dystonia),6,7 or a systemic metabolic defect leading to a hypermetabolic state.8,9
In 2 studies6,7 that examined 24-hour energy expenditure and physical activity using indirect calorimetry, total energy expenditure was 11%–14% higher in early to mid-stage HD cases compared with controls and was attributed to increased physical activity during waking hours. Similar studies have not been performed in pre-manifest HD. A study using a semiquantitative, open-ended questionnaire showed significantly higher daily caloric intake and lower BMI in 15 presymptomatic HD cases compared to 21 controls.8
We assessed the relationship between BMI and diet by administering a semiquantitative food frequency questionnaire (FFQ)10,11 to individuals participating in the Prospective Huntington At Risk Observational Study (PHAROS).12 Our aims were to compare participants with and without expanded CAG to determine 1) whether there were differences in macronutrient intake (protein, carbohydrate, fat), total caloric intake, and BMI, and 2) the relationship between CAG repeat length, BMI, and caloric intake.
METHODS
Subjects.
All research participants were enrolled in PHAROS between July 1999 and January 2004.12 Institutional review boards at all participating sites approved the protocols and consent procedures. The aim of PHAROS is to identify the earliest clinical features with the highest specificity that predict manifest HD. At baseline, participants were between 26 and 55 years of age, and at risk for HD by virtue of having an affected parent or sibling. All opted to remain unaware of their HD gene carrier status at the time of enrollment. Details of the baseline assessment of these 1,001 individuals and blinding procedures have been published.12 A medical history, physical examination, and weight were obtained at each visit. Visits were scheduled every 9 months. Blood was obtained at baseline to measure the trinucleotide expansion (CAGn) of the HD gene. The central tenet of the PHAROS study stipulates that neither research participants, investigators, nor anyone else will ever be informed of the individual genetic research data. At each assessment, an independent rater at each site performed the motor component of the Unified Huntington’s Disease Rating Scale (UHDRS) and assigned a level of diagnostic confidence of HD based solely on this motor examination. A rating of 4 indicated ≥99% confidence of clinically definite HD based on the presence of an unequivocal unexplained extrapyramidal movement disorder. This rating has been shown to have good reliability.13
Dietary assessment.
A total of 675 individuals initially completed the National Cancer Institute FFQ, which has been shown to be reliable and valid.11 The FFQ was administered, on average, 30 months after baseline examination. Twenty-three individuals were excluded from the analyses presented here because they were identified as having clinically definite HD prior to or at the time of the FFQ, or because they were missing CAGn data. The analyses focus on this first dietary assessment and the BMI at that visit, or the closest visit if the FFQ was not completed at the visit. The closest visit was defined as 180 days prior to the FFQ or within 14 days following the FFQ. Seventy-nine percent of subjects completed the FFQ within 2 days of a visit, and 95% of subjects had a visit that fell within the specified window.
Statistical analyses.
All statistical analyses were conducted by the HSG Biostatistics Center at the University of Rochester using SAS v9. PHAROS baseline measures in individuals who completed the FFQ were compared with those who did not using t tests and χ2 tests as appropriate. Demographic characteristics and summary measures from the FFQ were compared in the CAG <37 (nonexpanded) and CAG ≥37 (expanded) groups12 using t tests and χ2 tests. Macronutrient intake (protein, carbohydrate, fat) was calculated from the FFQ. BMI and summary measures from the FFQ, including caloric intake and macronutrient categories, were divided into quartiles based on the entire sample, and the distribution of individuals with CAG <37 and CAG ≥37 within these quartiles was compared using Mantel-Haenszel tests for trend.
In individuals whose weight is stable, caloric intake (measured in kcal) should equal total energy expenditure (TEE). TEE is based on basal energy expenditure (BEE), the thermic energy of food, and physical activity. TEE is measured by indirect calorimetry. In this study, because indirect calorimetry was not performed, we considered the caloric intake an estimate of TEE. BEE was calculated for each individual using the Harris Benedict equation.14 Male BEE equals 66 + 13.7 (weight in kg) + 5 (height in cm) − 6.8 (age in years), and female BEE equals 65 + 9.6 (weight in kg) + 1.8 (height in cm) − 4.7 (age in years). TEE minus BEE in a weight-stable individual is a measure of physical activity; large differences suggest that a greater amount of energy is expended in physical activity rather than basal metabolism, digestion, or heat production. Among those who completed the FFQ, the mean (SD) of the change in weight between the FFQ and the following visit (mean 325 days) was 0.50 kg (4.68), p = 0.01. While this small increase in weight represents a significant difference in this large sample, the mean weight gain in kg for those with CAG <37 was 0.65 (SE 0.23) (p < 0.05) and among those with CAG ≥37 was 0.20 (SE 0.36); this difference was not significant. TEE-BEE will be referred to as estimated physical activity.
Logistic regression models were used to examine the relationship of BMI and dietary measures (macronutrient intake, total energy intake, BEE, and estimated physical activity) to genetic status (CAG <37 or CAG ≥37) as the outcome. The dietary measures and BMI were defined by quartiles, with the lowest quartile (quartile 1) serving as the reference. Covariates included age (≥50, 40–49, <40 years), gender, and education (>16, 13–16, ≤12 years). In separate analyses total motor score dichotomized (≤1, >1 units) and chorea score (0, >0) were included as additional covariates.
