

Abstract
The bacterial mechanosensitive channel MscS forms a homoheptamer of subunits composed of a transmembrane (TM) domain and a large cytoplasmic (CP) domain. Recent studies suggest that a lateral expansion of the TM domain, structural change in the CP domain, and TM-CP interactions are essential to open the channel. However, it has not been examined whether the CP domain undergoes structural changes during channel opening. The aim of this study was to estimate structural changes in the CP domain during channel opening using fluorescence resonance energy transfer (FRET) spectroscopy. To monitor changes in the horizontal diameter of the CP domain, four point mutants (A132C, F178C, L246C, and R259C), all of which had channel activity, were created and labeled with Alexa488 and Alexa568 for FRET analysis. The FRET efficiency of these mutants decreased when lysophosphatidylcholine was applied to open the channel, suggesting that the CP domain swells up when the channel opens. The degree of the decease in FRET efficiency after lysophosphatidylcholine treatment was smaller in the D62N/F178C mutant, which was deficient in the TM-CP interactions, than in the F178C mutant. These findings provide the first, to our knowledge, experimental evidence that the CP domain swells up during channel opening, and the swelling is mediated by the TM-CP interactions.
Introduction
Mechanosensitive (MS) channels are expressed in virtually all eukaryotic and prokaryotic cells. Bacterial MS channels protect bacteria from cell lysis upon hypoosmotic shock by acting as a safety valve (1). However, it remains largely unknown how MS channels convert mechanical stimuli into channel opening. MscL (mechanosensitive channel of large conductance) and MscS (mechanosensitive channel of small conductance) are bacterial MS channels and have been used to address this question because they are the only MS channels whose three-dimensional (3D) crystal structures have been determined (2,3). An overall picture of the gating transition in MscL has been described comprehensively in genetic (4,5), electrophysiological (6,7), structural (2,8,9), and computational studies (10).
MscS is located in the bacterial inner membrane, is directly activated by membrane tension (11), and is a ∼1 nS slightly anion-selective channel. This channel forms a symmetric homoheptamer (3). Each subunit contains three transmembrane (TM) helices (TM1, TM2, and TM3A-TM3B) and a large cytoplasmic (CP) domain (3). This channel has a central pore with a diameter of ∼11 Å. The third TM helix forms a narrow constriction of the channel. The CP domain forms a large CP vestibule, which has seven openings in the side (∼14 Å) and one in the bottom with a diameter of ∼8 Å. Initially, the crystal structure was considered to represent an open state because of the large diameter of the TM pore (3). Subsequently, several groups suggested that the crystal structure represents a “more nearly conducting” or “minimally conductive” state (12,13). Now it is plausible that the crystal structure represents a nonconductive state that corresponds to a closed state or an inactivated/desensitized state after opening (14–16). Two bulky hydrophobic rings comprising Leu-105 and Leu-109, located in TM3A, face the pore lumen to prevent water and ions from entering the channel pore, a process that has been termed “vapor lock” (14).
The channel activity of MscS is retained when the purified proteins are reconstituted into artificial liposomes (11), suggesting that the channel is directly activated by membrane tension. When asparagine, a hydrophilic amino acid, was substituted at the hydrophobic residues near either end of the TM1 or TM2 helix, the gating threshold increased and the viability upon hypoosmotic shock decreased (17). In particular, it was shown that the application of high pressure up to membrane lysis does not activate the double mutant I37N/L86N (17). These observations indicate that MscS senses the change in membrane tension through protein-lipid interactions at the ends of both the TM1 and TM2 helices.
This leads to a crucial question: After MscS senses the tension, how do the conformational changes associated with tension sensing lead to opening of the channel? TM1 and TM2 helices undergo a clockwise rotation during channel opening (18). Rotating and tilting of TM3A (19) or straightening of the TM3 helix (15) presumably breaks up the vapor lock and leads to pore expansion. However, the conformational changes in the TM domain may not directly lead to channel opening, because the TM and CP domains electrically interact mainly between Asp-62 and Arg-128 during channel opening (20), and between Asp-62 and Arg-131 in the open state (21). When one or two residues of these three residues are replaced with an electrically neutral or opposing amino acid, the gating threshold of these MscS mutants increases, suggesting that this interaction is crucial for channel opening (21). Asp-62 is located in the TM1-TM2 loop, and Arg-128 and Arg-131 are located at the end of TM3B (Fig. 1 A). This electrostatic interaction is termed the “TM-CP interaction”. The conformational changes in TM1 and TM2 helices caused by membrane tension (18) are highly likely to induce structural changes in the CP domain, because the CP domain is associated with TM1 and TM2 helices via the TM-CP interaction.
Figure 1.

Crystal structure of MscS. (A) The structure of MscS is shown with a ribbon model based on the data set of 2OAU in the Protein Data Bank. Ala-132, Phe-178, Leu-246, and Arg-259, which are residues introduced by point mutations in this study, are colored in blue, green, yellow, and red, respectively. Cyan residues show Asp-62, which interacts with Arg-128 and Arg-131 in the open state (21). Purple residues show Ile-37 and Leu-86, which take part in mechanosensing sites of MscS (17). All colored residues are depicted in a space-filled model. The putative location of the membrane is shown by dotted lines. (B) The structures of AF488 and AF568 are shown on approximately the same scale based on the chemical structures in Panchuk-Voloshina et al. (34).
The large CP domain is the most notable structural feature of MscS. Voltage-gated potassium channels, some types of transient receptor potential (TRP) channels, and hyperpolarization-activated cyclic nucleotide-modulated (HCN) channels also have large CP domains (22–25). It is thought that these CP domains allosterically influence channel functions; for example, the ligand-binding sites of HCN channels are located in the CP domain, and structural changes in this domain caused by binding of cyclic nucleotides activate the channel (26). It has been postulated that seven side portals of the CP domain of MscS act as a molecular sieve (3) or endow the channel with a slight anion preference (27), and structural changes in this domain regulate the transition from a closed to an open state and/or from an open to an inactivated state (21,28). Thus, the structural changes in the CP domain are hypothesized to be crucial for channel opening (29). However, the structural changes that occur in the CP domain during channel opening, and the relationship between the change and the gating transition of the channel have not been uncovered.
