

Abstract
Dielectrophoretic field-flow-fractionation (DEP-FFF) was applied to several clinically relevant cell separation problems, including the purging of human breast cancer cells from normal T-lymphocytes and from CD34+ hematopoietic stem cells, the separation of the major leukocyte subpopulations, and the enrichment of leukocytes from blood. Cell separations were achieved in a thin chamber equipped with a microfabricated, interdigitated electrode array on its bottom wall that was energized with AC electric signals. Cells were levitated by the balance between DEP and sedimentation forces to different equilibrium heights and were transported at differing velocities and thereby separated when a velocity profile was established in the chamber. This bulk-separation technique adds cell intrinsic dielectric properties to the catalog of physical characteristics that can be applied to cell discrimination. The separation process and performance can be controlled through electronic means. Cell labeling is unnecessary, and separated cells may be cultured and further analyzed. It can be scaled up for routine laboratory cell separation or implemented on a miniaturized scale.
Modern cell separation techniques1,2 have been fundamental to many advances in cell biology, molecular genetics, biotechno-logical production, clinical diagnostics, and therapeutics. The most common of these techniques, centrifugation,2 electrophoresis,3 and both fluorescence- (FACS)4 and magnetic-activated-cell sorting (MACS),5-6 take advantage of differences in cell density, electrical charge, and immunological surface markers. Using these methods, investigators and clinicians are able to deplete particular cell populations (e.g., purge tumor cells from stem cell transplants) or enrich them (e.g., purify CD34+ stem cells from human blood).
As these technologies have reached maturity, however, it has become more difficult to make fundamental improvements in separation resolution, cell purity, sample size, and device cost and portability. Therefore, novel physical methods by which different cell types may be discriminated and selectively manipulated are desirable. Besides offering additional avenues for cell identification, such methods should, ideally, allow us to enhance cell separation, exploit integrated microfluidic methods, separate microliter-size samples, reduce cost, and develop portable separation devices. To this end, cell dielectric properties have been explored through dielectrophoresis (DEP) and other AC electro-kinetic effects7-15 for developing cell separation techniques.16-20
Dielectrophoretic forces occur on cells when a nonuniform electrical field interacts with field-induced electrical polarization.8,15 Depending on the dielectric properties of the cells relative to their suspending medium, these forces can be positive or negative, directing the cells toward strong or weak electrical field regions, respectively.8,15,21 Because cells of different types or in distinct biological states have different dielectric properties,12,17,22-24 differential DEP forces can be applied to drive their separation into purified cell subpopulations.14,16-20,25-26
A recently developed technique, dielectrophoretic-field-flow fractionation (DEP-FFF), has been demonstrated to exhibit a high and electrically controllable discrimination for cell separation.18-20 27 DEP forces produced by microelectrodes are used to levitate cells in a thin chamber to equilibrium heights where sedimentation forces balance the vertical DEP force components (Figure 1). A carrier fluid moves through the chamber and establishes a hydrodynamic velocity profile causing cells of different dielectric and density properties levitated to different equilibrium heights to be transported through the chamber at different velocities and thereby separated.
Figure 1.
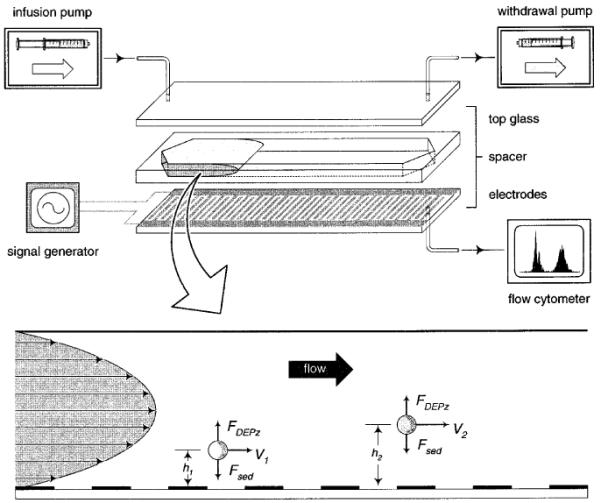
DEP-FFF principle and experimental setup. A thin, rectangular chamber was constructed with microfabricated, interdigitated electrodes on its bottom wall. Different cell types were levitated to different equilibrium heights under the influence of the opposing DEP (FDEPz) and sedimentation (Fsed) forces. With a flow-velocity profile established in the chamber from the injection syringe pump, the cells at different heights (h2 > h1) were carried through the chamber at different velocities (V2 > V1) and thereby separated. Cells exited the chamber from the bottom outlet port and were detected and counted by an on-line flow cytometer.
Here we report the application of the DEP-FFF method to several problems relevant to basic research and clinical settings. These are the separation of breast cancer cells from normal T-lymphocytes and from CD34+ hematopoietic stem cells, the separation of the major leukocyte subpopulations, and the enrichment of leukocytes from blood. We analyze the separation criteria, describe how the applied DEP-field conditions can be chosen so as to vary the DEP-FFF separation discrimination, and discuss the applicability of the DEP-FFF technique to various cell separation problems including those for which microfluidic devices would be most suitable.
THEORETICAL BACKGROUND
As shown in Figure 1, dielectrophoretic levitation forces generated by the interdigitated microelectrode array are used to levitate cells to different equilibrium heights in the chamber. It was demonstrated previously18-20 that the equilibrium heights are dependent on the cell dielectric polarization factor αDEP, the density difference (ρm -ρc) between the cells and the suspending medium, and the applied DEP signal amplitude U, and are given by
| (1) |
where d0 and d1 are constants dependent on electrode geometry and dielectric permittivity of the suspending medium. The dielectric polarization factor αDEP is a function of the applied field frequency f. Under the influence of a parabolic flow profile, the velocity of a cell located at a height heq from the chamber bottom surface is given by
| (2) |
where H and W are the chamber thickness and width, respectively, and Fr is the fluid flow rate. kr (<1) is a coefficient that characterizes a retardation effect that occurs when particles are close to the chamber wall.
