

Abstract
ROMK (Kir1.1) potassium channels are closed by internal acidification with a pKa of 6.7 ± 0.01 in 100 mM external K and a pKa of 7.0 ± 0.01 in 1 mM external K. Internal acidification in 1 mM K (but not 100 mM K) not only closed the pH gate but also inactivated Kir1.1, such that realkalization did not restore channel activity until high K was returned to the bath. We identified a new putative intersubunit salt bridge (R128-E132-Kir1.1b) in the P-loop of the channel near the selectivity filter that affected the K sensitivity of the inactivation process. Mutation of either R128-Kir1.1b or E132-Kir1.1b caused inactivation in both 1 mM and 100 mM external K during oocyte acidification. However, 300 mM external K (but not 200 mM Na + 100 mM K) protected both E132Q and R128Y from inactivation. External application of a modified honey-bee toxin, tertiapin Q (TPNQ), also protected Kir1.1 from inactivation in 1 mM K and protected E132Q and R128Y from inactivation in 100 mM K, which suggests that TPNQ binding to the outer mouth of the channel stabilizes the active state. Pretreatment of Kir1.1 with external Ba prevented Kir1.1 inactivation, similar to pretreatment with TPNQ. In addition, mutations that disrupted transmembrane helix H-bonding (K61M-Kir1.1b) or stabilized a selectivity filter to helix-pore linkage (V121T-Kir1.1b) also protected both E132Q and R128Y from inactivation in 1 mM K and 100 mM K. Our results are consistent with Kir inactivation arising from conformational changes near the selectivity filter, analogous to C-type inactivation.
Introduction
Renal outer medullary potassium (ROMK) channels (Kir1.1) are gated by internal pH, closing in response to acidification and reopening after alkalization (1–7). Acid-induced closure of Kir1.1 (pH gating) probably occurs by hydrophobic side-chain occlusion of the permeation path at the inner transmembrane helix bundle crossing at L160-Kir1.1b (8–12). However, in cyclic-nucleotide-gated channels (13), and in Kir2 (14), closure of the inner transmembrane (TM) helix bundle alone was insufficient to block permeation, implying the existence of another gate, perhaps at the selectivity filter.
In addition to pH gating, Kir1.1 also undergoes an inactivation process when intracellular pH is reduced in the presence of low (1 mM) external K (1–7), but the mechanism for this inactivation remains unclear. A possible site (6,15,16) suggested for this inactivation is the selectivity filter. This would be similar to C-type inactivation in Shaker (17–30) and slow inactivation in KcsA (31–33), both of which involve conformational changes at the selectivity filter.
Since Kir1.1 inactivation is not voltage-dependent and requires pH-gate closure, there must be communication between the pH gate at the bundle crossing and the inactivation site. Previous studies have implicated H-bonding between inner and outer transmembrane helices as a crucial step in the Kir inactivation process (2). Hence, we reinvestigated the K61-Kir1.1b locus to evaluate possible links between helix H-bonding and salt bridges near the selectivity filter that might be involved in inactivation. Mutations at V121-Kir1.1b were also examined, because this site at the cytoplasmic side of the selectivity filter has been implicated in Kir1.1 inactivation (5,16).
We constructed a homology model of Kir1.1b (ROMK2) based on the closed-state coordinates of KirBac1.1 (8) and identified a new intersubunit salt bridge at R128-E132 in Kir1.1b, near the selectivity filter. This bridge would correspond to R147-E151 in Kir1.1a (ROMK1), R148-D152 in Kir2.1,and R155-E159 in Kir3.4. In addition, the intersubunit bridge R128-E132-Kir1.1b is linked to E118-Kir1.1b (homologous to E71-KcsA). Bridging between these three residues, E118-R128-E132, may be required for stabilization of the Kir1.1b selectivity filter, and disruption of this bridging may precipitate inactivation.
Precedence for salt-bridge stabilization of the inward rectifier selectivity filter comes from studies of the R148-E138 salt bridge in Kir2.1, where disruption of this ion pair altered both selectivity and permeation (34). Other studies have identified R148-Kir2.1 as essential for the protective effect of high external [K] on Kir2.1 inactivation (35).
Methods
Mutant construction and expression of channels
Point mutations in Kir1.1b (ROMK2; EMBL/GenBank/DDBJ accession No. L29403) were engineered with a polymerase chain reaction QuikChange mutagenesis kit (Stratagene, La Jolla, CA), using primers synthesized by Integrated Data Technologies (Coralville, IA). Nucleotide sequences were checked on an Applied Biosystems (Carlsbad, CA) 3100 DNA sequencing machine at the University of Chicago Cancer Research Center.
Plasmids were linearized with Not I restriction enzyme and transcribed in vitro with T7 RNA polymerase in the presence of the GpppG cap using mMESSAGE mMACHINE kit (Ambion, Austin, TX). Synthetic cRNA was dissolved in water and stored at −70°C before use. Stage V-VI oocytes were obtained by partial ovariectomy of female Xenopus laevis (NASCO, Ft. Atkinson, WI), anesthetized with tricaine methanesulfonate (1.5 g/L, adjusted to pH 7.0). Oocytes were defolliculated by incubation (on a Vari-Mix rocker (Thermo Scientific, Waltham, MA)) in Ca-free modified Barth's solution (82.5 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 5 mM HEPES, adjusted to pH 7.5 with NaOH) containing 2 mg/ml collagenase type IA (C9891, Sigma Chemical, St. Louis, MO) for 90 min, and (if necessary) for another 90 min in a fresh enzyme solution at 23°C. Oocytes were injected with 0.5–1 ng of cRNA and incubated at 19°C in twice-diluted Leibovitz medium (Life Technologies, Grand Island, NY) for 1–3 days before measurements were made.
Whole-cell experiments
Whole-cell currents and conductances were measured in intact oocytes using a TEVC (TEVC; model CA-1, Dagan, Minneapolis, MN) with 16 command pulses of 30 ms duration between −200 mV and +100 mV, centered around the resting potential.
Oocytes expressing Kir1.1 or mutants of Kir1.1 were bathed in permeant acetate buffers to control their internal pH, as previously described (1,36,37). The relation between intracellular and extracellular pH was calculated from a previous calibration with Kir1.1 oocytes: pHi = 0.595 × pHo + 2.4 (36). There was no external pH dependence of single-channel conductance or gating for the channels used in this study, as determined in separate experiments with impermeant external buffers.
