

Abstract
The membrane lipids of archaea are characterized by unique isoprenoid biochemistry, which typically is based on two core lipid structures, sn-2,3-diphytanylglycerol diether (archaeol) and sn-2,3-dibiphytanyldiglycerol tetraether (caldarchaeol). The biosynthetic pathway for the tetraether lipid entails unprecedented head-to-head coupling of isoprenoid intermediates by an unknown mechanism involving unidentified enzymes. To investigate the isoprenoid ether lipid biosynthesis pathway of the hyperthermophilic archaeon, Archaeoglobus fulgidus, its lipid synthesis machinery was reconstructed in an engineered E. coli strain in an effort to demonstrate, for the first time, efficient isoprenoid ether lipid biosynthesis for the production of the intermediate, digeranylgeranylglyceryl phosphate (DGGGP). The biosynthesis of DGGGP was verified using a LC/MS/MS technique and was accomplished by cloning and expressing the native E. coli gene for IPP isomerase (idi), along with the A. fulgidus genes for G1P dehydrogenase (egsA) and GGPP synthase (gps), under the control of the lac promoter. The A. fulgidus genes for GGGP synthase (GGGPS) and DGGGP synthase (DGGGPS), under the control of the araBAD promoter, were then introduced and expressed to enable DGGGP biosynthesis in vivo. This investigation established roles for four A. fulgidus genes in the isoprenoid ether lipid pathway for DGGGP biosynthesis and provides a platform useful for identification of subsequent, currently unknown, steps in tetraether lipid biosynthesis proceeding from DGGGP, which is the presumed substrate for the head-to-head coupling reaction yielding unsaturated caldarchaeol.
Keywords: Archaeoglobus fulgidus, isoprenoid, ether lipid, DGGGP
1. INTRODUCTION
Isoprenoid compounds embody a large family of biological species including carotenoids, sterols, quinones, prenols, dolichols, and hormones. Many of these isoprenoid compounds have significant commercial roles as pigments, fragrances, vitamins, and antibiotics (Barkovich and Liao, 2001; Khosla and Keasling, 2003; Lee and Schmidt-Dannert, 2002; Sacchettini and Poulter, 1997). Whereas almost all organisms synthesize isoprenoid compounds as secondary metabolites, the Archaea necessarily require higher capacity pathways for biosynthesis of their distinguishing isoprenoid membrane lipids. In addition, archaea are hypothesized to carry out novel isoprenoid biochemistry, in particular, the head-to-head condensation of isoprenoid intermediates (Coolbear and Threlfall, 1989; Kates, 1992; Koga and Morii, 2007; Langworthy, 1985). Therefore, the production of industrially valuable isoprenoid compounds may be advanced by studying, and perhaps utilizing, the highly developed isoprenoid lipid biosynthesis machinery of archaea.
The membrane lipids of archaea exhibit distinct ether-linked isoprenoid chemistry (Kates, 1992; Koga et al., 1993; Langworthy, 1985). Although core membrane lipid structures can vary slightly among archaeal species, the basic and most common structural forms are the glycerol diether lipid and the glycerol tetraether lipid (Figure 1). The glycerol diether core lipid structure, 2,3-di-O-phytanyl-sn-glycerol, also called archaeol, includes two fully saturated C20 isoprenoid chains. The tetraether core lipid, 2,3-di-O-dibiphytanyl-sn-diglycerol, also known as caldarchaeol, appears to be constructed from two archaeol molecules linked covalently at the reduced hydrocarbon chain ends to give a bipolar, macrocyclic tetraether structure containing two C40 isoprenoid chains. These isoprenoid ether lipids, which constitute the lipid component of archaeal membranes, are a distinguishing evolutionary feature of the archaea.
Figure 1.
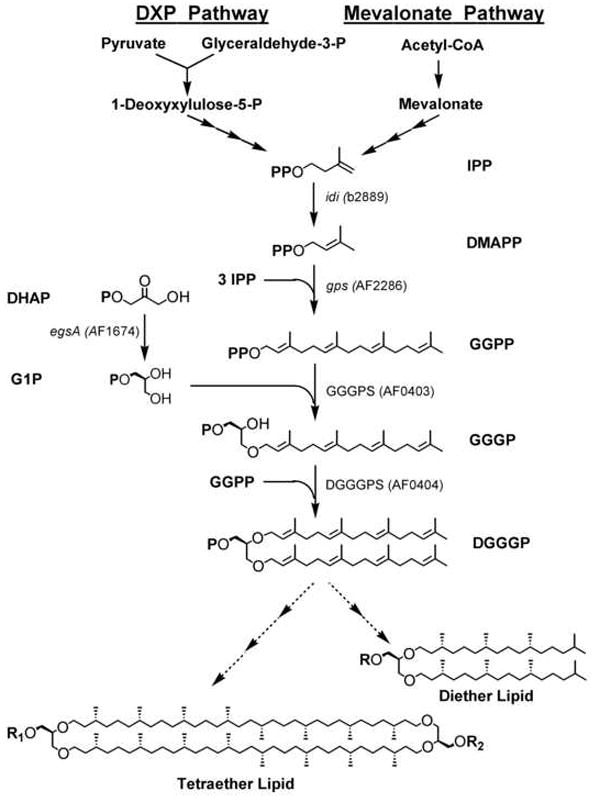
The isoprenoid biosynthetic pathway in E. coli (via deoxyxylulose phosphate (DXP) pathway) and in archaea (via mevalonate pathway) for ether lipid biosynthesis. Indicated genes were cloned and expressed in E. coli TOP10.
In isoprenoid biosynthesis, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) serve as the basic building blocks. In the archaeal ether lipid biosynthesis pathway (Figure 1), the path to IPP proceeds via mevalonate, also observed in most eukaryotes. However, most bacteria including Escherichia coli, operate the deoxyxylulose phosphate (DXP) pathway to IPP (Barkovich and Liao, 2001; Khosla and Keasling, 2003). For the synthesis of archaeol (Koga and Morii, 2007), IPP is first isomerized to DMAPP by IPP isomerase (idi). DMAPP undergoes chain elongation to form geranylgeranyl diphosphate (GGPP), catalyzed by the multifunctional enzyme, GGPP synthase (gps). The glycerol backbone is provided by sn-glycerol-1-phosphate (G1P), the enantiomer of the glycerol backbone in bacterial and eukaryotic membrane lipids, which is formed from the conversion of dihydroxyacetone phosphate (DHAP) by glycerol-1-phosphate dehydrogenase (egsA, G1PDH). In the first committed step of ether lipid biosynthesis, the C20 side chain, GGPP, is attached to G1P to form geranylgeranylglyceryl phosphate (GGGP), catalyzed by GGGP synthase (GGGPS). The attachment of the second side chain to GGGP gives digeranylgeranylglyceryl phosphate (DGGGP), catalyzed by DGGGP synthase (DGGGPS). Remaining steps to the final diether lipids include side chain reduction by DGGGP reductase and the addition of polar headgroups. The genes responsible for completing the biosynthetic route to the final tetraether lipids remain unidentified, including those hypothesized to encode enzymes for the novel head-to-head coupling of the diether lipid, the reduction to the biphytanyl side chains, the formation of cyclopentane rings on the biphytanyl side chain, and the addition of polar headgroups (Koga and Morii, 2007).