Thus, by choosing appropriate DEP field and fluid flow conditions based on eqs 1 and 2, cells having different dielectric and density properties can be levitated to different heights above the electrode surface and thereby be caused to move at different velocities under the influence of the flow profile. Typically, DEPFFF is operated under a single frequency field (f in eq 1). The optimal separation frequency can be determined theoretically by calculating18-20 the frequency dependencies of cell velocities on the basis of dielectric and density properties of cell types to be separated and choosing the frequency at which maximum velocity difference occurs. Alternatively, the separation frequency can be identified empirically after experimentally determining the DEPFFF responses of individual cell populations at different frequencies. DEP field conditions used in this work were chosen through this latter approach. As described18-20 previously, stable DEP levitation of cells is possible only in the frequency region where the cell dielectric polarization factor αDEP is negative. For mammalian cells suspended in a medium having an electrical conductivity ~10 mS/m, negative αDEP occurs in the frequency range up to ~100 kHz. Thus, we first measured the DEP-FFF responses of individual cell populations in the frequency ranges up to 100 kHz and then chose the separation frequencies at which the cell populations to be separated demonstrated large differences in the DEP-FFF elution times.
For separation of some cell mixtures, DEP signals having a frequency modulation were used. Under a single-frequency condition, efficient separations typically occurred in the frequencies where DEP forces exerted on the cells of one type exhibited a wide distribution from being negative to positive because of the inherent heterogeneity in cell dielectric properties.18,20 Thus, the cells under negative DEP forces were levitated to different heights while those under positive DEP effects were trapped at the electrode elements. This lead to a broadening of the cell elution fractograms. We found experimentally that sweeping the signal frequency could decrease the number of the cells being trapped on the electrode elements and lead to more compact elution fractograms. A comprehensive analysis of the single-frequency and swept-frequency protocols has been provided elsewhere.45
EXPERIMENTAL SECTION
Cell Preparation
Human breast cancer MDA-435 cells were cultured in MEM supplemented with 10% fetal bovine serum under standard tissue culture conditions.12,22 Hematopoietic CD34+ stem cells were prepared from mobilized peripheral blood samples using a MACS system (Miltenyi Biotech), yielding >90% pure CD34+ cells. Leukocyte subpopulations (T-, B-lymphocytes, monocytes, and granulocytes) were derived from buffy coat preparations (Gulf Coast Regional Blood Bank, Houston, TX) using density-gradient centrifugation, MACS sorting, and erythrocyte lysis, as described previously.24 To allow for flow cytometry detection of the cells eluted from the DEP-FFF chamber, CD34+ cells, B-lymphocytes, and monocytes were labeled with PE- or FITC-conjugated CD34, CD3, and CD14 antibodies (Becton Dickinson, San Jose, CA), respectively, by incubating the cell suspension with the antibody solution (volume ratio 5:2) for 30 min at 4 °C in the dark. Labeled cells were then washed once and resuspended in an isotonic buffer (8.5% w/v sucrose plus 0.3% w/v dextrose) having an electrical conductivity of 10 mS/m, which was adjusted by adding culture medium. Appropriate cell populations were then mixed in the sucrose buffer. Cell mixtures included MDA-435 cells with T-lymphocytes, MDA-435 cells with CD34+ stem cells, B-lymphocytes with monocytes. The final cell concentration was ~1.2 × 106/mL.
For DEP-FFF enrichment of leukocytes from blood, human blood was taken from healthy volunteers and stained with PECD45 antibody solutions to label leukocytes. The cell samples were then diluted in the isotonic sucrose buffer to achieve the final cell concentration of ~5 × 106/mL.
DEP-FFF System Setup
The DEP-FFF chamber and experimental setup are shown in Figure 1. Interdigitated microelectrodes having 50-μm width and spacing were fabricated on 50 × 50 mm glass substrates using standard photolithography. A Teflon spacer, which was cut in the center to provide a separation channel (H 0.42 × W 25 × L 388 mm), was sandwiched between a top glass plate and a long electrode plate (consisting of eight electrode substrates in series). The microelectrodes were connected in parallel to a lab-built PA05-based power amplifier (Apex Micro-technology, Tucson, AZ). The top and bottom plates were drilled with 1.6-mm-diameter holes to fit inlet and outlet PEEK tubing (0.0625-in o.d., 0.010-in i.d., Upchurch Scientific, Oak Harbor, WA). An infusion syringe pump (Daigger, Wheeling, IL) was connected to the chamber through an injection valve (Rheodyne model 7010, Rheodyne, Cotati, CA) equipped with a 50-μL loop to provide continuous flow of the carrier medium in the chamber. A second syringe pump was connected to the top outlet port of the chamber and was operated to withdraw the cell-free portion of the carrier fluid that constituted as much as 95% of the total flow through the chamber. Cells exited the chamber through the bottom outlet port and were fed directly to the injection needle of a flow cytometer (BRYTE HS, Bio-Rad, Hercules, CA) for detection, bypassing the normal cytometer sample handling fluidics.
