

Abstract
The involvement of matrix metalloproteinase-9 (MMP-9) activities in the development of abnormal water diffusion in the brain after cardiac arrest is not fully understood. We used magnetic resonance imaging to determine the correlation between MMP-9 activity and the mechanism of abnormal water diffusion after global cerebral ischemia (GCI)-induced brain damage in C57black6 mice. We induced GCI in mice by occluding both carotid arteries for 60 min, then allowing reperfusion. We labeled a short DNA that targets mmp-9 mRNA activity [phosphorothioate-modified oligodeoxynucleotide (sODN)-mmp9] or a control probe without intracellular target (sODN-Ran) with iron-based MR contrast agent [superparamagnetic iron oxide nanoparticle (SPION)-mmp9 or SPION-Ran] or fluorescein isothiocyanate (FITC)-sODN-mmp9 or FITC-sODN-Ran; we then delivered these probes by intracerebroventricular infusion or intraperitoneal injection within 3 h of reperfusion. At low dose (120 pmol/kg) the SPION-mmp9 probe was retained at significant levels in the striatum and cortex of living brains 10 h after GCI. Probe retention was validated by similar elevation of mmp-9 mRNA and antigens in postmortem samples taken from regions that exhibited GCI-induced hyperintensity in diffusion-weighted imaging, and a significant reduction in apparent diffusion coefficient (rADC, p = 0.0006, n = 12). At a higher dose (120 nmol/kg), the FITC-sODN-mmp9 probe revealed significant knockdown of MMP-9 activity, per zymography, and a reversal of striatal rADC (p = 0.004, n = 6). These observations were not duplicated in the control group. We conclude that expression of mmp-9 mRNA is associated with abnormal ADC after GCI.
Introduction
Disruption of the blood–brain barrier (BBB) contributes the formation of vasogenic edema in the CNS after stroke, traumatic brain injury, multiple sclerosis, and certain other neurological disorders. Our understanding of abnormal water diffusion as measured by hyperintense diffusion-weighted imaging (hDWI) comes from studies involving a stroke model (Moseley et al., 1993; Montaner et al., 2003). Increasingly, however, reports of BBB leakage, cell death, and MMP-9 activities from studies using global cerebral ischemia (GCI) or cardiac arrest models have also emerged (Mossakowski et al., 1994; Li et al., 2005; Lee et al., 2007). It is known that matrix metalloproteinases (MMPs), a family of proteolytic enzymes, degrade all components of the extracellular matrix within 24 h of the onset of cerebral ischemia, when they are activated from pro-MMP-9 (gelatinase B, EC.3.4.24.35) or pro-MMP-2 after oxidative stress or cerebral ischemia. Activated MMP-9 (88 kDa) remains elevated in neurons for as long as 20 h, with activation peaking at 6 h after incident onset (Leone et al., 2000; Gu et al., 2002; Zozulya et al., 2008). Gene knockdown using a short interfering RNA with sequence antisense to the mRNA has been effectively used to inhibit MMP-9 expression in cell culture conditions (Kao et al., 2003; W. Liu et al., 2006) and to inhibit invasion by glioma and angiogenesis (Girolamo et al., 2004; Gondi et al., 2004). The results of these studies therefore indicate that this technique may be of value for validating gene function in neurological disorders (Horstmann et al., 2003).
Methods to investigate the mechanisms that underlie gene activities, especially at the level of the DNA or RNA, have typically depended on tissue biopsy of affected organs or postmortem testing. Because these conventional assays require the removal of tissue, the ability to assess gene activation to evaluate the efficacy of potential therapies, for instance, gene or stem cell approaches, has been significantly limited. As an alternative, we have developed several brain probes that target cerebral gene transcripts and can be detected in contrast-enhanced magnetic resonance imaging (MRI). As part of a long-term goal to explore in greater detail the feasibility of MRI technologies to study gene action at the level of mRNA in live animal brains, we have designed a probe that targets mmp-9 mRNA. This work is driven by the hypothesis that MMP-9 activities mediate cerebral abnormal water diffusion after GCI.
The brain probe we have developed comprises short polymers of oligodeoxynucleotide (ODN), 16–26 bases (5∼8 kDa) in length, with sequences complementary to an mRNA transcript (P. K. Liu et al., 2008). To make it biologically active, the backbone of the ODN is phosphorothioate-modified such that it develops resistance to nucleases in the cytoplasm. The Watson–Crick base pairing mechanism mediates the targeting or binding of ODN to mRNA, allowing the brain probe to report gene activities when used at a low dose and with proper labels. The same base pairing mechanism also inhibits translation at high dose (Wahlestedt et al., 1993; Wang et al., 2003). In this communication, we tested the hypothesis that phosphorothioate-modified ODN (sODN)-mmp9 could delineate the mechanism of hDWI in living brains.
Materials and Methods
Brain damage in living brains using a global cerebral ischemia model
All of the procedures and animal care practices we used adhered strictly to Association for Assessment and Accreditation of Laboratory Animals, Society for Neuroscience, and institutional guidelines for experimental animal health, safety, and comfort. Male C57black6 mice (n = 35, 23 ± 3 g, Taconic Farm) were anesthetized with ketamine (80 mg/kg, i.p.) and xylazine (12 mg/kg, i.p.). The mice were kept at 36 ± 1°C. Cerebral ischemia was induced by 60 min of bilateral carotid artery occlusion (BCAO) and MRI acquisition for DWI/apparent diffusion coefficient (ADC) and R2* (1/T2*) maps was performed as described (C. H. Liu et al., 2007b,c, 2008). After GCI, the mice were given 5% dextrose-enriched Ringer's solution (1 ml, s.c.), daily, for 1 week.
We conducted diffusion-weighted MR imaging on a total of 35 mice at various time points after reperfusion, from 1 h and up to 6 d after BCAO, using an MRI protocol previously described (C. H. Liu et al., 2007a). We measured (bilaterally) the cortical and striatal volume of metabolic disturbance (VMD) in each animal. For each animal, MRI scans were separated by at least 7 h; that is, no scan was conducted on the same animal less than 7 h after the previous, to avoid undue exposure to anesthesia during the recovery period. In animals that received fluorescein isothiocyanate (FITC)-sODN, we acquired DWI and calculated ADC changes. In animals that received superparamagnetic iron oxide nanoparticle (SPION)-sODN by intracerebroventricular infusion (C. H. Liu et al., 2007a,b,c), however, we acquired DWI only and did not analyze ADC changes because the uptake of SPION causes transverse relaxation time (T2) signal reduction, which interferes with ADC and VMD calculations.
Protocols for in vivo MRI at 9.4T
Iron assessment and R2* imaging.
The total cumulative scan time for each animal was ∼30 min (0.5 h). Animals were anesthetized by gas anesthesia (2% isofluorane in O2/air mixture) before and during MRI. The MRI protocols included: (1) RARE tripilot acquisition sequence for localization (2) Serial, two-dimensional gradient echo fast imaging (GEFI) with repetition time (TR)/echo time (TE) = 500/3, 4, 6, 8, 10 ms, 117 × 117 μm2, 20 0.5 mm contiguous slices, flip angle = 30, number of average = 2.
Gadolinium-enhanced MRI.
For detection of BBB leakage, animals were scanned before injection of gadolinium (Gd)-DTPA (Magnevist; Shering), using a T1-weighted three-dimensional spin echo protocol (TR/TE = 400/11 ms, 120 × 120 × 500 μm2, number of average = 4). Gd-DTPA was administered to the jugular vein (0.1 mmol/kg, i.v.) and the mouse was imaged within 10 min. Extensive, enhanced T1 signal compared with a pre-Gd scan was considered evidence of leakage; that is, an increase in the T1 signal identified leaky areas of the brain.