Whole-cell conductances, measured with the TEVC, provide a simple measure of aggregate channel activity, since at constant [K], internal pH had no effect on single-channel conductance or channel kinetics until the channel abruptly closed (Po = 0) as a result of intracellular acidification (36). All whole-cell conductances were normalized to the maximum whole-cell conductance for that oocyte to compensate for differences in expression efficiency between wild-type and mutant channels.
The composition of the bath for the whole-cell experiments was 50 mM KCl, 50 mM K acetate, 1 mM MgCl2, 2 mM CaCl2, and 5 mM HEPES. In low-K solutions, NaCl and Na acetate replaced KCl and K acetate, respectively. In the high-K and high-Na solutions, additional KCl or NaCl was added to achieve final cation concentrations of 300 mM K or 100 mM K + 200 mM Na. Although the latter solutions were hyperosmotic, the oocytes functioned normally for at least 1 h. None of the oocytes in these experiments had significant chloride currents. Tertiapin-Q trifluoroacetate salt (TPNQ) was obtained from Sigma Aldrich (T1567). Unless otherwise noted, the concentration of TPNQ used in these experiments was 500 nanomolar. All experiments described were conducted at room temperature (21 ± 2°C) on Kir1.1b (ROMK2) (or mutants of Kir1.1b) expressed in Xenopus oocytes.
The rate of exchange of the external bath was determined by measuring the membrane potential during a rapid increase in external K (4). These results demonstrated that 98% of the bath solution could be replaced within 10 s, which is also confirmed by the rapid phase of the final conductance increase (Fig. S2 in the Supporting Material).
Homology modeling
To facilitate interpretation of the electrophysiological results, we made a homology model of the Kir1.1b based on the x-ray crystal structure of the closed state of KirBac1.1 (1P7B (8)). Model building and refinement were carried out using the CHARMM molecular modeling program (38) and the CHARMM Param22 force field (39). An alignment of Kir1.1b with other K channels is shown in Fig. S1.
Results
Low K inactivation of Kir1.1
We studied inactivation in wild-type and mutant Kir1.1 using TEVC oocytes. Consistent with previous studies (6), Kir1.1 was strongly gated by internal (but not external) pH. Cytoplasmic acidification produced abrupt channel closure with an effective pKa of 6.7 ± 0.01 in 100 mM external K (Fig. 1, blue line) and an effective pKa of 7.0 ± 0.01 in 1 mM external K (Fig. 1, brown line).
Figure 1.
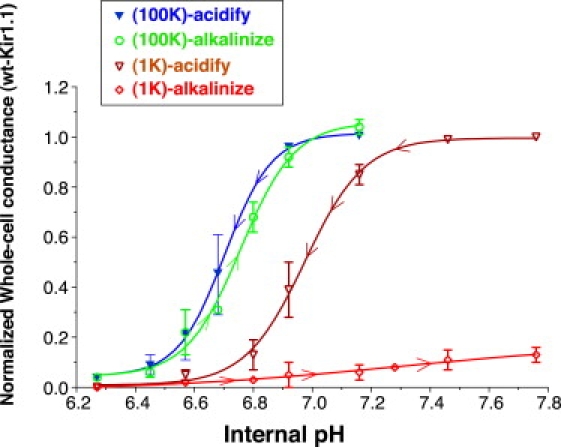
Steady-state titration data for wt Kir1.1 acidification (blue line) and 90-min realkalization (green line) in 100 mM external K compared to acidification (brown line) and 90-min realkalization (red line) in 1 mM external K. Internal pH was controlled with permeant acetate buffers and whole-cell conductances were normalized to their maximal values. Changes in extracellular pH alone (in the absence of acetate) had no significant effect on Kir1.1 activity.
Realkalization in 100 mM K returned Kir1.1 activity to its control level (Fig. 1, green line). However, the same realkalization in 1 mM external [K] failed to restore Kir1.1 activity after 90 min at pHi = 7.8 (Fig. 1, red line), although channel activity eventually returned to its initial level after 3 h at pHi = 8. Kir inactivation required both low external K and low internal pH, but was independent of which was applied first. Using a protocol that was the reverse of that shown in Fig. 1, cytoplasmic acidification followed by reduction of extracellular [K] to 1 mM inactivated Kir1.1 to an equivalent extent (Fig. S2).
TPNQ rescue of Kir1.1 inactivation
In addition to steady-state pH titrations (Fig. 1), we studied Kir inactivation using a rapid acidification-realkalization protocol (Fig. 2). In 100 mM external K, Kir1.1 completely recovered after a transient acidification, with a time constant of 92 ± 5 s (Fig. 2, cyan line). However, Kir1.1 recovery after the same acidification in 1 mM external K required at least 3 h at pHi = 8 (Fig. 2, green line). Based on this observation, we operationally defined Kir inactivation as <20% channel recovery after 30 min in pHi = 8.
Figure 2.
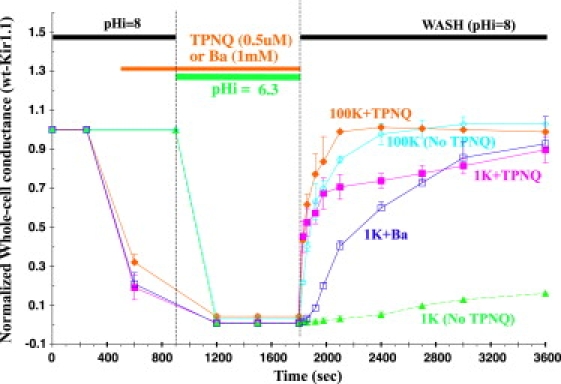
Wild-type Kir1.1 inactivation in 1 mM external K (green dashed line), and protection from inactivation in 1 mM K by either 500 nM TPNQ (magenta line) or 1 mM Ba (dark blue line). High (100 mM) external K prevents inactivation in both the absence (cyan line) and presence (orange line) of TPNQ during internal acidification from pHi = 8 to pHi = 6.3. External K was maintained constant throughout the protocol at either 1 mM or 100 mM, and whole-cell conductances were normalized to their initial values. In the Ba experiments, conductance was measured at a holding potential near −100mV.
Prior exposure to 500 nM external TPNQ (40–42) protected Kir1.1 from inactivation during acidification in low K, such that realkalization in 1 mM K restored 90% of channel activity in 30 min (Fig. 2, magenta line), compared to a 12% recovery for channels in 1 mM K with no exposure to TPNQ (Fig. 2, green line). As a control, realkalization in both 100 mM external K and 500 nM TPNQ allowed complete recovery of channel activity with a time course (Fig. 2, orange line) that was not significantly different from recovery in 100 mM K alone (Fig. 2, cyan line).