DEP-FFF Operation Protocol
The DEP-FFF chamber was first loaded with sucrose buffer. An aliquot of the cell mixture (50 μL) was then introduced into the inlet port of the chamber through the injection valve, as described previously.26,28,29 A DEP signal (4 V p-p) at 10 kHz was applied to the electrodes during sample injection so that cells were levitated in the chamber by DEP forces and thereby prevented from adhering to the bottom surface of the chamber. Different operation protocols were then applied for separating different cell mixtures after sample injection. The protocols for separating MDA-435 cells from T-lymphocytes consisted of the following steps:
Prior to the application of the fluid flow, a 10 kHz field was applied for 10 min to allow the cells to reach their equilibrium height positions. As described previously,19 cell sedimentation time depends on cell size, density difference between the cells and the suspending medium, the viscosity of the medium, and the sedimentation distance. Our calculation based on these parameters demonstrated that all the cell types studied here would sediment to a total chamber height of 0.42 mm in less than 7 min.
A flow velocity profile was established in the chamber by starting the injection and withdrawal syringe pumps at rates of 2 and 1.6 mL/min, respectively. The DEP field was switched to 40 kHz (or swept between 15 and 35 kHz at a cycle period of 5 s). This condition was maintained for 5 or 7 min so that all the T-lymphocytes were eluted from the chamber and identified and counted by the on-line flow cytometer.
The DEP field was changed to 5 kHz so that the previously retained MDA-435 cells were now levitated, eluted from the chamber, and detected by the flow cytometer.
For separating other cell mixtures, different DEP field conditions were applied during the second segment of the protocol and are summarized in Table 1. Fluid flow conditions were chosen according to two factors. First, as in our earlier findings,18-20 we noted that the fluid flow rate as high as several milliliters per minute influenced the time when the cells were eluted but not relative widths of the elution peaks of the cells. Thus, it was desirable to operate at a high flow rate to reduce the separation time. Second, the separated cells were detected by the flow cytometer, which has a maximum operational flow rate of ~0.4 mL/min. This sets an upper limit on the total flow rate since it is required that all eluted cells go through the cytometer. On the basis of these considerations and the observed DEP-FFF responses of individual cell populations, we have chosen a total flow rate of 2 mL/min for most of our separation studies. The flow rate at the withdrawal syringe pump was varied for different studies to maximize the cell concentration going through the cytometer. The only exception was for the separation of leukocytes from blood where a total flow rate of 0.5 mL/min was used because of the relatively high cell concentrations in the initial cell mixture. In all the studies reported here, we experimentally verified that no cells were present in the withdrawal syringe after separation experiments.
Table 1.
DEP-FFF Separation Performance Summary
| experimental systems (protocol)a | cell types | purity after separationd (%) | total-cell recoveryf (%) | separation time (min) |
|---|---|---|---|---|
| MDA-435 vs T-lymphocytes (2:3)b | MDA-435 | 99.2 | 69 | 11 |
| (40 kHz, 5 min; 2/1.6 ml/min)a | T-lymphocytes | 92 | ||
| MDA-435 vs T-lymphocytes (2:3)b | MDA-435 | 98 | 75 | 11 |
| (15-35 kHz, 5 min; 2/1.6 ml/min)a | T-lymphocytes | 92 | ||
| MDA-435 vs CD34+ cells (1:1)b | MDA-435 | 96 | 70 | 14 |
| (40 kHz, 7 min; 2/1.9 ml/min)a | CD-34+ | 99.5 | ||
| monocytes vs B-lymphocytes (1:2)b | monocytes | 94 | 73 | 15 |
| (20-40 kHz, 7 min; 2/1.9 ml/min)a | B-lymphocytes | 92 | ||
| enrichment of leukocytes from blood | leukocytes | 5e | 55 | 25 |
| (10 kHz, 25 min; 0.5/0.4 ml/min)c | erythrocytes | 99.99 |
Single frequency or swept frequency (cycle period, 5 s) of the DEP field used during the 2nd segment of the protocol (see DEP-FFF Operation Protocol for the meaning of the 2nd segment). The two flow rates correspond to the infusion and withdrawal syringe pumps at the chamber inlet and outlet ends, respectively.
The ratio between the two cell populations in initial mixtures.
The enrichment of leukocytes from blood used a DEP field at 10 kHz for 25 min.
Purity after separation was determined by the flow cytometry for the corresponding elution peaks.
Leukocyte to erythrocyte ratio was increased from 1:700 to 1:19 after DEP-FFF enrichment.
Cell recovery was defined as the ratio of the total cell number detected by flow cytometry at the DEP-FFF chamber outlet end to the targeted total cell number that was calculated on the basis of the cell concentration and the injection loop volume. We noted that some cells were temporarily trapped at the inlet (and outlet) tubing between the separation chamber and the injection valve (and the cytometer) during the DEP-FFF process.
RESULTS AND DISCUSSION
Separation of Breast Cancer Cells from Normal T-lymphocytes
The isolation and enumeration of cancer cells circulating in peripheral blood is potentially an important screening tool for early detection of cancer and allows for the genetic and biochemical characterization of cancer cells for diagnosis and prognosis.28-30 Current methods, which can detect one cancer cell per ~106 mononuclear cells, involve separation of mononuclear cells from the blood, enrichment of cancer cells, and finally flow cytometric28,29 or PCR detection.30 To demonstrate the usefulness of the dielectrophoretic approach to this problem, we investigated the DEP-FFF separation of cultured, human breast cancer MDA-435 cells from T-lymphocytes, which constitute approximately 80% of peripheral blood mononuclear cells.
Cell dielectric properties and DEP behaviors depend sensitively on the frequency of the applied electrical field.7,8,16,17 Therefore, to establish suitable cell-separation conditions, we measured the DEP-FFF responses of breast cancer cells and T-lymphocytes separately as a function of frequency. Figure 2 shows cell elution fractograms for the two populations. While both cell types exhibited narrow, single elution peaks at 5 kHz, the fractogram for the breast cancer cells quickly broadened as the field frequency was increased above 10 kHz. At 20 kHz, only ~35% of the breast cancer cells were eluted; the rest were retained in the chamber by positive DEP forces. These cells were held at electrode edges by positive DEP forces and eluted from the chamber when we reduced the applied field frequency to 5 kHz. In contrast, the elution peak width for T-lymphocytes changed more gradually, and almost all were eluted at frequencies up to 50 kHz. These differences suggested that the two cell types should be separable at frequencies between 20 and 50 kHz.
