

Abstract
In the healthy adult brain, neurogenesis normally occurs in the subventricular zone (SVZ) and hippocampal dentate gyrus (DG). Cerebral ischemia enhances neurogenesis in neurogenic and non-neurogenic regions of the ischemic brain of adult rodents. The present study demonstrated that post-insult treatment with an HDAC inhibitor, sodium butyrate (SB), stimulated the incorporation of bromo-2′-deoxyuridine (BrdU) in the SVZ, DG, striatum, and frontal cortex in the ischemic brain of rats subjected to permanent cerebral ischemia. SB treatment also increased the number of cells expressing polysialic acid-neural cell adhesion molecule (PSA-NCAM), nestin, GFAP, phospho-CREB, and brain-derived neurotrophic factor (BDNF) in various brain regions after cerebral ischemia. Furthermore, extensive co-localization of BrdU and PSA-NCAM was observed in multiple regions after ischemia, and SB treatment upregulated protein levels of BDNF, phospho-CREB and GFAP. Intraventricular injection of K252a, a TrkB receptor antagonist, markedly reduced SB-induced cell proliferation detected by BrdU and Ki67 in the ipsilateral SVZ, DG and other brain regions, blocked SB-induced nestin expression and CREB activation, and attenuated the long-lasting behavioral benefits of SB. Together, these results suggest that HDAC inhibitor-induced cell proliferation, migration and differentiation require BDNF-TrkB signaling and may contribute to SB’s long-term beneficial effects after ischemic injury.
Keywords: HDAC inhibitors, cerebral ischemia, neurogenesis, BrdU, BDNF, K252a
Introduction
Neurogenesis is the process of forming integrated neurons from progenitor cells, which includes cell proliferation, migration, and differentiation in the adult brain (Eriksson et al., 1998; Kornack and Rakic, 2001). In adult rodents, this process occurs mostly in the subventricular zone (SVZ) of the rostral lateral ventricle, and the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) (Gage, 2000; Alvarez-Buylla et al., 2002). The neural stem cells in the SVZ migrate into the olfactory bulb via the rostral migratory stream, and then differentiate into interneurons; new neurons in the SGZ migrate into the adjacent DG granule cell layer (Pencea et al., 2001a; Gould et al., 1999). Neurogenesis is regulated by a number of signaling pathways. For example, phospho-cAMP response element-binding protein (p-CREB) is known to play a prominent role in the proliferation, differentiation, and survival of neuronal precursor cells (Nakagawa et al., 2002; Giachino et al., 2005; Kitagawa, 2007). p-CREB directly regulates the expression of brain-derived neurotrophic factor (BDNF), which also enhances cell survival and differentiation of SVZ progenitor cells in vitro and increases the number of newborn cells in vivo (Barnabé-Heider and Miller, 2003; Sairanen et al., 2005). Indeed, infusion of BDNF into the rat lateral ventricle up-regulates the number of proliferating cells and the expression of its receptor, TrkB (Pencea et al., 2001b).
Stroke induces rapid neuronal loss and neurological deficits following ischemic insult. It has been suggested that the replacement of new neurons contributes to the self-repair system of cerebral ischemic injury (Arvidsson et al., 2002; Yamashita et al., 2006). Previous studies reported that, in rats, stroke increased cell proliferation and neurogenesis in the SVZ and hippocampal DG (Arvidsson et al., 2002; Jin et al., 2001; Parent et al., 2002; Zhang et al., 2004). However, neurogenesis after cerebral ischemia or targeted apoptosis has also been detected in non-neurogenic regions such as the striatum and cortex (Arvidsson et al., 2002; Gu et al., 2000; Magavi et al., 2000; Senatorov et al., 2004). Furthermore, ischemia-induced migration of neuroblasts from the SVZ into the injured striatum has also been reported (Yamashita et al., 2006).
Histone protein modification such as acetylation and deacetylation plays a key role in regulating gene expression during the processes of cell proliferation and differentiation. Inhibitors of histone deacetylases (HDACs)—such as sodium butyrate (SB), valproic acid (VPA), and trichostatin A (TSA)—induce neuronal differentiation in primary rat cortical cultures, presumably by inducing the neurogenic basic helix-loop-helix (bHLH) transcription factor NeuroD (Hsieh et al., 2004). Moreover, chronic treatment of adult rats with VPA stimulates hippocampal neurogenesis (Hao et al., 2004). We recently reported that post-insult treatment with HDAC inhibitors robustly reduced infarct volume, cell death, neuroinflammation, and improved neurological performance in rats subjected to middle cerebral artery occlusion (MCAO) (Ren et al., 2004; Kim et al., 2007). The present study investigated the possibility that post-insult treatment with SB or TSA might be associated with cell proliferation and up-regulation of neural progenitor cells after stroke-induced brain injury. We also studied whether SB-induced changes in cell proliferation, migration, differentiation, and behavioral benefits require activation of BDNF-TrkB signaling.
Materials and Methods
Permanent middle cerebral artery occlusion (pMCAO)
All experiments were approved by the National Institutes of Health (NIH) Animal Care and Use Committee in accordance with the NRC Guide for the Care and Use of Laboratory Animals. Male Sprague Dawley rats (Charles River Laboratories, Charles River, CA; 250–300 gm) were anesthetized with 3% isoflurane in a 70% to 30% mixture of N2O to O2 and underwent pMCAO as described previously (Kim et al., 2007). Briefly, the left common carotid artery and external carotid artery were isolated and ligated with a 4-0 suture. A nylon thread was inserted into the left internal carotid artery and advanced to the Circle of Willis. The thread was left in place until the rats were sacrificed. Sham-operated control surgery was performed in an identical manner without perturbation of the carotid artery. Body temperature was maintained at 37.0–37.5°C with a heating pad.
Drug treatment and 5-bromo-2'-deoxyuridine (BrdU) labeling
Rats were treated once daily with subcutaneous injections of either SB (300 mg/kg), TSA (0.2 mg/kg) (both from Sigma, St. Louis, MO) or vehicle, starting immediately after pMCAO, and lasting for the indicated time period. To label dividing cells, rats were intraperitoneally injected with BrdU (Sigma, 50 mg/kg body weight) twice daily, at an eight-hour interval, from Day 3 to Day 7 after ischemia, and sacrificed on Day 7. Alternately, rats were injected with BrdU from Day 5 to Day 14 after ischemia, and sacrificed on Day 14. A schematic diagram showing the treatment regimen is shown in Supporting Fig. 1 (Fig. S1).
Experimental design and stereotaxic injection
Stereotaxic surgery was performed two hours prior to pMCAO. Rats were anesthetized with 1.5% isoflurane, 50% N2O and 30% O2 inhalation, and then placed in a stereotaxic surgical apparatus (David Kopf Instruments, Tujunga, CA). A Hamilton syringe (Hamilton, Reno, NV) needle was inserted into the left lateral ventricle (coordinates from the bregma: anterior-posterior −0.3 mm, medio-lateral (left) −1.2 mm, and dorsoventral −3.6 mm from the meninges). Five µl of K252a (Sigma), a blocker of the BDNF TrkB receptor, (0.5 mM dissolved in 1% DMSO) was constantly infused into the left lateral ventricle at a rate of 1µl/min. The dose of K252a was selected based on previous studies (Xu et al., 2007). Treatment groups consisted of 1) sham-operated (1% DMSO with Sham surgery); 2) vehicle-treated (1% DMSO with pMCAO); 3) SB-treated (1% DMSO + 300 mg/kg SB with pMCAO); 4) K252a + SB-treated (K252a + 300 mg/kg SB with pMCAO); and 5) K252a-treated (K252a with pMCAO). Each group comprised seven to 10 animals per experiment.
Immunohistochemistry
Rats were sacrificed with CO2 exposure and then transcardially perfused with phosphate buffered saline (PBS, pH 7.4). Brain tissues were quickly immersed in dry ice-precooled isopentane and stored in a −80°C freezer. Coronal brain sections (20 µm) [corresponding, bregma −3.0 to −1.2 mm (SVZ) and bregma −4.52 to −3.14 (DG)] were cut with a cryostat and fixed with 4% paraformaldehyde. To detect newly generated BrdU-positive cells, the sections were incubated for 30 min in 2N HCl to denature the DNA. Thereafter, endogenous peroxidase was blocked by incubation with 0.3% H2O2 for 45 minutes, and nonspecific binding was inhibited by incubating the sections in buffer (10% normal serum, 0.2% Triton X-100 in PBS) for one hour.
The brain sections were incubated with primary antibodies at 4°C overnight, and then incubated with secondary antibodies in a humidified chamber for one hour at room temperature. Brain sections were double-labeled with anti-BrdU, and an antibody to detect a cell type specific marker. The primary antibodies used were as follows: anti-rat BrdU (Accurate Chemicals, Westbury, NY), mouse anti-NeuN (Chemicon, Temecula, CA), mouse anti-glial fibrillary acidic protein (GFAP) (Sigma), rabbit anti-BDNF (Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-polysialic acid-neural cell adhesion molecule (PSA-NCAM) (Chemicon), mouse anti-nestin (Abcam, Cambridge, MA), rabbit anti-Ki67 (Chemicon), rabbit anti-phospho-CREB (Upstate Biotechnology, Lake Placid, NY), and anti-rabbit acetylated H3 (Upstate). The secondary antibodies used were anti-rat Alexa Flour 555 (Invitrogen, Eugene, Oregon), anti-mouse FITC-conjugated IgG (Santa Cruz Biotechnology), and anti-rabbit Alexa Flour 488 (Invitrogen). Other secondary antibodies were obtained from Jackson Immunoresearch (West Grove, PA) and were used at 1:200 dilutions. Sections were rinsed three times for 10 minutes with PBS, mounted onto superfrost slides, and coverslipped with Fluorescent Mounting Medium (Vector Laboratories, Inc., Buringame, CA).
