

Abstract
The expression of the methylated DNA-binding protein MeCP2 increases during neuronal development, which suggests that this epigenetic factor is crucial for neuronal terminal differentiation. We evaluated dendritic and axonal development in embryonic day-18 hippocampal neurons in culture by measuring total length and counting branch point numbers at 4 days in vitro, well before synapse formation. Pyramidal neurons transfected with a plasmid encoding a small hairpin RNA (shRNA) to knockdown endogenous Mecp2 had shorter dendrites than control untransfected neurons, without detectable changes in axonal morphology. On the other hand, overexpression of wildtype (wt) human MECP2 increased dendritic branching, in addition to axonal branching and length. Consistent with reduced neuronal growth and complexity in Rett syndrome (RTT) brains, overexpression of human MECP2 carrying missense mutations common in RTT individuals (R106W or T158M) reduced dendritic and axonal length. One of the targets of MeCP2 transcriptional control is the Bdnf gene. Indeed, endogenous Mecp2 knockdown increased the intracellular levels of BDNF protein compared to untransfected neurons, suggesting that MeCP2 represses Bdnf transcription. Surprisingly, overexpression of wt MECP2 also increased BDNF levels, while overexpression of RTT-associated MECP2 mutants failed to affect BDNF levels. The extracellular BDNF scavenger TrkB-Fc prevented dendritic overgrowth in wt MECP2-overexpressing neurons, while overexpression of the Bdnf gene reverted the dendritic atrophy caused by Mecp2-knockdown. However, this effect was only partial, since Bdnf increased dendritic length only to control levels in mutant MECP2-overexpressing neurons, but not as much as in Bdnf-transfected cells. Our results demonstrate that MeCP2 plays varied roles in dendritic and axonal development during neuronal terminal differentiation, and that some of these effects are mediated by autocrine actions of BDNF.
Keywords: MeCP2, Rett, dendrite, axon, pyramidal neuron, BDNF, hippocampus
Introduction
Neurodevelopmental disorders caused by genetic disruptions or chemical/physical insults during brain development affect neuronal terminal differentiation, i.e. axon and dendrite growth and branching, as well as synaptogenesis, thus impairing synaptic transmission and plasticity leading to altered network connectivity (Chechlacz and Gleeson, 2003; Johnston et al., 2001). Reduced dendritic length and branching have been demonstrated in many disorders associated with mental retardation (Kaufmann and Moser, 2000). One disorder associated with impaired brain development is Rett syndrome (RTT), a disease that affects nearly 1:15,000 females worldwide (Hagberg et al., 1985). Early development appears normal in individuals with RTT until approximately 6-18 months of age, when physical, motor and social-cognitive behavior enter a period of regression (Hagberg, 2002; Percy and Lane, 2005). Indeed, the neuropathology of RTT reveals several areas of abnormal brain development. Head circumference is decelerated, and postmortem observations of RTT brains demonstrate a reduction in brain weight (Armstrong et al., 1999; Jellinger et al., 1988; Schultz et al., 1993). Microscopically, brain autopsy material from RTT patients revealed impaired dendritic growth and complexity of pyramidal cells in the frontal and motor cortices, as well as in the subiculum (Armstrong et al., 1995), and reduced levels of MAP-2, a dendritic protein involved in microtubule stabilization (Kaufmann et al., 2000; Kaufmann et al., 1995; Kaufmann et al., 1997). These observations support the notion that RTT is a disorder caused by impaired dendritic, axonal and eventually synapse formation and maturation, leaving afflicted individuals in a state of arrested brain development (Armstrong, 2002; Johnston et al., 2001; Johnston et al., 2003; Kaufmann et al., 2005; LaSalle, 2004; Naidu, 1997; Philippart, 2001).