Data processing for MR image alignment, T2* (or R2*) map calculation, and region of interest analysis.
For voxelwise and region-of-interest comparison, images were automatically and manually aligned using 12 degrees of freedom; 3 each for rotation, translation, inflation, and skew. We fine-tuned the image alignment by visual comparison to the template images, focusing on obvious landmarks such as the corpus callosum and outlines of the ventricles. We constructed R2* maps from the aligned images by pixelwise linear fitting for sets of images with the same TR and incremental TEs, as determined by the equation M = M0 × exp (−TE/T2*). The presence of SPION in the tissue is reflected by elevated R2* (or reduced T2*) (Boxerman et al., 1995). We constructed R2* maps using an in-house software. We followed the work of Paxinos and Franklin (2001) to outline the regions of interest (ROIs). We assumed that the cell population and its organization within a particular ROI remained largely unchanged during the experimental timeframe of 10 h, such that we could compare the ROI analysis of brain regions with similar structural composition between the experimental and control animal groups on the day of the experiment. As part of the ROI analysis, we extracted R2* values (MRVision) from the brain regions depicted in Figure 2 and statistically computed the mean and SEM from the averaged values in each group of animals (see Statistical analysis). After the MRI scanning protocol, we excised postmortem brain samples for immunohistochemical analysis of probe uptake or antigen expression.
Figure 2.

MRI of MMP-9 gene activity in vivo after GCI. A, MRI protocol. Phosphorothioate-modified ODN with randomized sequence (sODN-Ran) or antisense sequence to mmp-9 mRNA (sODN-mmp9) was linked to SPION-NA and delivered at the time indicated according to the experimental protocol; we intracerebroventricularly infused SPION-mmp9 or SPION-Ran (40 μg Fe/kg) at 3 h of reperfusion, and acquired MRI scans for R2* maps (for SPION retention) and DWI. B, R2* subtraction maps of SPION-mmp9: individual R2* maps of SPION-mmp9 minus the averaged SPION-Ran maps obtained from three mice in this study (multislice images for each animal displayed in rows). C shows group average R2* maps obtained from animals infused with SPION-mmp9 (top row) and SPION-Ran (bottom row). D depicts outlines of ROIs for statistical analyses performed according to the work of Paxinos and Franklin (2001). All are rostral view. E, Bar graphs from ROI analysis that quantitatively compares the difference between SPION retention at 10 h of reperfusion after GCI (60 min) in animals that received SPION-mmp9 (open bars, N = 3) and SPION-Ran (closed bars, N = 3), *p < 0.05 by t test.
Immunohistochemistry of MMP-9 antigen.
Immunohistochemical staining of vascular endothelial cells by Cy3-griffonia simplicifolia lectin I, cell nuclei by Hoechst and mouse MMP-9 antigen by rabbit polyclonal antibodies against MMP-9 (ab38898, AbCam) was followed by FITC-anti-rabbit IgG for the secondary antibodies, as previously described (C. H. Liu et al., 2007a; You et al., 2007).
Preparation of biotinylated sODN and SPION-NeutrAvidin.
Activated SPION-NeutrAvidin (SPION-NA) was stored in an amber-colored bottle at 4°C, at a concentration of 4 mg of iron per milliliter of sodium citrate buffer (C. H. Liu, 2008). We synthesized 5′-biotin-labeled antisense sODN: sODN-mmp9 (5′ TAC ATG AGC GCT TCC GGC AC 3′) to mmp-9 mRNA (accession no. AY902320) of the mouse (Zechel et al., 2002); the sequence of sODN-Ran has been previously reported (C. H. Liu et al., 2008). Single-stranded ODNs were protected from nonspecific nuclease by phosphorothioate modification of all nucleotide backbones. All sODN were purified using PAGE and delivered by intracerebroventricular infusion (C. H. Liu et al., 2007b), and MMP-9 zymography was performed as described (Gursoy-Ozdemir et al., 2004). The R2* maps were constructed from five 0.5 mm MR slices (+1.3 to −1.2 mm, bregma) acquired in each animal.
Statistical analysis.
We performed a power calculation, using the average and SE of the data obtained from the first set of animals, to calculate the number of animals required in each group to achieve 90% power for a p value of 0.02; for this calculation we used in-house software [W. W. La Morte, Sample Size Calculation in Animal Research; Boston University Medical Center, Institutional Animal Care and Use Committee Newsletter (http://www.stat.uiowa.edu/∼rlenth/Power/)]. Unless the power calculation called for a greater number of animals, each study was repeated with three animals in each treatment group. If the minimum number of animals was >10, we revised both our hypothesis and our protocol. We computed the mean and SEM from the averages in each group of animals, and compared the statistical significance of these values using a t test (one-tailed, type II or equal variant) or contingency tests (Fisher's exact test for p values, GraphPad Prism IV, GraphPad Software). A p value of <0.05 was statistically significant.
Results
GCI in C57black6 mice leads to oxidative stress (Huang et al., 2000), a condition causing metabolic disturbance that manifests as decreased intracellular pH and adenosine triphosphate concentration as well as reactive species of oxygen and nitrogen. Brain damage induced by the GCI model, most often, neuronal death by DNA fragmentation within a few days after the procedure (Barone et al., 1993; P. K. Liu et al., 1996; Fujii et al., 1997; C. H. Liu et al., 2007a), seldom produces necrosis and has no ischemic core or penumbra at the damage site. However, increased levels of intracellular calcium and glutamate after GCI can cause abnormal water diffusion in the brain, which can be detected by MR as an hDWI. We observed that cortical and striatal hDWI was accompanied by a reduction in apparent diffusion coefficient (rADC), to less than two SEM below the average ADC of the normal brain (Fig. 1A, top panels). This model also produces leakage of the BBB in the cortex and striatum, as shown by Evans blue extravasation (C. H. Liu et al., 2008); the BBB in the striatum remained leaky near the location where we measured abnormal water diffusion for 2–3 months (Fig. 1A, bottom panels). We hypothesized that abnormal water diffusion in striatal regions showing significant rADC is a sign of metabolic disturbance and could serve as the initial parameter for detection of tissue damage. To quantitatively compare the severity of brain damage after GCI, we attempted to measure the volume of abnormal water diffusion from MRI. To do so, we outlined areas of the brain that exhibited ADC values below the threshold ADC, that is, two SEM below the average ADC in normal mice, or having a ADC at <2.5% of the distribution curve (one tail) of normal ADC (C. H. Liu et al., 2007a). Figure 1A shows an example of striatal ADC with highlighted regions of metabolic disturbance from two representative animals. After identifying these areas of low ADC, we determined the VMD by adding the product of the area below the threshold ADC to the thickness of the MRI slice (1 mm). We calculated the average VMD by incorporating measurements acquired from at least four mice at each time point, which thus allowed us to define a temporal VMD profile in the cortex and striatum (Fig. 1B,C). The hippocampus, although a region vulnerable to cell death (P. K. Liu et al., 1996; Huang et al., 2000), did show small VMD in one third of mice (n = 12) examined for rADC; vulnerability to cell death in the hippocampus after GCI is not always exhibited as hDWI/rADC (C. H. Liu et al., 2007a). We observed that, at all time points, VMD was highest immediately after reperfusion. This peak was followed by a gradual drop in VMD between 3–8 h of reperfusion (Fig. 1B,C). In the striatum, VMD rose gradually after 9 h, plateaued between 9 and 48 h, then dropped again in volume beyond 2 d. We observed the same peak in the cortex, but cortical VMD peaked anywhere between 9 h and 5 d after reperfusion. We also observed that brain regions with significant VMD after 60 min BCAO and reperfusion could appear either unilaterally (n = 2, or 25%) or bilaterally (n = 6, or 75%) in a single series of experiments (Fig. 1A). Indeed, Gd-enhanced MRI suggests that the hemisphere exhibiting brain damage matches that where VMD is noted in the striatum several weeks later (Fig. 1A). Because ventriculomegaly and tissue damage in the striatum developed after hDWI, we hypothesized that mmp-9 mRNA expression differed in the cortex and the striatum.