Single optical images or z series stacks of 0.5 to 2 µm slice thickness were taken using a Zeiss LSM 510 confocal laser microscope (Carl Zeiss, Oberkochen, Germany). Image analysis was performed in corresponding areas of indicated brain regions for each animal group using LSM 510 Browser imaging software and Adobe Photoshop (Adobe Systems, Mountain View, CA). Fig. S2A is a schematic diagram illustrating the extent of damage caused by pMCAO in the ipsilateral brain hemisphere, the site of K252a intracerebral injection and the areas used for immunohistochemical examinations in the SVZ, anterior SVZ (aSVZ), striatum and frontal cortex, as shown in the boxes. Fig. S2B shows the boxed area of the ipsilateral DG subgranular zone used for immunohistochemical analysis.
Western blotting analysis
For Western blotting analysis of ischemic brain hemispheres, animals were sacrificed and brains were homogenized in ice-cold lysis buffer as previously described (Kim et al., 2007). Total protein amounts (20 µg/lane) were separated on a 4–12% SDS-polyacrylamide gel (Invitrogen, Carlsbad, CA), transferred to a polyvinylidene difluoride membrane, and then incubated with primary antibodies overnight at 4°C: rabbit anti-BDNF (1:200, Santa Cruz Biotechnology), rabbit anti-phospho-CREB (ser 133) (1:200, Upstate), mouse anti-GFAP (1:10,000, Sigma), and mouse anti-Bcl-2 (1:200, Santa Cruz Biotechnology). After incubation with secondary antibodies conjugated to horseradish peroxidase (1:1,500, Santa Cruz Biotechnology), protein bands were detected by an ECL kit (GE Health Care, Piscataway, NJ) and exposed to hyperfilm (GE Health Care). Densitometric analysis of the blots was performed with the image analysis program UN-SCAN-IT gel version 5.1 (Silk Scientific, Orem, UT).
Behavioral studies
Behavioral tests were performed by investigators who were blinded to the experimental groups, as described previously (Kim et al., 2007). The tests were conducted seven or 14 days after pMCAO. Before ischemic onset, all rats were able to perform the tests and exhibited no abnormalities. Briefly, for the rotarod test, an accelerating rotarod, 4–40 rpm (San Diego Instruments, Inc., San Diego, CA) was used to assess the motor coordination of rats by measuring their retention time on the rotating rod. Neurological deficit severity scores included three tests where each animal was suspended by the tail, three tests in an open field, pinna reflex, and visual placement test. Each test was scored as 1 for normal and 0 for abnormal, producing a summed severity of injury score graded on a scale of 0 to 8 (maximum deficit score = 0, normal = 8).
Quantitative analysis and statistics
Corresponding brain coronal sections including the SVZ, DG, frontal cortex, and striatum were selected for comparison using a rat atlas (Paxinos and Watson, 1982). BrdU (+) cell counting was performed by confocal microscopy using a 25× objective lens (field size, 343 × 343 µm). Measurements in the areas of the SVZ, DG, and others were made using three to five sections per animal (each group, n = 3–4 rats). Analysis of BrdU and double-labeled cells in the ipsilateral brain regions were made using confocal z-series stacks obtained using 25× and 40× objectives. All data were expressed as mean ± SEM and analyzed using ANOVA and Student’s t-tests. Western blotting (n = 3–4) and behavioral assessment (n = 5–10) results were analyzed by ANOVA with post-hoc test. Differences of p<0.05 were considered statistically significant.
Results
Effects of HDAC inhibitors on cell proliferation in the ischemic brain of pMCAO rats
Cerebral ischemia in rats was induced by pMCAO. Rats then received an injection of BrdU (50 mg/kg, i.p., twice daily), a cell proliferation marker, from Day 3 to Day 7, or from Day 5 to Day 14. The HDAC inhibitors SB (300 mg/kg), TSA (0.2 mg/kg), or their vehicle were injected (s.c.) once daily, starting immediately after ischemia. Animals were sacrificed on Day 7 or Day 14, about eight hours after the last injection. Days 7 and 14 were chosen because enhanced BrdU incorporation into DNA has been reported to occur at these time points in brain regions including the SVZ after cerebral ischemia (Jin et al., 2001; Zhang et al., 2004; Lee et al., 2006).
We first investigated whether treatment with SB increased cell proliferation in the SVZ and hippocampal DG, two classic neurogenic brain regions where cerebral ischemia is known to induce neurogenesis (reviewed in Lichtenwalner and Parent, 2006). As expected, the labels of BrdU immunoreactivity were enhanced in the SVZ and DG of the ipsilateral ischemic brain hemisphere of vehicle-treated rats on Day 7 after pMCAO, compared with the sham-operated control (Fig. 1A & B). Notably, treatment of pMCAO rats with SB or TSA—both of which are structurally dissimilar HDAC inhibitors—further enhanced BrdU labels in both brain areas. A similar, but less pronounced, increase was observed after SB treatment for 14 days (Fig. 1C & D).
Fig. 1. Post pMCAO-insult treatment of rats with SB increased the number of BrdU (+) cells in the SVZ and DG of the ischemic hemisphere.

Brain tissues were analyzed by double immunostaining with BrdU (red) and NeuN (green, a mature neuronal marker).
(A & B) Seven days post pMCAO treatment. (a) Sham-operated, (b) vehicle-treated, pMCAO, (c) SB-treated, pMCAO, (d) TSA-treated, pMCAO. Arrows identify BrdU (+) cells. Scale bar = 50 µm. The vehicles for SB and TSA were saline and DMSO respectively; no significant differences were found in stimulating cell proliferation.
(C, D & E) Representative data analyzed from 3–4 animals per group, 14 days post pMCAO treatment. (a) Sham, (b) vehicle, (c) SB. Arrows identify BrdU (+) cells. Scale bar = 50 µm. Fewer BrdU (+) cells were found in the contralateral hemisphere than in the ipsilateral hemisphere (E).
(F & G) Quantified results of the number of BrdU (+) cells in the ipsilateral SVZ on day 7, and DG on Day 14 post-pMCAO, respectively. Data are mean ± SEM (n = 3–4 animals per group). **p<0.01, ***p<0.001, between indicated groups. SVZ: subventricular zone, LV: lateral ventricle. DG: dentate gyrus, H: hilus, SGZ: subgranular zone, GCL: granular cell layer.
In contrast, little BrdU labeling was detected in the contralateral DG of vehicle or SB-treated pMCAO rats (Fig. 1E). Quantification of the number of BrdU-labeled cells in the ipsilateral SVZ 7 days after pMCAO revealed an increase in vehicle-treated rats and further robustly increased in SB and TSA-treated rats (Fig. 1F). Moreover, the number of BrdU-positive cells in the ipsilateral DG 14 days after pMCAO was increased about 2.5-fold by vehicle, and 5.5-fold by SB treatment (Fig. 1G). Double-labeling with BrdU and NeuN, a mature neuronal marker, revealed little overlap in the expression of these two markers at both time points. These results suggest that the newborn cells in the SVZ and DG have not yet differentiated into mature neurons at this time, consistent with previous reports (Liu et al., 1998; Kawai et al., 2004). The restoration of the loss of NeuN-positive cells in the SVZ of SB-treated rats could be due, at least in part, to the neuroprotective effects of this drug.
To confirm that SVZ neuroblasts were increased after pMCAO, we performed confocal double immunohistochemistry with BrdU and PSA-NCAM. PSA-NCAM is expressed in the cytoplasm of newborn migrating cells in the rodent brain, and plays an important role in cell development and migration of SVZ neural precursors to the olfactory bulb (Bonfanti, 2006). pMCAO enhanced levels of PSA-NCAM immunostaining in the ipsilateral brain SVZ, and treatment with SB further increased the immunostaining levels after seven days (Fig. 2A & B). Notably, extensive co-localization of PSA-NCAM and BrdU was observed in the SVZ of SB-treated rats. A similar increase and colocalization of PSA-NCAM and BrdU was found in the anterior SVZ (aSVZ) after SB treatment (Fig. 2C). In contrast, BrdU/PSA-NCAM immunostaining in the contralateral SVZ and aSVZ showed little BrdU/PSA-NCAM expression from SB-treated pMCAO animals, suggesting that BrdU/PSA-NCAM expression may be triggered by ischemic injury (Fig. 2A and C). Quantified results of PSA-NCAM- and BrdU/PSA-NCAM-positive cells in the ipsilateral SVZ and aSVZ of sham, vehicle- and SB-treated pMCAO rats are shown in Fig. S3A–D. Quantified results of PSA-NCAM- and BrdU/PSA-NCAM-positive cells in the ipsilateral SVZ and aSVZ region showed that post-insult SB treatment significantly increased the number of PSA-NCAM and BrdU/PSA-NCAM-labeled cells, suggesting that SB treatment stimulates cell proliferation, neurogenesis and migration to the ischemic injury site. The aSVZ is connected to the rostral migrating system through which newborn cells migrate to the olfactory bulb (Alvarez-Buylla et al., 2002; Kornack and Rakic, 2001; Iwai et al., 2003).