Although the magenta curve of Fig. 2 depicts application of TPNQ before oocyte acidification, similar results were obtained when TPNQ was applied after oocyte acidification, following closure of the bundle-crossing pH gate. A 10-min acidification followed by 1 min of TPNQ at continued low pH overlaps the magenta line of Fig. 2, indicating that TPNQ is actually more effective at preventing inactivation at low pH, probably because TPNQ binding affinity is greatly increased at low pH (43).
External barium was as effective as 500 nM TPNQ at preventing Kir1.1 inactivation in 1 mM K. Application of 1 mM Ba followed by acidification and realkalization restored 93 ± 11% of Kir1.1 channel activity in 30 min, as measured by normalized whole-cell conductance (Fig. 2, dark blue line). The speed of conductance recovery after barium was slower than with TPNQ, but this could be attributed to residual barium block during washout. Barium was also effective at preventing Kir1.1 inactivation when applied to channels whose bundle-crossing pH gate was already closed by internal acidification. Consequently, both TPNQ and Ba protected Kir1.1 from inactivation, independent of the pH-gate state.
In addition to blocking Kir1.1 inactivation, external TPNQ could also reverse existing inactivation. As indicated in Fig. 3, application of 500 nM TPNQ for varying periods of time reversed the inactivation arising from the low-K, low-pH protocol. Rapid jumps in whole-cell conductance (Fig. 3), coincident with TPNQ washout, reflect the fraction of Kir1.1 channels that were reactivated by progressively longer exposure to TPNQ. The slow phase of the whole-cell conductance increase after TPNQ (Fig. 3, cyan and orange lines) was not significantly different from the slow conductance increase in the absence of TPNQ (Fig. 3, green line) and probably represents the baseline recovery from inactivation in 1 K solutions.
Figure 3.
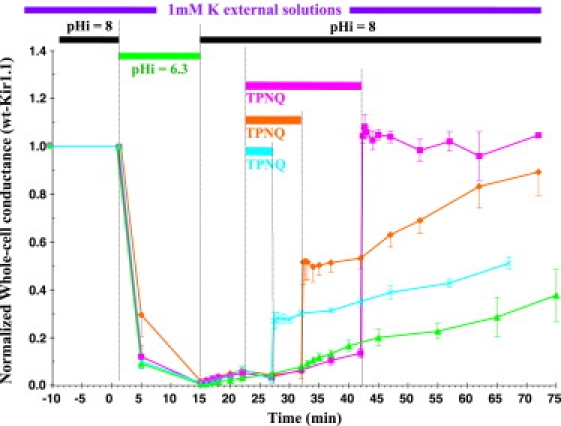
Rescue of wt Kir1.1 from inactivation by 500 nM external TPNQ. Oocytes were maintained in 1 mM [K] and inactivated by reducing internal pH from 8 to 6.3, using permeant acetate buffers. Kir1.1 remained inactivated during realkalization to pHi = 8, but could be reactivated by exposure to external TPNQ. The fraction of channels rescued from inactivation increased progressively with 5 min (cyan line), 10 min (orange line), and 20 min (magenta line) exposure to TPNQ.
TPNQ reversal of Kir1.1 inactivation increased with the duration of TPNQ application, where 50% of channel activity was restored after 13 min of exposure. In separate experiments, 500 nM external TPNQ had no effect on fully activated Kir1.1 in oocytes clamped at an internal pH of 8.0. Hence, TPNQ prevents and reverses inactivation but does not itself activate Kir1.1.
Modifying a salt bridge near the selectivity filter alters sensitivity to inactivation
Our working hypothesis is that an intersubunit salt bridge (R128-E132) near the selectivity filter (Fig. 4) stabilizes the active state of Kir1.1b, analogous to the R148-E138 bridge of Kir2.1 (34). We believe that the intersubunit bridge R128-E132 of Kir1.1b is further stabilized by intrasubunit links between E118 and R128 on each of the four subunits (Fig. 4).
Figure 4.
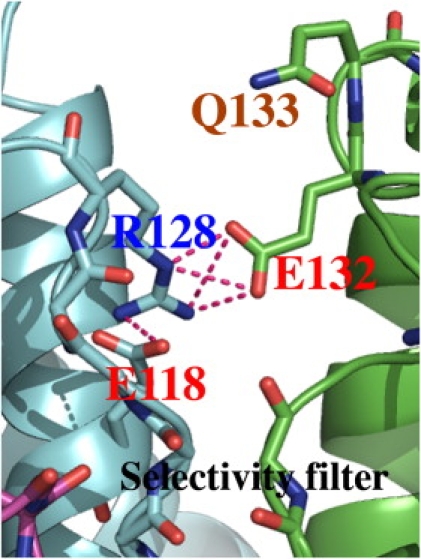
Kir1.1b intersubunit salt bridge R128-E132 and the additional link to E118 on the same subunit as R128. Two (of four) adjacent subunits in the region of the selectivity filter are depicted as cyan and green ribbons.
Since all mutations of the highly conserved E118-Kir1.1b were nonfunctional, we concentrated on evaluating the role of the intersubunit salt bridge, R128-E132, by making the mutations E132Q, E132D, E132N, E132K, E132L, R128Y, and R128E. Three of these mutants, E132K, E132L, and R128E, did not produce significant currents, but the other mutants (E132Q, E132D, E132N, and R128Y) expressed as well as wt Kir1.1.
Specifically, both E132Q and R128Y inactivated by >90% after internal acidification in 100 mM external K, as indicated by an absence of channel recovery during realkalization (Fig. 5, orange and purple lines). This is in sharp contrast to wt Kir1.1, which inactivated in 1 mM external K, but not in 100 mM K (Fig. 1). The E132D mutant, which conserves side-chain charge but is farther from R128 than the native E132, behaved in a manner similar to that of wt Kir1.1(Table 1). In contrast, the E132N mutant, with an uncharged side chain at 132, behaved like the uncharged E132Q and inactivated in both 1 mM and 100 mM K (Fig. S3).
Figure 5.

Intersubunit salt-bridge mutants E132Q (orange line) and R128Y (purple line) inactivate in 100 mM external K (as well as in 1 mM K), as evidenced by their failure to recover whole-cell conductance during 30 min of realkalization. Internal pH was controlled with permeant acetate buffers, and whole-cell conductances were normalized to their maximal values.
Table 1.