Figure 2.
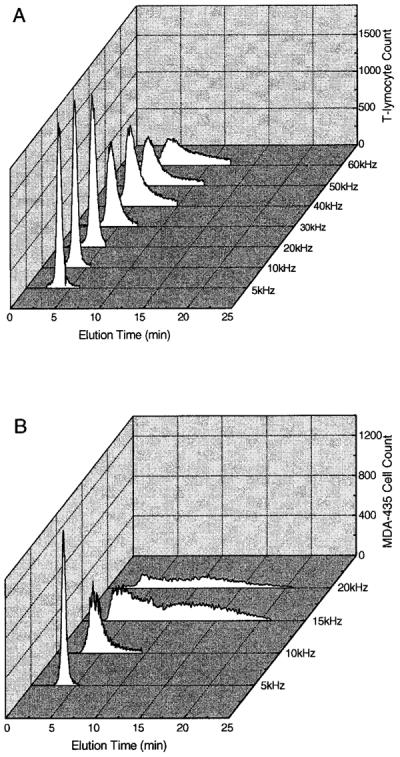
Frequency dependency of DEP-FFF elution fractograms for (A) T-lymphocytes and (B) human breast cancer MDA-435 cells obtained by the on-line flow cytometer. Compared with T-lymphocytes, MDA-435 cells exhibited rapidly broadening elution fractograms as frequencies increased above 10 kHz. Note the frequency scale difference. Cells were suspended at 1.2 × 106/mL in an isotonic sucrose/dextrose buffer having an electrical conductivity of 10 mS/ m. The applied voltage was 4 V p-p. The injection and withdrawal syringe pumps were operated at 2 and 1.6 mL/min, respectively.
To examine this possibility under demanding conditions, we prepared a cell mixture at a ratio of 2:3 (breast cancer/T lymphocyte) at a concentration of 1.2 × 106 cells/mL and applied DEP field frequencies around 30 kHz. As the DEP-FFF fractograms in Figure 3 show, the mixture was separated in 11 min and purities above 92% for the two populations and a total cell recovery of ~70% were achieved (Table 1). The separation occurred because, at frequencies around 30 kHz, T-lymphocytes were levitated well above the chamber bottom wall by DEP forces and transported under the influence of the fluid flow. Meanwhile, breast cancer cells were either barely levitated (and, therefore, carried slowly by the slower-moving fluid near the chamber bottom wall) or were trapped at the electrodes and immobilized by positive DEP forces. After T-lymphocytes were eluted from the chamber, the DEP field was switched to 5 kHz and breast cancer cells were then levitated and quickly eluted.
Figure 3.
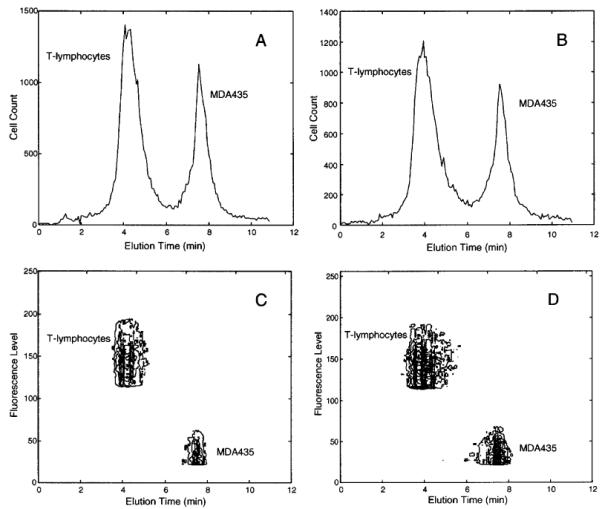
DEP-FFF fractograms showing the separation of human breast cancer MDA-435 cells from T-lymphocytes by a DEP field (A) at 40 kHz and (B) swept between 15 and 35 kHz, followed by a 5 kHz field (see Method and Materials for details). The corresponding contour plots (C and D) show fluorescence level vs elution time for cells exiting the DEP-FFF chamber. To allow identification, the T-lymphocytes were florescently labeled with PE-conjugated CD3 antibodies. Cell suspension, DEP signal voltage, and fluid-flow conditions were the same as for Figure 2.
Purging Breast Cancer Cells from CD34+ Hematopoietic Stem Cells
Treatment of advanced cancers often requires autologous, hematopoietic progenitor cell transplantation. For this to succeed, all cancer cells must be removed from the bone marrow or mobilized peripheral blood that will constitute the transplant. Currently, the most widely used technique for removing these cells is MACS in which CD34+ stem cells are positively selected and cancer cells are washed away and thereby excluded.31-32 Disadvantages of this method are the need for antibody-activated magnetic beads, a long incubation time for adequate cell labeling, and difficulty of removing the labels after sorting. DEP-FFF as a stand-alone or adjunct method for preparing stem-cell transplants eliminates the need for labeling by exploiting the intrinsic cell properties and overcomes these disadvantages.
As a demonstration, we used a mixture of cultured breast cancer cells and CD34+ stem cells at a ratio of 1:1. On the basis of the frequency dependencies of DEP-FFF responses of the two cell populations (data not shown), a DEP field of 40 kHz was applied for DEP-FFF separation. The fractograms (not shown) were similar to those shown in Figure 3. As was the case for T-lymphocytes, CD34+ cells were levitated higher than breast cancer cells and, therefore, into a faster moving part of the velocity flow profile where they were eluted more quickly than the breast cancer cells. DEP-FFF separation resulted in a CD34+ stem cell fraction having purity above 99.5% (Table 1).