Figure 1.
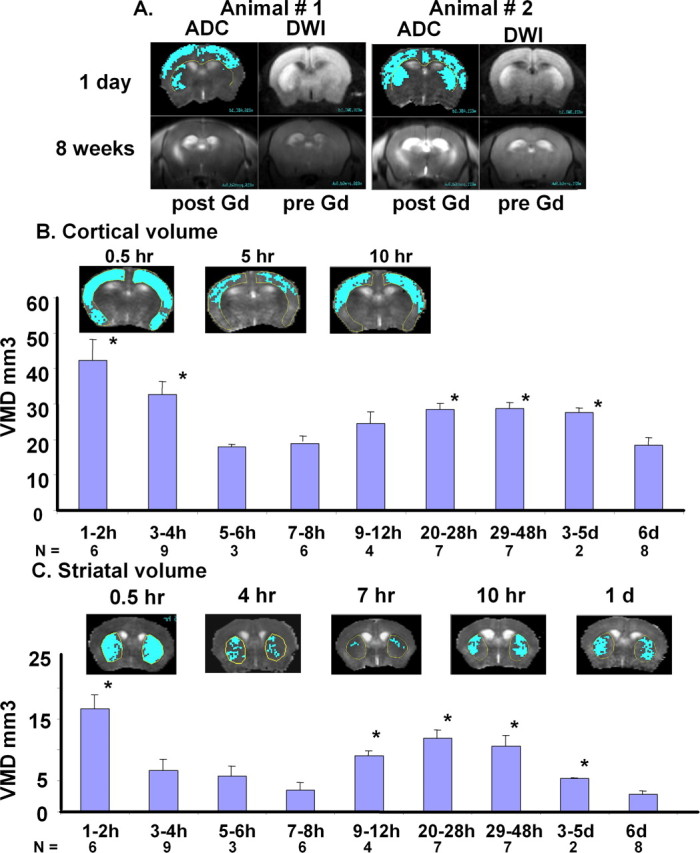
A, A rostral view of regions of metabolic disturbance as depicted by DWI or ADC in two representative mice at 1 d (first row), compared with Gd-enhanced MRI acquired at 8 weeks (second row). ADC maps with highlighted areas correspond to regions of significant metabolic disturbance (ADC drop below threshold) and predict disruption of the BBB at 8 weeks (post- and pre-Gd image of Gd-MRI, second rows). Our previous studies have shown that BBB leakage varied (25% of the animals during the first 3 h of reperfusion after 60 min BCAO using Gd-DTPA), but all animals developed BBB leakage by 4–12 weeks (Liu et al., 2008). B, C, Temporal changes in the VMD of the cortex and striatum. Cerebral metabolic disturbance is defined as a region with ADC below the threshold ADC of normal mice (2 SEM below average ADC in normal mice: striatum threshold was 4.69 × 10−4 mm2/s; cortex threshold was 5.41 × 10−4 mm2/s). The areas of metabolic disturbance were obtained from multiple MR slices (representative slide shown above the B for cortex and C for striatum), and VMD was determined by summation of these areas in the corresponding brain regions and multiplying by the thickness of each MRI slice (1 mm). The VMD value at each time point was compared with the VMD at the end point at 6 d by t test (*p < 0.03). The corresponding number of animals (N) used at each time period is included at the bottom of the bar graph. We did observe 4 of 12 of the mice had significant rADC below the threshold (supplemental material, available at www.jneurosci.org) in the hippocampus; we did not include them in the volume calculation for metabolic disturbance.
What did the SPION-mmp9 probe and MRI report after GCI in mice?
We aimed to compare expression of the mmp-9 mRNA transcript in the cortex and striatum in living brains using MRI. We conjugated sODN-mmp9 (and sODN-Ran as a control) to SPION-NA via biotin and NA linkage (C. H. Liu et al., 2007a,b,c). The SPION-mmp9 and SPION-Ran probes (Fe = 40 μg or SPION = 120 pmol per kg) were delivered by intracerebroventricular infusion to the left lateral cerebral ventricle of the brain (C. H. Liu et al., 2007c) in two groups of mice after GCI (Fig. 2A); we chose the intracerebroventricular route of delivery to avoid variations in probe uptake caused by BBB disruption. We computed individual subtraction maps (Fig. 2B) from MR images acquired 10 h after reperfusion in the experimental and control groups to assess SPION probe retention. We observed localized elevation of R2* values with stronger retention in the striatal regions in each of three mice. Group averaged R2* maps were computed (as presented in Fig. 2C) and, again, showed greater retention in the striatum after SPION-mmp9, indicating that probe concentration was greater in ischemic brains that received SPION-mmp9 than in those that received SPION-Ran (Fig. 2C, corresponding R2* scale to the right).
Although the probe was infused into the left ventricle, we found no significant difference between the R2* values in the left and right hemispheres of individual animals at 10 h of reperfusion, whether in the SPION-mmp9 (between cortices, p = 0.09; between striata, p = 0.18) or the SPION-Ran (p > 0.25 in both cortex and striatum) group. Statistical analysis of regional SPION retention showed that retention of SPION-mmp9, compared with SPION-Ran, was significantly elevated in both the striatum and cortex at 10 h of reperfusion (Fig. 2E). Still, the elevation of SPION-mmp9 retention was greater in the striatum than in the cortex. SPION-Ran retention was not significantly different from baseline measures in either sham-operated (SO) or normal animals. SPION-mmp9 retention in GCI animals showed that striatal mmp-9 mRNA expression was twofold greater than that of the control group, and the elevation in cortical mmp-9 mRNA was less than twofold in live brains. The data prompted us to determine whether MMP-9 activity and sODN-mmp9 targeted mmp9 mRNA were in fact elevated after GCI. We aimed to determine whether or not changing MMP-9 expression would alter rADC or VMD in the striatum.
GCI in mice induces mmp-9 mRNA expression in regions with hDWI
In this ex vivo hybridization assay, we delivered FITC-sODN-mmp9 or FITC-sODN-Ran by intraperitoneal injection 1 h after GCI. DWI was conducted the next day, and postmortem samples were collected immediately after scanning. We chose this protocol because the transient BBB leakage that occurs immediately after GCI allows cross-over of small molecules such as Evans Blue, GD-DTPA, or FITC-sODN (5–8 kDa) (C. H. Liu et al., 2008). We noted the presence of FITC-sODN-mmp9 in three of four GCI-treated mice given FITC-sODN-mmp9. Two representative mice with bilateral and unilateral hDWI/rADC are shown in Figure 3, A and C, respectively. Histological examination for the presence of FITC-sODN was accomplished using the hippocampus as the reference point. Figure 3B shows the histological correlation between hDWI/rADC and the presence of FITC-sODN-mmp9/mRNA hybrid in both hemispheres of the same brain. We observed retention of FITC-ODN-mmp9 in the parenchyma surrounding the microvessels (red), and extends to cells at least 50 μm away from the vessels (Fig. 3B); the data suggested that leakage started in the vascular endothelia. In another mouse (Fig. 3C,D), FITC-sODN-mmp9 was present in the cytoplasm of granule cells in the subgranular zone of the dentate gyrus, where MRI detected hDWI/rADC (Fig. 3D, square insert). However, we observed no mmp-9 mRNA expression in the region where DWI/ADC appeared normal (Fig. 3D, right panel). Similarly, we did not observe probe retention in any of the four GCI mice administered FITC-sODN-Ran (Fig. 3E), or in the SO group (Fig. 3F), although the SO group shows similar green background (Fig. 3F) as in the hemisphere without hDWI (Fig. 3D, right panel). These findings are consistent with the results of the SPION-Ran probe and MRI shown in Figure 2. This is true although the mice treated with GCI and FITC-sODN-Ran did show hDWI with elevated MMP-9 activity (Fig. 3G,H). The four mice that underwent SO (Fig. 3I) showed no MMP-9 expression. In addition, we observed a loss of Hoechst staining in the dentate gyrus of the hippocampus in one mouse (Fig. 3D, circle in the left panel). The data suggest that a loss of nuclear DNA 1 d after GCI may signal apoptotic cell death in the dentate gyrus, where hDWI/rADC was detected in vivo. We concluded that BBB leakage allowed translocation of FITC-sODN from the microvasculature to the cerebral parenchyma, and that the sODN sequences had intracellular specificity to discriminate mRNA targets; the exception, however, were those mice that received FITC-sODN-Ran, the control probe with no intracellular target. Having observed that mmp-9 mRNA levels were elevated in hDWI regions after GCI, we sought evidence that MMP-9 antigen would also be elevated in similar hDWI regions.