Finally, CAG repeat length was correlated with BMI, caloric intake, and estimated physical activity in the CAG ≥37 group with multiple linear regression using the quartile values (1, 2, 3, 4) as continuous variables and adjusting for age, gender, and education. In addition, for BMI we adjusted for caloric intake, and for caloric intake we adjusted for BMI. We also calculated the predicted 5-year probability of clinical onset of HD15,16 using a CAG-based model developed using data from close to 3,000 individuals. The above linear regression was carried out using the predicted 5-year probability of onset as an outcome variable.
All analyses were repeated, excluding all individuals who had more than 10 items missing on the dietary interview, and women with caloric intake <550 kcal or >3,500 kcal per day, and men with caloric intake <650 kcal or >4,000 kcal per day.
RESULTS
The PHAROS study enrolled 1,001 research participants. The FFQ was completed by 675 individuals (441 CAG <37 and 233 CAG ≥37 [1 missing CAG]), but was not completed by 326 individuals. The FFQ was completed on average 30 months after the baseline visit (range 0–74 months). Completers and noncompleters did not significantly differ in age, gender, ethnicity, or percentage with expanded CAG repeat (data not shown). Noncompleters had significantly fewer years of education (14.4 [2.5] vs 15.1 [2.6]), and were significantly more likely to report depressive symptoms (2.5 [3.6] vs 1.9 [2.6]) on the Beck Depression Inventory II. They also scored significantly lower (more functionally impaired) on the total functional capacity scale of the UHDRS17 (12.9 [0.4] vs 13.0 [0.2]) and on each of the cognitive measures on the UHDRS. The chorea and bradykinesia scores were significantly higher among noncompleters.
Seven individuals who completed the FFQ were rated a 4 (meets clinical criteria for HD) at the time of their baseline examination and were excluded from this analysis. An additional 15 individuals were rated a 4 prior to or at the time of the FFQ and were excluded from the analysis, for a total of 22 individuals. One individual who did not have CAG repeat data was also excluded.