The majority of RTT cases are associated with mutations in a gene located on the X chromosome that encodes methyl-CpG-binding protein 2 (MeCP2) (Amir et al., 1999; Bienvenu et al., 2000; Van den Veyver and Zoghbi, 2001; Wan et al., 1999). MeCP2 is a nuclear protein that binds DNA specifically to A/T rich sites in close proximity to methylated CpG (cytidine-phospodiester-guanosine) islands. After binding to these methylated CpG sites, MeCP2 recruits the mSin3a co-repressor, which contains histone deacetylase complexes, thereby altering the structure of genomic DNA and regulating the transcription of specific target genes (Jones et al., 1998; Klose et al., 2005; Nan and Bird, 2001; Nan et al., 1997; Nan et al., 1998). DNA methylation is a common mechanism of silencing genes during cellular differentiation, and MeCP2 may play a critical role in neuronal terminal differentiation by reading this epigenetic code. Furthermore, MeCP2 expression in cortical neurons increases during brain development (Akbarian et al., 2001; Cohen et al., 2003, Jung, 2003 #21226; LaSalle et al., 2001; Shahbazian et al., 2002; Zoghbi, 2003). The developmental increase in its expression strongly suggests that MeCP2 plays a critical role in neuronal terminal differentiation, i.e. the development of axons, dendrites and dendritic spines leading to proper synapse formation and maturation (Cassel et al., 2004; Kaufmann et al., 2005; Kishi and Macklis, 2004; Matarazzo et al., 2004; Matarazzo and Ronnett, 2004; Mullaney et al., 2004). Missense mutations in MECP2 identified in RTT patients cluster in the methyl-binding domain (MBD) and transcriptional repressor domain (TRD) of the protein, suggesting that they represent loss-of-function mutations. Indeed, many of these mutations on the MBD of MeCP2 (like R106W and T158M used in the present studies; see below), reduce the affinity of MeCP2 for methylated DNA, thus impairing its ability to localize to heterochromatin and repress transcription in gene reporter assays (Kudo et al., 2001; Kudo et al., 2003).
To extend our knowledge of its role in neuronal terminal differentiation in the context of RTT, we manipulated MeCP2 expression levels in primary cultures of embryonic day-18 hippocampal neurons, a well-established experimental model for studies of neuronal differentiation (Banker and Goslin, 1998; Craig and Banker, 1994). At the time of plating, dissociated neurons were transfected with expression cDNA plasmids encoding either: (1) a small hairpin RNA (shRNA) interfering sequence to knockdown endogenous Mecp2 expression; (2) wildtype (wt) human MECP2; (3) one of two of the most common missense mutations in MECP2 found in RTT patients, R106W or T158M (www.mecp2.org.uk). Quantitative analyses of dendritic and axonal morphology (total length and number of branch points) were performed after 4 days in vitro, when a single defined axon and several dendrites are well defined (Dotti et al., 1988), and before synapse formation (Ziv and Smith, 1996). Considering that Bdnf (the gene coding for the neurotrophin brain-derived neurotrophic factor, BDNF) is a prominent gene shown to be regulated by MeCP2 (Abuhatzira et al., 2007; Chahrour et al., 2008; Chang et al., 2006; Chen et al., 2003; Klein et al., 2007; Martinowich et al., 2003; Ogier et al., 2007; Wang et al., 2006), we estimated intracellular BDNF protein levels by quantitative immunocytochemistry. Lastly, we tested whether Bdnf overexpression or extracellular BDNF scavenging with TrkB-Fc could revert the morphological effects of manipulations of MeCP2 levels. Our results demonstrate that MeCP2 plays varied roles in dendritic and axonal development during neuronal terminal differentiation, and that some of these effects are mediated by autocrine actions of BDNF.
Material and Methods
Cell Culture of Dissociated Hippocampal Neurons
All animal procedures followed national and international ethical guidelines, and were reviewed and approved by the IACUC at UAB on an annual basis. Hippocampal neurons were cultured from embryonic day-18 (E18) Sprague-Dawley rat embryos (Charles River, Wilmington, MA) as previously described (Moore et al., 2007). Briefly, both hippocampi were dissected and neurons were re-suspended in Neurobasal medium containing B-27 supplement with penicillin–streptomycin and L-glutamine (InVitrogen; Carlsbad, CA). Dissociated neurons were plated on glass cover slips coated with poly-L-lysine (Sigma Aldrich, St. Louis, MO) and cultured in Neurobasal medium (B-27 supplement, penicillin–streptomycin, L-glutamine) at 37°C in a 5% CO2 incubator.