Effect of salt-bridge mutation on inactivation
| Channel type | External K (mM) | TPNQ (nM) | Inactivation∗ |
|---|---|---|---|
| wt Kir1.1b, E132D, Q133E, V121N | 1 | 0 | YES |
| 100 | 0 | Absent | |
| 1 | 500 | Absent | |
| 100 | 500 | Absent | |
| E132Q, E132N, R128Y | 1 | 0 | YES |
| 100 | 0 | YES | |
| 300 | 0 | Absent | |
| 1 | 500 | YES | |
| 100 | 500 | Absent | |
| E132Q-Q133E | 1 | 0 | YES |
| 100 | 0 | Absent | |
| 1 | 500 | YES | |
| 100 | 500 | Absent | |
| (K61M, V121T, K61M-E132Q, V121T-E132Q V121S, V121Q) | 1 | 0 | Absent |
| 100 | 0 | Absent | |
| 1 | 500 | Absent | |
| 100 | 500 | Absent | |
Inactivation resulting from a transient internal acidification to pHi = 6.3.
Introducing a negative charge at Q133 to form the double mutant E132Q-Q133E (Fig. S3) prevented the high-K inactivation seen with E132Q alone (Fig. 5). In E132Q-Q133E, the negative charge at Q133E compensates for removal of the negative charge by E132Q, causing the double mutant to behave in a manner similar to that of the wild-type. In our homology model of E132Q-Q133E, the Glu side-chain of Q133E takes the place of E132 and reestablishes an intersubunit salt bridge to R128. The effects of these and other mutations are summarized in Table 1.
E132Q channel inactivation could be prevented by 300 mM extracellular K (Fig. 6, purple tracing). This rescue of E132Q activity by very high K was not the result of increased ionic strength or changes in osmolarity, because 200 mM Na + 100 mM K was ineffective at preventing inactivation (Fig. 6, red line). A similar rescue of inactivation by 300 mM K was also observed with the R128Y mutant.
Figure 6.
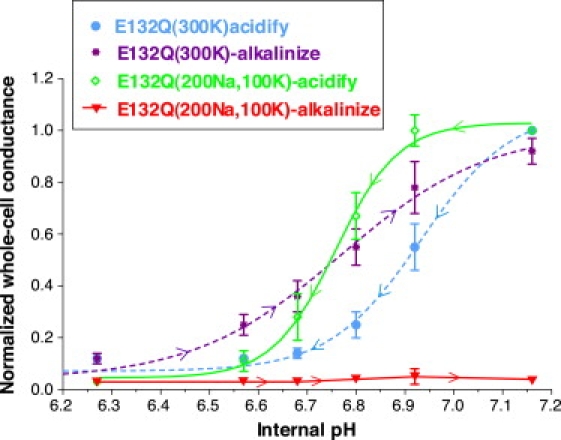
Inactivation of E132Q can be rescued by high (300 mM) bath K (purple and blue dashed lines), but not by 200 mM Na plus 100 mM K bath (green and red lines). Internal pH was controlled with permeant acetate buffers, and whole-cell conductances were normalized to their maximal values.
Finally, the E132Q mutant had a 24 ± 1% lower permeability ratio (PRb/PK) and a 56 ± 4% lower inward conductance ratio (GRb/GK) than wt Kir1.1 channels. However the PK/PNa ratio remained high.
The extracellular blocker TPNQ protects E132Q and R128Y from high-K inactivation
The salt-bridge mutants E132Q and R128Y were subjected to the same inactivation protocol used for wt Kir1.1(Fig. 2), during which external K was kept constant at either 1 mM or 100 mM, and whole-cell conductances were normalized to their initial values (Fig. 7).
Figure 7.
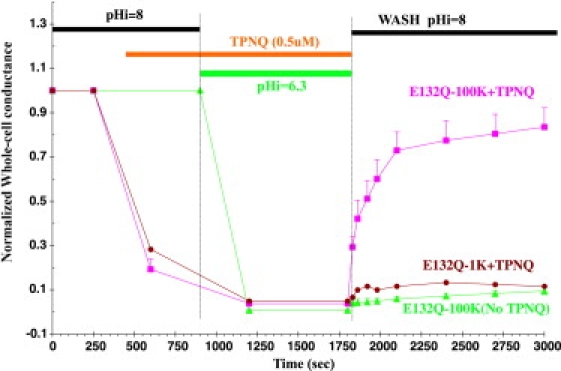
E132Q was inactivated in 100 mM K (green line) by oocyte acidification from pHi 8 to pHi 6.3 using permeant acetate buffers. TPNQ (500 nM) protected E132Q from inactivation in 100 mM external K (magenta line), but not from inactivation in 1 mM external K (brown line). External K was maintained constant at either 1 mM or 100 mM throughout the protocol, and whole-cell conductances were normalized to their initial values.
Both E132Q (Fig. 7, green line) and R128Y (Fig. S4) were inactivated after rapid acidification in both 1 mM and 100 mM external K. This is consistent with steady-state inactivation of these mutants after acidification in 100 mM external K (Fig. 5). External TPNQ (500 nM) protected E132Q (Fig. 7), E132N (Fig. S3), and R128Y (Fig. S4) from inactivation in 100 mM external K. However, TPNQ protected neither E132Q (Fig. 7), R128Y (Fig. S4), nor E132Q-Q133E (Fig. S3) from inactivation in 1 mM external K. For comparison, the same concentration of TPNQ protected wt Kir1.1 from inactivation in 1 mM external K (Fig. 2, magenta line).
K61M-Kir1.1b prevents inactivation
Replacement of a lysine with methionine (K61M-Kir1.1b) shifted the pKa of the channel from 6.7 ± 0.01 to 5.4 ± 0.1 (44) and prevented inactivation in 1 mM external K (Fig. 8, red line), possibly by disrupting H-bonding with the inner helix (2) (see Fig. S5, A and B). The absence of inactivation in K61M was not the result of preventing pH gate closure, since in this experiment, internal pH was reduced below the pKa of K61M.
Figure 8.
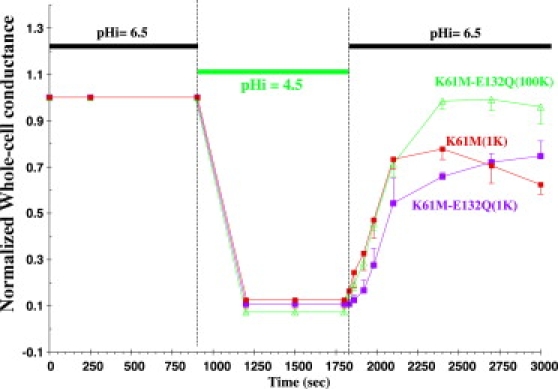
K61M-Kir1.1b mutation blocks inactivation in 1 mM external K (red line) and prevents E132Q inactivation in both 1 mM (purple line) and 100 mM (green line) external K. Internal oocyte pH was controlled with permeant acetate buffers. Lower internal pH was used because the K61M mutants have lower pKa values relative to wt Kir1.1b (see text). External K was maintained constant at either 1 mM or 100 mM throughout the protocol, and whole-cell conductances were normalized to their initial values.