Fig. 2. SB treatment markedly increased the number of cells expressing PSA-NCAM in the SVZ on Day 7 post pMCAO.

Brain tissues were analyzed by double immunostaining with BrdU (red) and PSA-NCAM (green).
(A) (a) Sham, (b) vehicle, (c) SB, (d) SB in the contralateral SVZ.
(B) Most BrdU (+) cells showed colocalization with PSA-NCAM in the SVZ (yellow). (a) BrdU (+) cells in the SB-treated group, (b) PSA-NCAM (+) cells in the SB-treated group, (c) merged image.
(C) SB treatment increased PSA-NCAM immunostaining in the ipsilateral aSVZ. (a) Sham, (b) vehicle, (c) SB, (d) SB in the contralateral aSVZ.
(D) Nestin, a neuroblast marker, was increased by SB treatment in the ipsilateral SVZ on Day 14 post pMCAO. (a) Sham, (b) vehicle, (c) SB.
(E) (a) BrdU (+) cells in the SB-treated group, (b) nestin (+) cells in the SB group, (c) merged image.
(F) Enlarged images from the box in Fig. 2E (c). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU/PSA-NCAM or BrdU/nestin. Scale bar = 50 µm. SVZ: subventricular zone, aSVZ: anterior subventricular zone, LV: lateral ventricle.
We further examined the effects of SB on the expression of nestin, a neural stem cell marker for neural precursors with radial glia-like morphology and putative progenitors with limited self-renewal capacity (Kempermann et al., 2004). On Day 14 after SB treatment, the immunoreactivity of nestin was increased in the SVZ (Fig. 2D). Some SB-induced nestin labels co-localized with the BrdU labels (Fig. 2D–F).
In addition, levels of BrdU incorporation were examined in other brain areas, notably the striatum and frontal cortex. The striatum has been shown to generate new neurons after stroke or apoptotic insult, and the process could originate either from the SVZ or from progenitors residing in the parenchyma (Arvidsson et al., 2002; Senatorov et al., 2004). We found that BrdU labels were markedly increased in the ischemic striatum on Day 7 after pMCAO, compared with sham-operated controls, and that this increment was further potentiated by post-insult treatment with SB or TSA (Fig. 3A). Neither SB nor TSA-induced BrdU labels colocalized with NeuN labels in the striatum, although the severe loss of striatal NeuN-expressing cells was largely prevented by SB or TSA treatment. In addition to increasing NeuN-labeling, SB treatment also increased the overall expression of PSA-NCAM-positive cells in the ischemic striatum; furthermore, a substantial fraction of SB-induced BrdU-positive cells were co-localized with PSA-NCAM (Fig. 3B). In contrast, little enhancement of BrdU/PSA-NCAM was detected in the contralateral striatum derived from SB-treated pMCAO rats, compared with the sham group (Fig. 3Bd). Quantified results of PSA-NCAM- and BrdU/PSA-NCAM-positive cells in the ipsilateral striatum are shown in Fig. S3E & F. Interestingly, SB or TSA treatment potentiated ischemia-induced increases in striatal cells expressing GFAP, a glial cell marker, seven days after ischemic insult (Fig. 3C). Some striatal BrdU-positive cells induced by SB showed co-localization with GFAP after seven or 14 days of treatment (Fig. 3C & D). Finally, SB treatment also upregulated nestin-positive cells in the striatum, compared with sham-operated or vehicle-treated controls, and some co-localizations of BrdU and nestin were noted (Fig. 3E). Quantified results confirmed that SB treatment enhanced the increase in the number of BrdU-positive cells in the ipsilateral striatum 7 days after pMCAO (Fig. 3F). Quantified results of GFAP- and nestin-positive cells in the striatum of sham, vehicle- and drug-treated pMCAO rats are shown in Fig. S4A–C.
Fig. 3. SB or TSA treatment robustly increased BrdU, PSA-NCAM, GFAP, and nestin-positive immunostaining in the ipsilateral striatum on Days 7 or 14 after pMCAO.

(A) BrdU (+) cells and NeuN (+) cells did not show colocalization on Day 7 after pMCAO. (a) Sham, (b) vehicle, (c) SB, (d) TSA in the corresponding areas of the striatum. BrdU (red), NeuN (green). Arrows identify BrdU (+) cells.
(B) BrdU and PSA-NCAM were both extensively present on Day 7 after pMCAO. (a) Sham, (b) vehicle, (c) SB in the corresponding areas, (d) SB in the contralateral striatum. BrdU (red), PSA-NCAM (green), colocalized BrdU and PSA-NCAM (yellow). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and PSA-NCAM.
(C) GFAP (+) cells were markedly increased by SB or TSA on Day 7 after pMCAO. (a) Sham, (b) vehicle, (c) SB, (d) TSA. BrdU (red), GFAP (green), colocalized BrdU and GFAP (yellow). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and GFAP.
(D) SB treatment increased GFAP (+) cells on Day 14 following cerebral ischemia. (a) Sham, (b) vehicle, (c–f) SB. BrdU (red), GFAP (green), colocalized BrdU and GFAP (yellow). Arrows identify GFAP (+) cells. Arrowheads identify cells with colocalized expression of BrdU and GFAP. (d–f) enlarged area indicated in (c).
(E) Nestin was potentiated by SB treatment on Day 14 after pMCAO. (a) Sham, (b) vehicle, (c) SB. BrdU (red), nestin (green), colocalized BrdU and nestin (yellow). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and nestin. Stri: striatum. Scale bar = 50 µm.
(F) Quantification of BrdU (+) cells in the ipsilateral striatum of Sham-operated, Vehicle-treated and SB-treated rats on day 7 following pMCAO. Data are mean ± SEM (n = 3–4 animals per group). **p<0.01, ***p<0.001, between indicated groups.
Because differentiation of endogenous multipotent precursors into neurons has been reported in the cerebral cortex of rodents after brain injury (Gu et al., 2000; Magavi et al., 2000), we examined the effects of SB and TSA on cell proliferation and expression of cell markers in the frontal cortex of pMCAO rats. Treatment with SB or TSA for seven days markedly enhanced BrdU incorporation in the frontal cortex, compared with the sham-operated or vehicle-treated control, while restoring the loss of NeuN-positive cells that resulted from cerebral ischemia (Fig. 4A). SB treatment for 14 days, but not seven days, induced some co-expression of BrdU and NeuN, suggesting the occurrence of neurogenesis (Fig. 4A & B). Quantification of BrdU-positive cells revealed that SB increased the number by approximately four-fold compared with vehicle-treated control (Fig. 4C). Treatment with SB or TSA for seven days elicited some increase in nestin-positive cells in the ischemic frontal cortex (Fig. 4D). BrdU labeling was also found to be adjacent to nestin labeling, and some co-localizations were detected, notably in the TSA-treated group. SB or TSA treatment robustly increased GFAP-expressing cells that showed some co-localization with BrdU on Day 14 (Fig. 4E). Quantified data of GFAP-, BrdU-, nestin-and BrdU/nestin-positive cells in the frontal cortex are presented in Fig. S4D–G.
Fig. 4. SB treatment robustly increased BrdU, NeuN, PSA-NCAM, GFAP, and nestin-positive immunostaining in the frontal cortex of the ischemic hemisphere on Day 7 or 14 after pMCAO.

(A) Double immunostaining of BrdU (+) and NeuN (+) cells on Day 7 after stroke. (a) Sham, (b) vehicle, (c) SB, (d) TSA in the corresponding areas of the frontal cortex. BrdU (red), NeuN (green). Arrows identify BrdU (+) cells.
(B) Day 14 after cerebral ischemia. BrdU was coexpressed with NeuN in some cells. (a) Sham, (b) vehicle, (c) SB, (d–f) enlarged area indicated in (c). BrdU (red), NeuN (green), colocalized BrdU and NeuN (yellow). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and NeuN.
(C) Quantification of BrdU (+) cells in the ipsilateral frontal cortex of Sham-operated, Vehicle-treated and SB-treated rats on day 7 following pMCAO. Data are mean ± SEM (n = 3–4 animals per group). ***p<0.001, between indicated groups.
(D) Immunostaining of BrdU (+) and nestin (+) cells on Day 7 following cerebral ischemia. (a) Sham, (b) vehicle, (c) SB, (d) TSA. BrdU (red), nestin (green). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and nestin. Fcx: frontal cortex, Scale bar = 50 µm. Data obtained from representative confocal microscopy sections of rat brains (n = 3–4, each group).
(E) Double immunostaining of BrdU (+) and GFAP (+) cells on Day 14 after ischemia. (a) Sham, (b) vehicle, (c) SB, (d) TSA. BrdU (red), GFAP (green), colocalized BrdU and GFAP (yellow). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and GFAP.