Transfections of Cultured Neurons
Primary hippocampal neuron cultures were transfected using electroporation (Gresch et al., 2004). Approximately 30min after isolation of both hippocampi, 2,000,000 dissociated neurons were centrifuged and re-suspended in 100μl of rat neuron Nucleofector solution (Amaxa Biosystems; Cologne, Germany). Three micrograms of cDNA per expression plasmid was added in a 2mm electroporation cuvette. After electroporation following the manufacturer protocol, Neurobasal medium was added and neurons were plated at a density of 50,000 to 70,000 cells per well in a 12-well plate at 37°C in 5% CO2 for 4 days in vitro (div).
Expression cDNA plasmids encoding small hairpin RNA (shRNA) interfering sequences were obtained from Origene (Rockville, MD). shRNA interfering sequences consist of a 29-base pair target gene-specific sequence, a 7-base pair loop, followed by a 29-base pair reverse complementary sequence. The expression plasmids encoding shRNA sequences are under the control of the human U6 promoter. The MECP2-specific shRNA sequence AATGAGACAGCAGTCTTATGCTTCCAGAA (sequence #2), reduced MeCP2 protein levels by 65%, estimated by quantitative Western blot analyses of PC12 cells co-transfected with human wt MECP2 and shRNA plasmids (Supplemental Fig. 1A). Consistently, MeCP2 expression levels were 55% lower in hippocampal neurons transfected with the same shRNA plasmid (sequence #2) than in untransfected neighboring neurons, as estimated by quantitative MeCP2 immunofluorescence (see below and Supplemental Fig. 1B, D). Another shRNA sequence, TCAATAACAGCCGCTCCAGAGTCAGTAGT, which did not affect MeCP2 expression, was used as a control for off-target effects.
To overexpress MeCP2 protein levels, expression cDNA plasmids encoding human wildtype (wt) MECP2 were used as previously described (Kudo, 1998; Kudo et al., 2001; Kudo et al., 2003). These plasmids represent the originally isolated form of MeCP2, where the start methionine is in exon 2, known as MeCP2β (Kriaucionis and Bird, 2004) or MeCP2A (Mnatzakanian et al., 2004). Expression plasmids encoding two of the most common missense mutations in MECP2 (R106W and T158M) were constructed by site directed mutagenesis using PCR, where wt MECP2 was used as a template (Kudo et al., 2001). MeCP2 was tagged with enhanced green fluorescent protein (eGFP), which allowed the identification of transfected cells. Finally, the MeCP2-eGFP construct was cloned into an expression vector under control of the cytomegalovirus (CMV) promoter. These expression plasmids produced a twofold increase in MeCP2 protein expression compared to untransfected neighboring neurons, as measured by quantitative MeCP2 immunofluorescence (Supplemental Figure 1C, D). A cDNA plasmid expressing BDNF-GFP under the control of the CMV promoter was used to overexpress Bdnf in cultured neurons (kindly provided by Dr. Kojima); this GFP-tagged BDNF retains biological activity (Kojima et al., 2001).
Immunofluorescence
After 4 days in vitro, hippocampal neurons were fixed for 20min with 4% paraformaldehyde in 100mM phosphate buffer (PB). Cells were permeabilized on ice for 10min with 4% formaldehyde and 0.25% Triton, washed three times with 100mM PB saline (PBS), and then blocked with 10% bovine serum albumin for 1hr at 37°C. Primary and secondary antibodies were diluted in PBS and 3% normal horse serum and incubated overnight at 4°C, or for 2hrs at room temperature. Cover slips were washed 3 times for 10min and once with Milli-Q water, before they were dried and mounted with Vectashield and DAPI (Vector Laboratories, Burlingame, CA). The following antibodies were used: rabbit anti-MeCP2 (Millipore Corporation, Billerica, MA); rabbit anti-BDNF (Sigma); anti-mouse cascade blue; and anti-rabbit Texas red (Santa Cruz Biotechnology; Santa Cruz, CA). Mouse anti-actin and anti-α-tubulin were obtained from the Developmental Studies Hybridoma Bank under the auspices of NIH-NICHD and maintained by the Department of Biological Sciences, University of Iowa.