As part of a larger effort to elucidate the pathway for tetraether lipid biosynthesis, the goal of this study was to utilize lipid synthesis genes from the hyperthermophilic archaeon, Archaeoglobus fulgidus, to construct an E. coli strain capable of isoprenoid ether lipid biosynthesis from simple precursor metabolites. The E. coli strain constructed in this work demonstrates the operation of a pathway of four A. fulgidus genes in a heterologous host to achieve production of the ether lipid biosynthesis intermediate, DGGGP, and provides a platform for the study of subsequent enzymatic steps in tetraether lipid biosynthesis. To achieve DGGGP production, the IPP isomerase gene from E. coli, in addition to the A. fulgidus genes that encode GGPP synthase, G1P dehydrogenase, GGGP synthase, and DGGGP synthase were cloned and expressed in the E. coli strain. Previous studies have confirmed the expression and in vitro activity of recombinant GGPP synthase and GGGP synthase from A. fulgidus in E. coli hosts (Payandeh et al., 2006; Wang et al., 1999). This study confirms the presence of a G1P dehydrogenase and a DGGGP synthase in A. fulgidus and, to the best of our knowledge, demonstrates for the first time a bacterial strain that has been metabolically engineered for archaeal ether lipid biosynthesis.
2. MATERIALS AND METHODS
2.1. Strains, Plasmids, and Culture Conditions
Archaeoglobus fulgidus VC-16 (DSM 4304) was grown anaerobically at 83 °C at pH 7 in a minimal medium, as described by Rohlin et al. (2005). A. fulgidus genomic DNA was isolated using a purification kit (Promega) for use as PCR gene templates for amplification of egsA, GGGPS, and DGGGPS. The E. coli strains DH5α (Stratagene) and TOP10 (Invitrogen) were used for plasmid constructions, and E. coli TOP10 was used for all gene expression studies and for DGGGP production. For routine cultivation, E. coli was grown in Luria-Bertani medium at 37 °C on a rotary shaker at 200 rpm. The expression vector, pBAD, was obtained from Invitrogen. The plasmids, pCW2 (containing idi from E. coli), pCW3 (containing gps from A. fulgidus), and pCW14 (containing crtB and crtI from Agrobacterium aurantiacum) were described previously (Wang et al., 1999; Wang et al., 2000). Strains and plasmids used in this study are listed in Table 1.
Table 1.
Strains and plasmids used in this study for DGGGP biosynthesis
| Strain/Plasmid | Description | Source/Ref. |
|---|---|---|
| Strains | ||
| A. fulgidus VC-16 | Wild-type | DSM 4304a |
| E. coli TOP10 | [F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(araleu)7697 galU galK rpsL (Strr) endA1 nupG] | Invitrogen |
| E. coli DH5α | [F- φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rk-, mk+) phoA supE44 λ- thi-1 gyrA96 relA1] | Stratagene |
| Plasmids | ||
| pBAD | ParaBAD expression vector, low copy; Ampr | Invitrogen |
| pCW2 | pACYC184∷idi; Tetr | Wang et al., 1999 |
| pCW3 | pCL1920∷gps; Specr | Wang et al., 1999 |
| pCW14 | pJF118EH∷crtB, crtI; Ampr | Wang et al., 2000 |
| pDL10 | pCL1920∷gps, egsA; Specr | This study |
| pDL11 | pCL1920∷idi, gps, egsA; Specr | This study |
| pDL6 | pBAD∷GGGPS, DGGGPS; Ampr | This study |
Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH
2.2. Plasmid Construction
All plasmid constructions were performed according to manufacturer-supplied protocols and established methods (Sambrook et al., 2001). The plasmid, pDL11, which contains the genes idi, gps, and egsA, was constructed by inserting idi and egsA into pCW3, which is derived from pCL1920. The parent plasmid, pCL1920 (specr), is a low-copy plasmid which features an expression system controlled by the lac promoter. The gene, idi, was amplified from pCW2 by PCR using KOD DNA polymerase (Novagen) with the primers 5′-GTTAGTCGACAGGAGAAATTATGCAAACGGAAC-3′ and 5′-GTGTTCTAGATTATTTCAGCTGGGTAAATGC-3′ and was ligated between the SalI and XbaI restriction sites on pCW3. The gene, egsA (AF1674), was amplified by PCR from A. fulgidus genomic DNA using the primers 5′-CAGGATCCGAAGAGGTGGTAGCATGAGA-3′ and 5′-GTGGTACCTTA(ATG)6AGCCACGCCTGTCTTCTCCAC-3′. The egsA gene, encoding G1P dehydrogenase, was designed to express a C-terminal 6×His-tag and was inserted between the BamHI and KpnI restriction sites on pCW3.
The genes encoding GGGP synthase (AF0403) and DGGGP synthase (AF0404) are located adjacent to each other on the A. fulgidus genome, and thus one set of primers were designed to amplify both genes together. To construct pDL6, these genes were amplified by PCR using the primers 5′-GTGAGCTCCGATGGAGAAAGTGGAGACAC-3′ and 5′-GTGGTACCCTA(ATG)6AAACGCCCCTGCAAAAAATCC-3′, and the resulting product was ligated between the SacI and KpnI restrictions sites on pBAD. The parent plasmid, pBAD (ampr), features a highly regulated expression system controlled by the araBAD promoter. As a result of these plasmid designs, the recombinant GGGP synthase is produced with a N-terminal 6×His-tag, whereas the DGGGP synthase is produced with a C-terminal 6×His-tag.
2.3. Lycopene-Production Assay for Expression of idi and gps in vivo
To demonstrate idi and gps expression in E. coli, a simple in vivo assay was employed by co-expressing crtB and crtI, resulting in a lycopene-producing E. coli strain. The plasmid pDL10, containing gps only, or pDL11, containing both gps and idi, was transformed into an E. coli TOP10 strain containing the plasmid, pCW14, which harbors crtB and crtI. The resulting strain produced lycopene in vivo at 37 °C which was easily observed by formation of distinct red-colored colonies on LB agar plates.