Role of HDAC inhibition and BDNF-TrkB signaling in SB-induced cell proliferation
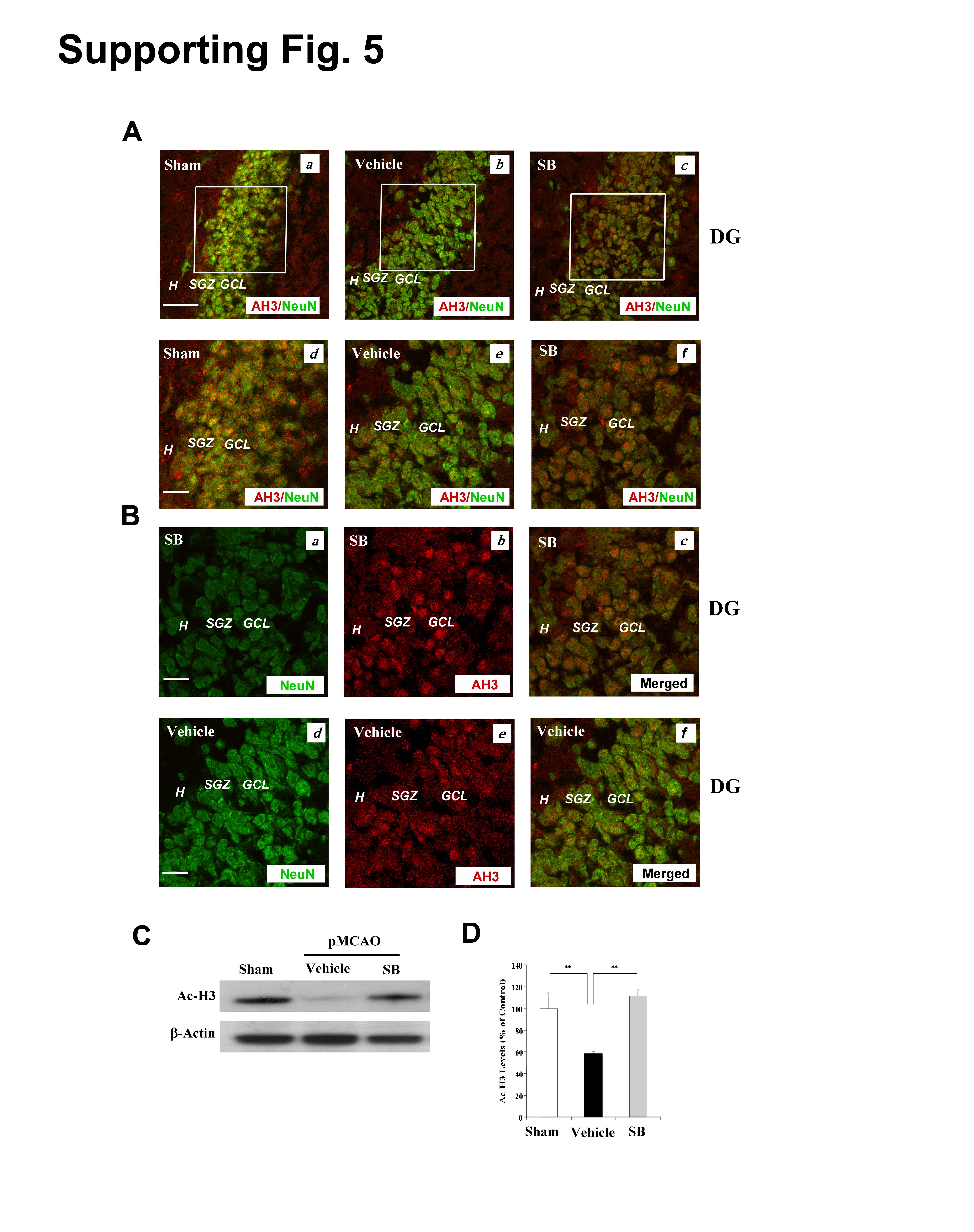
Because SB is an HDAC inhibitor, we assessed whether SB-induced cell proliferation correlates with HDAC inhibition. On Day 14 after pMCAO, immunoreactive levels of acetylated histone-H3 (AH3) were decreased in the DG of vehicle-treated controls, and this loss was prevented by SB treatment (Fig. S5A). Much of the AH3 immunostaining was localized in cells that expressed the neuronal marker, NeuN (Fig. S5A & B). Western blotting analysis showed that acetylation levels of histone H3 were robustly decreased in the ipsilateral brain hemisphere, and this reduction was prevented by SB treatment (Fig S5C & D), similar to our previous report (Kim et al., 2007). Because histone hyperacetylation caused by HDAC inhibition may result in chromatin relaxation and changes in gene expression, we used Western blotting to examine protein molecule levels related to cell proliferation and survival in the ischemic brain hemisphere. Levels of both high and low molecular weight forms of BDNF in the ischemic hemisphere were reduced by pMCAO on Day 7; these decrements were blocked by SB treatment (Fig. S6A & B). Similar effects were observed for p-CREB levels (Fig. S6A & C). GFAP protein levels were enhanced by pMCAO and further increased by SB treatment in these ischemic animals (Fig. S6A & D). In contrast, little change in Bcl-2 or β-actin levels were noted under the treatment conditions.
BDNF is known to be involved in the neurogenesis, migration, maturation, and survival of newborn cells after ischemic insult (Benraiss et al., 2001). Immunohistochemical images of BDNF showed that BDNF expressing cells were down-regulated in the ipsilateral SVZ, and that trezatment with SB or TSA raised BDNF levels above the sham-operated control (Fig. 5A). Similar effects for SB were found in the aSVZ and frontal cortex of the ischemic hemisphere (Fig. 5B & C). In addition, studies have consistently shown that the neurophysiological effects of BDNF are mediated by activation of its cell-surface receptor TrkB. In order to elucidate the role of BDNF-TrkB signaling in SB-induced cell proliferation, we injected K252a, an antagonist of Trk receptors, notably TrkB, into the left lateral ventricle two hours prior to pMCAO to block TrkB activity. SB-enhanced BDNF immunostaining in both the aSVZ and frontal cortex was markedly reduced by co-treatment with K252a; alone, K252a had little effect (Fig. 5B & C). Quantification of BDNF-positive cells in the SVZ, aSVZ and frontal cortex in sham, vehicle- and drug-treated pMCAO rats confirmed this notion (Fig. 5D–F). In the SVZ and DG, SB-induced BrdU labeling on Day 7 was also robustly suppressed by treatment with K252a (Fig. 6A & B). Quantification of the number of BrdU-positive cells in the SVZ revealed that SB treatment increased the number by nearly four-fold, compared with the vehicle-treated control, and that most of this increase was suppressed by co-treatment with K252a (Fig. 6C).
Fig. 5. BDNF immunoreactivity was markedly down-regulated in the vehicle-treated group, but up-regulated in SB- or TSA-treated rats in the ipsilateral SVZ, aSVZ, and frontal cortex on Day 7 after pMCAO.

SB-induced BDNF up-regulation in the aSVZ and frontal cortex was blocked by co-treatment with K252a.
(A) (a) Sham, (b) vehicle, (c) SB, (d) TSA. BDNF (green). Scale bar = 50 µm. Results are from representative brain sections (n = 3–4 per group).
(B) (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BDNF (green). Scale bar = 50 µm.
(C) (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BDNF (green). Scale bar = 50 µm. Experimental conditions are described in the Methods. SVZ: subventricular zone, LV: lateral ventricle, aSVZ: anterior subventricular zone, Fcx: frontal cortex.
(D), (E) & (F) Quantified results of BDNF (+) cells in the SVZ, aSVZ and frontal cortex under the experimental conditions shown in (A)–(C). Data are mean ± SEM and were analyzed from 4 rats per group, **p< 0.01, ***p< 0.001, between indicated groups.
Fig. 6. Intracerebral injection of K252a into the lateral ventricle robustly suppressed BrdU labels in the SVZ and DG of the ipsilateral hemispheres on Day 7 following cerebral ischemia.

(A) (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a groups in the SVZ. BrdU (red), NeuN (green). Arrows identify BrdU (+) cells.
(B) (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a groups in the DG. BrdU (red), NeuN (green). Arrows identify BrdU (+) cells. Scale bar = 50 µm.
(C) Quantification of the number of BrdU-labeled cells from the ipsilateral SVZ in sham operated, vehicle, SB, K252a + SB, and K252a-treated rats on Day 7 following pMCAO. Data are mean ± SEM., **p< 0.01, ***p<0.001, comparison between the indicated groups. Data were analyzed from 3–4 animals per group. SVZ: subventricular zone, LV: lateral ventricle. H: hilus, SGZ: subgranular zone, GCL: granular cell layer.
BrdU incorporation could also reflect a DNA repair process, as opposed to cell proliferation. We therefore performed immunohistochemical staining of Ki67, a nuclear protein tightly associated with cell mitosis (Scholzen and Gerdes, 2000), and found that the expression of Ki67 and BrdU were similarly regulated and co-localized in the ischemic SVZ and striatum of pMCAO rats treated with SB for seven days; in addition, SB-induced Ki67 immunoreactive levels were blocked by pre-treatment with K252a (Fig. S7A & B). Co-labeling with BrdU and Ki67 in consecutive sections from the ischemic striatum confirmed their co-localization (Fig. S7C). Furthermore, BrdU and Ki67 labeling were co-upregulated and co-localized in the SVZ, aSVZ, striatum, frontal cortex, and DG after seven days of TSA treatment (Fig. S7D). TSA treatment elicited little immunostaining of BrdU/Ki67 in the contralateral SVZ and striatum of pMCAO rats (Fig. S7E & F). Quantified results of BrdU-, Ki67- and BrdU/Ki67-positive cells in the ipsilateral SVZ and striatum of sham, vehicle- and drug-treated pMCAO rats are shown in Fig. S8A–I. The BrdU labels in the SVZ were also co-localized with AH3 in SB-treated pMCAO rats on Day 7, and K252a treatment blocked SB-induced increases in BrdU/AH3 (Fig. S9A). K252a had similar effects on BrdU/AH3 labels in the SB-treated DG (Fig. S9B), and double labeling using consecutive sections confirmed their co-localization in this brain region (Fig. S9C).