The fluorescence resonance energy transfer (FRET) technique is a powerful tool for monitoring the change in the distance between two adjacent fluorophores using the spectral overlap of the donor emission and acceptor absorption, and is used to estimate protein-protein interactions and protein conformational changes (30,31). The diameter of a symmetrical multimeric membrane protein can be measured by randomly labeling with donor and acceptor fluorophores at a specific residue in two adjacent subunits (9). If the diameter increases, the distance between the two adjacent subunits will increase and result in a decrease in the FRET efficiency, because the FRET efficiency depends on the distance between the donor and acceptor fluorophores.
In this study we examined the structural changes that occur in the CP domain of MscS during channel opening using a FRET technique. By monitoring the change in the diameter of several levels of the CP domain, we were able to reconstruct a 3D view of the structural change that occurred during channel opening. This demonstrates that the CP domain of MscS swells up during channel opening and is presumably mediated by the electrostatic interaction between TM and CP domains.
Materials and Methods
Strains and plasmids
Escherichia coli strain MJF455 (ΔmscL::Cm, ΔyggB) was used for purification, hypoosmotic shock experiments, and patch-clamp experiments. E. coli strain TOP10 (Invitrogen, Carlsbad, CA) was used for site-directed mutagenesis. Wild-type MscS with a polyhistidine-tag at the C-terminus was cloned into the pB10b vector (32). All mutants in this study were created using the megaprimer method (5), and confirmed by DNA sequencing with the CEQ 2000XL DNA Analysis System (Beckman Coulter, Fullerton, CA).
Protein purification, fluorescence labeling, and membrane reconstitution
MscS was purified according to a protocol modified from that originally described by Sukharev (11). Cells were grown in 450 mL of Luria Broth medium at 37°C. When the cells reached an OD600 of ∼0.5, expression of MscS was induced by adding isopropyl beta-D-thiogalactopyranoside to a final concentration of 1 mM. After an additional 3 h of growth, the cells were harvested, suspended in 8 mL of sonication buffer (50 mM K-Pi, 5 mM MgSO4, 1 mM DTT, 1 mM Pefabloc, pH 7.2), and disrupted with an ultrasonicator (model 450 sonifier; Branson Ultrasonics, Danbury, CT). The sonicated cell suspension was centrifuged for 1 h at 100,000 g. The pellet was resuspended in extraction solution (Ni-NTA buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0), 28 mM imidazole, 0.5 mM EGTA, 0.5 mM EDTA, 3% octylglucoside (OG), 4 mM Pefabloc, pH 8.0). After insoluble materials were removed by centrifugation for 1 h at 100,000 g, the supernatant was loaded onto a preequilibrated Ni-NTA column (QIAGEN, Valencia, CA). The specimen was washed with 10 volumes of wash buffer (Ni-NTA buffer, 200 mM imidazole, 3.3 mM ATP, 5 mM MgSO4, 1% OG, pH 8.0) and elution was performed with elution buffer (Ni-NTA buffer, 500 mM imidazole, 1% OG, pH 8.0). Fractions of 1 mL were collected. Protein quantification was done by the Bradford method. The yield of purified His-tagged protein ranged from 5 to 15 μM.
Two types of fluorescently labeled MscS (double- or acceptor-labeled MscS) were made. For double-labeled MscS, the purified protein was reacted for 1 h on ice with two and eight equivalents of AlexaFluor488-maleimide (AF488, as a donor) and AlexaFluor568-maleimide (AF568, as an acceptor) (Invitrogen), respectively. For acceptor-labeled MscS, the purified protein was reacted for 1 h on ice with eight equivalents of AF568. The excess dyes were separated by gel filtration on a PD-10 desalting column (GE Healthcare, Buckinghamshire, UK) equilibrated with Pi-OG buffer (20 mM K-Pi, 1% OG, pH 7.2), and the proteins were concentrated by ultrafiltration (Amicon Ultra; Millipore, Billerica, MA). The concentration of AF488 or AF568 was calculated from the absorbance at 495 or 578 nm, respectively, and the MscS concentration was determined by protein quantification using the Bradford method. The extinction coefficients of AF488 and AF568 were 71,000 and 91,300 cm−1M−1, respectively. The labeling ratio was estimated by dividing the AF488 or AF568 concentration by the MscS concentration. Before gel filtration, the proteins mixed with fluorophores were loaded on a 12% sodium dodecyl sulfate polyacrylamide gel. The gel pattern was visualized with an ultraviolet lamp and stained with Coomassie brilliant blue (CBB).
Each type of protein was mixed with 3 mg of OG-dissolved asolectin at a protein/lipid ratio of 1:150–250 (w/w). The mixture was dialyzed for 36 h against 500 mL of dialysis buffer (100 mM NaCl, 0.2 mM EDTA, 0.02% NaN3, 5 mM Tris-HCl, pH 7.2) with three changes in the presence of detergent-absorbing beads (Calbiosorb; Calbiochem, La Jolla, CA) to accomplish the reconstitution of MscS in asolectin liposomes. For microscopy observations, giant liposomes (∼10 μm) were prepared by the freezing-thawing procedure (freezing at −80°C and thawing at room temperature).
Spectrum analysis and FRET efficiency determination
The emission spectra of each sample by excitation at 488 and 543 nm (RF5300, Shimadzu, Kyoto, Japan) were obtained every 5 min before and after application of 200 μM lysophosphatidylcholine (LPC). The LPC was dissolved in methanol to make a 10 mM stock. It was then mixed with sample solution that contained MscS liposomes before the emission spectra were measured.
The measured spectra of double-labeled MscS were a mixture of FRET signal (the acceptor emission excited by the donor emission due to energy transfer), the acceptor bleed-through (the acceptor emission excited directly by 488 nm light), and the background signal from the buffer. The acceptor bleed-through (∼6% of the measured spectrum) and the background signal (<1%) were subtracted from the measured spectrum of double-labeled MscS according to a previously described procedure (33) to calculate the FRET efficiency. The application of drugs sometimes affects the emission intensity of fluorescence dyes. LPC (200 μM) increased the acceptor emission of acceptor-labeled MscS excited at 543 nm (27% ± 3% increase in F178C (n = 5), and 18% ± 1% increase in L246C (n = 3)), whereas no significant change in the acceptor emission of A132C or L246C was observed (n = 3–5). These effects were also compensated for according to the procedure (33). FRET efficiencies were calculated using the following equation:
| (1) |
where E is the FRET efficiency; ID and IA are the compensated fluorescence intensities of AF488 and AF568, respectively (ID and IA are an integration of fluorescence intensities 505–535 nm and 590–620 nm); and ΨD and ΨA are fluorescence quantum yields of AF488 and AF568, respectively (ΨD = 0.6, and ΨA = 0.37) (34).