Demographic characteristics and summary measures from the FFQ in 435 individuals with CAG <37 and the 217 individuals with CAG ≥37 who completed the FFQ (n = 652), had CAGn data, and did not meet clinical criteria for HD are presented in table 1. The total UHDRS motor score and the chorea score were significantly higher among those with an expanded CAG repeat. We examined the difference between TEE (estimated using total caloric intake) and BEE as a measure of estimated physical activity. Due to the presence of outliers for the dietary variables, the distributions of these variables by quartile were compared for individuals with CAG <37 and CAG ≥37 (table 2). Caloric intake was significantly higher in the CAG ≥37 group (p for trend = 0.01). BMI and estimated physical activity were marginally different, while BEE did not differ significantly. Carbohydrate intake was significantly higher in the expanded CAG repeat group (p for trend = 0.02).
Table 1 Demographics of subjects who completed the Food Frequency Questionnaire by CAG repeat length
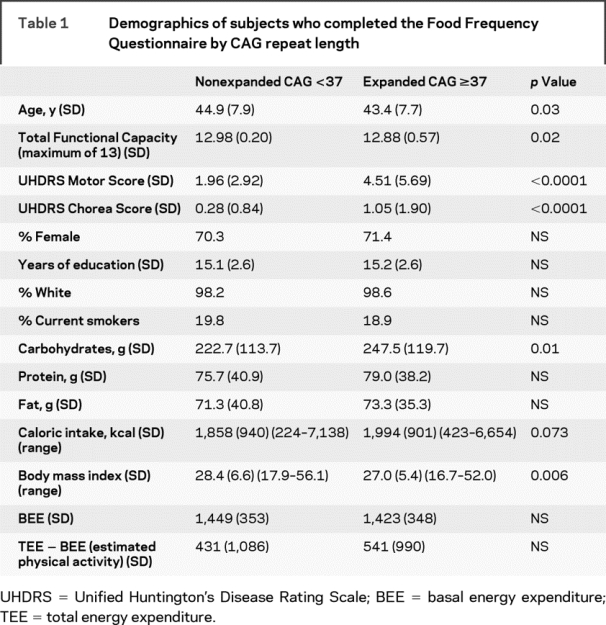
Table 2 Distribution of dietary variables and body mass index for subjects with CAG <37 and CAG ≥37
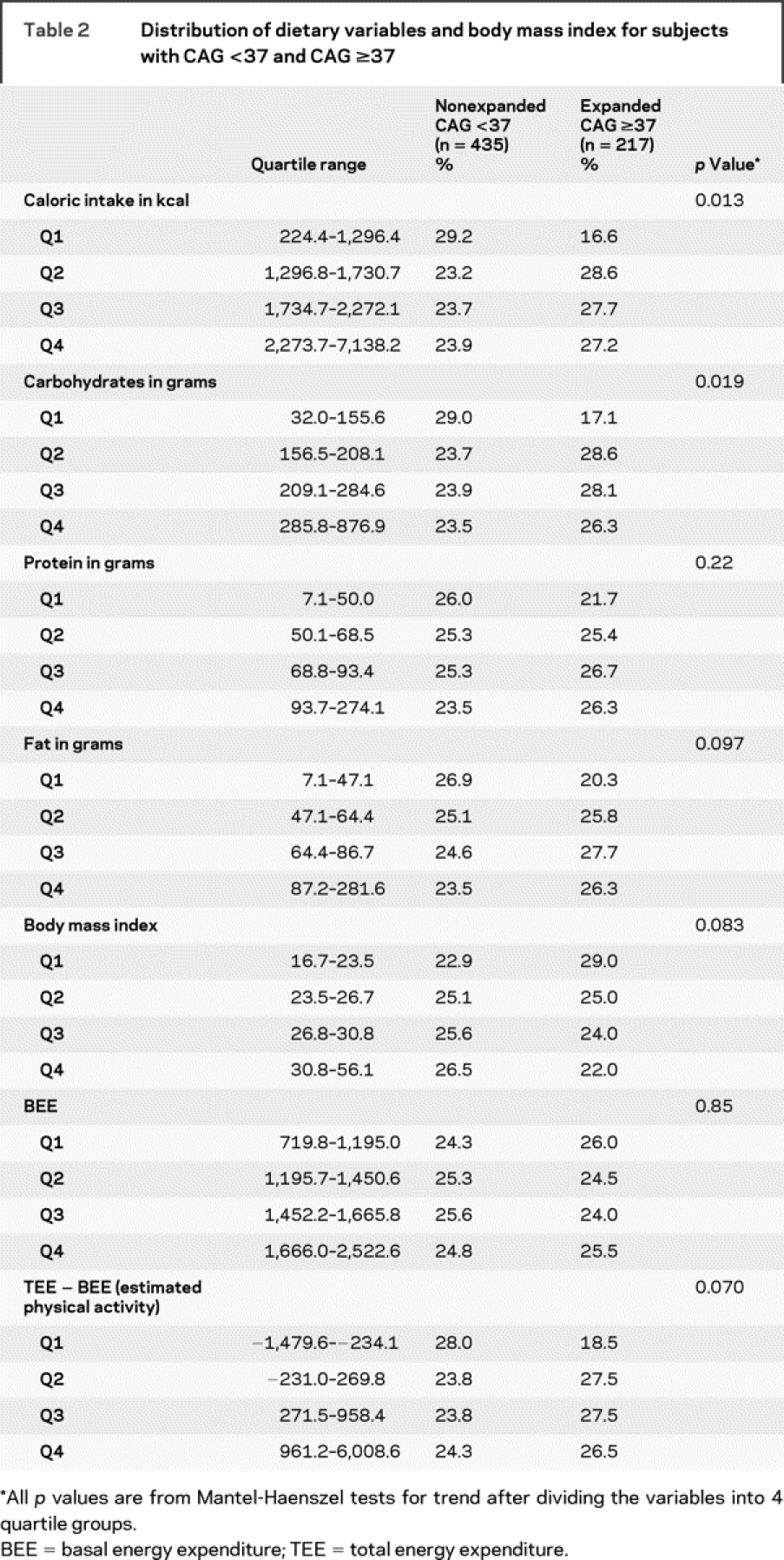
Unadjusted and adjusted odds ratios (OR) are presented for the relationship between caloric intake, BMI, and estimated physical activity, with group membership (CAG <37 or CAG ≥37) as the outcome (table 3). In both unadjusted and adjusted analyses, the odds of membership in the CAG ≥37 group were increased approximately twofold for each quartile (second, third, and fourth) of caloric intake compared with the lowest quartile (first) (reference) (p for trend 0.05). The highest odds were for the second quartile compared to the reference, and the odds decreased with increasing caloric intake (figure). When total motor score was included as a covariate, this relationship was attenuated only in the highest quartile (fourth). Higher estimated physical activity was associated with an increased odds of membership in the CAG ≥37 group after adjustment for age, gender, and education (p for trend 0.02). When total motor score from the UHDRS was included as a covariate, the relationship between total estimated physical activity and group membership was attenuated, as would be expected since total motor score is a measure of physical activity. The association of BMI and genetic status did not reach significance. Adjusting for the dichotomized chorea score (0 vs >0) produced results that were similar to those produced by adjustment of the dichotomized motor score (data not shown).
Table 3 Unadjusted and adjusted odds ratios (ORs) comparing subjects with CAG ≥37 to subjects with CAG <37
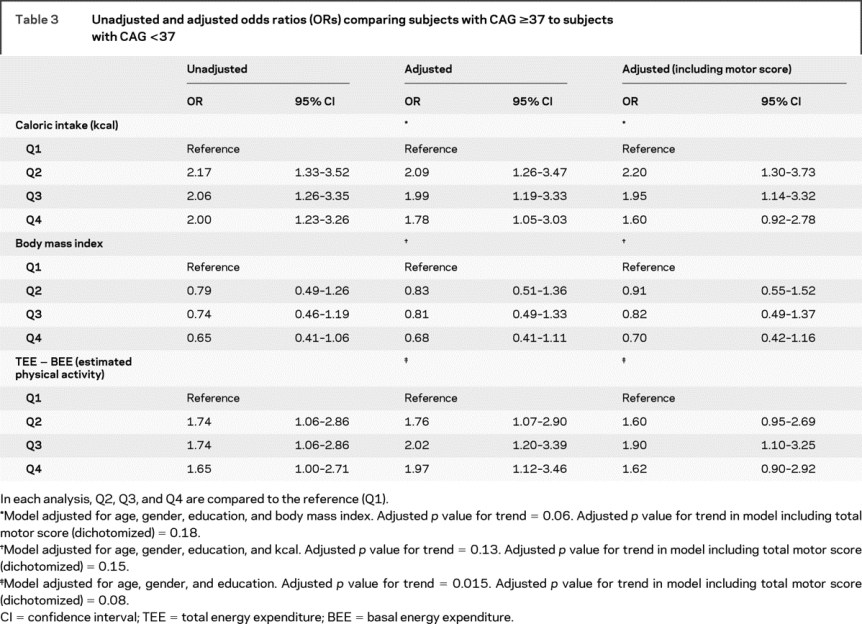

Figure Adjusted odds of CAG ≥37 group membership in second, third, or fourth quartile compared to the lowest quartile
Q1 is the reference for each analysis.