Activation of CREB through serine 133 phosphorylation has been shown to be involved in neurogenesis in the DG after cerebral ischemic stroke in rodents (Zhu et al., 2004). We found that, compared with sham-operated or vehicle-treated pMCAO rats, immunostaining of phospho-CREBSer133 was enhanced in both the SVZ and DG following treatment with SB on Day 7 (Fig. S10A & B), and this occurred concurrently with potentiated BrdU labeling. These SB-induced effects were blocked by K252a co-treatment. Moreover, phospho-CREBSer133 and BrdU showed co-localization, notably in the SVZ and DG (Fig. S10A–C). SB-induced nestin immunostaining in the frontal cortex was also suppressed by K252a co-treatment on Day 7 after pMCAO (Fig. S10D). SB or TSA treatment for 7 days produced only marginal effects in BDNF immunostaining in the contralateral SVZ, or p-CREB immunostaining in the contralateral DG subgranular zone, respectively, compared with sham-operated control (data not shown).
Role of BDNF-TrkB signaling in SB-induced long-term behavioral benefits in pMCAO rats
Previous studies from our laboratory showed that treatment with SB or other HDAC inhibitors improved performance in motor, sensory, and reflex tests conducted relatively soon after ischemic onset (Kim et al., 2007). The results of the present study indicate that the decrease in retention time on an accelerating rotarod in vehicle-treated pMCAO rats was completely prevented by SB treatment measured two weeks after pMCAO (Fig. 7A). The severe impairments in motor, sensory, and reflex performance detected on an eight-point behavioral test two weeks after ischemia were also largely reversed in SB-treated pMCAO rats (Fig. 7B). When SB and K252a were administered together, K252a significantly suppressed the beneficial effects of SB on the rotarod and eight-point tests performed seven days after ischemia (Fig. 7C & D), despite the fact that K252a alone had minimal behavioral effects. These results suggest that BDNF-TrkB signaling is involved in SB-induced long-term behavioral improvement after cerebral ischemia.
Fig. 7. Long-term behavioral benefits of SB in pMCAO rats were sensitive to K252a treatment.

(A & B) Post-insult SB treatment largely prevented ischemia-induced neurological deficits as determined on Day 14 after pMCAO by rotarod (A) and an eight-point behavioral test (B). Data were analyzed from sham-operated animals (n = 5), vehicle (normal saline)-treated (n = 6), and SB-treated animals (n = 6). Data are mean ± SEM. *p< 0.05, and ***p<0.001 compared with the vehicle group.
(C & D) K252a attenuated the beneficial effects of SB as determined by rotarod (C) and eight-point behavioral test (D) on Day 7 after pMCAO. Data were obtained from sham-operated animals (n = 7), vehicle (normal saline)-treated (n = 9), SB-treated (n = 9), K252a + SB-treated (n = 9), and K252a-treated animals (n = 10). Data are mean ± SEM. **p< 0.01, and ***p<0.001, comparison between the indicated groups.
Discussion
This study had several notable results. First, we demonstrated that treatment with the HDAC inhibitors SB or TSA markedly expanded the population of proliferating cells detected by confocal microscopy of BrdU and Ki67-immunostaining in the SVZ and DG—both of which are traditional neurogenic areas—of the ipsilateral ischemic brain hemisphere of rats that underwent pMCAO (Fig. 1 & Fig. S7). Second, we found that BrdU and NeuN were not co-localized in either the SVZ or DG on Day 7 and Day 14 after treatment with HDAC inhibitors. These results suggest that the newborn cells had not yet differentiated into mature neurons at these times, consistent with the report that transient MCAO-induced newborn cells do not express mature neuronal markers, such as NeuN and Hu, up to three weeks after injury (Jin et al., 2001). Third, we found that pMCAO also enhanced cell proliferation in injured brain areas, including the striatum and frontal cortex (Fig. 3, Fig. 4 & Fig. S7), similar to previous reports (Jin et al., 2003, Yamashita et al., 2006); treatment with SB or TSA markedly enhanced BrdU-staining in both brain regions and largely restored their loss of NeuN-expressing cells resulting from pMCAO. It should be noted that some BrdU-positive cells overlapped with NeuN-expressing cells in the frontal cortex of ischemic rats treated with SB (Fig. 4). Finally, cells expressing GFAP or nestin, a neuroblast marker, were also increased in the striatum and frontal cortex following injury, and further upregulated by treatment with SB or TSA (Fig. 3 & Fig. 4). A small number of cells co-expressed of BrdU with GFAP or nestin.
In addition, this study found that ischemia-induced increases in cells expressing PSA-NCAM were robustly further increased by SB treatment in the SVZ, aSVZ, and striatum, and extensive co-localization of PSA-NCAM and BrdU was observed (Fig. 2 & Fig. 3). These findings are remarkable for several reasons. PSA-NCAM is a neuroblast marker with multiple functions including migration, differentiation, survival, and plasticity. For example, PSA-NCAM knockout mice have a smaller olfactory bulb and less cell migration from the SVZ (Tomasiewicz et al., 1993; Chazal et al., 2000), and enzymatic removal of PSA-NCAM from the mouse SVZ causes premature differentiation (Petridis et al., 2004). Although cell migration has not been studied in this report, our results are in line with the view that SB treatment stimulates the migration of PSA-NCAM-positive neuroblasts from the SVZ to the injured striatum. In light of its multiple neuronal roles, the robust increases seen in PSA-NCAM may have important neurophysiological consequences. Taken together, the results of this study strongly suggest that the HDAC inhibitors SB and TSA stimulate ischemia-induced cell proliferation and neurogenesis in the SVZ, DG, striatum, and frontal cortex.
VPA—an anticonvulsant and mood-stabilizing drug frequently used to treat mood disorders—is another HDAC inhibitor that has been shown to induce hippocampal neurogenesis following treatment of normal rats for six weeks (Hao et al., 2004). It also stimulates neuronal differentiation of adult hippocampal neuronal progenitors through induction of NeuroD (Hsieh et al., 2004). However, it is well documented that HDAC inhibitors—including SB, VPA, and TSA—are neuroprotective in both in vitro and in vivo experimental settings, including cerebral ischemia in rodents (Endres et al., 2000; Ren et al., 2004; Kim et al., 2007). Chromatin remodeling via HDAC inhibition has been demonstrated to confer resistance to multiple insults in the central nervous system (reviewed in Langley et al., 2005). Therefore, it is possible that the effects of HDAC inhibitors observed in this report were in part due to their ability to protect ischemia-induced newborn cells from premature death.
We found that SB treatment was associated with marked upregulation of AH3 in the DG and SVZ (Fig. S5 & Fig. S9), compared with the vehicle control. Immunostaining revealed co-localization of AH3 with NeuN and BrdU. These results together with the western data (Fig. S5) suggest that HDAC inhibition did occur under our experimental conditions. Previous studies from our laboratory found that HDAC inhibition activates BDNF promoter IV and increases BDNF mRNA levels in dissociated rat cortical neurons (Yasuda et al., 2009). Here, we showed that BDNF protein levels were decreased in the ischemic brain, but this reduction was restored to normal levels by treatment with SB (Fig. 5 & Fig. S6). Two protein bands for BDNF were found by immunostaining (Fig. S6), presumably reflecting the detection of both pro-BDNF and mature BDNF (Matsumoto et al., 2008); both bands were augmented by SB, compared with vehicle control. Neuroprotective effects of BDNF in the neonatal brain against hypoxic-ischemic injury are supported by a report noting that intracerebrovascular administration of BDNF decreased infarct size and caspase-3 activation via the BDNF-TrkB signaling pathway (Han and Holtzman, 2000).
The key role that BDNF plays in neurogenesis has been increasingly recognized. Administration of BDNF into the lateral ventricle of adult rats leads to new neurons in the striatum, septum, thalamus, and hypothalamus (Pencea et al., 2001b), while BDNF knockout suppresses adult hippocampal neurogenesis (Lee et al., 2002). Injection of adenoviral BDNF into the ventricular zone substantially augments both neostriatal and olfactory neuronal recruitment in the adult rat brain (Benraiss et al., 2001). Moreover, neurogenesis induced by antidepressant treatment is mediated, at least in part, through potentiation of BDNF-TrkB signaling (D’sa and Duman, 2002). The role of BDNF-TrkB signaling in mediating HDAC inhibitor-induced cell proliferation, migration, and differentiation in the ischemic brain of pMCAO rats is supported by several of our observed findings, namely: (1) intraventricular injection of K252a decreased the number of BrdU or Ki67-positive cells induced by SB treatment in the ipsilateral SVZ, DG, and striatum (Fig. 6, Fig. S7 & Fig. S8); (2) K252a injection blocked SB-induced nestin expression in the ipsilateral frontal cortex (Fig. S10D); and (3) SB treatment up-regulated p-CREBSer133 in the ipsilateral SVZ and DG (Fig. S10A–C). It is interesting to note that p-CREB, an important transcription factor downstream of BDNF-TrkB signaling, has been shown to mediate neurogenesis in the adult DG after focal ischemia (Zhu et al., 2004). SB-induced upregulation of p-CREB in the SVZ and DG was blocked by treatment with K252a (Fig. S10). Furthermore, K252a also suppressed SB-induced BDNF upregulation in the aSVZ and frontal cortex (Fig. 5). Finally, the long-lasting behavioral effects of SB in pMCAO rats were also significantly attenuated by pre-treatment with K252a (Fig. 7). It has been suggested that the interaction of BDNF and TrkB induces migration of SVZ neuroblasts via activation of PI 3-kinase, MAP-kinase and p-CREB signaling pathways, in addition to promoting differentiation and survival of olfactory bulb interneurons (Chiaramello et al., 2007). On the other hand, it was recently reported that BDNF does not stimulate SVZ neurogenesis in rodents (Galvão et al., 2008). The difference between their and our results in SVZ cell proliferation/neurogenesis could arise from the fact that SVZ in the brain of normal rodents was employed in the previous report, while we studied SVZ in the ischemic brain where endogenous BDNF levels were up-regulated by SB treatment.