Separation of the Major Leukocyte Subpopulations
Purification of major leukocyte subpopulations (i.e., T- and B-lymphocytes, monocytes, and granulocytes) is important in many clinical and biomedical applications. The differential diagnosis of bacterial, viral and parasitic infections and of mononucleosis and leukemia, for example, requires the enumeration of leukocyte subtypes. In research, purified leukocyte subpopulations are required for the study of molecular signaling between leukocyte subpopulations by the interleukins and for the analysis of immunological capacities of distinctive cell types.33-34
The major leukocyte subpopulations have significantly different dielectric properties.24 To determine the feasibility of purifying them by DEP-FFF, we mixed T- (or B-) lymphocytes with monocytes, T- (or B-) lymphocytes with granulocytes, and monocytes with granulocytes. To illustrate the DEP-FFF separations, we will consider the mixture of B-lymphocytes and monocytes, which was typical of all the blood cell mixtures. To obtain these cells, B-lymphocytes and monocytes were purified from a leukocyte-enriched buffy-coat preparation using the MACS method.24 The DEP-FFF elution times for B-lymphocytes and monocytes differed greatly with applied DEP field between 25 and 45 kHz (data not shown). This suggested that a swept frequency field between 25 and 45 kHz would be a suitable separation condition, and this was applied for DEP-FFF to a cell mixture (1:2 for monocytes/B-lymphocytes) at a total concentration of 1.2 × 106 cells/mL. A typical DEP-FFF fractogram obtained under these conditions is shown in Figure 4. DEP-FFF separation resulted in monocyte and B-lymphocyte fractions having purities of 94 and 92%, respectively (Table 1).
Figure 4.
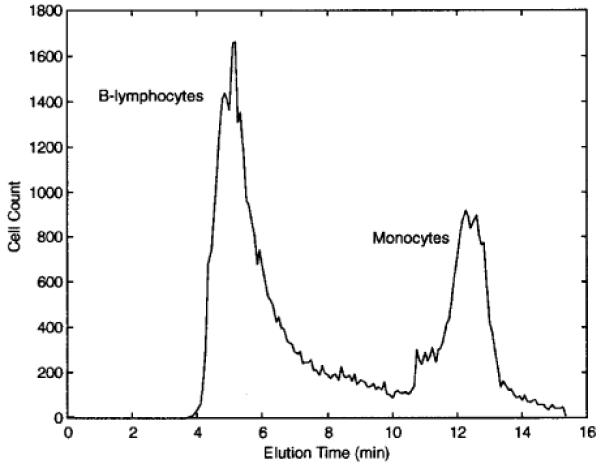
DEP-FFF fractogram showing the separation of monocytes from B-lymphocytes. The injection and withdrawal syringe pumps were operated at 2 and 1.9 mL/min, respectively. Identification of monocytes and B-lymphocytes by flow cytometry was made possible by prelabeling them with PE-CD14 and FITC-CD19 antibodies, respectively. The cell suspension and DEP field conditions were the same as in Figure 3, except that the DEP field was swept between 20 and 40 kHz.
Enrichment of Leukocytes from Blood
Separation of erythrocytes and leukocytes from blood, typically performed using centrifugation or filtration,35 is a basic requirement for many biomedical procedures such as erythrocyte transfusion.36 We therefore attempted to use DEP-FFF to enrich leukocytes (and erythrocytes) from blood diluted 1:1000 in a sucrose buffer. On the basis of the DEP-FFF characteristics of erythrocyte and leukocyte subpopulations (data not shown), a DEP field at 10 kHz was applied for DEP-FFF. A typical fractogram is shown in Figure 5; leukocytes were enriched 35-fold and erythrocyte purity was increased from 99.8% in the blood sample to >99.99% in the final erythrocyte fraction. Again, we believe that the differential cell velocities exploited in the DEP-FFF separation arose from levitation-height differences between leukocytes and erythrocytes.
Figure 5.
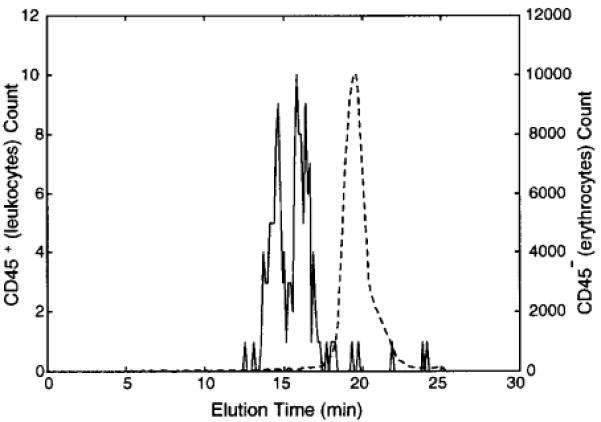
Flow cytometric cell count for leukocytes (CD45+, solid line) and erythrocytes (CD45-, dashed line) as a function of elution time during DEP-FFF enrichment of leukocytes from blood. More than 95% of leukocytes eluted between 15 and 17.5 min. The leukocyte/ erythrocyte ratio increased from 1:700 to 1:19, a 35-fold enrichment. The DEP-FFF was operated under a 10 kHz DEP field with the injection and withdrawal syringe pumps operated at 0.5 and 0.4 mL/ min, respectively.