When all analyses were repeated excluding individuals who had 10 or more missing data points or had extreme caloric intakes, the logistic regression results were similar (data not shown).
Macronutrient assessment.
There was no difference in the distribution of macronutrients (protein, carbohydrates, and fat) between the CAG ≥37 and CAG <37 groups after adjustment for age, gender, education, and total caloric intake (data not shown).
Relationship between CAG repeat length, BMI, kcal, and estimated physical activity.
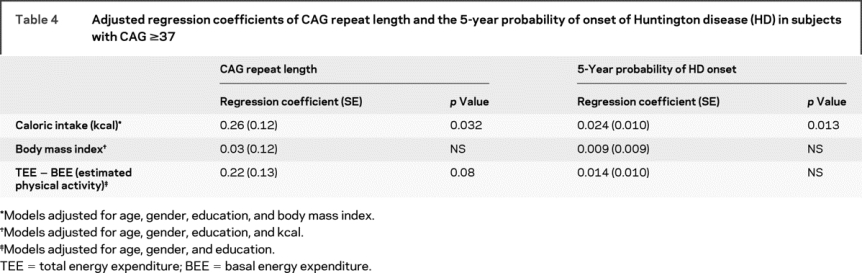
For the expanded CAG repeat length group (CAG ≥37), caloric intake was significantly correlated with both CAG repeat length and estimated 5-year probability of HD onset after adjustment for age, gender, education, and BMI (table 4). There was an estimated 0.26 CAG unit increase for each unit increase in kcal quartile. Similarly, we estimate an increase of 2.4% in the 5-year probability of onset for each unit increase in kcal quartile. BMI was not correlated with CAG repeat length after adjustment for age, gender, education, and caloric intake.
Table 4 Adjusted regression coefficients of CAG repeat length and the 5-year probability of onset of Huntington disease (HD) in subjects with CAG ≥37
DISCUSSION
This is the largest study of dietary factors in individuals at risk for HD who were unaware of their genetic status. Individuals with expanded CAG (≥37) reported a similar caloric intake yet had a lower BMI compared with those without expanded CAG (<37). In the CAG ≥37 group, higher caloric intake was related to both higher CAG repeat length and higher 5-year probability of onset of HD after adjustment for relevant covariates. This linear relationship was not demonstrated with BMI. We did not find any difference in macronutrient intake after adjustment for covariates in the CAG ≥37 group compared to CAG <37. One small study that utilized a 3-day diary reported increased carbohydrate intake among individuals with an expanded CAG repeat,5 as we also report in unadjusted analysis (table 1). Longitudinal studies will be helpful in determining whether there are changes in macronutrients and energy requirements as some individuals in this cohort develop HD.
Weight loss among HD gene carriers may be due to excessive involuntary movements, difficulty with swallowing, or malabsorption. We cannot exclude the possibility of increased energy expenditure secondary to movement; however, we believe that we considerably reduced the possibility that increased energy expenditure would be due solely to subtle involuntary movement by eliminating 22 individuals who received a clinical research diagnosis of HD at baseline or at any time up to the administration of the FFQ. In a study of 517 patients with early stage HD, BMI decrease over a 3-year period was related to expanded CAG repeat length, but not to any specific component of the UHDRS (motor, cognitive, or behavioral).18 When we adjusted for total motor score and chorea alone, the relationship between caloric intake and membership in the CAG ≥37 category was diminished but did not disappear.
Using the Huntington Study Group database of manifest HD cases compared to age-matched controls (1:5), we reported significantly lower BMI in 361 symptomatic HD cases at the earliest stage of the illness (Total Functional Capacity >11 [0–13; 13 normal], chorea or dystonia score ≤2, and duration <4 years), suggesting that differences in body composition may occur early among symptomatic individuals.4 BMI was lower than reported in the current study of pre-manifest individuals, although scores on the chorea and dystonia scores were similar, suggesting that the individuals in the previous study were more advanced. Because the participants in this study were not aware of their genetic status, any increase in caloric intake could not be attributed to that information. In addition, if swallowing difficulty accounted for weight loss, one might expect a decrease rather than an increase in caloric intake in the CAG ≥37 group.
Limitations of the current study include the fact that the dietary assessments were self reports, and there was no opportunity to validate dietary intake. In addition, all surveys were filled out privately. There were no corrections or queries for missing data. One-third of the participants did not complete the FFQ. These individuals performed significantly worse on the cognitive portion of the UHDRS and reported more depressive symptoms and functional complaints; however, the proportion of carriers of an expanded CAG repeat did not significantly differ between those who completed the survey and those who did not. We did not have specific questions about dysphagia. We were only able to approximate TEE because indirect calorimetry was not obtained. We also had no measure of reported physical activity to determine to what extent the TEE was due to physical activity. Indirect calorimetry studies and precise measurement of physical activity in this population of pre-manifest individuals would be valuable.