2.4. Protein Expression and Purification of G1P Dehydrogenase and GGGP Synthase
For expression of G1P dehydrogenase, pDL11 was transformed into E. coli TOP10. A 300 ml culture was grown at 37 °C in Luria-Bertani (LB) medium supplemented with 50 μg/ml spectinomycin. When the optical density reached 0.5 (OD600), the TOP10/pDL11 culture was induced with 1 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG). Similarly, for expression of GGGP synthase and DGGGP synthase, pDL6 was transformed into E. coli TOP10 and was grown in 300 ml of LB medium and 200 μg/ml ampicillin with incubation at 37 °C. The TOP10/pDL6 culture was induced with 0.2% arabinose when the optical density reached 0.5 (OD600). After an additional 4 hours of incubation, cell cultures were harvested by low-speed centrifugation for 20 minutes at 4 °C.
Cell pellets were resuspended in 15 ml of buffer A (20 mM tris-HCl, pH 8, 500 mM KCl, 0.1% triton X-100) and disrupted by passage through a French pressure cell at 16,000 psi. Cell lysate was centrifuged at low-speed for 20 minutes to remove cellular debris and then subjected to heat treatment at 70 °C for 20 minutes. The membrane fraction, isolated by ultracentrifugation at 200,000×g for 1 hour, was solubilized in buffer A containing 2% triton X-100.
Purification of His-tagged recombinant proteins was achieved by affinity chromatography with Ni-NTA resin (Qiagen) and elution with buffer A, also containing 250 mM imidazole. Elution fractions were concentrated with 10 kDa molecular weight cut-off centrifugal filters (Millipore). Protein concentrations were determined by the Bradford protein assay (Bio-Rad).
Expression of recombinant proteins in whole cell lysate and in purified fractions was verified by western blot according to manufacturer-supplied protocols (Invitrogen, 2003). The antibody used was a Penta-His HRP conjugate (Qiagen), and protein bands were visualized by chemiluminescent detection using the Super Signal West Pico detection kit (Pierce) and exposure to film.
2.5. Enzymatic Assay for G1P Dehydrogenase
The enzyme activity of G1P dehydrogenase was confirmed by a biochemical enzyme assay measuring NADH oxidation, similar to methods previously described (Noguchi et al., 1998). In an enzymatic reaction to produce G1P, a 0.75 ml reaction mixture contained 800 μM dihydroxyacetone phosphate (DHAP) and 300 μM NADH as substrates, 6 μg/ml G1P dehydrogenase from a purified protein fraction, and a buffer consisting of 50 mM sodium phosphate buffer at pH 7 and 500 mM KCl. Enzyme reactions were conducted at incubation temperatures of 60 °C and 37 °C and were followed spectrophotometrically at 340 nm to monitor consumption of NADH. One unit of enzyme activity is defined as 1 μmol NADH oxidized per min.
2.6. Radioassays for GGGP Synthase
A highly sensitive in vitro enzyme assay employing a radiolabeled substrate was used to detect recombinant GGGP synthase activity using methods similar to that previously described (Hemmi et al., 2004; Nemoto et al., 2003). In a 50 μl reaction mixture, the initial reaction mixture included 2.5 μCi [1-3H]GGPP (American Radiolabeled Chemicals), 100 μM unlabeled GGPP (Echelon Biosciences), 500 μM α-glycerol phosphate (Sigma-Aldrich), 0.5 mg/ml GGGP synthase, 50 mM sodium phosphate buffer at pH 7, 500 mM KCl, 0.5% n-octyl-β-D-glucopyranoside, 1 mM DTT, and 5 mM MgCl2. Reaction mixtures were incubated at either 60 °C or 37 °C for 2 hours. Reaction products were extracted 3 times with 200 μl of 1-butanol saturated with water. After solvent evaporation, the extracted products were dephosphorylated enzymatically by methods similar to those previously described (Fujii et al., 1982), by treatment with acid phosphatase, which contained in a final volume of 50 μl: 100 mM sodium acetate at pH 5, 0.5% n-octyl-β-D-glucopyranoside, and 5 units/ml of acid phosphatase (Sigma-Aldrich). The reaction mixture was incubated overnight at 37 °C. Dephosphorylated products were extracted 3 times with 200 μl of hexane, solvent was evaporated, and dried extract was redissolved in methanol.
Using thin-layer chromatography (TLC) analysis, extracted radiolabeled product was spotted on MKC18F reversed-phase TLC plates (Whatman) and developed in a solvent system of acetone-water (9:1). Spots were visualized by spraying with En3hance (Perkin-Elmer), incubated at -80 °C for 24 hours, and exposed to film. A standard of geranylgeraniol (GG) (Sigma-Aldrich), visualized with iodine, was used as reference.
Alternatively, radiolabeled products were analyzed by high performance liquid chromatography (HPLC) (Agilent 1090 HPLC) using a reversed-phase column (YMC-ODS-AQ, 150 × 2.0 mm, 3 μm particle size). Injection volumes of 10 μl per sample were loaded onto the column and eluted isocratically at a flow rate of 0.2 ml/min with a mobile phase of methanol-water (90:10) for 20 minutes. The tritium radioactivity was detected by an online scintillation counter (Beta-RAM) after mixture with a continuous flow of ScintiVerse (Fisher Scientific) at a scintillant to eluate flow rate ratio of 7:1.
2.7. DGGGP Biosynthesis in vivo and LC/MS/MS Analysis
For DGGGP biosynthesis in vivo, both pDL11 and pDL6 were transformed into the E. coli strain, TOP10. Five hundred ml cultures were grown in LB medium supplemented with 200 μg/ml ampicillin and 50 μg/ml spectinomycin. Cultures were placed on a rotary shaker and incubated at 37 °C. When cultures reached an optical density of 0.5 (OD600), they were induced by addition of 100 μM IPTG and 0.02% arabinose. Cells were harvested after 24 hours by low-speed centrifugation at 4 °C. Lipids were extracted from the cells using 30 ml of a mixture of butanol–75 mM ammonium hydroxide–ethanol (4:5:11) by a method previously demonstrated for efficient extraction of GGPP and FPP (Tong et al., 2005). As described above, product was dephosphorylated enzymatically by acid phosphatase, extracted with hexane, and re-dissolved in methanol.