Cell Dielectric Separation Criteria
The DEP-FFF method exploits cell dielectric and density properties as the basis of separation.18-20 In the frequency range used for the separations described here, cell dielectric properties are determined by the extent to which the applied field penetrates the cell interior via the capacitance of the plasma membrane. At low frequencies, the field penetration is small and the entire applied field appears across the poorly conducting membrane. Thus, cells are less polarizable than the suspending medium and tend to be repelled from strong electrical field regions by negative DEP forces. In our electrode configuration, this causes levitation. At higher frequencies, the field penetrates the plasma membrane into the cell interior, which is more conductive than the suspending medium under our conditions. Cells become more polarizable than the medium, the DEP forces become positive, and cells are attracted to the strong field regions and immobilized at the electrodes.7,8,11,16 Take breast cancer MDA-435 cells and T-lymphocytes as an example. Because of differences in cell membrane morphology and composition, T-lymphocytes have a mean membrane capacitance approximately half that of breast cancer cells,17,23 and the frequency range in which sufficient field penetration occurred for the DEP force to become positive was therefore higher for T-lymphocytes. Thus, in the frequency range from 15 to 35 kHz, T-lymphocytes were strongly levitated by negative DEP forces and were transported quickly through the chamber under the influence of the fluid flow. Breast cancer cells, however, were only weakly levitated into the slow-moving fluid close to the chamber bottom wall or were trapped by positive DEP forces at the electrodes. The differential velocities for T-lymphocytes and the breast cancer cells resulted in the DEP-FFF separation of the two populations. It follows that cell membrane dielectric properties, determined by membrane composition and morphological structures,14,18,21-23 were the DEPFFF separation criteria exploited here.
Over the last 15 years, the dielectric properties8-12,14,16,17,22-24,37-41 of many normal and cancerous cell types have been shown to be distinct and to depend not only on cell type but also on the biological state. For this reason, we have introduced the concept of cell dielectric phenotype.16-17 The dielectric phenotypes of T-lymphocytes allow them to be distinguished from breast cancer cells, monocytes, and granulocytes, and leukemia cells can be distinguished from normal leukocytes.23 Other examples include significant membrane dielectric alterations accompanying temperature-sensitive transformation of rat kidney cells;12 drug-induced differentiation in leukemia cells;22 mitogenic stimulation of human lymphocytes;23 fertilization of rabbit oocytes;38 and changes in cell environment such as exposure to heavy metals,39 organic toxins,40 or hypo-osmotic media.41 These examples suggest that by exploiting such dielectric phenotypes, the DEP-FFF technique may be applied for discrimination, characterization, and separation of cell subpopulations in many biomedical problems.
DEP-FFF Operational Parameters
DEP-FFF is quite distinct from conventional FFF in which hydrodynamic lift forces make an important contribution to performance. Under our conditions, cell levitation heights were determined by the balance between DEP and sedimentation forces and were not influenced by the flow-rate-dependent hydrodynamic lifting forces.
DEP-FFF separation depends on the balance of DEP levitation and sedimentation forces acting on cells in the starting mixtures. Sedimentation forces can be controlled by varying the density and osmolarity of the suspending medium. In general, the density of the medium should be less than that of the cells, and the combined solute concentrations in the medium should be chosen to maintain the desired osmolarity. Further, by adjusting the conductivity of the medium and the voltage and frequency of the applied electrical field, DEP forces experienced by different cell types can be fine-tuned to allow small differences in cell-dielectric phenotypes to be sensitively exploited. These parameters may be predicted16,18-20 from the dielectric and density properties of the cells in the starting mixture and can be chosen to ensure that cell levitation height differentials are maximized.18,19 Thus, the dielectric polarization factor, αDEP, may first be calculated as a function of the applied frequency based on the mean cell dielectric parameters of the individual cell populations.16,17,20 Then, the frequency-dependent cell levitation heights and corresponding DEP-FFF velocities may be calculated18-20 using eqs 1 and 2. Optimal separation frequencies may then be identified as the frequencies at which the largest velocity differences are found for the cell populations to be separated. If preferred, optimal electrical field parameters may also be discovered empirically by scanning the applied field over a range of frequencies. For example, for the electrical conductivity (10 mS/m) of the suspending medium used here, DEP frequencies may be varied between 1 and 100 kHz to achieve separation of most mammalian cell types because, above this frequency range, cells exhibit positive dielectrophoretic forces and are trapped at the electrode elements.
The cells being studied here have a minimum characteristic dimension of 6 or 7 μm. Brownian motion for such ªlargeº particles is negligible and plays little or no role in determining cell-levitation heights. Thus, the diffusion/dispersion process of cells is not an important factor that influences the cell distribution in the vertical dimension or leads to zone broadening. Rather, zone broadening, in our case, reflects limitations in how tightly cells were initially focused in the flow direction (injection mode, relaxation, etc.) and inherent cell dielectric and density heterogeneities.