These results are consistent with the notion that SB-induced cell proliferation, migration, and differentiation in brain areas injured by ischemia may contribute to long-term behavioral improvement. Most notably, because there are no reliable treatments currently available for acute stroke, HDAC inhibitors—especially SB—should be clinically evaluated for their potential use in stroke patients.
Supplementary Material
{kind=link}
(A) p-CREBSer133 in the SVZ: (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BrdU (red), p-CREBSer133 (green), colocalized BrdU and p-CREBSer133 (yellow). Arrowheads identify cells with colocalized expression of BrdU and p-CREBSer133 (yellow).
(B) phospho-CREBSer133 in the DG. (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BrdU (red), p-CREBSer133 (green), colocalized BrdU and p-CREBSer133 (yellow). Arrowheads identify cells with colocalized expression of BrdU and p-CREBSer133 (yellow). Scale bar = 50 µm. Representative images from each group are shown (n = 4–6 rats per group).
(C) Consecutive confocal image planes (z series) of SB-treated animals showing colocalization of BrdU (+) and p-CREBSer133 (+) cells in the ipsilateral DG on Day 7 after pMCAO. Values show relative distance in the z direction from the confocal plane (0 µm). BrdU (red, top row), p-CREBSer133 (green, middle row), merged image (yellow, bottom row). The two markers nicely colocalize in the inner layer of the SGZ of the ipsilateral DG. Images are obtained from representative sections (n = 3–4 rats per group). Experimental conditions are described in the legend to Fig. 6. SVZ: subventricular zone, LV: lateral ventricle, H: hilus, SGZ: subgranular zone, GCL: granular cell layer of the DG.
(D) Confocal images of nestin-immunopositive cells in the rat ipsilateral frontal cortex seven days after pMCAO. SB treatment in rats undergoing induced stroke increased nestin (+) cell labeling compared with vehicle treated controls. However, K252a treatment attenuated this effect. (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a in the corresponding areas of frontal cortex. Nestin (green). Scale bar = 50 µm. Data were analyzed from 4–6 animals per group. Experimental conditions are described in the legend to Fig. 6.
{kind=link}
(A) shows the typical infarct area (as shown in the shaded area) in a brain section after pMCAO, and the regions of the SVZ, aSVZ, striatum and frontal cortex (as shown in boxes) used for immunohistochemical examinations. Also shown is the site of stereotaxic injection of K252 (the lateral ventricle). (B) shows the area in the hippocampal dentate gyrus used for immunohistochemical examinations. Abbreviations: FR COX, frontal cortex; P COX, periatal cortex; CC, corpus callosum; DG, dentate gyrus.
{kind=link}
Experimental conditions are as described in the legends to Fig. 2–Fig. 4. Data are mean ± SEM and were analyzed from 4 animals in each group. *p<0.05, **p<0.01, ***p<0.001, between the indicated groups.
{kind=link}
Experimental conditions are as described in the legends to Fig. 2–Fig. 4. Data are mean ± SEM and were analyzed from 4 animals in each group. *p<0.05, **p<0.01, ***p<0.001, between the indicated groups.
{kind=link}
Most AH3 (+) cells were colocalized with NeuN (+) cells.
(A) (a) Sham, (b) vehicle, (c) SB. Scale bar = 50 µm. (d–f) enlarged areas indicated in (a–c), respectively. Scale bar = 25 µm. AH3 (red), NeuN (green), colocalized AH3 and NeuN (yellow). Data obtained from representative confocal microscopy sections of rat brains (n = 3–4, each group).
(B) (a) SB-treated NeuN (+) cells (green). (b) SB-treated AH3 (+) cells (red). (c) Merged image of AH3 (+) and NeuN (+) cells (yellow). (d) Vehicle-treated NeuN (+) cells (green). (e) Vehicle-treated AH3 (+) cells (red). (c) Merged image of AH3 (+) and NeuN (+) cells (yellow). Scale bar = 25 µm. Experimental conditions are as described in the methods. H: hilus, SGZ: subgranular zone, GCL: granular cell layer.
(C) Western blots of AH3 in the ischemic brain hemispheres of sham-operated-vehicle-treated and SB-treated pMCAO rats. β-actin was used as the loading control.
(D) Quantification of the results shown in (C). Data are mean ± SEM (n = 3–4 animals per group). **p<0.01, between indicated groups.
{kind=link}
(A) Western blots of BDNF, p-CREBSer133, GFAP, and Bcl-2 in sham-operated, vehicle-treated and SB-treated rats (n = 3–4 per group) on Day 7 after pMCAO. β-actin was used as the loading control.
(B–D) Quantified analysis of Western blotting data of BDNF, p-CREBSer133, and GFAP, respectively, from (A). Data are mean ± SEM, *p < 0.05, **p< 0.01, ***p< 0.001, comparison between the indicated groups. Experimental conditions are described in the Methods.
{kind=link}
(A) (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a in the ischemic SVZ. BrdU (red), Ki67 (green), colocalized BrdU and Ki67 (yellow). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and Ki67 (n = 4–6 rats per group).
(B) Confocal image of BrdU and Ki67 labeled cells showing colocalization in the same nucleus of the ischemic striatum region after SB treatment. (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BrdU (red), Ki67 (green), colocalized BrdU and Ki67 (yellow). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and Ki67. Most BrdU (+) cells also show Ki67 colocalization (n = 4–6 rats per group). Experimental conditions are described in the legend to Fig. 6.
(C) Consecutive confocal image planes (z series) of SB-treated animals showing colocalization of BrdU (+) and Ki67 (+) cells in the striatum. Values show relative distance in the z direction from the confocal plane (0 µm). BrdU (red, top row), Ki67 (green, middle row), merged image (yellow, bottom row). Data are obtained from representative sections (n = 3–4 rats per group).
(D) TSA treatment enhanced BrdU and Ki67 labeling of cells in the ipsilateral SVZ, aSVZ, striatum, frontal cortex, and DG on Day 7 after pMCAO. (a) SVZ, subventricular zone, LV, lateral ventricle, (b) aSVZ, anterior subventricular zone, (c) Stri, striatum, (d) Fcx, frontal cortex. (e) H, hilus, SGZ, subgranular zone, GCL, granule cell layer of the dentate gyrus. BrdU (red), Ki67 (green), colocalized BrdU and Ki67 (yellow). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and Ki67. Scale bar = 50 µm. (f–h) SVZ of TSA-treated group. (f) Arrows identify BrdU (+) cells (red). (g) Arrows identify Ki67 (+) cells (green). (h) Arrowheads identify cells with colocalized expression of BrdU and Ki67 (merged image, yellow). (i–k) DG of TSA-treated group. (i) Arrows identify BrdU (+) cells (red). (j) Arrows identify Ki67 (+) cells (green). (k) Arrowheads identify cells with colocalized expression of BrdU and Ki67 (merged image, yellow). SVZ: subventricular zone, LV: lateral ventricle, H: hilus, SGZ: subgranular zone, GCL: granular cell layer.
(E) BrdU/Ki67 immunostaining in the contralateral SVZ of the TSA-treated group. BrdU (red), Ki67 (green). SVZ: subventricular zone, LV: lateral ventricle.
(F) BrdU/Ki67 immunostaining in the contralateral striatum of the TSA-treated group. BrdU (red), Ki67 (green). Stri: striatum.
{kind=link}
Experimental conditions are as described in the legend to Fig. 6. Data are mean ± SEM and were analyzed from 4 animals in each group. *p<0.05, ***p<0.001, between the indicated groups.
{kind=link}
(A) (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BrdU (red), acetylated H3 (AH3) (green). Arrows identify BrdU (+) cells, (n = 4–6 rats per group). Scale bar = 50 µm. (f–g) Enlargement of the indicated area in (c) showing colocalization in the SB-treated ipsilateral SVZ region. (f) BrdU (red), (g) AH3 (green), (h) merged image (yellow).
(B) (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BrdU (red), acetylated H3 (AH3) (green), colocalized BrdU and AH3 (yellow). Arrowheads identify cells with colocalized expression of BrdU and AH3 (yellow). Scale bar = 50 µm. Representative images from each group are shown (n = 4–6 rats per group).
(C) Consecutive confocal image planes (z series) of SB-treated animals showing colocalization of BrdU (+) and AH3 (+) cells in the ipsilateral DG after pMCAO. Values show relative distance in the z direction from the confocal plane (0 µm). BrdU (red, top row), AH3 (green, middle row), merged image (yellow, bottom row). Results are obtained from representative sections (n = 3–4 rats per group). SVZ: subventricular zone, LV: lateral ventricle, H: hilus, DG: dentate gyrus, SGZ: subgranular zone, GCL: granular cell layer of the DG.