These data add to the converging evidence from both murine and human HD of a heightened metabolic (procatabolic) state that may occur prior to the development of the overt motor manifestations of HD.9,19,20 A metabolomic (small molecule metabolite) profile consisting of changes in fatty acid breakdown products, increased nucleic acid breakdown, and a dysregulation of amino acid metabolism was associated with a procatabolic state in both murine models and human HD compared with controls.20 In murine models and humans, the metabolic signatures differed between the pre-manifest and clinically manifest states, suggesting a change with disease progression. A study of 15 pre-manifest HD cases using 1H NMR spectroscopy showed that low plasma levels of branched-chain amino acids distinguished pre-manifest HD cases from controls, and these levels were correlated with weight loss and CAG repeat length.8
Weight loss may predate the motor manifestations in HD transgenic mice (R6/2 and N17182Q). It has been postulated that reduction in HAP-1, which binds more strongly to mutant Huntingtin than normal huntingtin protein and is a feeding stimulatory protein, may be linked to weight loss in HD.9 HAP-1 knockout mice show both weight loss and hypothalamic degeneration.21
Whether changes in TEE and BMI are strictly pre-manifest or occur in the setting of subtly emerging illness is unclear. In Parkinson disease (PD), weight loss in the setting of increased caloric intake occurred 2 to 4 years prior to diagnosis22 and increases with disease progression. No difference in BMI was seen among men who participated in the Harvard Alumni study who developed incident PD (n = 106) when BMI was examined prior to college and at baseline assessment; however, those who lost 0.5 kg per decade between college and baseline were at increased risk for development of PD.23 In another population-based study, BMI >23 between ages 25–59 was associated with a twofold increased risk of PD.24 These studies suggest that while high absolute BMI in midlife may be associated with increased risk of PD, reduction in BMI over time may also be a risk for incident PD.
The correlation of caloric intake, but not BMI, with higher CAG repeat length and increased estimated probability of onset of HD within 5 years suggests that individuals may be consuming more calories to maintain their weight during the pre-manifest period but eventually may not be able to compensate and preserve energy balance. Early to mid-stage HD cases provided with sufficient caloric intake in a controlled setting were capable of maintaining a positive energy balance. However, under free living conditions, repeated 24-hour dietary recalls were more variable in these patients with HD than controls and variability increased with disease stage (Total Functional Capacity),7 which may explain why some patients with HD lose weight. The relationship between dietary intake and measures of disease severity indicators is expected to be clarified with longitudinal assessment of the PHAROS cohort.
AUTHOR CONTRIBUTIONS
Statistical analyses performed by Drs. Zhao and Oakes and S. Eberly.
ACKNOWLEDGMENT
The authors thank their committees, consultants, and sponsors, as well as the PHAROS research participants and their families.
DISCLOSURE
Dr. Marder has received grants from the National Institutes of Health, the Parkinson Disease Foundation, and research support through Columbia University from Amarin Neuroscience LTD, Boehringer-Ingelheim, and Neurosearch. Dr. Zhao has received grants from the National Institutes of Health. S. Eberly reports no disclosures. Dr. Tanner has received research support from the National Institutes of Health, the Michael J. Fox Foundation, the Parkinson’s Disease Foundation, the Department of Defense, a group of current and former welding manufacturers, and James and Sharron Clark. Dr. Tanner has received honoraria from Lundbeck Pharmaceuticals. Dr. Oakes has received grants from the National Institutes of Health, has served as a Special Government Employee for the FDA, and has received compensation from Link Medicine, Neurogen Corporation, and Pfizer Inc. for professional services unrelated to the study described in this article. Dr. Shoulson has received research support through the University of Rochester from Amarin Neuroscience LTD, Cephalon Inc., CHDI Foundation Inc., Department of Defense, the National Institutes of Health (NHGRI, NINDS), Neurogen Corporation, and Prestwick Pharmaceuticals. He has received consulting honoraria from Alnylam, American Neurological Association, Archives of Neurology, Campbell Alliance, CombinatoRx Inc., EMD Serono Inc., EnVivo Pharm, Impax Laboratories, Intec Pharmaceutical, Johnson & Johnson, Link Medicine, Massachusetts General Hospital, Medtronic, Michael J. Fox Foundation, Movement Disorder Society, Neurogen Corporation, Omeros Corporation, Parkinson’s Disease Foundation, Prana Biotechnology, RJG Foundation, Sanofi-Aventis, University of California Irvine, University of Iowa, University of Pennsylvania, and Xenoport. US Government Sponsors: National Institutes of Health (# 2 R01 HG002449-06), including support from the National Human Genome Research Institute and the National Institute of Neurological Disorders and Stroke; Foundation Sponsors: High Q Foundation (New York, NY), Huntington’s Disease Society of America (New York, NY), Hereditary Disease Foundation (Santa Monica, CA), Huntington Society of Canada (Kitchener, Ontario), and the Fox Family Foundation (New Jersey).