Comparison with Other Cell Separation Methods
DEPFFF is quite distinct from conventional FFF in which hydrodynamic lift forces make an important contribution to performance. Under our conditions, cell-levitation heights were determined by the balance between DEP and sedimentation forces and were not influenced by the flow-rate-dependent hydrodynamic lifting forces. Generally speaking, separation techniques can be divided into bulk (including centrifuge-based sedimentation and MACS) and single-cell (such as FACS) methods. DEP-FFF is a bulk cell-sorting method that can be applied to many separation problems. Cell labeling is not necessary, making it particularly attractive to applications where labels for targeted populations are not available or labels would interfere with cell uses after separation. Cells separated by DEP-FFF are viable and can be cultured for further studies. Furthermore, physical properties exploited by DEP-FFF play little or no role in other cell identification/separation methods so that the technique may be used in conjunction with other methods to achieve improved discrimination between cells. For example, populations from DEP-FFF separation may be further analyzed for their genetic characteristics by PCR or sorted by FACS. Like most bulk techniques, DEP-FFF can be applied to cell separations where sterile conditions are necessary. Finally, the DEP-FFF method is applicable to a wide range of sample sizes and can be scaled up for routine laboratory cell separation, handling several million cells at a time or, at the other extreme, implemented on miniaturized scale. This makes the method ideal as a sample preparation step for integrated microfluidic analysis devices such as micro total analysis systems.26,42-44 Indeed, DEPFFF may become the method of choice in such applications because most current cell separation techniques cannot be readily implemented for microscale sample preparation needs. As an example, the DEP-FFF enrichment of leukocytes from blood could be applied in conjunction with micro PCR in an integrated microfluidic device to allow molecular amplification/analysis of a target-cell subpopulation in blood. Microfluidic applications of DEP-FFF will dramatically reduce the apparatus size, cost, and operational complexity associated with cell separation. To this end, we have built a prototype DEP-FFF device having a separation channel of 0.2 × 1 × 50 mm and demonstrated that it had similar separation performance to the system used in this work. With mass-produced microelectrodes, a DEP-FFF system could be made for less than 1/100 the cost of a typical FACS sorter. Thus, while cell separation with current methods can be achieved mainly in well-equipped biological laboratories, DEP-FFF based microfluidic diagnostic devices could become accessible at the point-of-care in doctors' offices and available in rural settings or in developing nations.
ACKNOWLEDGMENT
We are grateful to J. Noshari and C. Joyce for cell culture and to T. Anderson for help in constructing the DEP-FFF chamber. We thank Drs. G. De Gasperis, M. Cristofanilli, and X. J. Wang for valuable discussions. Special thanks are due to Dr. M. Donato and J. Lauppe in the Department of Bone Marrow Transplantation for providing leukapheresis samples as well as helpful advice in handling and detecting CD34+ cells. This work is supported in part by a research contract from the Electronics Technology Office of the Defense Advanced Research Program Agency (NRaD Contract N66001-97-C-8608 under DARPA Order E934), by an NIH grant R01 DK51065-01 from the National Institute of Diabetes and Digestive and Kidney Disease, and by an Advanced Research Project grant from the Higher Education Coordinating Board of the State of Texas.
References
- (1).Orfao A, Ruiz-ArguÈelles A. Clinical Biochem. 1998;29:5–9. doi: 10.1016/0009-9120(95)02017-9. [DOI] [PubMed] [Google Scholar]
- (2).Bauer J. J. Chromatogr. B. 1999;722:55–69. doi: 10.1016/s0378-4347(98)00308-9. [DOI] [PubMed] [Google Scholar]
- (3).Golovanov MV. In: Cell Electrophoresis. Bauer J, editor. CRC Press; Boca Raton, FL: 1994. pp. 181–196. [Google Scholar]
- (4).Villas BH. Cell Vision. 1998;5:56–61. [PubMed] [Google Scholar]
- (5).Handgretinger R, Lang P, Schumm M, Taylor G, Neu S, Koscielnak E, Niethammer D, Klingebiel T. Bone Marrow Transplant. 1998;21:987–93. doi: 10.1038/sj.bmt.1701228. [DOI] [PubMed] [Google Scholar]
- (6).Mavrou A, Colialexi A, Tsangaris GT, Antsaklis A, Panagiotopoulou P, Tsenghi C, Metaxotoy C. In Vivo. 1998;12:195–200. [PubMed] [Google Scholar]
- (7).Pethig R. Crit. Rev. Biotechnol. 1996;16:331–348. [Google Scholar]
- (8).Fuhr G, Zimmermann U, Shirley SG. In: Electromanipulation of cells. Zimmermann U, Neil GA, editors. CRC Press; Boca Raton, FL: 1996. pp. 259–328. [Google Scholar]
- (9).Arnold WM, Zimmermann U. J. Electrost. 1988;21:151–188. [Google Scholar]
- (10).HoÈlzel R, Lamprecht I. Biochim. Biophy. Acta. 1992;1104:195–200. doi: 10.1016/0005-2736(92)90150-k. [DOI] [PubMed] [Google Scholar]