Product was first subjected to HPLC analysis using the method described above, however, using UV detection at 210 nm. The effluent from the column was fed directly into a 4000 QTRAP hybrid triple quadrupole linear ion trap mass spectrometer (AppliedBiosystems). Mass spectrometry was operated in the quadrupole three (Q3) scan mode and the enhanced product ion (EPI) scan mode. The positive ion electrospray ionization method was employed based upon the structure and chemical nature of the analytes, as well as the ability of the analytes to form lithiated ion adducts (D’Alexandri et al., 2006). For optimal signal detection, a lithium iodide solution was mixed into the LC effluent through the provided “T” connection on the source to obtain a final in-source concentration of 2 mM lithium iodide. During chromatography, the third quadrupole of the mass spectrometer (Q3) was scanned from 250 to 700 m/z. The declustering potential was 160, the entrance potential was 10, and the collision cell exit potential was 15. These values were tuned during sequential chromatography runs using standard geranylgeraniol (GG) to obtain the maximal signal. The ESI source parameters were set as follows: curtain gas (CUR), 20; electrospray voltage (IS), 5500; temperature, 400 °C; gas one (GS1), 35; and gas two (GS1), 45. Due to the strong intensity of the signal, tuning of the source parameters was not performed. For MS/MS analysis, EPI scans were performed using the above parameters with addition of collision energy (CE) at 160 or 40, for DGGG and GGG, respectively, and a collisionally activated dissociation (CAD) gas value of “high”. Data analysis was performed using the manufacturer-provided data analysis software, Analyst 1.4.2 (AppliedBiosystems).
3. RESULTS AND DISCUSSION
3.1. Identification of Genes for Ether Lipid Biosynthesis in A. fulgidus
With the goal of engineering E. coli to produce the pathway intermediate, DGGGP, the A. fulgidus genome was examined for genes involved in isoprenoid ether lipid biosynthesis. A. fulgidus is a particularly good source for genetic material as expression of its genes in E. coli readily results in soluble proteins, which have been shown to retain significant activity at 37 °C (Schröder et al., 2004; Vadas et al., 1999).
A. fulgidus ether lipid biosynthesis genes primarily were identified through homology searches using previously identified genes from other archaea. Internet-based resources such as the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov), The Institute for Genomic Research (TIGR, http://www.tigr.org), and the Kyoto Encyclopedia for Genes and Genomes (KEGG, http://www.genome.jp/kegg) aided in the assignment of A. fulgidus open reading frames hypothesized to function in the ether lipid biosynthesis pathway. Further confirmation was achieved using the Basic Local Alignment Search Tool (BLAST) from NCBI.
To identify the gene for G1P dehydrogenase, the egsA gene from Methanothermobacter thermoautotrophicus was used to identify its homolog in the A. fulgidus genome. The putative protein encoded by open reading frame (ORF) AF1674 displays 39% identity and 58% similarity in its amino acid sequence to the M. thermoautotrophicus egsA gene, which previously had been expressed in E. coli and its function confirmed as a G1P dehydrogenase (Han et al., 2002; Nishihara et al., 1999; Noguchi et al., 1998).
GGGP synthase from A. fulgidus recently has been crystallized by Payandeh et al. (2006). The function for homologous GGGP synthases from Methanothermobacter marburgensis and Thermoplasma acidophilum was established by expression of these GGGP synthase genes and characterization of the kinetic properties of the resulting recombinant enzymes (Nemoto et al., 2003; Soderberg et al., 2001). Using BLAST, the GGGP synthase homolog in A. fulgidus was verified as the gene at ORF AF0403 which shares 24% identity and 45% similarity compared to its homolog in T. acidophilum.
The identification of the DGGGP synthase from A. fulgidus was based on the homologous enzyme in Sulfolobus solfataricus, which previously has been recombinantly expressed and characterized by Hemmi et al. (2004). The putative A. fulgidus DGGGP synthase shares 30% identity and 52% similarity to the S. solfataricus DGGGP synthase. Its adjacent genomic location to the GGGP synthase supported the notion that the gene at ORF AF0404 also is involved in ether lipid biosynthesis. The amino acid sequence of the putative DGGGP synthase was predicted to contain 7 transmembrane spanning helices, indicating that it is an integral membrane protein.
3.2. Expression of A. fulgidus Ether Lipid Biosynthesis Genes in E. coli
In order to reconstruct the ether lipid biosynthesis pathway in E. coli, the previously identified A. fulgidus genes were introduced and expressed in the E. coli strain, TOP10. In the isoprenoid pathway in E. coli, IPP is isomerized to DMAPP, followed by a series of condensation reactions to form GGPP (Figure 1). The synthesis of GGPP in E. coli, however, occurs at exceedingly low levels, as an intermediate of the multifunctional octaprenyl diphosphate synthase involved in ubiquinone biosynthesis (Wang et al., 1999). To increase carbon flux towards GGPP, the A. fulgidus GGPP synthase gene (gps) together with the E. coli IPP isomerase gene (idi) were introduced into E. coli on a multi-copy plasmid (Figure 1). Wang et al. (1999) previously showed that the expression of both genes in an engineered E. coli strain resulted in the improved production of astaxanthin, a carotenoid normally not synthesized by E. coli. To test whether idi and gps are expressed and functional, the expression plasmid was transformed into E. coli TOP10 containing a compatible plasmid carrying the Agrobacterium aurantiacum crtB and crtI genes required for lycopene biosynthesis. Lycopene, a red-colored carotenoid, was easily produced by E. coli expressing both idi and gps, resulting in intensely red-colored colonies (Supplementary Figure 1). Moderate production of lycopene was indicated by pink-colored colonies as a result of the expression of A. fulgidus gps alone in the strain, suggesting that the native E. coli idi expression is limiting. White colonies were observed from a control strain without idi and gps.
The existing plasmid containing idi and gps also contained the A. fulgidus G1P dehydrogenase gene (egsA). To monitor the expression of G1P dehydrogenase in E. coli, the A. fulgidus egsA gene was amended with a 6×His-tag. Because the A. fulgidus GGGP synthase and DGGGP synthase genes comprise one putative transcriptional unit, both genes were introduced into the E. coli TOP10 strain on a compatible expression plasmid. Both recombinant GGGP synthase and DGGGP synthase were modified with a 6×His-tag at the N- and C- termini, respectively. The parent plasmids were chosen based on their ability to regulate and control protein expression tightly in case the desired gene products or biosynthetic products were toxic to the cell. Following induction by IPTG and arabinose to induce gene expression from both plasmids, crude cell lysate was separated into soluble and membrane fractions and analyzed by western blot using an anti-His antibody. Expression of G1P dehydrogenase (G1PDH) and GGGP synthase (GGGPS) was clearly indicated by western blot analysis of the crude cell lysate, showing signals corresponding to their anticipated molecular weights of 39.4 kDa and 30.4 kDa, respectively (Figure 2, lane 1). Next, both enzymes were enriched from the soluble fraction using Ni affinity chromatography and analyzed by western blot, confirming the identity of the protein bands detected in the crude cell lysate (Figure 2, lane 2, lane 3). The signal for DGGGP synthase, which was expected at 33.6 kDa, was not detected in the membrane fraction nor the soluble fraction (data not shown), suggesting that this enzyme was either not expressed or expressed at extremely low levels below detection limits.