APPENDIX
The following members of the Huntington Study Group are investigators in Prospective Huntington At-Risk Observational Study (PHAROS) and authors of this report. Steering Committee—University of Rochester, NY: Ira Shoulson, MD (principal investigator); Massachusetts General Hospital, Charleston, MA: Anne Young, MD, PhD (co-principal investigator); University of Rochester, NY: Karl Kieburtz, MD, MPH (director, Clinical Trials Coordination Center), David Oakes (chief biostatistician), Elise Kayson, MS, RNC (project coordinator), Hongwei Zhao, ScD, M. Aileen Shinaman, JD (HSG executive director), Megan Romer, MS; Johns Hopkins University, Baltimore, MD: Kevin Biglan, MD (medical monitor); Massachusetts General Hospital, Charleston: Steven Hersch, MD, PhD, Jack Penney, MD†; Columbia University Medical Center, New York, NY: Karen Marder, MD, MPH; University of Iowa: Jane Paulsen, PhD; Indiana University School of Medicine, Indianapolis: Kimberly Quaid, PhD; Lily Corporate Center, Indianapolis, IN: Eric Siemers, MD; The Parkinson’s Institute, Sunnyvale, CA: Caroline Tanner, MD. Participating Investigators and Coordinators—Hereditary Neurological Disease Centre (HNDC), Wichita, KS: William Mallonee, MD, David Palmer, MD†, Greg Suter, BA; University of Kansas Medical Center, Kansas City: Richard Dubinsky, MD, Gary Gronseth, MD, R. Neil Schimke, MD, Carolyn Gray, RN; Hennepin County Medical Center/Minneapolis, MN: Martha Nance, MD, Scott Bundlie, MD, Dawn Radtke, RN; Ohio State University, Columbus: Sandra Kostyk, MD, PhD, George W. Paulson, MD, Karen Thomas, DO, Nonna Stepanov, MD, Corrine Baic, BS; Wake Forest University School of Medicine, Winston Salem, NC: James Caress, MD, Francis Walker, MD, Vicki Hunt, RN; Hotel-Dieu Hospital-CHUM, Montreal, QC: Sylvain Chouinard, MD, Guy Rouleau, MD, PhD, Hubert Poiffaut, RN, Brigitte Rioux†; Emory University School of Medicine, Atlanta, GA: Claudia Testa, MD, PhD, Timothy Greenamyre, MD, PhD, Joan Harrison, RN; University of California, San Diego (UCSD), LaJolla: Jody Corey-Bloom, MD, PhD, David Song, MD, Guerry Peavy, PhD, Jody Goldstein, BS; University of Iowa: Jane Paulsen, PhD, Henry Paulson, MD, Robert L. Rodnitzky, MD, Ania Mikos, BA, Becky Reese, BS, Laura Stierman, BS, Katie Williams, BA, Lynn Vining, RN, MSN; Columbia University Medical Center, New York, NY: Karen Marder, MD, MPH, Elan Louis MD, MSc, Carol Moskowitz, RN; Indiana University School of Medicine, Indianapolis: Kimberly Quaid, PhD, Joanne Wojcieszek, MD, Melissa Wesson, MS; University of Washington & VA Puget Sound Health Care System, Seattle: Ali Samii, MD, Thomas Bird, MD, Hillary Lipe, ARNP; Medical College of Wisconsin, Milwaukee: Norman Reynolds, MD, Karen Blindauer, MD, Jeannine Petit, ANP; University of Rochester, NY: Peter Como, PhD, Frederick Marshall, MD, Timothy Counihan, MD, Kevin Biglan, MD, Carol Zimmerman, RN; Oregon Health & Science University, Portland: Penelope Hogarth, MD, John Nutt, MD, Pamela Andrews, BS, CCRC; Massachusetts General Hospital, Charleston: Steven Hersch, MD, PhD, Leslie Shinobu, MD, PhD, Diana Rosas, MD, Yoshio Kaneko, BA, Sona Gevorkian, MS, Paula Sexton, BA, CCRA; Mayo Clinic Scottsdale, AZ: John Caviness, MD, Charles Adler, MD, PhD; University of California Davis, Sacramento: Vicki Wheelock, MD, David Richman, MD, Teresa Tempkin, RNC, MSN; Brown University (Memorial Hospital of Rhode Island), Pawtucket, RI: Chuang-Kuo Wu, MD, PhD, Hubert Fernandez, MD, Joseph H. Friedman, MD, Margaret Lannon, RN, MS; Colorado Neurological Institute, Englewood: Lauren Seeberger, MD, Christopher O’Brien, MD, Sherrie Montellano, MA; University of Michigan, Ann Arbor: Ninith Kartha, MD, Sharin Sakurai, MD, PhD, Susan Hickenbottom, MD, PhD, Roger Albin, MD, PhD, Kristine Wernette, RN, MS; Washington University, St. Louis, MO: Brad Racette, MD, Joel S. Perlmutter, MD, Laura Good, BA; UCLA Medical Center, Los Angeles, CA: George Jackson, MD, PhD, Susan Perlman, MD, Shelley Segal, MD, Russell Carroll, MA, Laurie Carr, BS; University of Alberta, Edmonton: Wayne Martin, MD, Ted Roberts, MD, Marguerite Wieler, BSC, PT; University of British Columbia, Vancouver: Blair Leavitt, MD, Lorne Clarke, MD, CM, Lynn Raymond, MD, PhD, Joji Decolongon, MSC, Vesna Popovska, MD, Elisabeth Almqvist, RN, PhD; Baylor College of Medicine, Houston, TX: William Ondo, MD, Madhavi Thomas, MD, Tetsuo Ashizawa, MD, Joseph Jankovic, MD; University of South Florida, Tampa: Robert Hauser, MD, Juan Sanchez-Ramos, MD, PhD, Karen Price, MA, Holly Delgado, RN; University of Calgary, Alberta: Sarah Furtado, MD, PhD, Anne Louise LaFontaine, MD, Oksana