LPC did not affect the emission from the loss-of-function (LOF) mutant I37N/D62N/L86N/F178C (n = 3), suggesting that changes in FRET efficiencies induced by LPC in other MscS mutants were most likely dependent on the closed-to-open transition.
Hypoosmotic shock assay
Survival rates of E. coli after hypoosmotic shock were examined using a method described previously (1,5,17,21). Expression was induced by the addition of isopropyl beta-D-thiogalactopyranoside (1 mM) to MJF455 cells grown to OD600 = 0.15 in minimal medium supplemented with 500 mM NaCl and 100 μg/mL ampicillin. After incubation for 1 h, the cells were diluted 1/20 in prewarmed minimal medium with or without 500 mM NaCl (hypoosmotic shock). After a 5-min shock, each sample was spread on a plate and cultured overnight. The ratio of the number of colony-forming units of cells that experienced the hypoosmotic shock (Ndown) to those that did not experience the shock (Ncontrol) was used to calculate the survival rates (Ndown/Ncontrol). The survival rate was normalized to that of wild-type MscS. The assay was carried out three times on each mutant.
Patch clamp recordings
Giant E. coli spheroplasts expressing wild-type MscS were prepared as described previously (35), and patch-clamp recordings were made from these cells in the excised patch mode (21). The pipette potential was held constant at +20 mV. The pipette solution contained 200 mM KCl, 90 mM MgCl2, 10 mM CaCl2, and 5 mM HEPES (pH 6.0), whereas the bath solution contained an additional 0.3 M sucrose to stabilize the spheroplasts. Channel currents were amplified with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) and filtered at 2 kHz. Current recordings were digitized at 5 kHz with a Digidata 1322A interface using pCLAMP 9 software (Axon). A negative pressure was applied by syringe-generated suction through the patch-clamp pipette and measured with a pressure gauge (PM 015R; World Precision Instruments, Sarasota, FL). LPC (10 μL, 10 mM) was added to the bath solution (∼500 μL) for patch-clamp recordings to reach a final concentration of 200 μM after diffusion, which caused 5–23 min of lag time.
Fluorescence observation and acceptor photobleaching and electron paramagnetic resonance measurements
Fluorescence images were acquired with a conventional inverted microscope (TE2000-U; Nikon, Tokyo, Japan) equipped with a ×100 1.45 NA PlanApo oil-immersion objective lens and a 75 W Xe light source, and a Cascade 512F EMCCD camera (Roper Scientific, Tucson, AZ). AF488 was excited through a 480/30 nm excitation filter and fluorescence was collected through a 542/27 bandpass filter; AF568 was excited through a 540/25 nm excitation filter and collected through a 590 nm longpass filter. Acceptor photobleaching was performed with a 543 nm He-Ne laser beam (5 mW; Melles Guirot, Carlsbad, CA) that entered the microscope through a back port and was focused on a small spot (∼10 μm) of a giant liposome for ∼10 s. Electron paramagnetic resonance (EPR) measurements and analyses were performed as described previously (36).
Data are presented as mean ± SE unless otherwise noted.
Results
By collecting several cross sections of an object, one can usually obtain a 3D reconstruction of that object. In this study, we estimated the diameter of four cross sections in the membrane plane of the CP domain of MscS in both the closed and open states to directly explore the structural change that occurs in the domain when the channel opens. We determined the diameter of each cross section by monitoring the intersubunit distance using the FRET method. To specifically label the residues of interest with fluorophores of a FRET pair, we created four point mutants because the MscS protein contains no cysteine residues. Residues located in the upper (Ala-132), middle (Phe-178 and Leu-246), and lower (Arg-259) parts of the CP domain (Fig. 1) were substituted with cysteine and labeled with AF488 and AF568.
The channel activity of these mutants was tested by means of a hypoosmotic shock assay because single amino acid substitution can sometimes affect the channel activity (17). Although E. coli knockout mutants of MscS and MscL cannot avoid cell lysis upon hypoosmotic shock from 500 mM NaCl to zero NaCl concentration, E. coli expressing the wild-type and all four cysteine mutants of MscS survived against the same hypoosmotic shock with approximately the same survival rate (Fig. 2 A). We also confirmed the mechanically activated channel currents of these mutants by patch clamping (data not shown). These findings indicate that all of these mutants maintain the channel function.
Figure 2.
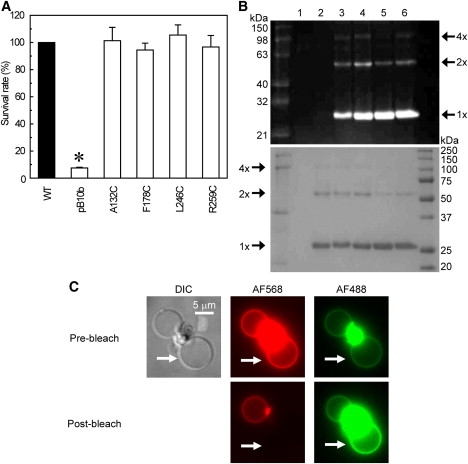
Characterization of four cysteine mutants of MscS. (A) Effects of hypoosmotic shock on cells expressing mutant MscS. The survival rate was normalized to that of the wild-type (mean ± SE). The asterisk indicates that the survival rate is significantly different from that of the wild-type (p < 0.05 by t-test). (B) Proteins labeled with Alexa dyes were loaded on an SDS-PAGE gel and illuminated with an ultraviolet lamp (upper) or stained with CBB (lower). Lane 1, empty vector; lane 2, wild-type MscS; lanes 3–6, cysteine mutants of MscS (A132C, F178C, L246C, and R259C). (C) Fluorescence images of giant liposomes containing F178C MscS labeled by AF488 and AF568. The upper panels show a differential interference contrast image of giant liposomes, and a fluorescence image of AF568 and AF488 before bleaching. The lower panels show the fluorescence images of AF568 and AF488 after bleaching in a liposome indicated by white arrows.