Figure 2.
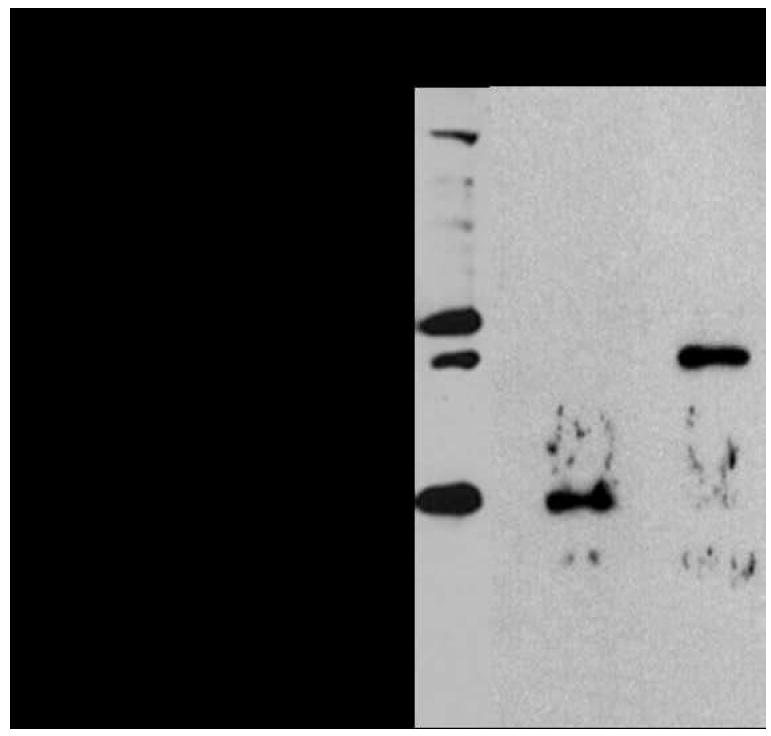
Western blot analysis with an anti-His antibody confirmed expression of His-tagged G1P dehydrogenase (39.4 kDa) and GGGP synthase (30.4 kDa) in E. coli. Lane 1: Crude cell lysate of E. coli TOP10/pDL11/pDL6. Lane 2: Affinity chromatography fraction of E. coli TOP10/pDL6 lysate containing His-tagged GGGP synthase. Lane 3: Affinity chromatography fraction of E. coli TOP10/pDL11 lysate containing His-tagged G1P dehydrogenase.
3.3. Recombinant G1P Dehydrogenase and GGGP Synthase are Functionally Active in vitro
Many A. fulgidus enzymes that have been expressed in E. coli have been shown to fold correctly and to be active catalytically (Lim et al., 2004; Schröder et al., 2004; Vadas et al., 1999). A prerequisite for demonstrating ether lipid biosynthesis in E. coli is to demonstrate activity of the recombinant A. fulgidus enzymes. G1P dehydrogenase activity was confirmed by spectrophotometrically measuring the dihydroxyacetone phosphate-dependent oxidation of NADH (Supplementary Figure 2). Under the reaction conditions described above (see Materials and Methods), G1P dehydrogenase was determined to have a specific activity of 4 U/mg at 60 °C. At 37 °C, well below the optimal A. fulgidus growth temperature, G1P dehydrogenase still exhibited significant activity of 0.6 U/mg, suggesting that the enzyme can function at the moderate temperatures necessary for application in vivo in an E. coli strain. The A. fulgidus G1P dehydrogenase activity determined at 60 °C is similar to the recombinant G1P dehydrogenase activity from the hyperthermophile, Aeropyrum pernix, also measured at 60 °C (Han et al., 2002). However in this study, reaction conditions were not explored to find optimum activity, therefore the activity of A. fulgidus G1P dehydrogenase likely is much higher when measured at its physiological optimum temperature and salt concentration.
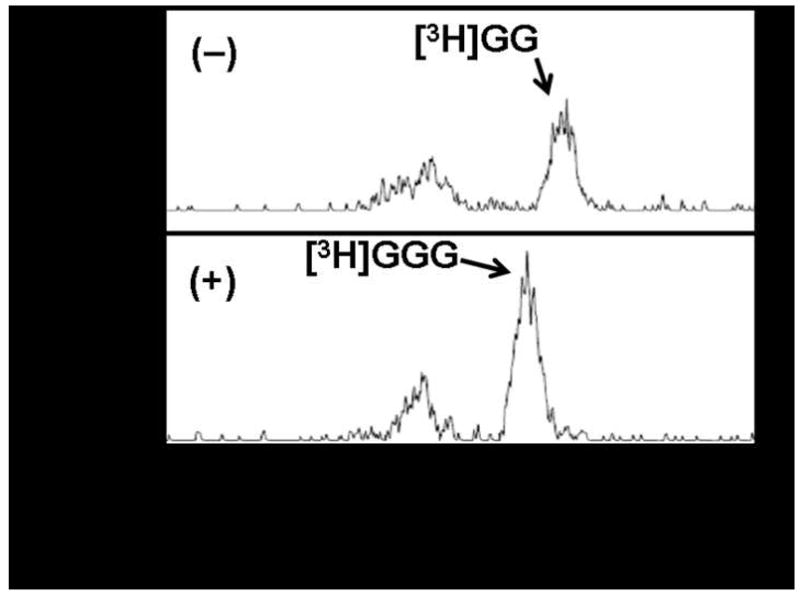
GGGP synthase, which catalyzes the first committed step in ether lipid synthesis, was assayed using [3H]GGPP and G1P as substrates. The radiolabeled product, [3H]GGGP, was analyzed by reversed-phase TLC and visualized using autoradiography. To facilitate identification of substrates and products for analysis, GGPP and GGGP were dephosphorylated to yield GG and GGG, respectively. After 2 hours at an incubation temperature of 60 °C or 37 °C, [3H]GGPP was completely converted into a radioactive product, presumably [3H]GGGP, in the presence of GGGP synthase, but not in its absence (Figure 3). The nonradiolabeled standard for GG migrated under the test conditions with an Rf value of 0.32, identical to the Rf value of [3H]GG. In contrast, the presumed product, [3H]GGG, separated with an Rf value of 0.42. The relative mobilities of these compounds observed by TLC agree with results reported by Hemmi et al. (2004). Other unidentified spots on the TLC plate were unknown reaction byproducts.