Figure 3.

A modified ex vivo hybridization assay to determine the presence of intracellular mRNA in a total of 12 mice (Cui et al., 1999). The protocol is the same as in Figure 2A, except FITC-sODN was delivered. We delivered FITC-labeled sODN (6 nmol/kg, i.p.) to the mice at 1 h of reperfusion, because the BBB is open in 75% of GCI-treated mice (Liu et al., 2008). One day after GCI, we obtained DWI and prepared postmortem samples from the region where hDWI is identified in the MRI (A, C, E, G). The sODN-mmp9 sequence has been shown to bind to mmp-9 mRNA of the mouse (Zechel et al., 2002). Samples were counterstained with Hoechst for nuclei and Cy3-griffonia simplicifolia lectin I for the vessel endothelia (B, D–F). The presence of enhanced FITC stain indicates the location of probe retention. H and I show detection of MMP-9 antigen (red) and endothelial cells (green), using the same antibodies as in Figure 4 (labeling was reversed to show difference from hybrids). Arrows show the regions where photography was taken. CPU, Caudate putamen.
GCI in mice induces MMP-9 activity in regions with hDWI
Based on the observation above, we hypothesized that MMP-9 protein was expressed in the mouse brain in response to GCI insult. Knowing that VMD plateaus and is significant in both the cortex and striatum at 1 d, we acquired DWI 1 d after GCI (n = 5) and SO (n = 2), then obtained postmortem brain samples for immunohistochemistry immediately after MRI data acquisition was completed (Fig 4A). Samples were stained with rabbit anti-MMP-9 antibodies, costained with Hoechst for nuclei (purple), and Cy3-griffonia simplicifolia lectin I for endothelial cells (red). As Figure 4 shows, the mice did exhibit MMP-9 immunoreactivity (green) in the brain after GCI (Fig 4C–F). Regions without hDWI in the same mice showed less or no expression of MMP-9 protein. No significant MMP-9 activities were observed in tissue taken from GCI mice without antibodies (Fig. 4G) or in SO mice (data not shown). We observed that MMP-9 antigen is localized in the cytoplasm and around the nuclei; both the neurons and endothelia expressed MMP-9 (Fig. 4H). The results of the immunohistochemistry assay of MMP-9 antigen were consistent with mmp-9 mRNA in vivo hybridization, as measured by MRI.
Figure 4.

Immunohistochemistry of MMP-9 antigen in C57black6 after 60 min GCI (n = 5). Protocol is shown in A. Regional metabolic disturbance was identified by hDWI 1 d after GCI (rostral view of one animal is shown in B). Postmortem brain tissue sections were obtained after MRI and assayed for MMP-9 antigen using rabbit polyclonal anti-MMP-9 IgG followed by secondary FITC-anti-rabbit IgG, and each sample was counter-stained for endothelia using Cy-3-griffonia simplicifolia lectin I and Hoechst stain for nuclei (C–F). The same GCI mouse without primary antibodies is shown (G). MMP-9 antigen is present in endothelia (H). No detection of anti-MMP-9 IgG is shown when the antibodies are deleted (I).
Gene targeting of mmp-9 mRNA by translation blocking
Our gene targeting strategy, which uses a short nucleic acid complementary to a specific region of the target mRNA, can correlate gene activities and their functions by hybridizing to the target mRNA (P. K. Liu et al., 1994; C. H. Liu et al., 2007c), either subjecting the mRNA to degradation or by blocking protein translation (gene expression/activity). Either of these mechanisms will reduce translation of the target mRNA and eventually reduce the activities of the target gene (Wahlestedt et al., 1993; Cui et al., 1999). We hypothesized that sODN-mmp9, after hybridization to mmp-9 mRNA (Zechel et al., 2002), would inhibit MMP-9 activities. We sought to test this hypothesis by infusing sODN-mmp9 as a short inhibitory DNA (siDNA) at high dose (120 nmol/kg) after GCI. We also included sODN-Ran as a control. To bypass the BBB and establish effective concentration, we used an intracerebroventricular route of delivery and sought to quantitatively evaluate MMP-9 activities 10 h after GCI using zymography.
For these experiments, we included additional control groups: mice that underwent either SO or GCI but received no intracerebroventricular infusion. We collected tissue samples for MMP-9 zymography from the brain areas between the hippocampus and the olfactory bulbs (using the intracerebroventricular infusion site as the reference point), the same areas where we had observed hDWI (Fig. 1). For each assay, we used 20 μg of protein from each mouse; we found similar levels of actin protein in all samples from all animals (Fig. 5A). We included as a positive control a C57black6 mouse brain sample acquired after traumatic brain injury (TBI), which expressed a higher MMP-9 activity level than did the GCI sample (both without intracerebroventricular infusion) and served as a marker for mouse MMP-9 activity. We also included a positive control of human origin. In all but one animal (asterisk) that received GCI and sODN-mmp9, we observed no MMP-9 activation (Fig. 5B); similarly, no MMP-9 activity was noted in SO animals that did not receive GCI and the intracerebroventricular infusion (Fig. 5C). Because we detected MMP-9 activity in animals that experienced GCI or TBI without intracerebroventricular infusion as positive controls, it is quite clear that it is not the intracerebroventricular infusion that causes MMP-9 activity. Furthermore, we observed that GCI with nontargeting sODN-Ran did not reduce MMP-9 activities. Altogether, in repeated observations, MMP-9 activities were present in seven of eight animals from the GCI and SPION-Ran group (87.5%), and in only two of 10 mice treated with GCI and sODN-mmp9 (20%). The total numbers of animals used in these knockdown experiments exceeded or equaled the required number indicated by power analysis (Table 1). The results in the GCI plus sODN-mmp9 group were significantly different from those measured in the GCI group that received no intracerebroventricular infusion (p < 0.04), as well as in the GCI sODN-Ran group (p < 0.01, Fisher's exact test). There was no significant difference between the GCI animals with and without sODN-Ran (p > 0.69). As such, it is clear that the effect of sODN-mmp9 on post-GCI MMP-9 activities differed significantly compared with all of the control groups we measured. We did not compare BBB leakage because it could not be quantified in mice that received intracerebroventricular infusion. Having observed no gene knockdown of MMP-9 activities 24 h after delivery (data not shown), we concluded that the gene knockdown effect of sODN-mmp9 on MMP-9 activation is transient. Nevertheless, the data indicate that sODN-mmp9 hybridizes to its target mRNA.
Figure 5.