Suchowersky, MD, Mary Lou Klimek, RN, MA; Centre for Addiction and Mental Health, Markham, Ontario: Rustom Sethna, MD, Mark Guttman, MD, Sandra Russell, BSW, RSW, Sheryl Elliott, RN; North Shore University Hospital, Manhasset, NY: Marc Mentis, MB, CHB, Andrew Feigin, MD, Marie Cox, RN, BSN, Barbara Shannon, RN; University of Alabama at Birmingham: Alan Percy, MD, Leon Dure, MD, Donna Pendley, RN, Jane Lane, RN, BSN; University of Virginia, Charlottesville: Madaline Harrison, MD, Elke Rost-Ruffner, RN, BSN; UMDNJ Robert Wood Johnson Medical Center, Stratford, NJ: William Johnson, MD; University of Pennsylvania, Philadelphia: Amy Colcher, MD, Andrew Siderowf, MD, Mary Matthews, RN; Institute for Neurodegenerative Disorders, New Haven, CT: Danna Jennings, MD, Kenneth Marek, MD, Karen Caplan, MSW; Albany Medical College, NY: Stewart Factor, DO, Donald Higgins, MD, Eric Molho, MD, Constance Nickerson, LPN, Sharon Evans, LPN, Diane Brown, RN†; Winnipeg Clinic, Winnipeg, Manitoba: Douglas Hobson, MD, Paul Shelton, MD, Shaun Hobson, RN; University of Miami, FL: Carlos Singer, MD, Nestor Galvez-Jimenez, MD, William Koller, MD†, Doris Martin, DDS, Kelly Lyons, PhD, Dinorah Rodriguez, RN; Rush Presbyterian-St. Luke’s Medical Center, Chicago, IL: Kathleen Shannon, MD, Cynthia Comella, MD, Jean Jaglin, RN, CCRC; University of Maryland School of Medicine, Baltimore: Karen Anderson, MD, William Weiner, MD, Kelly Dustin, RN, BSN; Johns Hopkins University, Baltimore, MD: Adam Rosenblatt, MD, Christopher Ross, MD, PhD, Deborah Pollard; Boston University, MA: Marie H. Saint-Hilaire, MD, Peter Novak, MD, J. Stephen Fink, MD, PhD†, Bonnie Hersh, MD, Melissa Diggin, MS, RN, Leslie Vickers, RN, MS; University of Connecticut, Hartford: Wallace Deckel, PhD, James Duffy, MD, Mary Jane Fitzpatrick, APRN. Participating NIH Authors—National Human Genome Research Institute, Bethesda, MD: Elizabeth Thomson, PhD; National Institute of Neurological Disorders and Stroke, Bethesda, MD. Event Monitoring Committee—Massachusetts General Hospital: Steven Hersch, MD, PhD (co-chair); Indiana University: Julie Stout, PhD (co-chair); James Calhoun; University of Iowa: William Coryell, MD, Cheryl Erwin, JD, PhD; Wake Forest University School of Medicine: Vicki Hunt, RN; Johns Hopkins University: Christopher Ross, MD, PhD; Minnesota Center for Health Care Ethics: Dorothy Vawter, PhD. Ethics Committee—Chicago-Kent College of Law: Lori Andrews, JD, Debbie Bury, James Calhoun; Massachusetts General Hospital: Steven Hersch, MD, PhD (chair); Wake Forest University School of Medicine: Vicki Hunt, RN, Carl Leventhal, MD; Indiana University School of Medicine: Kimberly Quaid, PhD; University of Rochester: Aileen Shinaman, JD; Minnesota Center for Health Care Ethics: Dorothy Vawter, PhD; Columbia University: Nancy Wexler, PhD. Biostatistics and Clinical Trials Coordination Center—University of Rochester: Alicia Brocht, BA, Susan Daigneault, Karen Gerwitz, BS, Connie Orme, BA, Ruth Nobel, Victoria Ross, MA, Mary Slough, Arthur Watts, BS, Joe Weber, BS, Christine Weaver, Elaine Julian-Baros. Genetic/Environmental Modifiers Committee—Massachusetts General Hospital: Anne Young, MD, PhD (chair); Columbia University Medical Center: Karen Marder, MD (co-chair); Indiana University School of Medicine: Tatiana Foroud, PhD; Massachusetts General Hospital: James Gusella, PhD; Massachusetts Institute of Technology: David Housman, PhD; Massachusetts General Hospital: Marcy MacDonald, PhD; Boston University: Richard Myers, PhD; The Parkinson’s Institute: Caroline Tanner, MD; Massachusetts General Hospital: Rudolph Tanzi, PhD. Independent Monitoring Committee—Columbia University: Stanley Fahn, MD; Indiana University: Michael Conneally, PhD; Columbia University: Weiu-Yann Tsai, PhD. Scientific Advisory Committee—New York Hospital Department of Neurology: Flint Beal, MD; Massachusetts Institute of Technology: David Housman, PhD; Johns Hopkins University: Christopher Ross, MD, PhD; Massachusetts General Hospital: Rudolph Tanzi, PhD, Anne Young, MD, PhD; University of California, Irvine: Claudia Kawas, MD; University of California Los Angeles: Marie Francoise-Chesselet, MD, PhD. DNA Oversight Committee—Indiana University Medical Center: Michael Conneally, PhD; University of Minnesota/Minnesota VA Medical Center: Martha Nance, MD; University of California San Diego, Clifford Shults, MD†; The Parkinson’s Institute: Caroline Tanner, MD. Independent Rater Video Committee—Oregon Health & Science University: Penelope Hogarth, MD; Massachusetts General Hospital: Diana Rosas, MD; University of Rochester: Hongwei Zhao, ScD. †Deceased.