Figure 3.

Autoradiogram of TLC analysis showed conversion of [3H]GGPP to [3H]GGGP by GGGP synthase. The GGGP synthase activity assay was carried out at 60 °C and 37 °C in the absence (−) and presence (+) of GGGP synthase. Prior to TLC analysis, compounds were dephosphorylated.
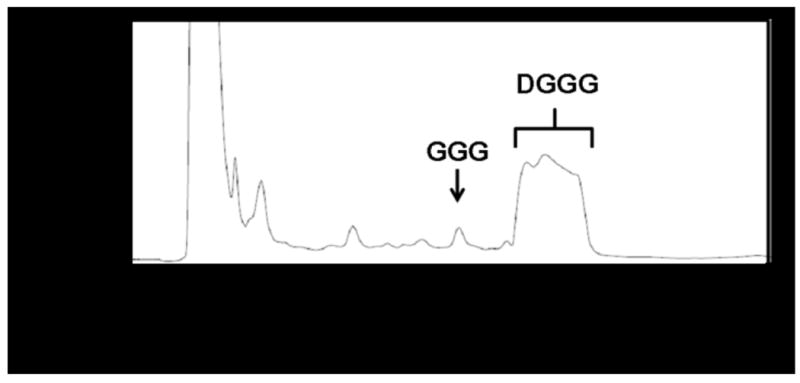
Reaction end products from the GGGP synthase assay were further analyzed by high performance liquid chromatography (HPLC) (Figure 4). In the presence of GGGP synthase, the retention time of the product was significantly shorter than that of [3H]GG (Figure 4) and the GG standard (data not shown). [3H]GG eluted with a characteristic retention time of 13.8 min, while the presumed [3H]GGG eluted at 12.5 min. The relative elution times for GG and GGG observed by HPLC analysis agree with their mobilities as determined by TLC analysis. The less intense peaks in each HPLC chromatogram at earlier times likely correspond to reaction byproducts also seen in the TLC analysis above. Thus, pathway reconstruction was demonstrated here through GGGP synthase.
Figure 4.
Chromatogram of GGGP synthase activity assay performed at 60 °C in the absence (−) and presence (+) of GGGP synthase. End products were analyzed by reversed-phase HPLC.
3.4. Reconstruction of the Ether Lipid Biosynthesis Pathway in E. coli Through DGGGP
E. coli previously has been metabolically engineered to produce carotenoids as well as other terpenoids (Farmer and Liao, 2000; Lee and Schmidt-Dannert, 2002; Martin et al., 2003; Reiling et al., 2004; Wang et al., 1999). Our goal was to reconstruct the ether lipid biosynthesis pathway leading to the DGGGP intermediate. The activities for all A. fulgidus enzymes could be confirmed directly or indirectly in this study with the exception of the putative membrane-bound DGGGP synthase. To establish whether E. coli containing four ether lipid biosynthesis genes from A. fulgidus is capable of producing DGGGP, lipid analysis was performed to detect DGGGP. Prior to lipid analysis, cells were induced and grown for an additional 24 hours. Lipids were extracted and dephosphorylated followed by mass analysis by LC/MS/MS, which entailed reversed-phase HPLC analysis coupled to electrospray ionization mass spectrometry (ESI–MS). Compounds were ionized by electrospray using a lithium ion cationization technique previously shown to promote ionization of uncharged polyprenols, compounds similar in molecular structure to archaeal ether lipids. This ionization technique results in the formation of lithiated ion adducts [M + Li]+, thus the molecular mass of compounds analyzed by this ESI(Li+)–MS method includes the mass of a lithium ion (D’Alexandri et al., 2006).
First, analysis by HPLC with UV detection produced a chromatogram that revealed several peaks which included two smaller peaks eluting at 7.0 min and 10.5 min in addition to a larger, broad peak at a retention time of 12.5–14.5 min (Figure 5A). Next, ESI–MS analysis of the eluant at 10.5 min produced a mass spectrum that displayed a lithium ion adduct peak at m/z 371.5, which was identified as GGG (Figure 5B, top). Subsequent MS/MS analysis revealed two peaks, one with the molecular mass of GGG at m/z 371.3, and a second fragment ion peak at m/z 99.0 corresponding to the glycerol backbone resulting from GGG fragmentation (Figure 5B, bottom). The mass peak at m/z 618.0 and other minor peaks in the spectrum also were observed in the lipid extracts of the control E. coli strain lacking the A. fulgidus ether lipid biosynthesis pathway genes, and thus were not analyzed further.
Figure 5.
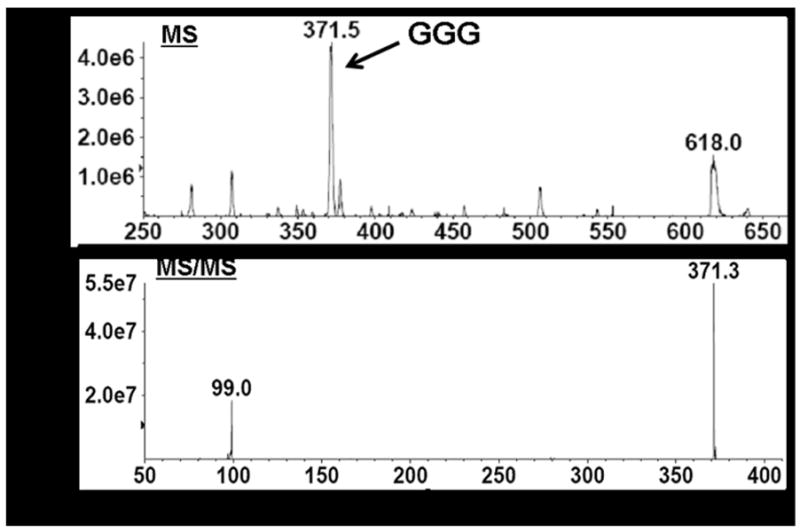
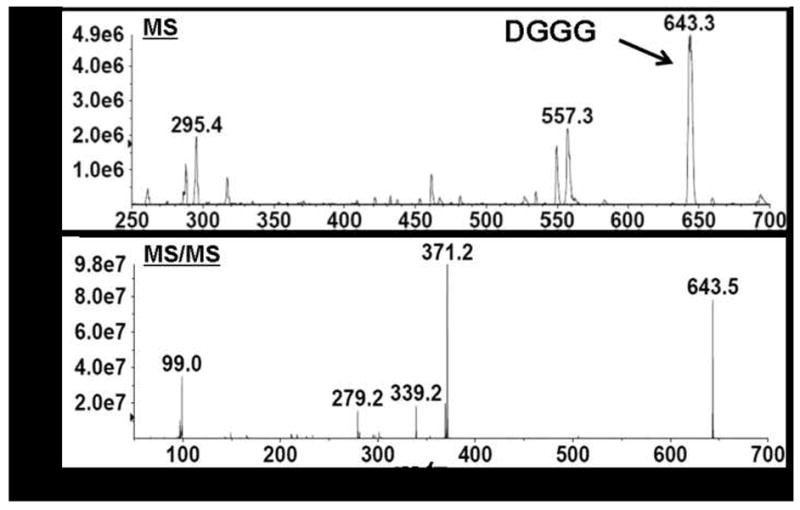
LC/MS/MS analysis of crude, dephosphorylated lipid extracts from E. coli TOP10/pDL11/pDL6 confirmed DGGGP biosynthesis in vivo.