{kind=link}
Acknowledgements
This study was supported by the Intramural Research Program, NIMH, NIH. We thank Dr. Carolyn Smith of NINDS, NIH for technical assistance with the confocal microscope study. We are grateful for the assistance of Ioline Henter of NIMH and the NIH Fellows Editorial Board for their review of this manuscript.
Footnotes
Additional Supporting information may be found in the online version of this article
References
- Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J. Neurosci. 2002;22:629–634. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neural replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Barnabé-Heider F, Miller FD. Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J. Neurosci. 2003;23:5149–5160. doi: 10.1523/JNEUROSCI.23-12-05149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benraiss A, Chmielnicki E, Lerner K, Roh D, Goldman SA. Adenoviral brain derived neurotrophic factor induces both neostriatal and olfactory neuronal recruitment from endogenous progenitor cells in the adult forebrain. J. Neurosci. 2001;21:6718–6731. doi: 10.1523/JNEUROSCI.21-17-06718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfanti L. PSA-NCAM in mammalian structural plasticity and neurogenesis. Prog. Neurobiol. 2006;80:129–164. doi: 10.1016/j.pneurobio.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Chazal G, Durbec P, Jankovski A, Rougon G, Cremer H. Consequences of neural cell adhesion molecule deficiency on cell migration in the rostral migratory stream of the mouse. J. Neurosci. 2000;20:1446–1457. doi: 10.1523/JNEUROSCI.20-04-01446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaramello S, Dalmasso G, Bezin L, Marcel D, Jourdan F, Peretto P, Fasolo A, De Marchis S. BDNF/TrkB interaction regulates migration of SVZ precursor cells via PI3-K and MAP-K signalling pathways. Eur. J. Neurosci. 2007;26:1780–1790. doi: 10.1111/j.1460-9568.2007.05818.x. [DOI] [PubMed] [Google Scholar]
- D’sa C, Duman RS. Antidepressants and neuroplasticity. Bipolar Disord. 2002;4:183–194. doi: 10.1034/j.1399-5618.2002.01203.x. [DOI] [PubMed] [Google Scholar]
- Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, Lipski A, Jaenisch R, Moskowitz MA, Dirnagl U. DNA methyltransferase contributes to delayed ischemic brain injury. J. Neurosci. 2000;20:3175–3181. doi: 10.1523/JNEUROSCI.20-09-03175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson P, Perfilieva E, Björk-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult hippocampus. Nature. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Galvão RP, Garcia-Verdugo JM, Alvarez-Buylla A. Brain-derived neurotrophic factor signaling does not stimulate subventricular zone neurogenesis in adult mice and rats. J. Neurosci. 2008;28:13368–13383. doi: 10.1523/JNEUROSCI.2918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachino CG, De Marchis C, Giampietro G, Parlato R, Perroteau I, Schütz G, Fasolo A, Peretto P. cAMP response element-binding protein regulates differentiation and survival of newborn neurons in the olfactory bulb. J. Neurosci. 2005;25:10105–10118. doi: 10.1523/JNEUROSCI.3512-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Reeves AJ, Graziano MSA, Groos CG. Neurogenesis in the neocortex of adult primates. Science. 1999;286:548–552. doi: 10.1126/science.286.5439.548. [DOI] [PubMed] [Google Scholar]
- Gu W, Brännström T, Wester P. Cortical neurogenesis in adult rats after reversible photothrombotic stroke. J. Cereb. Blood Flow Metab. 2000;20:1166–1173. doi: 10.1097/00004647-200008000-00002. [DOI] [PubMed] [Google Scholar]
- Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J. Neurosci. 2000;20:5775–5781. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao YH, Creson T, Zhang L, Li P, Du F, Yuan P, Gould TD, Manji HK, Chen G. Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J. Neurosci. 2004;24:6590–6599. doi: 10.1523/JNEUROSCI.5747-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc. Natl. Acad. Sci. USA. 2004;101:16659–16664. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai M, Sato K, Kamada H, Omori N, Nagano I, Shoji M, Abe K. Temporal profile of stem cell division, migration, and differentiation from subventricular zone to olfactory bulb after transient forebrain ischemia in gerbils. J. Cereb. Blood Flow Metab. 2003;23:331–341. doi: 10.1097/01.WCB.0000050060.57184.E7. [DOI] [PubMed] [Google Scholar]
- Jin K, Minami M, Lan JQ, Mao XO, Batteur S, Simon RP, Greenberg DA. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc. Natl. Acad. Sci. USA. 2001;98:4710–4715. doi: 10.1073/pnas.081011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Sun Y, Xie L, Peel A, Mao XO, Batteur S, Greenberg DA. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol. Cell. Neurosci. 2003;24:171–189. doi: 10.1016/s1044-7431(03)00159-3. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takagi N, Miyake-Takagi K, Okuyama N, Mochizuki N, Takeo S. Characterization of BrdU-positive neurons induced by transient global ischemia in adult hippocampus. J. Cereb. Blood Flow Metab. 2004;24:548–555. doi: 10.1097/00004647-200405000-00009. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Jessberger S, Steiner B, Kronenberg G. Milestones of neuronal development in the adult hippocampus. Trends Neurosci. 2004;27:447–452. doi: 10.1016/j.tins.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang D-M. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J. Pharmacol. Exp. Thera. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- Kitagawa K. CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J. 2007;274:3210–3217. doi: 10.1111/j.1742-4658.2007.05890.x. [DOI] [PubMed] [Google Scholar]
- Kornack DR, Rakic P. The generation, migration, and differentiation of olfactory neurons in the adult primate brain. Proc. Natl. Acad. Sci. USA. 2001;98:4752–4757. doi: 10.1073/pnas.081074998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, Gensert JM, Beal MF, Ratan RR. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr. Drug Targets CNS Neurol. Disord. 2005;4:41–50. doi: 10.2174/1568007053005091. [DOI] [PubMed] [Google Scholar]
- Lee J, Duan W, Mattson MP. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 2002;82:1367–1375. doi: 10.1046/j.1471-4159.2002.01085.x. [DOI] [PubMed] [Google Scholar]
- Lee SR, Kim HY, Rogowska J, Zhao BQ, Bhide P, Parent JM, Lo EH. Involvement of matrix metalloproteinase in neuroblast cell migration from the subventricular zone after stroke. J. Neurosci. 2006;26:3491–3495. doi: 10.1523/JNEUROSCI.4085-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenwalner RJ, Parent JM. Adult neurogenesis and the ischemic forebrain. J. Cereb. Blood Flow Metab. 2006;26:1–20. doi: 10.1038/sj.jcbfm.9600170. [DOI] [PubMed] [Google Scholar]
- Liu J, Solway K, Messing RO, Sharp FR. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J. Neurosci. 1998;18:7768–7778. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magavi SS, Leavitt BR, Macklis JD. Induction of neurogenesis in the neocortex of adult mice. Nature. 2000;405:951–955. doi: 10.1038/35016083. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde Y-A. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nature Neurosci. 2008;11:131–133. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Kim JE, Lee R, Malberg JE, Chen J, Steffen C, Zhang YJ, Nestler EJ, Duman RS. Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J. Neurosci. 2002;22:3673–3682. doi: 10.1523/JNEUROSCI.22-09-03673.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann. Neurol. 2002;52:802–813. doi: 10.1002/ana.10393. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic Press; 1982. [Google Scholar]
- Pencea V, Bingaman KD, Freedman LJ, Luskin MB. Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp Neurol. 2001a;172:1–16. doi: 10.1006/exnr.2001.7768. [DOI] [PubMed] [Google Scholar]
- Pencea V, Bingaman KD, Wiegand SJ, Luskin MB. Infusion of brain-derived neurothophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J. Neurosci. 2001b;21:6706–6717. doi: 10.1523/JNEUROSCI.21-17-06706.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petridis AK, El Maarouf A, Rutishauser U. Polysialic acid regulates cell contact-dependent neuronal differentiation of progenitor cells from the subventricular zone. Develop. Dynamics. 2004;230:675–684. doi: 10.1002/dvdy.20094. [DOI] [PubMed] [Google Scholar]
- Ren M, Leng Y, Jeong M, Leeds PR, Chuang D-M. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J. Neurochem. 2004;89:1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- Sairanen M, Lucas G, Ernfors P, Castren M, Castren E. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J. Neurosci. 2005;25:1089–1094. doi: 10.1523/JNEUROSCI.3741-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J. Cell. Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Senatorov VV, Ren M, Kanai H, Wei H, Chuang D-M. Short-term lithium treatment promotes neuronal survival and proliferation in rat striatum infused with quinolinic acid, a model of Huntington’s disease. Mol. Psychiatry. 2004;9:371–385. doi: 10.1038/sj.mp.4001463. [DOI] [PubMed] [Google Scholar]
- Tomasiewicz H, Ono K, Yee D, Thompson C, Goridis C, Rutishauser U, Magnuson T. Genetic deletion of a neural cell adhesion molecule variant (N-CAM-180) produces distinct defects in the central nervous system. Neuron. 1993;11:1163–1174. doi: 10.1016/0896-6273(93)90228-j. [DOI] [PubMed] [Google Scholar]
- Xu J, Zhang Q-G, Li C, Zhang G-Y. Subtoxic N-methyl-D-aspartate delayed neuronal death in ischemic brain injury through TrkB receptor- and calmodulin-mediated PI-3K/Akt pathway activation. Hippocampus. 2007;17:525–537. doi: 10.1002/hipo.20289. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Ninomiya M, Hernández Acosta P, García-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J. Neurosci. 2006;26:6627–6636. doi: 10.1523/JNEUROSCI.0149-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang D-M. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol. Psychiatry. 2009;14:51–59. doi: 10.1038/sj.mp.4002099. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhang Z, Zhang C, Zhang L, Robin A, Wang Y, Lu M, Chopp M. Stroke transiently increases subventricular zone cell division from asymmetric to symmetric and increases neuronal differentiation in the adult rat. J. Neurosci. 2004;24:5810–5815. doi: 10.1523/JNEUROSCI.1109-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu DY, Lau L, Liu SH, Wei JS, Lu YM. Activation of cAMP-response-element-binding protein (CREB) after focal cerebral ischemia stimulates neurogenesis in the adult dentate gyrus. Proc. Natl. Acad. Sci. USA. 2004;101:9453–9457. doi: 10.1073/pnas.0401063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) p-CREBSer133 in the SVZ: (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BrdU (red), p-CREBSer133 (green), colocalized BrdU and p-CREBSer133 (yellow). Arrowheads identify cells with colocalized expression of BrdU and p-CREBSer133 (yellow).