The purity and dye labeling of MscS were evaluated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). SDS-PAGE of all four cysteine mutants showed that MscS monomer and dimmer were labeled with AF488 and AF568, whereas those of the wild-type were not (Fig. 2 B), indicating that these single cysteine mutants of MscS were purified and labeled specifically with Alexa dyes. The degree of dimerization of A132C and F178C was similar to that of wild-type, whereas that of L246C and R259C was slightly lower than the wild-type MscS (Fig. 2 B). The oligomeric behavior of cysteine mutant MscS proteins was approximately the same as that of wild-type MscS. Residues Phe-178 and Leu-246 are located in the inner lumen of the CP cage (Fig. 1 A), but F178C and L246C were labeled by Alexa dyes similarly to other residues (Ala-132 and Arg-259) exposed to the outside (Fig. 2 B). This can be accounted for by considering the results of recent molecular-dynamics simulations, which showed that the linker groups in Alexa dyes are flexible and the length of these dyes oscillates from 8 to 17 Å (37), supporting the idea that Alexa dyes in the compact form can pass through the side portals of the CP domain (∼14 Å; Fig. 1) to the interior and label the residues. The labeling ratio of the donor and the acceptor was kept approximately the same among all of the mutants to estimate the diameter of the four cross sections of the CP domain; the labeling ratio of the donor was ∼0.3 and that of the acceptor was ∼0.55 (see Table S1 in the Supporting Material). Monte Carlo simulation of the labeling of donors and acceptors in 9362 MscS channels under the conditions used here shows that 91% of MscS channels have more than one donor and acceptor, i.e., FRET can occur in these channels (see the Supporting Material).
Giant liposomes containing MscS labeled with AF488 and AF568 were observed using fluorescence microscopy to test the FRET between AF488 and AF568. The bright fluorescence of Alexa dyes was identified in the membrane of giant liposomes, showing that fluorescence-labeled MscS was incorporated into the lipid membrane. FRET between Alexa dyes was examined by acceptor photobleaching experiments. Photobleaching of AF568 by a 543 nm He-Ne laser beam resulted in an increase in the fluorescence intensity of AF488 (e.g., AF568 in a liposome indicated by a white arrow in Fig. 2 C was bleached, and the fluorescence of AF488 only in this liposome increased, showing FRET between dyes). Cysteine mutants of MscS labeled with Alexa dyes were reconstituted into asolectin liposomes and used for FRET analyses.
The negative pressure in the pipette, which is supposed to increase tension in the membrane, activates and subsequently inactivates MscS. LPC is known to induce spontaneous opening of MscS, and locks the channels in the fully open state in the absence of negative pressure in the pipette (16). To test whether our channel is activated by LPC without negative pressure, we performed patch-clamp recordings from the inside-out excised patch of E. coli giant spheroplasts expressing wild-type MscS, because detailed analyses of the action of LPC on MscS are lacking. Using 100–200 μM LPC, we observed channel activities of MscS without negative pressure within 15–25 min. The current grew gradually and the saturated level of MscS current was kept until the membrane seal broke (Fig. 3 A); the amplitude of the LPC-induced current was 1.3 (n = 3) times larger than that of the maximum current evoked by negative pressure. This difference could be accounted for by a partial inactivation observed in the MscS channels activated by the negative pressure, which is not seen in the channels activated by LPC (16). The single-channel conductance of MscS activated by LPC was 1.1 nS (n = 10), almost the same value as that of the channel activated by negative pressure (1.1 nS, n = 10), suggesting that the open state of MscS activated by LPC is identical to that of the channel activated by negative pressure (Fig. 3). No MscS channel-like current was observed in MscS-free (pB10b) spheroplasts with or without LPC (Fig. 3 A). The channel opening was induced by LPC in all mutants except for the I37N/D62N/L86N/F178C mutant (data not shown). Thus, LPC is suitable for investigating the open conformation of MscS by the FRET technique.
Figure 3.
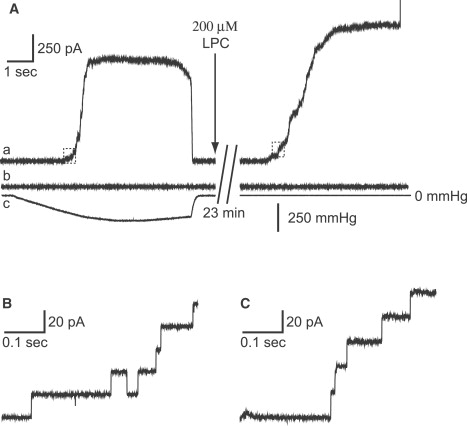
LPC and stress increases in the membrane activate MscS channels. The membrane current of wild-type MscS (trace a) and pB10b (trace b) in an inside-out patch configuration, and the negative pressure applied through a patch pipette (trace c) are shown. The outward current of wild-type MscS was evoked by the negative pressure (left panel) or by 200 μM LPC (right panel). Current traces in small boxes in the left and right traces in panel A are magnified in B and C, respectively. The membrane potential was +20 mV.
FRET analyses of changes in the emission spectra of AF488 and AF568 by LPC were performed. When 200 μM LPC were added, the intensity of AF488 emission of all four double-labeled mutants increased gradually (∼10 min), whereas that of AF568 decreased; these changes saturated in ∼20 min (Fig. 4 A). The measured raw spectrum of double-labeled MscS is a mixture of the FRET signal, the acceptor bleed-through, and the background signal from the buffer (the latter two were subtracted from the raw spectrum (e.g., see Fig. 4 B for F178)), and FRET efficiencies were calculated using Eq. 1. The FRET efficiencies of all four mutants decreased (p < 0.05) by 200 μM LPC (Fig. 4 C). The FRET efficiency decreased largely in A132C, F178C, and L246C from 0.40 ± 0.01 to 0.23 ± 0.02, 0.49 ± 0.02 to 0.18 ± 0.01, and 0.23 ± 0.01 to 0.09 ± 0.01, respectively, and it decreased less in R259C (from 0.30 ± 0.01 to 0.24 ± 0.01; n = 5–9). These results indicate that the CP domain swells up when MscS opens. The diameter of the CP domain increases at the middle and upper levels, and it increases less at the lower level (for details, see Discussion).
Figure 4.