MMP-9 activity knockdown using siDNA (sODN-mmp9). Phosphorothioate-modified ODN with randomized sequence (sODN-Ran) or antisense sequence to mmp-9 mRNA (sODN-mmp9) was infused at 1 and 3 h; all samples were collected at 10 h of reperfusion. Protocol is shown in A. B and C show two paired observations of MMP-9 activities in postmortem samples (20 μg of protein per lane) from bilateral frontal regions of individual animals, as assessed by zymography (You et al., 2007). The intracerebroventricular infusion was at 3 h reperfusion in B (sODN = 20 nmol/kg, brain extracts were obtained from left hemisphere) or at 1 h reperfusion in C (sODN = 120 nmol/kg, brain extracts were obtained from bilateral frontal brain at 1–2 bregma). Positive control (PC) samples showing human MMP-9/2 protein (Millipore Bioscience Research Reagents #CC073) and C57black6 mouse (TBI) were included as markers along with mouse controls (SO or CGI without intracerebroventricular infusion). D shows changes in cerebral rADC [mm2/s (× 10−4); three groups of mice are shown: mice with GCI but without intracerebroventricular infusion (open bars, n = 11), mice that received sODN-mmp9 (dark bars, n = 6; *p = 0.004, t test compared to mice with sODN-Ran), and sODN-Ran (gray bars, n = 6)]. E compares striatal VMD (mm3, mean ± SEM) in two groups of mice that received sODN-mmp9 (dark bar, 8.3 ± 1.3) or sODN-Ran (gray box, 12.8 ± 1.0) 1 d after BCAO (n = 6 each). *p = 0.004, t test. There was no significant difference between mice after BCAO with or without intracerebroventricular infusion of sODN-Ran (data not shown). The control antigen (β-actin) in B was stained with primary antibody mouse anti-β-actin (Sigma A-5441) followed by a secondary antibody: Cy5-conjugated donkey anti-mouse antibody (Jackson ImmunoResearch). Scanned with Typhoon 9400 (GE Healthcare). The control antigen in C was the same except the secondary antibody was HRP-conjugated sheep anti-mouse antibody (GE Healthcare detection: ECL Western blotting detection of reagents), exposed to x-ray film (Kodak MioMax MR film).
Table 1.
Statistical analysis for animals with MMP-9 activities
| Group (treatments) |
||||
|---|---|---|---|---|
| A(GCI, no ICV) | B(sham-operated, no ICV) | C(GCI sODN-Ran) | D(GCI sODN-mmp9) | |
| Total animals tested (N) | 3 | 1 | 8 | 10 |
| Power analysis for no. of animals between the groupsa | 104 (A vs C) | 8 (C vs D) | 3 (A vs D) | |
| Percentage of animals that show activated MMP-9 activities in zymograph | 100 | 0 | 87.5 | 20 |
| p values by Fisher's exact test (between zymograph groups) | 0.727 (A vs C) | 0.0076** (C vs D) | 0.035** (A vs D) | |
| VMD (mm3) | 37 ± 2 | 0 | 44 ± 3 | 27 ± 4* |
Data obtained from two separate experiments and a total of 22 mice at 10 h reperfusion for the zymographs in Figure 5, with the exception of DWI/ADC. *p = 0.04 (t test), **statistical significance (α < 0.05).
aPower analysis was performed as described in Materials and Methods.
Transient rADC reversal in MRI after MMP-9 knockdown
We aimed to demonstrate whether the level of MMP-9 activation impacts the development of hDWI/rADC after GCI. Our hypothesis was that a reduction in MMP-9 activity would impede the development of striatal metabolic imbalance by causing a reversal in ADC drop. We tested this hypothesis by determining the effect of siDNA to mmp-9 mRNA on the development of DWI/ADC after GCI. We delivered sODN-mmp9 via intracerebroventricular route 1 h after reperfusion and performed MR scans 1 d later to identify areas of hDWI/rADC. To quantitatively measure the effect of sODN-mmp9, we included data from all mice and did not exclude potential outliers in our statistical analysis. We observed a significant reversal in ADC drop in the striatum at 1 d in the group that received sODN-mmp9 (Fig. 5D), compared with the group that received sODN-Ran or no probe. We observed no change in cortical rADC at this time point in any group.
We measured total VMD in mice using siDNA for gene knockdown of mmp-9 mRNA. We observed a significant reversal of striatal ADC reduction and a decrease in VMD (30–40%) in the group that received sODN-mmp9 (Fig. 5, D,E, respectively), compared with two different control groups: GCI without intracerebroventricular infusion and GCI with sODN-Ran (Table 1). In repeated experiments using the same protocol (paired observations), five of six animals (83%) that received GCI and sODN-Ran exhibited hDWI similar to the control without sODN infusion at the same time points. We conclude that sODN-mmp9 may have multiple applications for MR imaging of in vivo target gene activities (Fig. 2) and ex vivo hybridization (Fig. 3), as well as gene knockdown (Fig. 5).
In conclusion, we have demonstrated that abnormal water diffusion (as measured by MRI and shown as hDWI or rADC) occurs at two time points after GCI: the first, at the release of BCAO (reperfusion), and the second, at 10 h of reperfusion. The second peak was found to last for at least 2 d (Fig. 1). Striatal hDWI with sufficient ADC reduction, measured as VMD, predicted BBB leakage at the late reperfusion time point. Elevated mmp-9 mRNA in the region with VMD could be detected by MRI in vivo using the SPION-mmp9 brain probe (Fig. 2), or by low-dose siDNA of FITC-sODN-mmp9 (Fig. 3), both at 10 h of reperfusion. MMP-9 antigen was expressed in neurons and endothelia within the region that exhibits hDWI at 1 d of reperfusion (Fig. 4). High-dose siDNA delivered at one to 3 h of reperfusion causes knockdown of MMP-9 activities and accompanies rADC reversal (Fig. 5). Most important, noninvasive delivery of FITC-siDNA (<10 kDa, i.p.) detected mmp-9 mRNA expression and minute BBB leakage after GCI (Fig. 3).
Discussion
The actions of genes in the brain cells regulate most activities in the normal and diseased brain, including brain functions related to learning, response to stress, and psychiatric and neurologic disorders, as well as general medical disorders such as cardiovascular disease, stroke and traumatic injury that can change brain energy metabolism and blood flow. Our data presented herein suggest that MMP-9 activities from neurons and endothelia can adversely affect the development of cerebral metabolic disturbance after GCI. Our data indicate that, by targeting mmp-9 mRNA, short DNA has multiple applications as siDNA and reporter of mmp-9 mRNA expression in living mouse brains.
Perturbations in neurovascular functional integrity are associated with upregulation of several genes that initiate cascades of injury. Upstream signals such as oxidative stress, together with neutrophil and/or platelet interactions with activated endothelium, can upregulate the mmp-9 gene, which has been suggested to play a role in the BBB disruption associated with many acute neurological conditions. Severe disruption of the BBB diminishes its protective capabilities, and thus causes the brain to become “leaky” (Gidday et al., 2005). In addition, attenuation of MMP-9 activity may be associated with a reduction in cell death after GCI (Lee et al., 2004). The regional consistency observed in rADC, elevation in levels of MMP-9 antigen, and retention of SPION-mmp9 in the striatum further strengthen the suggestion that MMP-9 activities may be involved in the development of initial metabolic imbalance after GCI.