The K61M mutation also protected E132Q from inactivation in both 1 mM K (Fig. 8, purple line) and 100 mM K (Fig. 8, green line). Similar results were obtained with K61M-R128Y, indicating that K61M could rescue even the highly unstable mutants (E132Q and R128Y) from inactivation.
V121T-Kir1.1b prevents inactivation
Since V121-Kir1.1b had been implicated in Kir1.1 inactivation (5,16), we applied the fast acidification-realkalization protocol to four mutants of V121-Kir1.1b. Three of the mutants, V121T, V121S, and V121Q, that had links between residue 121 and either the pore or the inner helix resisted inactivation (Fig. 9). However, the fourth such mutant, V121N, which like wt Kir1.1 had no polar linkages between 121 and the other helices, inactivated similarly to Kir1.1 (Fig. 9, dark blue and green lines).
Figure 9.
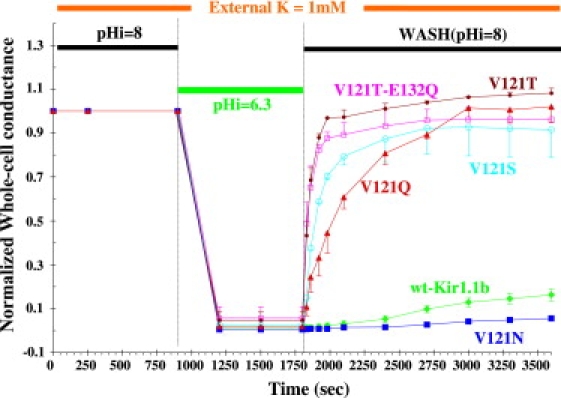
Mutation of the V121 residue to T (brown line), S (cyan line), or Q (red line) prevents inactivation in 1 mM external K, which occurred with both wt Kir1.1 (green line) and the V121N mutant (dark blue line). V121T-E132Q (magenta line) also prevented inactivation in 1 mM external K. As in previous protocols, oocyte pH was controlled with permeant acetate buffers. External K was maintained constant at 1 mM, and whole-cell conductances were normalized to their initial values.
Furthermore, V121T prevailed over the destabilizing effects of the E132Q salt-bridge mutation such that V121T-E132Q prevented inactivation in 1 mM external K (Fig. 9, magenta line), even though E132Q normally inactivated in 100 mM K (Fig. 7, green line). Steady-state pH titration of V121T-E132Q (Fig. 10) confirmed that V121T protected E132Q from inactivation. Inactivation block by V121T, V121S, and V121Q was not caused by altered pH gating, since the pKa's of V121T (6.6 ± 0.3), V121T-E132Q (6.7 ± 0.3), and wt Kir1.1b (6.7 ± 0.1) were not significantly different in 100 mM K.
Figure 10.
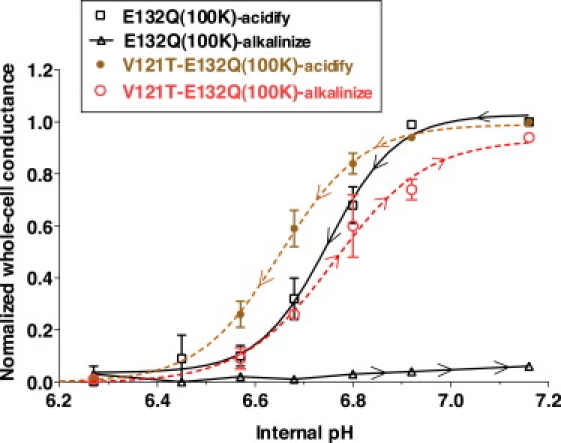
The V121T mutation prevents inactivation of E132Q in 100 mM K (red dashed line versus black line with triangles) without significantly altering the pKa during acidification (brown dashed line versus black line with squares). Internal oocyte pH was controlled with permeant acetate buffers. External K was maintained constant at 100 mM, and whole-cell conductances were normalized to their maximal values.
Discussion
Kir1.1 inactivation
In 100 mM external K, internal acidification closes Kir1.1 channels, and realkalization reopens them with a time constant of 92 ± 5 s. However, in 1 mM external K, the same protocol of acidification followed by realkalization fails to recover channel activity (Fig. 1), except at long times (3 h) or during elevation of external K. This type of low-pH, low-K inactivation is a characteristic of Kir1.1 (and possibly Kir4.1) that is related to, but distinct from, the pH gate at the bundle crossing of Kir1.1 (8–12).
A major assumption of these experiments was that changes in whole-cell conductance under constant K conditions are a good measure of aggregate channel gating. This assumption is based on observations in a previous study, where Kir1.1 pH gating occurred without a significant change in kinetics or single-channel conductance, until the moment of channel closure, when Po = 0 (36). Furthermore, there were no changes in Kir1.1 single-channel conductance or kinetics during recovery from inactivation, only an abrupt switch of single-channel open probability from Po = 0 to Po = 0.9.
We believe that inactivation is an intrinsic property of Kir1.1 channels and not the result of channel retrieval from the membrane. This was confirmed in separate experiments in which Kir1.1-eGFP (enhanced green fluorescent protein) constructs exhibited no significant change in membrane fluorescence after prolonged exposure to low K and low pH, indicating a constant Kir1.1 density at the oocyte surface. A further argument against the involvement of trafficking in inactivation is that whole-cell conductance rapidly increases after the final return to 100 mM external K (Fig. S2). For this to arise from channel insertion, a quiescent cytoplasmic pool of Kir1.1 channels would have to traffic to the plasma membrane in response to an external K signal, which is possible but highly unlikely.
Finally, we have elected not to focus on slight differences in the time course of whole-cell conductance recovery during realkalization (Figs. 2, 3, and 7–9, and Fig. S3 and Fig. S4), because the recovery times for noninactivated channels primarily reflect the buffering power and kinetics of oocyte realkalization rather than the molecular kinetics of the pH gate itself.
TPNQ protects Kir1.1 from inactivation
Pretreatment with the external blocker TPNQ protected Kir1.1 from low-K, low pH inactivation. TPNQ is a modified form of a small protein derived from honey bee venom that is known to bind to the external mouth of Kir1.1 (and GIRK1/4) and prevent ion flow between the external solution and the selectivity filter (40–42).