Quantitative Immunofluorescence
After 4 days in vitro, primary cultures were stained for BDNF or MeCP2 immunocytochemistry. Cells were incubated with primary antibodies overnight and an anti-Texas-red conjugated secondary antibody (Santa Cruz). Digital images were acquired using an Olympus IX70 epifluorescence microscope with a 100X (1.35 NA) oil-immersion objective, and using a Retiga 1300-cooled charge-coupled device (CCD) monochromatic camera (Qimaging, Surrey, British Columbia). Images were deconvolved using an empirical point-spread function of 0.1073μm in IPLab software (Scanalytics, BD Biosciences Bioimaging; Rockville, MD). To allow direct comparison of immunofluorescence intensities, the field of view always included more than one neuron and at least one of them was either transfected (i.e. expressing GFP for visualization) or untransfected (i.e. positive for DAPI, negative for GFP). Thus, BDNF or MeCP2 levels could be directly and quantitatively compared between transfected and untransfected neurons within the same field of view (neighboring cells). In rare circumstances, when a transfected cell was not in close proximity to an untransfected neuron, the closest untransfected neighboring cell was used for quantification. CCD camera exposure times were kept constant when taking images of separate fields of view.
Quantitative Analyses of Neuronal Morphology
Neuronal morphology was measured as described previously (Moore et al., 2007). After 4 days in vitro, embryonic hippocampal pyramidal neurons are polarized and extend a single axon and several dendrites that are highly branched. For quantitative morphological analyses, neurons were co-transfected with eGFP or stained with an anti-α-tubulin antibody. Digital images were acquired using an Olympus IX70 epifluorescence microscope with a 40X (1 NA) oil-immersion objective using a Retiga 1300-cooled CCD monochromatic camera (Qimaging). The length and branch points of the axons and dendrites were measured by tracing along each neuronal projection using ImageJ software (National Institutes of Health).
Statistical Analyses
Data were analyzed statistically using unpaired Student's t test or a one-way analysis of variance (ANOVA) followed by Tukey's procedure for multiple comparisons using Prism (GraphPad; San Diego, CA). P < 0.05 was considered significant. Data are presented as mean ± standard error of the mean (SEM). Compromise Power Analyses were performed to determine the statistical power given the number of observations (n), the mean, and the standard deviation of the mean (SD), using G*Power (Erdfelder et al., 1996). These power analyses yielded values of Statistical Power (1-β; where β is the Type-II error) larger than 0.95 (i.e., 95% confidence of accepting the null hypothesis when is true).
Results
We studied the role of MeCP2 on neuronal terminal differentiation using hippocampal neurons from 18 day-old rat embryos maintained in dissociated primary cell culture. Pyramidal neurons in this culture preparation undergo a well-characterized morphological differentiation of axons and dendrites (Banker and Goslin, 1998; Craig and Banker, 1994). Quantitative analyses of dendritic and axonal morphology in pyramidal neurons transfected with eGFP or stained with anti-α-tubulin antibodies were performed after 4 days in vitro, when a single defined axon and several dendrites are well defined (Supplemental Fig. 2) (Dotti et al., 1988). At this time, synapses have not formed yet (Ziv and Smith, 1996), allowing the isolation of MeCP2's role in neuronal differentiation from the well-known contribution of synaptically driven neuronal activity to this process (Cline and Haas, 2008). The morphological differentiation of dendrite and axons was estimated by measuring their total length and counting all their branch points in every neuron.