We established our GCI model in male C57black6 mice to investigate the expression of endogenous genes, cerebral oxidative stress, and brain damage. By virtue of a defect in the posterior communicating arteries of these animals, transient hypoxia/ischemia in this model induces GCI by restricting cerebral blood flow by at least 80% (Barone et al., 1993; Fujii et al., 1997; Murakami et al., 1998). Because early studies reported cell death in these animals (Barone et al., 1993; P. K. Liu et al., 1996), many other studies have used the same mouse strain and reported apoptotic neuronal death, BBB leakage, and gliosis in the striatum, cortex, and hippocampus (Fujii et al., 1997; Yang et al., 1997; Kitagawa et al., 1998; Murakami et al., 1998; Terashima et al., 1998; C. H. Liu et al., 2007a). Brain injury induced by this GCI model is not characterized by distinct core and penumbral regions, as is the case in stroke models involving focal occlusion of the middle cerebral artery. Ventriculomegaly observed in MR images acquired after GCI in two mice suggests that damage includes loss of brain cells (Fig. 1B). Some investigators believe this GCI model may simulate cerebral ischemia during cardiac arrest and cardiopulmonary resuscitation, because cerebral blood flow is partially reduced during BCAO. Rat studies of GCI that simulates cardiac arrest show that leakage of the BBB is biphasic in nature, with stable BBB leakage at 10–48 h of reperfusion (Fujioka et al., 1994; Mossakowski et al., 1994; Back et al., 2004). Our studies on VMD determined by using rADC (Fig. 1) and our data on ex vivo hybridization of FITC-sODN-mmp9 (Fig. 3) support these observations. However, our method using noninvasive MRI for volumetric measurement of the threshold ADC for VMD may have an advantage over such other methods involving invasive quantification to measure BBB leakage measurements. Regions that exhibit a drop in ADC below the threshold are thought to represent brain areas of enhanced metabolic disturbance, where potential BBB leakage may occur (Fig. 1A). We are certain that more studies will validate the potential application of noninvasive VMD to evaluate brain injury sites post-GCI.
By targeting mmp-9 mRNA, we have demonstrated the feasibility of detecting gene activities using MRI and manipulating gene action by changing VMD in the living brain after GCI. Our experiments have reliably shown that mmp-9 mRNA and protein are expressed in regions that exhibit metabolic disturbance, as detected by abnormal water content in MRI (hDWI), conventional immunohistochemistry, MMP-9 activation assay (zymography), and in vivo hybridization with an sODN-mmp9 probe coupled with ex vivo observation. We also demonstrated that gene knockdown by sODN-mmp9 after GCI reduces MMP-9 activities, and that this brain probe that reduces MMP-9 activities also significantly reverses striatal ADC drop, a parameter known to be associated with a reduction in brain damage (Mancuso et al., 2000). These observation are consistent with those of gene knockdown in acute neurological disorders in rodents (Wahlestedt et al., 1993; Zhang et al., 1999), although in our current study, we delivered the brain probe after GCI. By measuring VMD after blocking MMP-9 activities with the sODN-mmp9 brain probe, we observed a decrease in the severity of metabolic disturbance. Having made these observations, we found evidence to support the hypothesis that MMP-9 gene expression may play a role in the formation of striatal brain damage. This conclusion is consistent with results showing lesser levels of cell death after GCI in an MMP-9 deficient mouse strain (Lee et al., 2004).
An additional advantage of our method is that the probe was administered after the GCI episode, and as such more closely resembled a real clinical scenario than other methods that require delivery of brain probe before acute neurological disorder. The amount of iron needed in gene transcript MRI (120 pmol per kg body weight) has been demonstrated not to exacerbate GCI outcome (C. H. Liu et al., 2007a,b,c). Although a dose of sODN-mmp9 much higher than the tracking dose is required for gene knockdown, the MMP-9 knockdown protocol involves less or no iron oxide contrast agent. Iron oxide concentration as high as 8 nmol in human cells is considered safe (Bulte et al., 2001). Therefore, iron overload may not be an issue for manipulating MMP-9 activities. Indeed, we delivered repeated doses (as high as 10 nmol per kg once a week for 4 weeks via intraperitoneal route) and observed no adverse effect (C. H. Liu et al., 2008). Unique to our sODN-mmp9 probe, as reported here, is the potential application for mechanistic investigation using gene knockdown of mmp-9 mRNA in living mouse brains. By enabling such studies of mechanism, this probe represents a great advance for neuroscience. Our technique has potential for clinical application, especially for efforts to delineate gene expression and brain injury.
Other advantages of our probe are its applications for tracking intracellular mRNA transcripts at low dose and initiating gene knockdown at high dose. Both of these applications rely on the base pairing between nucleic acids of complementary sequences, as is evident by results obtained with a control probe of random sequence. Whereas the control reports no endogenous mRNA, no effect on MMP-9 activities, or change in rADC, the sODN-mmp9 probe is shown to reduce MMP-9 activity and create specific gene knockdown in live mouse brains after GCI, without affecting the expression of actin, which is known not to be affected by GCI (Cui and Liu, 2001). Other potential applications that have been reported include assessment of gene transcription activities, apoptotic cell death (marked by cells' inability to take up SPION-cfos), and cell typing (cells with specific gene transcript) during brain repair by gliosis and angiogenesis in animal models of acute neurological disorders (C. H. Liu et al., 2007a, 2008). Although the involvement of MMP-9 activities in the etiology of cell death is well established in stroke models, we know very little about brain injury after cardiac arrest. Elevated blood levels of MMP have been used to diagnose BBB disruption in humans (Clark et al., 1997; McGirt et al., 2002; Lynch et al., 2004), mice (Asahi et al., 2001; Huang et al., 2001; Magnoni et al., 2004) and rats (Pfefferkorn and Rosenberg, 2003); still, an MMP assay is less likely to offer information about a regional correlation between lesion and MMP-9 activity. We show here that MMP-9 antigen is expressed in the same location where metabolic disturbance is exhibited after GCI (Fig. 4), and our results agree with those that have been reported in the literature.
It is reasonable to suggest that elevated VMD, as detected by in vivo MRI, can serve as a metric of brain damage. We have observed here that GCI-induced VMD is significantly reduced when MMP-9 activity is temporarily blocked after GCI in C57black6 mice. We must conduct future studies to explore whether continuous and repeated dosing of sODN-mmp9 brings about an undesirable effect. Nevertheless, the short duration of action by antisense sODN-mmp9 may be more desirable than a longer lasting one, as MMP-9 activities may be required for angiogenesis and tissue repair (Zhao et al., 2006; Bove et al., 2007). Therefore, the balance of extended and repeated dosing will have to be carefully monitored as alternative approaches.
The knockdown effect of sODN-mmp9 on VMD is transient and never 100% effective; and results indicate that products of other gene transcripts may also be involved in the development of VMD or rADC. Therefore, the development of VMD after GCI is likely mediated by continuous expression of mmp-9 mRNA or multiple gene transcripts that can be investigated using the same strategies for different mRNA targets. It is important to note that sODN-mmp9 was administered in our studies after the GCI episode, eliminating the need for the preconditioning often used in therapies of cerebral ischemia. In conclusion, we have demonstrated the feasibility of using a short inhibitory nucleic acid to induce gene knockdown after vessel occlusion, and the feasibility of a novel MR contrast agent to trace sODN-mmp9 and allow MRI detection of endogenous MMP-9 activities in the striatum of living animal brains. We have further demonstrated that VMD is reduced and rADC reversed in the striatum after MMP-9 activity knockdown. The novel technique we have developed holds potential to revolutionize investigations of drug efficacy and therapeutic evaluation by alleviating reliance on methods that require postmortem tissue sampling.