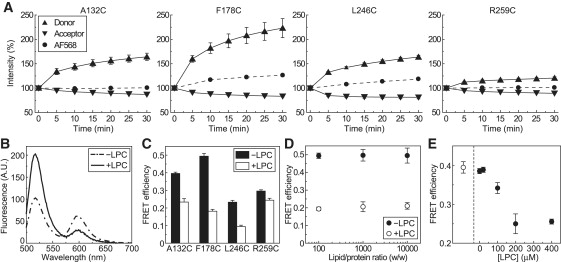
FRET efficiencies are decreased by LPC. (A) LPC (200 μM) induced time-dependent changes in the fluorescence intensities of four fluorescently labeled MscS mutants. The triangle (donor) and the upside-down triangle (acceptor) represent the integrated fluorescence intensity of the emission of AF488 and AF568 of double-labeled MscS, respectively. The circle (AF568) represents the integrated fluorescence intensity of the emission of AF568 of the acceptor-labeled MscS. LPC (200 μM) was applied at time 0 min. (B) The compensated-for spectral data of double-labeled MscS obtained by excitating the donor at 488 nm are shown. Dotted and solid lines show the spectra of the F178C mutant before (−LPC) and 30 min after (+LPC) addition of 200 μM LPC, respectively. (C) FRET efficiencies of the four mutants before (−LPC, black bars) declined significantly (p < 0.05) after exposure to 200 μM LPC for 30 min (+LPC, white bars). (D) FRET efficiencies of F178C are approximately the same at protein/lipid ratios of 1:100-1:10000 (w/w). The filled (−LPC) and open (+LPC) circles indicate the FRET efficiencies of F178C without and with 200 μM LPC, respectively. (E) LPC concentration dependence of the decreases in FRET efficiency. The open and filled circles represent the FRET efficiency of the A132C mutant before and 30 min after addition of different concentrations of LPC, respectively.
The FRET efficiencies of the F178C mutant MscS reconstituted into liposomes at various lipid/protein ratios were used to check the interchannel FRET, which is generally lipid/protein ratio-dependent. FRET efficiencies of F178C in control medium or in 200 μM LPC were not affected by the lipid/protein ratios in the range of 1:100–1:10000 (w/w) (n = 3–6; Fig. 4 D), and clustering of the fluorescently labeled channels at low lipid/protein ratios was not observed by epifluorescence microscopy or by total internal fluorescence microscopy. In addition, we did not detect the energy transfer when we took the purified fraction of A132C, F178C, L246C, or R259C MscS, separated the fraction in two parts, labeled them separately with the donor and acceptor dyes, mixed them, reconstituted them into liposomes, and measured FRET efficiencies. These results show that the energy transfer between interchannels would be negligible in the experimental conditions used here.
The LPC concentration dependence of the FRET efficiency decrease is shown in Fig. 4 E. The FRET efficiency of A132C without LPC was 0.40. It decreased with LPC concentration in a saturating manner; the saturating concentration was 200 μM, which decreased the FRET efficiency to 0.23. The concentration agrees with the observation by Vásquez et al. (16), assuming that asolectin is mainly composed of DOPC. MscS reconstituted liposomes were not ruptured by 200 μM LPC, as observed through a microscope (data not shown).
How does an increase in the membrane tension cause swelling of the CP domain? We examined a swelling of the CP domain of a mutant MscS that is deficient in TM-CP interactions. When Asp-62, a negatively charged residue located in the loop that connects TM1 and TM2 helices (Fig. 1), is replaced with a neutral (Cys or Asn) or a basic (Arg) amino acid (21), the threshold of membrane tension that opens MscS rises (21). We constructed a D62N/F178C mutant to explore the structural change that occurs in the CP domain when the TM-CP interaction is lost or reduced. The survival rate of the D62N/F178C mutant against hypoosmotic shock (81% ± 4%) was significantly lower than that of the wild-type (100%) and the F178C mutant (94% ± 5%), but was significantly higher than that of MscS/MscL knockout cells that harbored an empty vector (pB10b, 7% ± 1%, n = 3). The survival rate of the D62N/F178C mutant was almost the same as that of the D62N mutant (77% ± 3%, n = 3; Fig. 5 A), suggesting that the cysteine substitution does not affect the channel activity, and the D62N/F178C mutant raises its threshold for activation, as seen in the case of the D62N mutant. D62N/F178C MscS was purified, labeled using AF488 and AF568 randomly, and reconstituted into asolectin liposomes. The FRET efficiency of the D62N/F178C mutant without LPC was similar to that of the F178C. When the channel was opened by 200 μM LPC, the FRET efficiency of the D62N/F178C mutant decreased significantly (0.23 ± 0.01, n = 6), but was still higher than that of the F178C mutant (0.18 ± 0.01, n = 5, p < 0.05; Fig. 5 B). These results indicate that the intersubunit distance at the middle level of the CP domain (D62N/F178C) in the closed state is similar to that of F178C, whereas the degree of swelling of the CP domain of the D62N/F178C mutant during channel opening is less than that of F178C.
Figure 5.
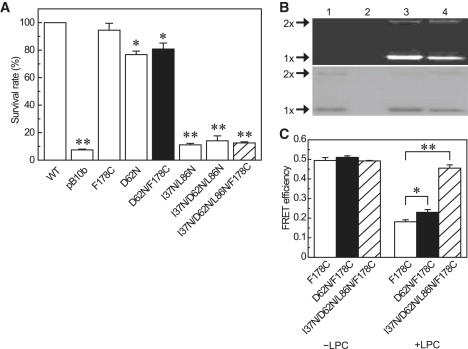
FRET efficiencies of D62N MscS are decreased by LPC to a lesser extent. (A) The survival rates of E. coli after hypoosmotic shock of wild-type, empty vector (pB10b), F178C, D62N, D62N/F178C (black bar), I37N/L86N, I37N/D62N/L86N, and I37N/D62N/L86N/F178C (hatched bar) are shown (mean ± SE, n = 3; ∗p < 0.05, ∗∗p < 0.01). (B) D62N/F178C and I37N/L86N, I37N/D62N/L86N were labeled with Alexa dyes, run on a SDS-PAGE gel, and detected using fluorescence imaging (upper) or stained with CBB (lower). Lane 1, wild-type MscS; lane 2, empty vector; lane 3, D62N/F178C; lane 4, I37N/D62N/L86N/F178C. (C) LPC decreased the FRET efficiency of F178C (open bar) and D62N/F178C to a lesser extent (black bar), whereas the FRET efficiency of I37N/D62N/L86N/F178C (hatched bar) was not changed by LPC (mean ± SE, n = 3–6; ∗p < 0.05, ∗∗p < 0.01).