- (11).Kaler KVIS, Jones TB. Biophys. J. 1990;57:173–182. doi: 10.1016/S0006-3495(90)82520-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Huang Y, Wang X-B, Becker FF, Gascoyne PRC. Biochim. Biophys. Acta. 1996;1282:76–84. doi: 10.1016/0005-2736(96)00047-8. [DOI] [PubMed] [Google Scholar]
- (13).Masuda S, Washizu M, Iwadare M. IEEE Trans. 1nd. Appl. 1987;23:474–480. [Google Scholar]
- (14).Markx GH, Huang Y, Zhou X-F, Pethig R. Microbiology. 1994;140:585–591. [Google Scholar]
- (15).Wang X-B, Hughes MP, Huang Y, Becker FF, Gascoyne PRC. Biochim. Biophys. Acta. 1995;1243:185–194. doi: 10.1016/0304-4165(94)00146-o. [DOI] [PubMed] [Google Scholar]
- (16).Becker FF, Wang X-B, Huang Y, Pethig R, Vykoukal J, Gascoyne PRC. Proc. Natl. Acad. Sci. U.S.A. 1995;92:860–864. doi: 10.1073/pnas.92.3.860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gascoyne PRC, Wang X-B, Huang Y, Becker FF. IEEE Trans. Ind. Gen. Appl. 1997;33:670–678. doi: 10.1109/28.585856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Huang Y, Wang X-B, Gascoyne PRC, Becker FF. Biophys. J. 1997;73:1118–1129. doi: 10.1016/S0006-3495(97)78144-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wang X-B, Vykoukal J, Becker FF, Gascoyne PRC. Biophys. J. 1998;74:2689–2701. doi: 10.1016/S0006-3495(98)77975-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yang J, Huang Y, Wang X-B, Becker FF, Gascoyne PRC. Anal. Chem. 1999;71:911–918. doi: 10.1021/ac981250p. [DOI] [PubMed] [Google Scholar]
- (21).Pethig R, Huang Y, Wang X-B, Burt JPH. J. Phys. D: Appl Phys. 1992;25:881–888. [Google Scholar]
- (22).Wang X-B, Huang Y, Gascoyne PRC, Becker FF, HoÈlzel R, Pethig R. Biochim. Biophys. Acta. 1994;1193:330–344. doi: 10.1016/0005-2736(94)90170-8. [DOI] [PubMed] [Google Scholar]
- (23).Huang Y, Wang X-B, Gascoyne PRC, Becker FF. Biochim. Biophys. Acta. 1999;1417:51–62. doi: 10.1016/s0005-2736(98)00253-3. [DOI] [PubMed] [Google Scholar]
- (24).Yang J, Huang Y, Wang X, Wang X-B, Becker FF, Gascoyne PRC. Biophys. J. 1999;76:3307–3314. doi: 10.1016/S0006-3495(99)77483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang X-B, Huang Y, Burt JPH, Markx GH, Pethig R. J. Phys. D: Appl. Phys. 1993;26:1278–1285. [Google Scholar]
- (26).Cheng J, Sheldon EL, Wu L, Uride A, Gerrue LO, Carrino J, Heller MJ, O'Connell JP. Nat. Biotechnol. 1998;16:541–546. doi: 10.1038/nbt0698-541. [DOI] [PubMed] [Google Scholar]
- (27).Markx GH, Rousselet J, Pethig R. J. Liq. Chromatogr. Relat. Technol. 1997;20:2857–2872. [Google Scholar]
- (28).Racilla E, Euhus D, Weiss A, Rao C, McConeil J, Terstappen LWMM, Uhr J. Proc. Natl. Acad. Sci. U.S.A. 1998;95:4589–4594. doi: 10.1073/pnas.95.8.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Terstappen LWMM, Rao C, Gross S, Kotelnikov V, Racilla E, Uhr J, Weiss A. Vox. Sang. 1998;74:269–274. doi: 10.1111/j.1423-0410.1998.tb05431.x. [DOI] [PubMed] [Google Scholar]
- (30).Peck K, Sher Y-P, Shih J-Y, Roffler SR, Wu C-W, Yang P-C. Cancer Res. 1998;58:2761–2765. [PubMed] [Google Scholar]
- (31).Cagnoni PJ, Shpall EJ. Blood Rev. 1996;10:1–7. doi: 10.1016/s0268-960x(96)90014-4. [DOI] [PubMed] [Google Scholar]
- (32).Mapara MY, KoÈrner IJ, Lentzsch S, Krahl D, Reichardt P, DoÈrken B. Exp. Hematol. 1999;27:169–175. doi: 10.1016/s0301-472x(98)00067-8. [DOI] [PubMed] [Google Scholar]
- (33).Stout RD. Curr. Opin. Immunol. 1993;5:398–403. doi: 10.1016/0952-7915(93)90059-2. [DOI] [PubMed] [Google Scholar]
- (34).Couraud PO. J. Leukocyte Biol. 1994;56:407–415. doi: 10.1002/jlb.56.3.407. [DOI] [PubMed] [Google Scholar]
- (35).HoÈgman CF. Vox. Sang. 1998;74:177–187. doi: 10.1111/j.1423-0410.1998.tb05419.x. [DOI] [PubMed] [Google Scholar]
- (36).Rosolski T, Matthey T, Frick U, Hachenberg T. Int. J. Artif. Organs. 1998;21:820–824. [PubMed] [Google Scholar]
- (37).Gimsa J, Marzalek P, Loewe U, Tsong TY. Biophys. J. 1991;60:749–760. doi: 10.1016/S0006-3495(91)82109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Arnold WM, Schmutzler RK, Al-Hasani S, Krebs D, Zimmermann U. Biochim. Biophys. Acta. 1989;979:142–146. doi: 10.1016/0005-2736(89)90535-x. [DOI] [PubMed] [Google Scholar]
- (39).Arnold WM, Geier BM, Wendt B, Zimmermann U. Biochim. Biophys. Acta. 1986;889:35–48. doi: 10.1016/0005-2736(87)90276-8. [DOI] [PubMed] [Google Scholar]
- (40).Arnold WM, Zimmermann U, Pauli W, Benzing M, Niehrs C, Ahlers J. Biochim. Biophys. Acta. 1988;942:83–95. doi: 10.1016/0005-2736(88)90277-5. [DOI] [PubMed] [Google Scholar]
- (41).Sukorukov VL, Arnold WM, Zimmermann U. J. Membr. Biol. 1993;132:27–40. doi: 10.1007/BF00233049. [DOI] [PubMed] [Google Scholar]
- (42).van den Berg A, Lammerink TSJ. In: Microsystem technologies in chemistry and life science. Manz A, Becker H, editors. Springer-Verlag; Heidelberg, Germany: 1998. pp. 21–50. Topics in Current Chemistry 194. [Google Scholar]
- (43).Wilding P, Kricka LJ, Cheng J, Hvichia G, Shoffner MA, Fortina P. Anal. Biochem. 1998;257:95–100. doi: 10.1006/abio.1997.2530. [DOI] [PubMed] [Google Scholar]
- (44).Weigl BH, Yager P. Science. Vol. 283. Washington, D.C.: 1999. pp. 346–347. [Google Scholar]
- (45).Yang J, Huang Y, Wang X-B, Becker FF, Gascoyne PRC. Biophys. J. 1999 doi: 10.1016/S0006-3495(99)77483-7. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]