1. Endogenous Mecp2 Knockdown Reduced Dendritic Length Without Affecting Axonal Morphology
In PC12 cells, Mecp2 knockdown using antisense oligonucleotides prevented neurite outgrowth induced by nerve growth factor (NGF) (Cusack et al., 2004). We first demonstrated that shRNA-mediated knockdown caused a 55% decrease in MeCP2 protein levels, as measured by Western blots from PC12 cells and quantitative immunocytochemistry of cultured hippocampal pyramidal neurons (p<0.05; Supplemental Fig. 1). After 4 days in vitro, Mecp2 knockdown caused a significant reduction of dendritic length (control shRNA sequence = 105.2 ± 7.54μm, n=5 vs Mecp2 shRNA = 50.43 ± 5.11μm, n=20; p<0.001; Fig. 1A, C), without affecting the number of dendritic branch points per neuron (control shRNA = 3 ± 0.55 branch points/neuron, n=5 vs. Mecp2 shRNA = 3.46 ± 0.37 branch points/neuron, n=20; p=0.573; Fig. 1A, C). On the other hand, neither axonal length (control shRNA = 139.7 ± 18.81μm length, n=5 vs. Mecp2 shRNA = 99.68 ± 24.89μm length, n=5; p=0.236) nor the number of axonal branch points were significantly affected by Mecp2 knockdown (shRNA control = 2.6 ± 0.51 branch points/neuron, n=5 vs. Mecp2 shRNA = 2.2 ± 0.73 branch points/neuron, n=5; p=0.668; Fig. 1A, B).
Figure 1. shRNA-mediated Knockdown of Endogenous Mecp2 Reduced Dendritic Length in Pyramidal Hippocampal Neurons, without Affecting Axonal Morphology.

A. Representative examples of 4 div hippocampal neurons transfected with a shRNA control sequence (left) or shRNA interfering sequence to knockdown endogenous Mecp2 (right). Neurons were co-transfected with eGFP (green) to perform quantitative morphological analyses (scale bar = 10μm). B. Population data on axonal length and branch points. C. Population data on dendritic length and branch points. In this and all subsequent figures, * indicates p<0.05, ** indicates p<0.01, and *** indicates p<0.001, from unpaired Student's t test or one-way ANOVA (see text for details). Additionally, in this and all subsequent figures, arrowheads indicate the position of the axons.
2. Overexpression of Wildtype Human MECP2 Increased Dendritic Branching, as well as Axonal Length and Branching
Quantitative immunocytochemistry of cultured pyramidal neurons demonstrated that expression plasmids using the CMV promoter caused a twofold increase in MeCP2 protein levels (p<0.05; Supplemental Fig. 1). These levels of immunolabeled MeCP2 reflect both the exogenously transfected protein plus the endogenously expressed MeCP2 (i.e. the endogenous Mecp2 gene was not manipulated). After 4 days in vitro, wt MECP2 overexpression did not affect dendritic length (GFP control = 108.5 ± 26μm, n=20 vs. wt MECP2 = 101.5 ± 10.6μm, n=18; p=0.05; Fig. 2A, C), but significantly increased the number of dendritic branch points (GFP control = 3.7 ± 0.53 branch points/neuron, n=20 vs. wt MECP2 = 6 ± 0.78 branch points/neuron, n=18; p=0.02; Figure 2A, C). On the other hand, both axonal length (GFP control = 147.1 ± 20μm, n=5 vs. wt MECP2 = 253.6 ± 10.79μm, n=5; p=0.002; Fig. 2A, B) and the number of branch points (GFP control = 2.4 ± 0.24 branch points/neuron, n=5 vs. wt MECP2 = 4.2 ± 0.37 branch points/neuron, n=5; p=0.004; Fig. 2A, B) were significantly increased after wt MECP2 overexpression.
Figure 2. The Overexpression of Wildtype MECP2 Increased Dendritic Branching and Axonal Growth, while the Overexpression of RTT-Associated MECP2 Mutations Reduced Dendritic and Axonal Development.

A. Representative examples of neurons transfected with either a control empty vector, a plasmid to overexpress wildtype human MECP2, or plasmids to overexpress to different missense MECP2 mutations (R106W or T158M) commonly found in Rett syndrome patients. Neurons were co-transfected with eGFP to perform quantitative morphological analyses (scale bar = 10μm). B. Population data on axonal length and branch points. C. Population data on dendritic length and branch points.