The double-mutant I37N/L86N was not activated by high negative pressures (17). By combining I37N/L86N with D62N, we constructed an I37N/D62N/L86N/F178C mutant, and determined whether the swelling of the CP domain was inhibited in I37N/D62N/L86N/F178C. The survival rate of the I37N/D62N/L86N/F178C mutant (13% ± 1%, n = 3) against hypoosmotic shock was as low as that of pB10b (Fig. 5 A), and the channel currents of this mutant were not seen with negative pressures or after 200 μM LPC treatment (data not shown), showing that I37N/D62N/L86N/F178C is a severe LOF mutant. The expression level of the I37N/D62N/L86N/F178C mutant was normal (Fig. 5 B). The mutant MscS was purified, labeled with AF488 and AF568 randomly, and reconstituted into asolectin liposomes. The FRET efficiency of the I37N/D62/L86N/F178C mutant (0.49, n = 3) was not changed in 200 μM LPC (0.46 ± 0.02, n = 3; Fig. 5 C), indicating that the I37N/D62N/L86N/F178C mutant served as a negative control.
Discussion
In this study, the FRET efficiencies of all four point mutants (A132C, F178C, L246C, and R259C) were decreased by 200 μM LPC (Fig. 4 C), indicating that the diameters of the cross sections of the heptagonal MscS at the level of Ala-132, Phe-178, Leu-246, and Arg-259 expanded, i.e., the CP domain of MscS swells up when MscS opens. According to our Monte Carlo simulation of energy transfers from AF488 to AF568 between all subunits, the distance ra between AF488 and AF568 was estimated (see the Supporting Material). Because the distance ra differs from the distance LC between the α-carbons of MscS labeled with fluorescence dyes in the adjacent subunits, the distance LC was calculated by considering the molecular size and the binding angle of Alexa dyes (see the Supporting Material). The estimated distance LC of all four mutants in the closed state was 1.4–1.7 times larger than that of the crystal structure of MscS, e.g., at Phe-178, 33.5 ± 0.5 Å (n = 5) from FRET efficiency and 24.6 ± 0.6 Å (mean ± SD, n = 7) from the crystal structure.
The intersubunit distance of F178C in the closed state was also estimated by using the electron-electron double resonance (ELDOR) method, an advanced EPR technique (38), to cover the shortcomings of the FRET method. For example, with the FRET method, the orientation factor κ2 cannot be determined in many cases, and can vary from 0 to 4 (it was assumed to be 2/3 in this study), making estimations of the distance inaccurate (39,40). The ELDOR method allows relatively quantitative measurements of the long-range distance between spin labels (20–80 Å) to be obtained, and is often used to measure the distance in various biomaterials (36,41) because the spin label MTSL is smaller (MW = 264.3) than Alexa dyes (MW = 720.66 and 880.92 for AF488 and AF568, respectively) and the dipole direction of the label for cysteine is fixed. The distance between MTSLs at Phe-178 was measured preliminarily to be 25.6 ± 0.6 Å (n = 3), which leads to an intersubunit distance of 21.3 Å (the size of the spin label was assumed to be ∼5 Å; see the Supporting Material). This distance was similar to the intersubunit distance at Phe-178 (24.6 ± 0.4 Å, mean ± SD, n = 7) in the crystal structure. However, the ELDOR method also has some shortcomings; for example, the samples are flash-frozen in liquid nitrogen, and LPC cannot be applied to a sample under such conditions. The ELDOR data were used as a supplementary guide in estimating the diameter of the CP domain of MscS in the closed state.
The CP domain is divided into three subdomains (42): a β-subdomain, a mixed αβ-subdomain, and a β-barrel (Fig. 1). The residues Ala-132, Phe-178, Leu-246, and Arg-259 are located in the β-subdomain, the linker between the β-subdomain and the mixed αβ-subdomain, the upper part of the mixed αβ-subdomain, and the lower part of the mixed αβ-subdomain, respectively. The distance LC was compensated for by the EPR value of 21.3 Å, and the diameters of heptagonal MscS at the four levels were estimated by using the FRET data under the assumption that κ2 and J(λ) were kept constant during the closed-to-open transition (Table 1), because this estimation should give a better picture of the transition from the closed to open state compared to that drawn from the FRET data alone. During channel opening, the CP domain expands at the β- and mixed αβ-subdomains, and this expansion was smaller at the lowest level (Fig. 6).
Table 1.
Putative estimation of the changes in the outer diameter of the heptagonal MscS
| MscS |
Diameter (Å, n = 5–9) |
||
|---|---|---|---|
| Control | 200 μM LPC | ΔD | |
| A132C | 56.5 ± 0.6 | 69.2 ± 2.0∗ | +12.8 ± 2.0 |
| F178C | 49.1 ± 1.4 | 76.3 ± 1.5∗ | +27.2 ± 2.0 |
| L246C | 70.5 ± 1.1 | 90.7 ± 1.8∗ | +20.2 ± 0.9 |
| R259C | 64.6 ± 0.6 | 69.3 ± 1.1† | +4.7 ± 0.7 |
Outer diameters were calculated from the distance LC between α-carbons in adjacent subunits.
p < 0.01.
p < 0.05.
Figure 6.

Model of the relationship between the swelling of the CP domain and gating of the MscS channel. Only two subunits, each consisting of TM1 and TM2 (boxes), TM3 (hatched box), and the CP domain (heavy line), are shown for simplicity. When the membrane tension increases, the TM domain expands via the lipid-protein interaction, followed by CP domain expansion via the TM-CP interaction. Ala-132, Phe-178, Leu-246, and Arg-259 are placed in the schematic diagram of the closed or open-state channel according to the results and estimations obtained in this work.