Address correspondence and reprint requests to Dr. Karen Marder, 630 W. 168th St. Unit 16, Columbia University College of Physicians and Surgeons, New York, NY 10032 ksm1@columbia.edu
*PHAROS co-investigators are listed in the appendix.
Disclosure: Author disclosures are provided at the end of the article.
Received September 2, 2008. Accepted in final form May 1, 2009.
REFERENCES
- 1.Morales LM, Estevez J, Suarez H, et al. Nutritional evaluation of Huntington disease patients. Am J Clin Nutr 1989;50:145–150. [DOI] [PubMed] [Google Scholar]
- 2.Hamilton JM, Wolfson T, Peavy GM, et al. Rate and correlates of weight change in Huntington’s disease. J Neurol Neurosurg Psychiatry 2004;75:209–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robbins AO, Ho AK, Barker RA. Weight changes in Huntington’s disease. Eur J Neurol 2006;13:e7. [DOI] [PubMed] [Google Scholar]
- 4.Djousse L, Knowlton B, Cupples LA, et al. Weight loss in early stage of Huntington’s disease. Neurology 2002;59:1325–1330. [DOI] [PubMed] [Google Scholar]
- 5.Trejo A, Tarrats RM, Alonso ME, et al. Assessment of the nutrition status of patients with Huntington’s disease. Nutrition 2004;20:192–196. [DOI] [PubMed] [Google Scholar]
- 6.Pratley RE, Salbe AD, Ravussin E, Caviness JN. Higher sedentary energy expenditure in patients with Huntington’s disease. Ann Neurol 2000;47:64–70. [PubMed] [Google Scholar]
- 7.Gaba AM, Zhang K, Marder K, et al. Energy balance in early-stage Huntington disease. Am J Clin Nutr 2005;81:1335–1341. [DOI] [PubMed] [Google Scholar]
- 8.Mochel F, Charles P, Seguin F, et al. Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS ONE 2007;2:e647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen A, Bjorkqvist M. Hypothalamic-endocrine aspects in Huntington’s disease. Eur J Neurosci 2006;24:961–967. [DOI] [PubMed] [Google Scholar]
- 10.Block G, Hartman AM, Naughton D. A reduced dietary questionnaire: development and validation. Epidemiology 1990;1:58–64. [DOI] [PubMed] [Google Scholar]
- 11.Block G, Woods M, Potosky A, Clifford C. Validation of a self-administered diet history questionnaire using multiple diet records. J Clin Epidemiol 1990;43:1327–1335. [DOI] [PubMed] [Google Scholar]
- 12.The Huntington Study Group PHAROS Investigators. At risk for Huntington disease: The PHAROS (Prospective Huntington At Risk Observational Study) cohort enrolled. Arch Neurol 2006;63:991–996. [DOI] [PubMed] [Google Scholar]
- 13.Hogarth P, Kayson E, Kieburtz K, et al. Interrater agreement in the assessment of motor manifestations of Huntington’s disease. Mov Disord 2005;20:293–297. [DOI] [PubMed] [Google Scholar]
- 14.Harris JA, Bennedict FG. A Biometric Study of Basal Metabolism in Man. Washington, DC: Carnegie Institute; 191.
- 15.Langbehn DR, Brinkman RR, Falush D, et al. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet 2004;65:267–277. [DOI] [PubMed] [Google Scholar]
- 16.Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry 2008;79:874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marder K, Zhao H, Myers RH, et al. Rate of functional decline in Huntington’s disease: Huntington Study Group. Neurology 2000;54:452–458. [DOI] [PubMed] [Google Scholar]
- 18.Aziz NA, van der Burg JM, Landwehrmeyer GB, et al. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 2008;71:1506–1513. [DOI] [PubMed] [Google Scholar]
- 19.Sathasivam K, Hobbs C, Mangiarini L, et al. Transgenic models of Huntington’s disease. Philos Trans R Soc Lond B Biol Sci 1999;354:963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Underwood BR, Broadhurst D, Dunn WB, et al. Huntington disease patients and transgenic mice have similar pro-catabolic serum metabolite profiles. Brain 2006;129:877–886. [DOI] [PubMed] [Google Scholar]
- 21.Li SH, Yu ZX, Li CL, et al. Lack of huntingtin-associated protein-1 causes neuronal death resembling hypothalamic degeneration in Huntington’s disease. J Neurosci 2003;23:6956–6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Zhang SM, Hernan MA, et al. Weight loss in Parkinson’s disease. Ann Neurol 2003;53:676–679. [DOI] [PubMed] [Google Scholar]
- 23.Logroscino G, Sesso HD, Paffenbarger RS, Jr., Lee IM. Body mass index and risk of Parkinson’s disease: a prospective cohort study. Am J Epidemiol 2007;166:1186–1190. [DOI] [PubMed] [Google Scholar]
- 24.Hu G, Jousilahti P, Nissinen A, et al. Body mass index and the risk of Parkinson disease. Neurology 2006;67:1955–1959. [DOI] [PubMed] [Google Scholar]