(A) Chromatogram from HPLC analysis at 210 nm. Peak assignment of GGG and DGGG was based on mass spectrometry identification.
(B) MS and MS/MS spectra of compound eluting at 10.5 min which was identified as GGG.
(C) MS and MS/MS spectra of compound eluting at 12.5–14.5 min which was identified as DGGG.
ESI–MS analysis of the broad peak eluting from the chromatography column at 12.5–14.5 min (Figure 5A) revealed several peaks (Figure 5C). Of all peaks, only the lithium ion adduct peak at m/z 643.3 was uniquely present in lipid extracts of the E. coli strain containing the A. fulgidus ether lipid biosynthesis pathway genes. All other minor peaks including those at m/z 295.4 and m/z 557.3 were also found in lipid extracts prepared from the control E. coli strain lacking the A. fulgidus genes. The molecular ion peak observed at m/z 643.3 corresponds to the lithium ion adduct of DGGG (Figure 5C, top). Subsequent MS/MS analysis allowed fragmentation of the molecular ion at m/z 643.3 which generated five peaks in the spectrum, the largest at m/z 643.5 confirming the identity of DGGG (Figure 5C, bottom). The other peaks in the spectrum corresponded to fragmentation products of DGGG. The peak at m/z 371.2 represents GGG resulting from the loss of one C20 isoprenoid chain, the peak at m/z 279.3 represents one C20 isoprenoid chain, and the peak at m/z 99.0 represents the glycerol backbone. The peak at m/z 339.2 could not be identified. These results established biosynthesis through DGGGP in the metabolically engineered E. coli strain.
Overall, our results establish that E. coli engineered to express IPP isomerase from E. coli, GGPP synthase, G1P dehydrogenase, GGGP synthase, and DGGGP synthase from A. fulgidus is capable of synthesizing DGGGP, a direct precursor for archaeal diether and tetraether lipid biosynthesis (Figure 1). At this point, direct quantitation of DGGGP from E. coli lipid analysis was not possible because a commercial DGGGP standard is unavailable. However, our HPLC and mass spectrometry methods clearly identified DGGGP production in the cell. Moreover, lipid extract from the engineered E. coli strain contained a small amount of the precursor GGGP in comparison to the relatively large amount of DGGGP formed (Figure 5A). Together with the finding from in vitro enzyme assays that [3H]GGPP is efficiently converted to GGGP (Figure 3, Figure 4), we predict that the engineered E. coli strain is capable of synthesizing sufficient amounts of DGGGP to be used as a substrate for future identification of genes downstream in the ether lipid biosynthesis pathway. Evidence of DGGGP biosynthesis also demonstrated that the membrane-bound DGGGP synthase from A. fulgidus, which was below the detection limit of the western blot analysis, is functionally active in vivo. This is, to the best of our knowledge, the first time that an archaeal lipid biosynthesis pathway has been reconstructed in E. coli.
4. CONCLUSION
An E. coli strain was engineered for isoprenoid ether lipid biosynthesis through DGGGP. The strain contained four genes from A. fulgidus involved in the formation of DGGGP from DMAPP. In addition, the strain was modified with the idi gene from E. coli to increase the carbon flux toward DMAPP formation. All genes were inserted on two multi-copy plasmids under controllable promoters. Lipid extracted from the engineered E. coli strain was confirmed to contain DGGG using LC/MS/MS analysis. This work provides a useful approach to generate DGGGP, the likely substrate for the head-to-head coupling reaction yielding unsaturated caldarchaeol. Therefore, the engineered E. coli strain and the analytical methods described here can be applied jointly as a tool for further elucidation of the novel pathway for tetraether lipid biosynthesis in archaea.