There are two hypotheses regarding the role of the β-barrel of the CP domain: 1), the subdomain forms a CP gate in the channel by moving apart upon channel opening (28); and 2), it enables organization of the channel and facilitates cycles of channel activity (42). The heptagonal diameter at the level of Arg-259 that located near the β-barrel expanded very slightly upon addition of 200 μM LPC (the diameter changed by 3.1 Å), suggesting that the β-barrel does not undergo a large conformational change during channel opening. The data presented here support the hypothesis that the β-barrel stabilizes the homoheptamer and facilitates the transition from the closed to open state like an S3 bundle of MscL (43).
The TM-CP interaction forms mainly between Asp-62 and Arg-128 or Arg-131 (20,21). Substitution for Asp-62 with a neutral charged amino acid (D62N) elevates the gating threshold of MscS (21). The degree of changes in FRET efficiency was smaller in the D62N/F178C mutant than in F178C (Fig. 5 C), which indicates that swelling of the CP domain of D62N/F178C MscS is reduced by the deficient TM-CP interaction. Thus, the β-subdomain and the upper part of the αβ-mixed subdomain undergo a large structural change, presumably due to the TM-CP interaction.
What is the role of the swelling of the CP domain in the channel opening? Two different roles have been proposed: 1) When the membrane tension increases, the TM domain expands via the lipid-protein interaction, which is followed by CP domain expansion via the TM-CP interaction and is associated with an influx of water and solutes to the vestibule of the CP domain (Fig. 6). This swelling of the CP domain may induce a structural change in the TM3 helix, resulting in breakage of the vapor lock as suggested by Akitake et al. (15). However, further studies are needed to test this idea. 2) When the stress in the membrane is kept at high levels, MscS opens and subsequently is inactivated. The electrophysiological analyses indicate that D62N MscS is inactivated more rapidly than wild-type MscS, and the CP domain presumably shrinks during the inactivation (21). The results presented here also support the idea that the CP domain shrinks due to the impaired TM-CP interaction and the inactivated D62N MscS (15). Consequently, it is postulated that conformational changes in the CP domain regulate the channel state of MscS, which may be achieved via structural changes in the TM3 helix.
Supporting Material
Additional text, equations, references, a table, and figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01025-X.
Supporting Material
Acknowledgments
We thank Dr. H. Hara (Bruker Biospin K.K., Japan) for ERP measurements and data analysis.
This work was supported by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (to H.T. and M.S.) and a grant from the Japan Space Forum (to H.T. and M.S.).
References
- 1.Levina N., Totemeyer S., Stokes N.R., Louis P., Jones M.A. Protection of Escherichia coli cells against extreme turgor by activation of MscS and MscL mechanosensitive channels: identification of genes required for MscS activity. EMBO J. 1999;18:1730–1737. doi: 10.1093/emboj/18.7.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang G., Spencer R.H., Lee A.T., Barclay M.T., Rees D.C. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 1998;282:2220–2226. doi: 10.1126/science.282.5397.2220. [DOI] [PubMed] [Google Scholar]
- 3.Bass R.B., Strop P., Barclay M., Rees D.C. Crystal structure of Escherichia coli MscS, a voltage-modulated and mechanosensitive channel. Science. 2002;298:1582–1587. doi: 10.1126/science.1077945. [DOI] [PubMed] [Google Scholar]
- 4.Levin G., Blount P. Cysteine scanning of MscL transmembrane domains reveals residues critical for mechanosensitive channel gating. Biophys. J. 2004;86:2862–2870. doi: 10.1016/S0006-3495(04)74338-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshimura K., Nomura T., Sokabe M. Loss-of-function mutations at the rim of the funnel of mechanosensitive channel MscL. Biophys. J. 2004;86:2113–2120. doi: 10.1016/S0006-3495(04)74270-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sukharev S., Betanzos M., Chiang C.S., Guy H.R. The gating mechanism of the large mechanosensitive channel MscL. Nature. 2001;409:720–724. doi: 10.1038/35055559. [DOI] [PubMed] [Google Scholar]
- 7.Sukharev S.I., Sigurdson W.J., Kung C., Sachs F. Energetic and spatial parameters for gating of the bacterial large conductance mechanosensitive channel, MscL. J. Gen. Physiol. 1999;113:525–540. doi: 10.1085/jgp.113.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perozo E., Kloda A., Cortes D.M., Martinac B. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 2002;9:696–703. doi: 10.1038/nsb827. [DOI] [PubMed] [Google Scholar]
- 9.Corry B., Rigby P., Liu Z.W., Martinac B. Conformational changes involved in MscL channel gating measured using FRET spectroscopy. Biophys. J. 2005;89:L49–L51. doi: 10.1529/biophysj.105.072009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gullingsrud J., Schulten K. Gating of MscL studied by steered molecular dynamics. Biophys. J. 2003;85:2087–2099. doi: 10.1016/s0006-3495(03)74637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sukharev S. Purification of the small mechanosensitive channel of Escherichia coli (MscS): the subunit structure, conduction, and gating characteristics in liposomes. Biophys. J. 2002;83:290–298. doi: 10.1016/S0006-3495(02)75169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spronk S.A., Elmore D.E., Dougherty D.A. Voltage-dependent hydration and conduction properties of the hydrophobic pore of the mechanosensitive channel of small conductance. Biophys. J. 2006;90:3555–3569. doi: 10.1529/biophysj.105.080432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vora T., Corry B., Chung S.H. Brownian dynamics investigation into the conductance state of the MscS channel crystal structure. Biochim. Biophys. Acta. 2006;1758:730–737. doi: 10.1016/j.bbamem.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Anishkin A., Sukharev S. Water dynamics and dewetting transitions in the small mechanosensitive channel MscS. Biophys. J. 2004;86:2883–2895. doi: 10.1016/S0006-3495(04)74340-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akitake B., Anishkin A., Liu N., Sukharev S. Straightening and sequential buckling of the pore-lining helices define the gating cycle of MscS. Nat. Struct. Mol. Biol. 2007;14:1141–1149. doi: 10.1038/nsmb1341. [DOI] [PubMed] [Google Scholar]