In our experiments, 500 nanomolar external TPNQ protected Kir1.1, E132D, and Q133E from inactivation during acidification in 1 mM external K (Fig. 2 and Table 1). Since the channels were exposed to low (1 mM) external K for 45 min before addition of TPNQ, TPNQ must be doing more than simply restricting K access to the filter. Moreover, TPNQ prevented inactivation regardless of whether it was applied before or after acid closure of the pH gate. This would imply that the locus of inactivation is not integral to the pH gate itself.
External application of TPNQ to inactivated channels rapidly reactivated Kir1.1 in direct proportion to the duration of TPNQ exposure (Fig. 3). The time course of inactivation reversal by TPNQ (Fig. 3) is consistent with the previously measured 39-s time constant for TPNQ washout (40). Since the Kir1.1 binding site for TPNQ is well established (40), we interpret these results as indicating that TPNQ can bind to closed, inactivated Kir1.1 channels and reverse inactivation, either by producing or preventing a conformational change at the outer mouth of the channel near the extracellular end of the selectivity filter.
Finally, prior application and removal of TPNQ had no effect on the pKa of the bundle-crossing pH gate. It is still possible that TPNQ transiently modifies the pH gate during binding; however, this could not be studied, since all measurable current is abolished while TPNQ is bound to the channel.
Pretreatment with external barium also prevented Kir1.1 inactivation, similar to pretreatment with TPNQ (Fig. 2). Since barium is known to bind strongly to the innermost S4 site of the selectivity filter (45), it could prevent inactivation by keeping the filter open under low K conditions. In summary, Kir1.1 inactivation was prevented by both the extracellular blocker TPNQ and the selectivity filter blocker Ba.
An intersubunit salt bridge stabilizes the Kir1.1 active state
Our homology model of Kir1.1 identified a putative intersubunit salt bridge (R128-E132) near the P-loop of Kir1.1b that might stabilize the open structure of the selectivity filter. We investigated six different mutations of this pair of amino acids. The mutations E132Q, E132N, and R128Y all yielded functional channels that inactivated in 100 mM as well as in 1 mM K (Table 1). These mutants could be protected from inactivation by elevating external K to 300 mM, consistent with stabilization of the open state by K.
TPNQ also protected these mutants from inactivation in 100 mM external K, but not in 1 mM external K (Table 1 and Fig. S3 and Fig. S4). This implies that the toxin is able to maintain the open state of the outer part of the pore in the face of either reduced external K or after elimination of the intersubunit salt bridge near the filter, but not when both of these destabilizing influences are present.
We do not believe that the increased sensitivity of E132Q, E132N, and R128Y to inactivation involves the pH gate at the bundle crossing (8,10), since none of these mutations significantly altered pH gating (Fig. 5). The simplest explanation is that E132Q, R128Y, and E132N destabilize the selectivity filter, allowing inactivation to occur in 100 mM external K as well as in 1 mM K.
K61M and V121T both rescue E132Q from inactivation
As shown in Fig. S5, A and B, the K61M-Kir1.1b mutation is believed to disrupt H-bonding between inner and outer transmembrane helices (2) and subsequently block the inactivation process. We confirmed that K61M prevented inactivation, not only in Kir1.1b, but in the bridge mutants E132Q and R128Y as well (Fig. 8). However, we were unable to identify an actual link between H-bonding at the bundle crossing and the selectivity filter, either experimentally or by inference from our homology model.
We also examined mutations at the V121 locus, which is 20 Å away from K61. The V121T mutation blocks both wt Kir1.1 inactivation and E132Q inactivation, even in 1 mM K (Fig. 9), similar to the K61M mutation. There are several possible mechanisms for the action of V121T. Since V121T increases the affinity of the channel for external Ba (15,46), V121T might increase the affinity of the S4 filter binding site for K. A greater K affinity at S4 might allow stabilization of the filter in low external K, thereby preventing inactivation (24,25,27).
However, we favor the alternative possibility that V121T, V121S, and V121Q prevent inactivation by stabilizing the selectivity filter via linkages between the cytoplasmic end of the filter and either the pore helix (V121T and V121S; see Fig. S6, A and B) or the inner helix (V121Q; see Fig. S6 C). The mutation V121N, which had no obvious links to either the pore helix or the inner TM helix (Fig. S6 D), did not protect against inactivation (Fig. 9).
Do structural changes at the selectivity filter gate mediate Kir1.1 inactivation?
The hallmark of Kir1.1 inactivation is a failure to recover channel activity after transient internal acidification in low external K. Based on the results of this study, we believe that Kir1.1 inactivation involves a structural change at or near the selectivity filter, precipitated by pH-gate closure in low extracellular K.
The K sensitivity of Kir1.1 inactivation is determined by the intersubunit link R128-E132-Kir1.1b, which is homologous to the link between R149-Kir3.1 and E159-Kir3.4 in the Kir3 heteromultimer (47). Comparison of channel pore sequences in Fig. S1 highlights this structural similarity. Functionally, the Kir3.1/Kir3.4(E145Q) mutant decreased both K/Rb and K/Na selectivity (47), whereas E132Q-Kir1.1b (homologous to Kir3.1/Kir3.4(E159Q) increased K/Rb selectivity and produced no apparent change in K/Na selectivity. This suggests that E132Q-Kir1.1b causes less of a distortion of the selectivity filter than E159Q-Kir3.4.
According to our homology model, R128-E132-Kir1.1b interacts with E118-Kir1.1b to form an intersubunit/intrasubunit triad (E118-R128-E132). The E118-R128 pair is homologous to the “bowstring” salt bridges E139-R149 in Kir3.1 and E145-R155 in Kir3.4 (47). However, since mutation of E118-Kir1.1b (corresponding to E71-KcsA, E139-Kir3.1, or E145-Kir3.4) was not tolerated by Kir1.1, we cannot assess the functional significance of mutating E118-Kir1.1b alone.
We also confirmed the importance of the lysine residue (K61-Kir1.1b) in the first transmembrane domain in the inactivation process. This is consistent with a recent interpretation that H-bonding between inner and outer helices at K61-Kir1.1b increases the likelihood of Kir1.1 inactivation (2). Conversely, polar linkages between V121T, V121S, or V121Q and the TM or pore helix stabilized the open state and prevented inactivation.
Our observations with TPNQ and barium implied that inactivation can be prevented either by stabilizing the outer mouth of the channel with a large toxin (TPNQ) or introducing a small blocker (Ba) at the S4 binding site of the selectivity filter. Furthermore, the protective effects of TPNQ and Ba occur independent of whether the pH gate is open or closed. This suggests that inactivation is not an integral part of the Kir1.1 pH gate.