(B) phospho-CREBSer133 in the DG. (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BrdU (red), p-CREBSer133 (green), colocalized BrdU and p-CREBSer133 (yellow). Arrowheads identify cells with colocalized expression of BrdU and p-CREBSer133 (yellow). Scale bar = 50 µm. Representative images from each group are shown (n = 4–6 rats per group).
(C) Consecutive confocal image planes (z series) of SB-treated animals showing colocalization of BrdU (+) and p-CREBSer133 (+) cells in the ipsilateral DG on Day 7 after pMCAO. Values show relative distance in the z direction from the confocal plane (0 µm). BrdU (red, top row), p-CREBSer133 (green, middle row), merged image (yellow, bottom row). The two markers nicely colocalize in the inner layer of the SGZ of the ipsilateral DG. Images are obtained from representative sections (n = 3–4 rats per group). Experimental conditions are described in the legend to Fig. 6. SVZ: subventricular zone, LV: lateral ventricle, H: hilus, SGZ: subgranular zone, GCL: granular cell layer of the DG.
(D) Confocal images of nestin-immunopositive cells in the rat ipsilateral frontal cortex seven days after pMCAO. SB treatment in rats undergoing induced stroke increased nestin (+) cell labeling compared with vehicle treated controls. However, K252a treatment attenuated this effect. (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a in the corresponding areas of frontal cortex. Nestin (green). Scale bar = 50 µm. Data were analyzed from 4–6 animals per group. Experimental conditions are described in the legend to Fig. 6.
(A) shows the typical infarct area (as shown in the shaded area) in a brain section after pMCAO, and the regions of the SVZ, aSVZ, striatum and frontal cortex (as shown in boxes) used for immunohistochemical examinations. Also shown is the site of stereotaxic injection of K252 (the lateral ventricle). (B) shows the area in the hippocampal dentate gyrus used for immunohistochemical examinations. Abbreviations: FR COX, frontal cortex; P COX, periatal cortex; CC, corpus callosum; DG, dentate gyrus.
Experimental conditions are as described in the legends to Fig. 2–Fig. 4. Data are mean ± SEM and were analyzed from 4 animals in each group. *p<0.05, **p<0.01, ***p<0.001, between the indicated groups.
Experimental conditions are as described in the legends to Fig. 2–Fig. 4. Data are mean ± SEM and were analyzed from 4 animals in each group. *p<0.05, **p<0.01, ***p<0.001, between the indicated groups.
Most AH3 (+) cells were colocalized with NeuN (+) cells.
(A) (a) Sham, (b) vehicle, (c) SB. Scale bar = 50 µm. (d–f) enlarged areas indicated in (a–c), respectively. Scale bar = 25 µm. AH3 (red), NeuN (green), colocalized AH3 and NeuN (yellow). Data obtained from representative confocal microscopy sections of rat brains (n = 3–4, each group).
(B) (a) SB-treated NeuN (+) cells (green). (b) SB-treated AH3 (+) cells (red). (c) Merged image of AH3 (+) and NeuN (+) cells (yellow). (d) Vehicle-treated NeuN (+) cells (green). (e) Vehicle-treated AH3 (+) cells (red). (c) Merged image of AH3 (+) and NeuN (+) cells (yellow). Scale bar = 25 µm. Experimental conditions are as described in the methods. H: hilus, SGZ: subgranular zone, GCL: granular cell layer.
(C) Western blots of AH3 in the ischemic brain hemispheres of sham-operated-vehicle-treated and SB-treated pMCAO rats. β-actin was used as the loading control.
(D) Quantification of the results shown in (C). Data are mean ± SEM (n = 3–4 animals per group). **p<0.01, between indicated groups.
(A) Western blots of BDNF, p-CREBSer133, GFAP, and Bcl-2 in sham-operated, vehicle-treated and SB-treated rats (n = 3–4 per group) on Day 7 after pMCAO. β-actin was used as the loading control.
(B–D) Quantified analysis of Western blotting data of BDNF, p-CREBSer133, and GFAP, respectively, from (A). Data are mean ± SEM, *p < 0.05, **p< 0.01, ***p< 0.001, comparison between the indicated groups. Experimental conditions are described in the Methods.
(A) (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a in the ischemic SVZ. BrdU (red), Ki67 (green), colocalized BrdU and Ki67 (yellow). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and Ki67 (n = 4–6 rats per group).
(B) Confocal image of BrdU and Ki67 labeled cells showing colocalization in the same nucleus of the ischemic striatum region after SB treatment. (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BrdU (red), Ki67 (green), colocalized BrdU and Ki67 (yellow). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and Ki67. Most BrdU (+) cells also show Ki67 colocalization (n = 4–6 rats per group). Experimental conditions are described in the legend to Fig. 6.
(C) Consecutive confocal image planes (z series) of SB-treated animals showing colocalization of BrdU (+) and Ki67 (+) cells in the striatum. Values show relative distance in the z direction from the confocal plane (0 µm). BrdU (red, top row), Ki67 (green, middle row), merged image (yellow, bottom row). Data are obtained from representative sections (n = 3–4 rats per group).
(D) TSA treatment enhanced BrdU and Ki67 labeling of cells in the ipsilateral SVZ, aSVZ, striatum, frontal cortex, and DG on Day 7 after pMCAO. (a) SVZ, subventricular zone, LV, lateral ventricle, (b) aSVZ, anterior subventricular zone, (c) Stri, striatum, (d) Fcx, frontal cortex. (e) H, hilus, SGZ, subgranular zone, GCL, granule cell layer of the dentate gyrus. BrdU (red), Ki67 (green), colocalized BrdU and Ki67 (yellow). Arrows identify BrdU (+) cells. Arrowheads identify cells with colocalized expression of BrdU and Ki67. Scale bar = 50 µm. (f–h) SVZ of TSA-treated group. (f) Arrows identify BrdU (+) cells (red). (g) Arrows identify Ki67 (+) cells (green). (h) Arrowheads identify cells with colocalized expression of BrdU and Ki67 (merged image, yellow). (i–k) DG of TSA-treated group. (i) Arrows identify BrdU (+) cells (red). (j) Arrows identify Ki67 (+) cells (green). (k) Arrowheads identify cells with colocalized expression of BrdU and Ki67 (merged image, yellow). SVZ: subventricular zone, LV: lateral ventricle, H: hilus, SGZ: subgranular zone, GCL: granular cell layer.
(E) BrdU/Ki67 immunostaining in the contralateral SVZ of the TSA-treated group. BrdU (red), Ki67 (green). SVZ: subventricular zone, LV: lateral ventricle.
(F) BrdU/Ki67 immunostaining in the contralateral striatum of the TSA-treated group. BrdU (red), Ki67 (green). Stri: striatum.
Experimental conditions are as described in the legend to Fig. 6. Data are mean ± SEM and were analyzed from 4 animals in each group. *p<0.05, ***p<0.001, between the indicated groups.
(A) (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BrdU (red), acetylated H3 (AH3) (green). Arrows identify BrdU (+) cells, (n = 4–6 rats per group). Scale bar = 50 µm. (f–g) Enlargement of the indicated area in (c) showing colocalization in the SB-treated ipsilateral SVZ region. (f) BrdU (red), (g) AH3 (green), (h) merged image (yellow).
(B) (a) Sham, (b) vehicle, (c) SB, (d) K252a + SB, (e) K252a. BrdU (red), acetylated H3 (AH3) (green), colocalized BrdU and AH3 (yellow). Arrowheads identify cells with colocalized expression of BrdU and AH3 (yellow). Scale bar = 50 µm. Representative images from each group are shown (n = 4–6 rats per group).
(C) Consecutive confocal image planes (z series) of SB-treated animals showing colocalization of BrdU (+) and AH3 (+) cells in the ipsilateral DG after pMCAO. Values show relative distance in the z direction from the confocal plane (0 µm). BrdU (red, top row), AH3 (green, middle row), merged image (yellow, bottom row). Results are obtained from representative sections (n = 3–4 rats per group). SVZ: subventricular zone, LV: lateral ventricle, H: hilus, DG: dentate gyrus, SGZ: subgranular zone, GCL: granular cell layer of the DG.