Footnotes
This work was supported by grants from the National Institutes of Health (R21DA024235, R01EB002066, R01NS045845, R21NS057556, R01NS047447) and the National Center for Research Resources (SP41RR14075). We appreciate Nichole Eusemann for editing and Dr. Joseph B. Mandeville for providing the in-house software for image registration (alignment). C.H.L. contributed MRI acquisition, data analysis, and animal care, and Z.Y. and M.W. performed MMP-9 zymography and MMP-9 immunohistology. C.-M.L. supplied sODN and siDNA, Y.R.K. and B.R.R. supplied SPION. P.K.L. designed sODN, conjugated, characterized, and delivered SPION-sODN, performed all surgical procedures for BCAO and probe delivery, and directed the project. C.H.L. and P.K.L. wrote this manuscript. All authors discussed the results and commented on this manuscript. Two patent applications have been submitted: (1) imaging nucleic acids (US serial no. 60/603,907 and PCT/US2005/029875); and (2) matrix metalloproteinase targeting nucleic acids (US serial no. 60/959,856).
The authors declare no competing financial interests.
References
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood–brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back T, Hemmen T, Schüler OG. Lesion evolution in cerebral ischemia. J Neurol. 2004;251:388–397. doi: 10.1007/s00415-004-0399-y. [DOI] [PubMed] [Google Scholar]
- Barone FC, Knudsen DJ, Nelson AH, Feuerstein GZ, Willette RN. Mouse strain differences in susceptibility to cerebral ischemia are related to cerebral vascular anatomy. J Cereb Blood Flow Metab. 1993;13:683–692. doi: 10.1038/jcbfm.1993.87. [DOI] [PubMed] [Google Scholar]
- Bove PF, Wesley UV, Greul AK, Hristova M, Dostmann WR, van der Vliet A. Nitric oxide promotes airway epithelial wound repair through enhanced activation of MMP-9. Am J Respir Cell Mol Biol. 2007;36:138–146. doi: 10.1165/rcmb.2006-0253SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxerman JL, Hamberg LM, Rosen BR, Weisskoff RM. MR contrast due to intravascular magnetic susceptibility perturbations. Magn Reson Med. 1995;34:555–566. doi: 10.1002/mrm.1910340412. [DOI] [PubMed] [Google Scholar]
- Bulte JW, Douglas T, Witwer B, Zhang SC, Strable E, Lewis BK, Zywicke H, Miller B, van Gelderen P, Moskowitz BM, Duncan ID, Frank JA. Magnetodendrimers allow endosomal magnetic labeling and in vivo tracking of stem cells. Nat Biotechnol. 2001;19:1141–1147. doi: 10.1038/nbt1201-1141. [DOI] [PubMed] [Google Scholar]
- Clark AW, Krekoski CA, Bou SS, Chapman KR, Edwards DR. Increased gelatinase A (MMP-2) and gelatinase B (MMP-9) activities in human brain after focal ischemia. Neurosci Lett. 1997;238:53–56. doi: 10.1016/s0304-3940(97)00859-8. [DOI] [PubMed] [Google Scholar]
- Cui J, Liu PK. Neuronal NOS inhibitor that reduces oxidative DNA lesions and neuronal sensitivity increases the expression of intact c-fos transcripts after brain injury. J Biomed Sci. 2001;8:336–341. doi: 10.1007/BF02258375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui JK, Hsu CY, Liu PK. Suppression of postischemic hippocampal nerve growth factor expression by a c-fos antisense oligodeoxynucleotide. J Neurosci. 1999;19:1335–1344. doi: 10.1523/JNEUROSCI.19-04-01335.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M, Hara H, Meng W, Vonsattel JP, Huang Z, Moskowitz MA. Strain-related differences in susceptibility to transient forebrain ischemia in SV-129 and C57black/6 mice. Stroke. 1997;28:1805–1810. doi: 10.1161/01.str.28.9.1805. discussion 1811. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Okuchi K, Sakaki T, Hiramatsu K, Miyamoto S, Iwasaki S. Specific changes in human brain following reperfusion after cardiac arrest. Stroke. 1994;25:2091–2095. doi: 10.1161/01.str.25.10.2091. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Girolamo F, Virgintino D, Errede M, Capobianco C, Bernardini N, Bertossi M, Roncali L. Involvement of metalloprotease-2 in the development of human brain microvessels. Histochem Cell Biol. 2004;122:261–270. doi: 10.1007/s00418-004-0705-x. [DOI] [PubMed] [Google Scholar]
- Gondi CS, Lakka SS, Dinh DH, Olivero WC, Gujrati M, Rao JS. Downregulation of uPA, uPAR and MMP-9 using small, interfering, hairpin RNA (siRNA) inhibits glioma cell invasion, angiogenesis and tumor growth. Neuron Glia Biol. 2004;1:165–176. doi: 10.1017/s1740925x04000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- Gursoy-Ozdemir Y, Qiu J, Matsuoka N, Bolay H, Bermpohl D, Jin H, Wang X, Rosenberg GA, Lo EH, Moskowitz MA. Cortical spreading depression activates and upregulates MMP-9. J Clin Invest. 2004;113:1447–1455. doi: 10.1172/JCI21227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S. Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke. 2003;34:2165–2170. doi: 10.1161/01.STR.0000088062.86084.F2. [DOI] [PubMed] [Google Scholar]
- Huang CY, Fujimura M, Chang YY, Chan PH. Overexpression of copper-zinc superoxide dismutase attenuates acute activation of activator protein-1 after transient focal cerebral ischemia in mice. Stroke. 2001;32:741–747. doi: 10.1161/01.str.32.3.741. [DOI] [PubMed] [Google Scholar]
- Huang D, Shenoy A, Cui J, Huang W, Liu PK. In situ detection of AP sites and DNA strand breaks bearing 3′-phosphate termini in ischemic mouse brain. FASEB J. 2000;14:407–417. doi: 10.1096/fasebj.14.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao YH, Guo WY, Wu YT, Liu KC, Chai WY, Lin CY, Hwang YS, Jy-Kang Liou A, Wu HM, Cheng HC, Yeh TC, Hsieh JC, Mu Huo Teng M. Hemodynamic segmentation of MR brain perfusion images using independent component analysis, thresholding, and Bayesian estimation. Magn Reson Med. 2003;49:885–894. doi: 10.1002/mrm.10440. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Yang G, Mabuchi T, Yagita Y, Hori M, Yanagihara T. Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: evaluation of the patency of the posterior communicating artery. J Cereb Blood Flow Metab. 1998;18:570–579. doi: 10.1097/00004647-199805000-00012. [DOI] [PubMed] [Google Scholar]
- Lee SR, Tsuji K, Lee SR, Lo EH. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci. 2004;24:671–678. doi: 10.1523/JNEUROSCI.4243-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Lok J, Rosell A, Kim HY, Murata Y, Atochin D, Huang PL, Wang X, Ayata C, Moskowitz MA, Lo EH. Reduction of hippocampal cell death and proteolytic responses in tissue plasminogen activator knockout mice after transient global cerebral ischemia. Neuroscience. 2007;150:50–57. doi: 10.1016/j.neuroscience.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone P, Janson CG, Bilaniuk L, Wang Z, Sorgi F, Huang L, Matalon R, Kaul R, Zeng Z, Freese A, McPhee SW, Mee E, During MJ, Bilianuk L. Aspartoacylase gene transfer to the mammalian central nervous system with therapeutic implications for Canavan disease. Ann Neurol. 2000;48:27–38. doi: 10.1002/1531-8249(200007)48:1<27::aid-ana6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Li ZP, Chen SQ, Wang SS, Huang WJ, Cheng JY, Wang W, Wang WT, Tan YX. [Expression of mRNA of matrix metalloproteinase and tissue inhibitor of matrix metalloproteinase after cardiopulmonary resuscitation for asphyxial cardiac arrest in rat] Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2005;17:548–551. [PubMed] [Google Scholar]