The protective effect of TPNQ on Kir1.1 is analogous to results with kaliotoxin on the chimeric KcsA-Kv1.3 channel, in which kaliotoxin binding prevented the filter from undergoing a conformational change associated with C-type inactivation (48,49). In addition, the K dependence of Kir1.1 inactivation is consistent with previously described characteristics of C-type inactivation in Shaker, including stabilization of the open state by external K (18–27,50,51). Furthermore, recent experiments involving a combination of electrophysiology, EPR, x-ray crystallography, and molecular dynamics confirm that C-type inactivation in KcsA involves a distortion of the selectivity filter (31–33). These experiments also indicated an important role for E71-KcsA (homologous to E118-Kir1.1b) in filter stabilization.
In summary, we have used homology modeling to identify what to our knowledge is a new intersubunit, P-loop salt bridge R128-E132-Kir1.1b, that affects the K sensitivity of inactivation in the Kir1.1 inward rectifier. Results of mutational analysis and blocker (TPNQ and Ba) experiments are consistent with Kir inactivation involving conformational changes at the selectivity filter, analogous to those underlying C-type inactivation (22,23,25,27,28,52).
Supporting Material
Six figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01150-3.
Supporting Material
Acknowledgments
The authors thank Janice Robertson, PhD, for expert assistance with the Kir1.1b homology model.
This work was supported by National Institutes of Health grants DK46950 (H.S.) and DK27847 (L.G.P.).
References
- 1.Doi T., Fakler B., Schultz J.H., Schulte U., Brandle U. Extracellular K+ and intracellular pH allosterically regulate renal Kir1.1 channels. J. Biol. Chem. 1996;271:17261–17266. doi: 10.1074/jbc.271.29.17261. [DOI] [PubMed] [Google Scholar]
- 2.Rapedius M., Fowler P., Shang L., Sansom M., Tucker S. H bonding at the helix-bundle crossing controls gating in Kir potassium channels. Neuron. 2007;55:602–614. doi: 10.1016/j.neuron.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rapedius M., Haider S., Browne K., Shang L., Sansom M. Structural and functional analysis of the putative pH sensor in the Kir1.1 (ROMK) potassium channel. EMBO Rep. 2006;7:611–616. doi: 10.1038/sj.embor.7400678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sackin H., Nanazashvili M., Li H., Palmer L.G., Walters D.E. External K activation of Kir1.1 depends on the pH gate. Biophys. J. 2007;93:L14–L16. doi: 10.1529/biophysj.107.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sackin H., Syn S., Palmer L.G., Choe H., Walters D.E. Regulation of ROMK by extracellular cations. Biophys. J. 2001;80:683–697. doi: 10.1016/S0006-3495(01)76048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sackin H., Vasilyev A., Palmer L.G., Krambis M. Permeant cations and blockers modulate pH gating of ROMK channels. Biophys. J. 2003;84:910–921. doi: 10.1016/S0006-3495(03)74908-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulte U., Hahn H., Wiesinger H., Ruppersberg J.P., Fakler B. pH-dependent gating of ROMK (Kir1.1) channels involves conformational changes in both N and C termini. J. Biol. Chem. 1998;273:34575–34579. doi: 10.1074/jbc.273.51.34575. [DOI] [PubMed] [Google Scholar]
- 8.Kuo A., Gulbis J., Antcliff J., Rahman T., Lowe E. Crystal stucture of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- 9.Phillips L.R., Enkvetchakul D., Nichols C. Gating dependence of inner pore access in inward rectifier K channels. Neuron. 2003;37:953–962. doi: 10.1016/s0896-6273(03)00155-7. [DOI] [PubMed] [Google Scholar]
- 10.Sackin H., Nanazashvili M., Palmer L.G., Krambis M., Walters D.E. Structural locus of the pH gate in the Kir1.1 inward rectifier channel. Biophys. J. 2005;88:2597–2606. doi: 10.1529/biophysj.104.051474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadja R., Smadja K., Alagem N., Reuveny E. Coupling G-βγ-dependent activation to channel opening via pore elements in inwardly rectifying potassium channels. Neuron. 2001;29:669–680. doi: 10.1016/s0896-6273(01)00242-2. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y., Sackin H., Palmer L.G. Localization of the pH gate in Kir1.1 channels. Biophys. J. 2006;91:2901–2909. doi: 10.1529/biophysj.106.087700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flynn G.E., Zagotta W.N. Conformational changes in S6 coupled to the opening of cyclic nucleotide-gated channels. Neuron. 2001;30:689–698. doi: 10.1016/s0896-6273(01)00324-5. [DOI] [PubMed] [Google Scholar]
- 14.Xiao J., Zhen X.G., Yang J. Localization of PIP2 activation gate in inward rectifier K channels. Nat. Neurosci. 2003;6:811–818. doi: 10.1038/nn1090. [DOI] [PubMed] [Google Scholar]
- 15.Dahlmann A., Li M., Gao Z., McGarrigle D., Sackin H. Regulation of Kir channels by intracellular pH and extracellular K: mechanisms of coupling. J. Gen. Physiol. 2004;123:441–454. doi: 10.1085/jgp.200308989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulte U., Weidemann S., Ludwig J., Ruppersberg J., Fakler B. K-dependent gating of Kir1.1 channels is linked to pH gating through a conformational change in the pore. J. Physiol. 2001;534:49–58. doi: 10.1111/j.1469-7793.2001.t01-1-00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armstrong C.M. Ionic pores, gates, and gating currents. Q. Rev. Biophys. 1975;7:179–210. doi: 10.1017/s0033583500001402. [DOI] [PubMed] [Google Scholar]