Acknowledgments
This work was supported by a NIH Metabolic Engineering Grant (5R01GM077627). The purchase of mass spectrometry instrumentation was supported by a National Center for Research Resources Grant (S10RR024605). The authors thank Dr. Catherine F. Clarke (Chemistry and Biochemistry Department, UCLA) for her valuable technical assistance and use of HPLC equipment. Assistance with mass spectrometry and HPLC also was provided kindly by Alek Dooley and Dr. Beth N. Marbois (Chemistry and Biochemistry Department, UCLA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barkovich R, Liao JC. Metabolic Engineering of Isoprenoids. Metab Eng. 2001;3:27–39. doi: 10.1006/mben.2000.0168. [DOI] [PubMed] [Google Scholar]
- Coolbear T, Threlfall DR. Biosynthesis of terpenoid lipids. In: Ratledge C, Wilkinson SG, editors. Microbial Lipids. Vol. 2. Academic Press; San Diego: 1989. pp. 115–254. [Google Scholar]
- D’Alexandri FL, Gozzo FC, Eberlin MN, Katzin AM. Electrospray ionization mass spectrometry analysis of polyisoprenoid alcohols via Li+ cationization. Anal Biochem. 2006;355:189–200. doi: 10.1016/j.ab.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Farmer WR, Liao JC. Improving lycopene production in Escherichia coli by engineering metabolic control. Nat Biotechnol. 2000;18:533–537. doi: 10.1038/75398. [DOI] [PubMed] [Google Scholar]
- Fujii H, Koyama T, Ogura K. Efficient Enzymatic Hydrolysis of Polyprenyl Pyrophosphates. Biochimica et Biophysica Acta. 1982;712:716–718. [PubMed] [Google Scholar]
- Han J, Kosugi Y, Ishida H, Ishikawa K. Kinetic study of sn-glycerol-1-phosphate dehydrogenase from the aerobic hyperthermophilic archaeon, Aeropyrum pernix K1. Eur J Biochem. 2002;296:969–976. doi: 10.1046/j.0014-2956.2001.02731.x. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Shibuya K, Takahashi Y, Nakayama T, Nishino T. (S)-2,3-Di-O-geranylgeranylglyceryl Phosphate Synthase from the Thermoacidophilic Archaeon Sulfolobus solfataricus. J Biol Chem. 2004;279:50197–50203. doi: 10.1074/jbc.M409207200. [DOI] [PubMed] [Google Scholar]
- Invitrogen. NuPAGE Technical Guide. Carlsbad, CA: 2003. [Google Scholar]
- Kates M. Archaebacterial lipids: structure, biosynthesis and function. Biochem Soc Symp. 1992;58:51–72. [PubMed] [Google Scholar]
- Khosla C, Keasling JD. Metabolic engineering for drug discovery and development. Nat Rev Drug Discovery. 2003;2:1019–1025. doi: 10.1038/nrd1256. [DOI] [PubMed] [Google Scholar]
- Koga Y, Morii H. Biosynthesis of Ether-Type Polar Lipids in Archaea and Evolutionary Considerations. Microbiol Mol Biol Rev. 2007;71:97–120. doi: 10.1128/MMBR.00033-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga Y, Nishihara M, Morii H, Akagawa-Matsushita M. Ether Polar Lipids of Methanogenic Bacteria: Structures, Comparative Aspects, and Biosyntheses. Microbiol Rev. 1993;57:164–182. doi: 10.1128/mr.57.1.164-182.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langworthy TA. Lipids of Archaebacteria. In: Woese CR, Wolfe RS, editors. Archaebacteria. Academic Press; New York: 1985. pp. 459–497. [Google Scholar]
- Lee PC, Schmidt-Dannert C. Metabolic engineering towards biotechnological production of carotenoids in microorganisms. Appl Microbiol Biotechnol. 2002;60:1–11. doi: 10.1007/s00253-002-1101-x. [DOI] [PubMed] [Google Scholar]
- Lim S, Schröder I, Monbouquette HG. A Thermostable Shikimate 5-dehydrogenase from the Archaeon Archaeoglobus fulgidus. FEMS Microbiol Lett. 2004;238:101–102. doi: 10.1016/j.femsle.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Martin VJ, Pitera DJ, Withers ST, Newman JD, Keasling JD. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol. 2003;21:796–802. doi: 10.1038/nbt833. [DOI] [PubMed] [Google Scholar]
- Nemoto N, Oshima T, Yamagishi A. Purification and Characterization of Geranylgeranylglyceryl Phosphate Synthase from a Thermoacidophilic Archaeon, Thermoplasma acidophilum. J Biochem. 2003;133:351–657. doi: 10.1093/jb/mvg083. [DOI] [PubMed] [Google Scholar]
- Nishihara M, Yamazaki T, Oshima T, Koga T. sn-Glycerol-1-Phosphate-Forming Activities in Archaea: Separation of Archaeal Phospholipid Biosynthesis and Glycerol Catabolism by Glycerophosphate Enantiomers. J Bacteriol. 1999;181:1330–1333. doi: 10.1128/jb.181.4.1330-1333.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi S, Maeda M, Masateru N, Koga Y, Sone N. Expression and Use of Methanobacterium thermoautotrophicum sn-Glycerol 1-Phosphate Dehydrogenase for the Assay of sn-Glycerol 1-Phosphate in Archaea. J Ferment Bioeng. 1998;3:266–270. [Google Scholar]
- Payandeh J, Fujihashi M, Gillon W, Pai EF. The Crystal Structure of (S)-3-O-Geranylgeranylglyceryl Phosphate Synthase Reveals an Ancient Fold for an Ancient Enzyme. J Biol Chem. 2006;281:6070–6078. doi: 10.1074/jbc.M509377200. [DOI] [PubMed] [Google Scholar]
- Reiling KK, Yoshikuni Y, Martin VJ, Newman J, Bohlmann J, Keasling JD. Mono and Diterpene Production in Escherichia coli. Biotechnol Bioeng. 2004;87:200–212. doi: 10.1002/bit.20128. [DOI] [PubMed] [Google Scholar]
- Rohlin L, Trent JD, Salmon K, Kim U, Gunsalus RP, Liao JC. Heat Shock Response of Archaeoglobus fulgidus. J Bacteriol. 2005;187:6046–6057. doi: 10.1128/JB.187.17.6046-6057.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchettini JC, Poulter CD. Creating Isoprenoid Diversity. Science. 1997;277:1788–1789. doi: 10.1126/science.277.5333.1788. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3. Cold Spring Harbor Laboratory Press; New York: 2001. [Google Scholar]
- Schröder I, Vadas A, Johnson E, Lim S, Monbouquette HG. A Novel Archaeal Alanine Dehydrogenase Homologous to Ornithine Cyclodeaminase and μ-Crystallin. J Bacteriol. 2004;186:7680–7689. doi: 10.1128/JB.186.22.7680-7689.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg T, Chen A, Poulter CD. Geranylgeranylglyceryl Phosphate Synthase: Characterization of the Recombinant Enzyme from Methanobacterium thermoautotrophicum. Biochemistry. 2001;40:14847–14854. doi: 10.1021/bi0111799. [DOI] [PubMed] [Google Scholar]
- Tong H, Holstein SA, Hohl RJ. Simultaneous determination of farnesyl and geranylgeranyl pyrophosphate levels in cultured cells. Anal Biochem. 2005;336:51–59. doi: 10.1016/j.ab.2004.09.024. [DOI] [PubMed] [Google Scholar]
- Vadas A, Monbouquette HG, Johnson E, Schröder I. Identification and Characterization of a Novel Ferric Reductase from the Hyperthermophilic Archaeon Archaeoglobus fulgidus. J Biol Chem. 1999;274:36715–36721. doi: 10.1074/jbc.274.51.36715. [DOI] [PubMed] [Google Scholar]
- Wang C, Oh M, Liao JC. Engineered Isoprenoid Pathway Enhances Astaxanthin Production in Escherichia coli. Biotechnol Bioeng. 1999;62:235–241. doi: 10.1002/(sici)1097-0290(19990120)62:2<235::aid-bit14>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Wang C, Oh M, Liao JC. Directed Evolution of Metabolically Engineered Escherichia coli for Carotenoid Production. Biotechnol Prog. 2000;16:922–926. doi: 10.1021/bp000124f. [DOI] [PubMed] [Google Scholar]