- 16.Vásquez V., Sotomayor M., Cordero-Morales J., Schulten K., Perozo E. A structural mechanism for MscS gating in lipid bilayers. Science. 2008;321:1210–1214. doi: 10.1126/science.1159674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nomura T., Sokabe M., Yoshimura K. Lipid-protein interaction of the MscS mechanosensitive channel examined by scanning mutagenesis. Biophys. J. 2006;91:2874–2881. doi: 10.1529/biophysj.106.084541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang W., Black S.S., Edwards M.D., Miller S., Morrison E.L. The structure of an open form of an E. coli mechanosensitive channel at 3.45 A resolution. Science. 2008;321:1179–1183. doi: 10.1126/science.1159262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards M.D., Li Y., Kim S., Miller S., Bartlett W. Pivotal role of the glycine-rich TM3 helix in gating the MscS mechanosensitive channel. Nat. Struct. Mol. Biol. 2005;12:113–119. doi: 10.1038/nsmb895. [DOI] [PubMed] [Google Scholar]
- 20.Sotomayor M., Schulten K. Molecular dynamics study of gating in the mechanosensitive channel of small conductance MscS. Biophys. J. 2004;87:3050–3065. doi: 10.1529/biophysj.104.046045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nomura T., Sokabe M., Yoshimura K. Interaction between the cytoplasmic and transmembrane domains of the mechanosensitive channel MscS. Biophys. J. 2008;94:1638–1645. doi: 10.1529/biophysj.107.114785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long S.B., Campbell E.B., Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 23.Moiseenkova-Bell V.Y., Stanciu L.A., Serysheva I.I., Tobe B.J., Wensel T.G. Structure of TRPV1 channel revealed by electron cryomicroscopy. Proc. Natl. Acad. Sci. USA. 2008;105:7451–7455. doi: 10.1073/pnas.0711835105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mio K., Ogura T., Sato C. Structure of six-transmembrane cation channels revealed by single-particle analysis from electron microscopic images. J. Synchrotron Radiat. 2008;15:211–214. doi: 10.1107/S0909049508004640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zagotta W.N., Olivier N.B., Black K.D., Young E.C., Olson R. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- 26.Johnson J.P.J., Zagotta W.N. The carboxyl-terminal region of cyclic nucleotide-modulated channels is a gating ring, not a permeation path. Proc. Natl. Acad. Sci. USA. 2005;102:2742–2747. doi: 10.1073/pnas.0408323102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sotomayor M., van der Straaten T.A., Ravaioli U., Schulten K. Electrostatic properties of the mechanosensitive channel of small conductance MscS. Biophys. J. 2006;90:3496–3510. doi: 10.1529/biophysj.105.080069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koprowski P., Kubalski A. C termini of the Escherichia coli mechanosensitive ion channel (MscS) move apart upon the channel opening. J. Biol. Chem. 2003;278:11237–11245. doi: 10.1074/jbc.M212073200. [DOI] [PubMed] [Google Scholar]
- 29.Grajkowski W., Kubalski A., Koprowski P. Surface changes of the mechanosensitive channel MscS upon its activation, inactivation, and closing. Biophys. J. 2005;88:3050–3059. doi: 10.1529/biophysj.104.053546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song S., Hanson M.J., Liu B.-F., Chylack L.T., Liang J.J.-N. Protein-protein interactions between lens vimentin and αB-crystallin using FRET acceptor photobleaching. Mol. Vis. 2008;14:1282–1287. [PMC free article] [PubMed] [Google Scholar]
- 31.Miyawaki A., Llopis J., Heim R., McCaffery J.M., Adams J.A. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- 32.Miller S., Bartlett W., Chandrasekaran S., Simpson S., Edwards M. Domain organization of the MscS mechanosensitive channel of Escherichia coli. EMBO J. 2003;22:36–46. doi: 10.1093/emboj/cdg011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Granier S., Kim S., Shafer A.M., Ratnala V.R.P., Fung J.J. Structure and conformational changes in the C-terminal domain of the β2-adrenoceptor: insights from fluorescence resonance energy transfer studies. J. Biol. Chem. 2007;282:13895–13905. doi: 10.1074/jbc.M611904200. [DOI] [PubMed] [Google Scholar]
- 34.Panchuk-Voloshina N., Haugland R.P., Bishop-Stewart J., Bhalgat M.K., Millard P.J. Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates. J. Histochem. Cytochem. 1999;47:1179–1188. doi: 10.1177/002215549904700910. [DOI] [PubMed] [Google Scholar]
- 35.Blount P., Sukharev S.I., Moe P.C., Martinac B., Kung C. Mechanosensitive channels of bacteria. Methods Enzymol. 1999;294:458–482. doi: 10.1016/s0076-6879(99)94027-2. [DOI] [PubMed] [Google Scholar]
- 36.Tateishi Y., Ariyoshi M., Igarashi R., Hara H., Mizuguchi K. Molecular basis for SUMOylation-dependent regulation of DNA binding activity of heat shock factor 2. J. Biol. Chem. 2009;284:2435–2447. doi: 10.1074/jbc.M806392200. [DOI] [PubMed] [Google Scholar]
- 37.Corry B., Jayatilaka D. Simulation of structure, orientation, and energy transfer between AlexaFluor molecules attached to MscL. Biophys. J. 2008;95:2711–2721. doi: 10.1529/biophysj.107.126243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hara H., Kawamori A., Astashkin A.V., Ono T. The distances from tyrosine D to redox-active components on the donor side of Photosystem II determined by pulsed electron-electron double resonance. Biochim. Biophys. Acta. 1996;1276:140–146. [Google Scholar]
- 39.Corry B., Jayatilaka D., Martinac B., Rigby P. Determination of the orientational distribution and orientation factor for transfer between membrane-bound fluorophores using a confocal microscope. Biophys. J. 2006;91:1032–1045. doi: 10.1529/biophysj.106.080713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corry B., Jayatilaka D., Rigby P. A flexible approach to the calculation of resonance energy transfer efficiency between multiple donors and acceptors in complex geometries. Biophys. J. 2005;89:3822–3836. doi: 10.1529/biophysj.105.069351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeschke G., Polyhach Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 2007;9:1895–1910. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]
- 42.Schumann U., Edwards M.D., Li C., Booth I.R. The conserved carboxy-terminus of the MscS mechanosensitive channel is not essential but increases stability and activity. FEBS Lett. 2004;572:233–237. doi: 10.1016/j.febslet.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 43.Anishkin A., Gendel V., Sharifi N.A., Chiang C.S., Shirinian L. On the conformation of the COOH-terminal domain of the large mechanosensitive channel MscL. J. Gen. Physiol. 2003;121:227–244. doi: 10.1085/jgp.20028768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.