- Liu CH, Huang S, Kim YR, Rosen BR, Liu PK. Forebrain ischemia-reperfusion simulating cardiac arrest in mice induces edema and DNA fragmentation in the brain. Mol Imaging. 2007a;6:156–170. [PMC free article] [PubMed] [Google Scholar]
- Liu CH, Kim YR, Ren JQ, Eichler F, Rosen BR, Liu PK. Imaging cerebral gene transcripts in live animals. J Neurosci. 2007b;27:713–722. doi: 10.1523/JNEUROSCI.4660-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CH, Huang S, Cui J, Kim YR, Farrar CT, Moskowitz MA, Rosen BR, Liu PK. MR contrast probes that trace gene transcripts for cerebral ischemia in live animals. FASEB J. 2007c;21:3004–3015. doi: 10.1096/fj.07-8203com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CH, You Z, Ren J, Kim YR, Eikermann-Haerter K, Liu PK. Noninvasive delivery of gene targeting probes to live brains for transcription MRI. FASEB J. 2008;22:1193–1203. doi: 10.1096/fj.07-9557com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PK, Salminen A, He YY, Jiang MH, Xue JJ, Liu JS, Hsu CY. Suppression of ischemia-induced fos expression and AP-1 activity by an antisense oligodeoxynucleotide to c-fos mRNA. Ann Neurol. 1994;36:566–576. doi: 10.1002/ana.410360405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PK, Hsu CY, Dizdaroglu M, Floyd RA, Kow YW, Karakaya A, Rabow LE, Cui JK. Damage, repair, and mutagenesis in nuclear genes after mouse forebrain ischemia-reperfusion. J Neurosci. 1996;16:6795–6806. doi: 10.1523/JNEUROSCI.16-21-06795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PK, Mandeville JB, Guangping Dai, Jenkins BG, Kim YR, Liu CH. Transcription MRI: a new view of the living brain. Neuroscientist. 2008;14:503–520. doi: 10.1177/1073858407309746. [DOI] [PubMed] [Google Scholar]
- Liu W, Rosenberg GA, Liu KJ. AUF-1 mediates inhibition by nitric oxide of lipopolysaccharide-induced matrix metalloproteinase-9 expression in cultured astrocytes. J Neurosci Res. 2006;84:360–369. doi: 10.1002/jnr.20895. [DOI] [PubMed] [Google Scholar]
- Lynch JR, Blessing R, White WD, Grocott HP, Newman MF, Laskowitz DT. Novel diagnostic test for acute stroke. Stroke. 2004;35:57–63. doi: 10.1161/01.STR.0000105927.62344.4C. [DOI] [PubMed] [Google Scholar]
- Magnoni S, Baker A, George SJ, Duncan WC, Kerr LE, McCulloch J, Horsburgh K. Differential alterations in the expression and activity of matrix metalloproteinases 2 and 9 after transient cerebral ischemia in mice. Neurobiol Dis. 2004;17:188–197. doi: 10.1016/j.nbd.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Mancuso A, Derugin N, Hara K, Sharp FR, Weinstein PR. Mild hypothermia decreases the incidence of transient ADC reduction detected with diffusion MRI and expression of c-fos and hsp70 mRNA during acute focal ischemia in rats. Brain Res. 2000;887:34–45. doi: 10.1016/s0006-8993(00)02963-2. [DOI] [PubMed] [Google Scholar]
- McGirt MJ, Lynch JR, Blessing R, Warner DS, Friedman AH, Laskowitz DT. Serum von Willebrand factor, matrix metalloproteinase-9, and vascular endothelial growth factor levels predict the onset of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2002;51:1128–1134. doi: 10.1097/00006123-200211000-00005. discussion 1134–1135. [DOI] [PubMed] [Google Scholar]
- Montaner J, Rovira A, Molina CA, Arenillas JF, Ribó M, Chacón P, Monasterio J, Alvarez-Sabín J. Plasmatic level of neuroinflammatory markers predict the extent of diffusion-weighted image lesions in hyperacute stroke. J Cereb Blood Flow Metab. 2003;23:1403–1407. doi: 10.1097/01.WCB.0000100044.07481.97. [DOI] [PubMed] [Google Scholar]
- Moseley ME, de Crespigny AJ, Roberts TP, Kozniewska E, Kucharczyk J. Early detection of regional cerebral ischemia using high-speed MRI. Stroke. 1993;24:I60–65. [PubMed] [Google Scholar]
- Mossakowski MJ, Lossinsky AS, Pluta R, Wisniewski HM. Abnormalities of the blood-brain barrier in global cerebral ischemia in rats due to experimental cardiac arrest. Acta Neurochir Suppl (Wien) 1994;60:274–276. doi: 10.1007/978-3-7091-9334-1_73. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Chan PH. The development of a new mouse model of global ischemia: focus on the relationships between ischemia duration, anesthesia, cerebral vasculature, and neuronal injury following global ischemia in mice. Brain Res. 1998;780:304–310. doi: 10.1016/s0006-8993(97)01217-1. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. London: Academic; 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Pfefferkorn T, Rosenberg GA. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke. 2003;34:2025–2030. doi: 10.1161/01.STR.0000083051.93319.28. [DOI] [PubMed] [Google Scholar]
- Terashima T, Namura S, Hoshimaru M, Uemura Y, Kikuchi H, Hashimoto N. Consistent injury in the striatum of C57BL/6 mice after transient bilateral common carotid artery occlusion. Neurosurgery. 1998;43:900–907. doi: 10.1097/00006123-199810000-00102. discussion 907–908. [DOI] [PubMed] [Google Scholar]
- Wahlestedt C, Golanov E, Yamamoto S, Yee F, Ericson H, Yoo H, Inturrisi CE, Reis DJ. Antisense oligodeoxynucleotides to NMDA-R1 receptor channel protect cortical neurons from excitotoxicity and reduce focal ischaemic infarctions. Nature. 1993;363:260–263. doi: 10.1038/363260a0. [DOI] [PubMed] [Google Scholar]
- Wang X, Lee SR, Arai K, Lee SR, Tsuji K, Rebeck GW, Lo EH. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med. 2003;9:1313–1317. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- Yang G, Kitagawa K, Matsushita K, Mabuchi T, Yagita Y, Yanagihara T, Matsumoto M. C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res. 1997;752:209–218. doi: 10.1016/s0006-8993(96)01453-9. [DOI] [PubMed] [Google Scholar]
- You Z, Yang J, Takahashi K, Yager PH, Kim HH, Qin T, Stahl GL, Ezekowitz RA, Carroll MC, Whalen MJ. Reduced tissue damage and improved recovery of motor function after traumatic brain injury in mice deficient in complement component C4. J Cereb Blood Flow Metab. 2007;27:1954–1964. doi: 10.1038/sj.jcbfm.9600497. [DOI] [PubMed] [Google Scholar]
- Zechel J, Gohil H, Lust WD, Cohen A. Alterations in matrix metalloproteinase-9 levels and tissue inhibitor of matrix metalloproteinases-1 expression in a transforming growth factor-beta transgenic model of hydrocephalus. J Neurosci Res. 2002;69:662–668. doi: 10.1002/jnr.10326. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Widmayer MA, Zhang B, Cui JK, Baskin DS. Suppression of post-ischemic-induced fos protein expression by an antisense oligonucleotide to c-fos mRNA leads to increased tissue damage. Brain Res. 1999;832:112–117. doi: 10.1016/s0006-8993(99)01459-6. [DOI] [PubMed] [Google Scholar]
- Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- Zozulya A, Weidenfeller C, Galla HJ. Pericyte-endothelial cell interaction increases MMP-9 secretion at the blood-brain barrier in vitro. Brain Res. 2008;1189:1–11. doi: 10.1016/j.brainres.2007.10.099. [DOI] [PubMed] [Google Scholar]