3. Overexpression of Missense MECP2 Mutations Common in Rett Syndrome Reduced Dendritic Length and Branching, as well as Axonal Length
Next, we transfected E18 hippocampal pyramidal neurons with expression plasmids encoding two different single-point missense mutations in human MECP2, R106W and T158M, which are located in the methyl-binding domain (MBD) of the MeCP2 protein and reduce its transcriptional repression activity (Kudo et al., 2001; Kudo et al., 2003). Of relevance to Rett syndrome, these are two of the most common missense mutations in MECP2 found in RTT patients (www.mecp2.org.uk). As in the previous experiments overexpressing wildtype human MECP2, the endogenous Mecp2 gene was not manipulated.
In contrast to the effects of wildtype MECP2, overexpression of either MECP2 mutation significantly reduced both dendritic length (GFP control = 108.5 ± 26μm vs. R106W = 43.26 ± 4.25μm, n=20; p=0.02; vs. T158M = 51.69 ± 7.7μm, n=20; p=0.04; Fig. 2A, C) and the number of dendritic branch points (GFP control = 3.7 ± 0.53 branch points/neuron, n=20 vs. R106W = 2.1 ± 0.34 branch points/neuron, n=20; p=0.01; vs. T158M = 2 ± 0.37 branch points/neuron, n=20; p=0.01; Fig. 2A, C). Axonal length was also significantly reduced in neurons overexpressing the R106W mutation (GFP control = 147.1 ± 20μm, n=5 vs. R106W = 81.67 ± 7.03μm, n=5; p=0.04; Fig. 2A, B), but not the T158M mutation (T158M = 84.78 ± 18.85μm, n=5; p=0.053; Fig. 2A, B). On the other hand, neither MECP2 mutation affected the number of axonal branch points (GFP control = 2.4 ± 0.24 branch points/neuron, n=5 vs. R106W = 2.8 ± 0.86 branch points/neuron, n=5; p=0.678; T158M = 1.8 ± 0.58 branch points/neuron, n=5; p=0.371; Fig. 2A, B).
4. Either Endogenous Mecp2 Knockdown or Overexpression of Wildtype MECP2 – but not of Rett-Associated MECP2 Mutations – Increased Intracellular BDNF Levels in Hippocampal Neurons
Considering that BDNF is a powerful modulator of dendritic, axonal and synaptic development (McAllister et al., 1999; Vicario-Abejon et al., 2002), the observation that Bdnf was a target of MeCP2 transcriptional repression (Chen et al., 2003; Martinowich et al., 2003) led to a so-called “BDNF hypothesis of Rett syndrome” (Amaral et al., 2007). Indeed, increased BDNF levels reverted some crucial RTT-like phenotypes in Mecp2 knockout mice (Chang et al., 2006; Ogier et al., 2007). However, whether MeCP2 represses or activates Bdnf gene transcription is still unclear (Cohen et al., 2008; Sun and Wu, 2006) and the specific mechanism/s and final BDNF protein levels may dependent on neuronal subtypes (Abuhatzira et al., 2007; Chahrour et al., 2008; Klein et al., 2007). In any case, deregulation of Bdnf transcription and BDNF protein levels by RTT-associated MECP2 mutations could have a profound impact on neuronal development.