- 18.Baukrowitz T., Yellen G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- 19.Baukrowitz T., Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K channel. Science. 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- 20.Choi K., Aldrich R.W., Yellen G. Tetraethylammonium blockade distinguishes two inactivating mechanisms in voltage-activated K channels. Proc. Natl. Acad. Sci. USA. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoshi T., Zagotta W., Aldrich R.W. Two types of inactivation in Shaker K channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 22.Kiss L., LoTurco J., Korn S. Contribution of the selectivity filter to inactivation in potassium channels. Biophys. J. 1999;76:253–263. doi: 10.1016/S0006-3495(99)77194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurata H., Fedida D. A structural interpretation of voltage-gated potassiuum channel inactivation. Prog. Biophys. Mol. Biol. 2006;92:185–208. doi: 10.1016/j.pbiomolbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y., Jurman M.E., Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- 25.Lopez-Barneo J., Hoshi T., Heinemann S., Aldrich R.W. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- 26.Molina A., Castellano A.G., Lopez-Barneo J. Pore mutations in Shaker K channels distinguish between the sites of tetraethylammonium blockade and C-type inactivation. J. Physiol. 1997;499:361–367. doi: 10.1113/jphysiol.1997.sp021933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molina A., Ortega-Saenz P., Lopez-Barneo J. Pore mutations alter closing and opening kinetics in Shaker K channels. J. Physiol. 1998;509:327–337. doi: 10.1111/j.1469-7793.1998.327bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yellen G. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- 29.Larsson H., Elinder F. A conserved glutamate is important for slow inactivation in K channels. Neuron. 2000;27:573–583. doi: 10.1016/s0896-6273(00)00067-2. [DOI] [PubMed] [Google Scholar]
- 30.Rasmusson R., Morales M., Wang S., Liu S., Campbell D. Inactivation of voltage-gated cardiac K channels. Circ. Res. 1998;82:739–750. doi: 10.1161/01.res.82.7.739. [DOI] [PubMed] [Google Scholar]
- 31.Cordero-Morales J., Cuello L., Perozo E. Voltage-dependent gating at the KcsA selectivity filter. Nat. Struct. Biol. 2006;13:319–322. doi: 10.1038/nsmb1070. [DOI] [PubMed] [Google Scholar]
- 32.Cordero-Morales J., Cuello L., Zhao Y., Jogini V., Cortes D.M. Molecular determinants of gating at the potassium channel selectivity filter. Nat. Struct. Mol. Biol. 2006;13:311–318. doi: 10.1038/nsmb1069. [DOI] [PubMed] [Google Scholar]
- 33.Cordero-Morales J., Jogini V., Lewis A., Vasquez V., Cortes D.M. Molecular driving forces determining potassium channel slow inactivation. Nat. Struct. Biol. 2007;14:1062–1069. doi: 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- 34.Yang J., Yu M., Jan Y.N., Jan L.Y. Stabilization of ion selectivity filter by pore loop ion pairs in an inwardly rectifying potassium channel. Proc. Natl. Acad. Sci. USA. 1997;94:1568–1572. doi: 10.1073/pnas.94.4.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shieh R. Mechanisms for the time-dependent decay of inward currents through cloned Kir2.1 channels expressed in Xenopus oocytes. J. Physiol. 2000;526:241–252. doi: 10.1111/j.1469-7793.2000.00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choe H., Zhou H., Palmer L.G., Sackin H. A conserved cytoplasmic region of ROMK modulates pH sensitivity, conductance, and gating. Am. J. Physiol. 1997;273:F516–F529. doi: 10.1152/ajprenal.1997.273.4.F516. [DOI] [PubMed] [Google Scholar]
- 37.Leipziger J., MacGregor G., Cooper G., Xu J., Hebert S. PKA site mutations of ROMK2 channels shift the pH dependence to more alkaline values. Am. J. Physiol. 2000;279:F919–F926. doi: 10.1152/ajprenal.2000.279.5.F919. (renal) [DOI] [PubMed] [Google Scholar]
- 38.Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 39.MacKerell A.D., Bashford D., Bellott M., Dunbrack R.L., Evanseck J.D. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 40.Jin W., Klem A., Lewis J., Lu Z. Mechanisms of inward-rectifier K channel inhibition by Tertiapin-Q. Biochemistry. 1999;38:14294–14301. doi: 10.1021/bi991206j. [DOI] [PubMed] [Google Scholar]
- 41.Jin W., Lu Z. A novel high-affinity inhibitor for inward-rectifier K channels. Biochemistry. 1998;37:13291–13299. doi: 10.1021/bi981178p. [DOI] [PubMed] [Google Scholar]
- 42.Jin W., Lu Z. Synthesis of a stable form of tertiapin: a high-affinity inhibitor for inward-rectifier K channels. Biochemistry. 1999;38:14286–14293. doi: 10.1021/bi991205r. [DOI] [PubMed] [Google Scholar]
- 43.Ramu Y., Klem A., Lu Z. Titration of tertiapin-Q inhibition of ROMK1 channels by extracellular protons. Biochemistry. 2001;40:3601–3605. doi: 10.1021/bi002584n. [DOI] [PubMed] [Google Scholar]
- 44.Sackin H., Nanazashvili M., Palmer L.G., Li H. Role of conserved glycines in pH gating of Kir1.1 (ROMK) Biophys. J. 2006;90:3582–3589. doi: 10.1529/biophysj.105.076653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang Y., MacKinnon R. The barium site in a potassium channel by X-ray crystallography. J. Gen. Physiol. 2000;115:269–272. doi: 10.1085/jgp.115.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou H., Chepilko S., Schutt W., Choe H., Palmer L.G. Mutations in the pore region of ROMK enhance Ba2+ block. Am. J. Physiol. 1996;271:C1949–C1956. doi: 10.1152/ajpcell.1996.271.6.C1949. [DOI] [PubMed] [Google Scholar]
- 47.Dibb K., Rose T., Makary S., Claydon T., Enkvetchakul D. Molecular basis of ion selectivity, block, and rectification of the inward rectifier Kir3.1/Kir3.4 K channel. J. Biol. Chem. 2003;278:49537–49548. doi: 10.1074/jbc.M307723200. [DOI] [PubMed] [Google Scholar]
- 48.Lange A., Giller K., Hornig S., Marin-Eauclaire M., Pongs O. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature. 2006;440:959–962. doi: 10.1038/nature04649. [DOI] [PubMed] [Google Scholar]
- 49.Zachariae U., Schneider R., Velisetty P., Lange A., Seeliger D. The molecular mechanism of toxin-induced conformational changes in a potassium channel: relation to C-type inactivation. Structure. 2008;16:747–754. doi: 10.1016/j.str.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 50.Loboda A., Melishchuk A., Armstrong C. Dilated and defunct K channels in the absence of K. Biophys. J. 2001;80:2704–2714. doi: 10.1016/S0006-3495(01)76239-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swenson R.P., Armstrong C.M. K channels close more slowly in the presence of external K and Rb. Nature. 1981;291:427–429. doi: 10.1038/291427a0. [DOI] [PubMed] [Google Scholar]
- 52.Gomez-Lagunas F. Shaker B K conductance in Na solutions lacking K ions: a remarkably stable non-conducting state produced by membrane depolarization. J. Physiol. 1997;499:3–15. doi: 10.1113/jphysiol.1997.sp021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.