To estimate intracellular BDNF protein levels in cultured hippocampal neurons after either rat Mecp2 knockdown or human MECP2 overexpression, we performed quantitative BDNF immunocytochemistry. As a benchmark for comparisons, transfection with a Bdnf-GFP expression plasmid under the control of the CMV promoter (Kojima et al., 2001) resulted in a 2.2-fold increase in BDNF immunofluorescence intensity, compared to an untransfected neighboring neuron (untransfected neurons 1649.83 ± 99 AU BDNF immunofluorescence intensity vs. BDNF-transfected neurons 3428 ± 62.72 AU BDNF immunofluorescence intensity; 2.19 ± 0.27 fold increase in immunofluorescence intensity, n=6; p=0.01; Fig. 3). Consistent with the function of a transcriptional repressor, endogenous Mecp2 knockdown caused a significant 1.5-fold increase in BDNF immunofluorescence levels (untransfected neurons 2140.83 ± 303.41 AU BDNF immunofluorescence intensity vs. shRNA Mecp2-transfected neurons 3150.5 ± 302.57 AU BDNF immunofluorescence intensity; 1.52 ± 0.08 fold increase in immunofluorescence intensity, n=6; p=0.002; Fig. 3). Surprisingly, overexpression of human wildtype MECP2 also resulted in a significant 1.4-fold increase in BDNF immunofluorescence levels (untransfected neurons 2120 ± 154.94 AU BDNF immunofluorescence intensity vs. wt MECP2 transfected neurons 2912.38 ± 139.22 AU BDNF immunofluorescence intensity; 1.41 ± 0.09 fold increase in immunofluorescence intensity, n=8; p=0.003; Fig. 3). These observations are consistent with BDNF mRNA levels observed in cultured cortical neurons from Mecp2 null mice in the absence of neuronal activity (i.e. in TTX) (Chen et al., 2003) or in cultured cortical neurons overexpressing wt MECP2 (Klein et al., 2007).
Figure 3. Mecp2 Knockdown or Wildtype MECP2 Overexpression Increased Intracellular BDNF Levels, while RTT-associated MECP2 Mutants Did Not Affect BDNF Levels.

A. Representative examples of BDNF immunostaining (red) in hippocampal neurons transfected with different expression plasmids (green). Neurons were transfected with expression plasmids to overexpress BDNF-GFP, wildtype MECP2 or MECP2 mutations tagged with GFP. Neurons were also transfected with an shRNA sequence to knockdown endogenous Mecp2 and eGFP (scale bar = 10μm). Images fields always included two or more transfected neurons (i.e. GFP positive) and untransfected neurons (i.e. GFP negative) to perform quantitative comparisons of BDNF immunofluorescence staining. B. Population data on BDNF immunofluorescence intensity normalized to untransfected neurons within the same fields of view.
We next examined whether Rett-associated MECP2 mutations affected intracellular BDNF protein levels. Overexpression of the R106W mutation did not change intracellular BDNF protein levels (untransfected neurons 3056.5 ± 211.6 AU BDNF immunofluorescence intensity vs. R106W-transfected neurons 3486.33 ± 258.5 AU BDNF immunofluorescence intensity; 1.18 ± 0.14 fold change in immunofluorescence intensity, n=6; p=0.310; Fig. 3). Likewise, the levels of BDNF protein were not affected by overexpression of the T158M mutation (untransfected neurons 2122.58 ± 96.72 AU BDNF immunofluorescence intensity vs. T158M-transfected neurons 1925.67 ± 112.77 AU BDNF immunofluorescence intensity; 0.92 ± 0.07 fold change in immunofluorescence intensity, n=12; p=0.232; Fig. 3). These observations demonstrate that in contrast to the overexpression of wildtype MECP2 or the knockdown of endogenous Mecp2, RTT-associated MECP2 mutations do not affect BDNF protein levels, suggesting that these mutations may represent a “toxic gain-of-function” and not a simple “loss-of-function”.
Considering the well-characterized effects of BDNF on dendritic and axonal growth (McAllister et al., 1999; Vicario-Abejon et al., 2002), and the changes in BDNF protein levels after Mecp2 knockdown or MECP2 overexpression (Fig. 3), we next tested whether the increased axonal and dendritic length and branching in wildtype MECP2-overexpressing neurons resulted from elevated BDNF levels. In addition, we setout to determine if Bdnf overexpression could rescue the neuronal atrophy observed after endogenous Mecp2 knockdown or expression of RTT-associated MECP2 mutations. In order to answer these questions, we first confirmed the consequences of Bdnf overexpression on axonal and dendritic length and complexity. It had been previously shown that the BDNF-GFP fusion protein encoded by an expression plasmid under control of the CMV promoter preserves biological activity (Kojima et al., 2001). Indeed, overexpression of BDNF-GFP in hippocampal pyramidal neurons for 4 days in vitro significantly increased dendritic length (GFP control = 108.5 ± 26μm, n=20 vs. BDNF-GFP = 267.7 ± 20.27μm, n=19; p<0.001; Supplemental Fig. 3A, C), and the number of dendritic branch points (GFP control = 3.7 ± 0.53 branch points/neuron, n=20 vs. BDNF-GFP = 5.8 ± 0.83 branch points/neuron, n=19; p=0.02; Supplemental Fig. 3A, C). On the other hand, BDNF-GFP overexpression only increased the number of axonal branch points (GFP control = 2.4 ± 0.24 branch points/neuron, n=5 vs. BDNF-GFP = 4.2 ± 0.37 branch points/neuron, n=5; p=0.02; Supplemental Fig. 3A, B), without affecting axonal length (GFP control = 147.1 ± 20μm, n=5 vs. BDNF-GFP = 155.9 ± 11.37μm, n=5; p=0.711; Supplemental Fig. 3A, B).
The soluble and membrane impermeable fusion protein TrkB-Fc inhibits BDNF-induced signaling by binding extracellular BDNF, thus preventing the activation of endogenous membrane-bound receptors (Shelton et al., 1995). Such “extracellular BDNF scavenging” prevents the effects of either exogenously applied or endogenously released BDNF (Amaral and Pozzo-Miller, 2007; Chen et al., 1999; Figurov et al., 1996; Kang et al., 1997; McAllister et al., 1997; Shimada et al., 1998; Tyler and Pozzo-Miller, 2001; Tyler et al., 2006). Application of TrkB-Fc (20μg/mL) to GFP-expressing control neurons for 4 days in vitro did not affect neither dendritic length/branching (54.5±4.88μm of dendritic length; 2.35 ± 0.47 branch points; n=17), or axonal length/branching (143.14 ± 9.79μm of axonal length; 2.0 ± 0.44 branch points, n=5; p>0.05 vs. GFP; Supplemental Fig. 3A-C), suggesting that release of endogenous BDNF is limited during this initial developmental period (i.e. before synapse formation). On the other hand, TrkB-Fc (20μg/mL) prevented the increase in dendritic length (TrkB-Fc + BDNF-GFP = 43.56 ± 6.5μm, n=9; p<0.001 vs. BDNF-GFP see above) and number of branch points (TrkB-Fc + BDNF-GFP = 0.11 ± 0.11 branch points/neuron, n=9; p<0.001 vs. BDNF-GFP see above) caused by BDNF-GFP expression (Supplemental Fig. 3A-C). In addition, TrkB-Fc (20μg/mL) prevented the increase in the number of axonal branch points in BDNF-GFP expressing neurons (TrkB-Fc + BDNF-GFP = 0.6 ± 0.25 branch points/neuron, n=5; p<0.001 vs. BDNF-GFP see above; Supplemental Fig. 3A-C).
Having confirmed that BDNF-GFP overexpression increases, and TrkB-Fc decreases, dendritic and axonal development in hippocampal neurons, we setout to determine whether the observed changes in neuronal morphology caused by wildtype human MECP2 overexpression or endogenous Mecp2 knockdown, as well as by expression of RTT-associated MECP2 mutations resulted from misregulated BDNF levels.
5. The BDNF Scavenger TrkB-Fc Prevented the Increase in Dendritic Branching Caused by Wildtype Human MECP2 Overexpression
Scavenging extracellular BDNF with TrkB-Fc allowed testing whether axonal and dendritic overgrowth caused by MECP2 overexpression was mediated by increased BDNF levels. Indeed, TrkB-Fc (20μg/mL) prevented the increase in the number of dendritic branch points observed in neurons overexpressing wt MECP2 (wt MECP2 + TrkB-Fc = 2.6 ± 0.58 branch points/neuron, n=17 vs. wt MECP2 = 6 ± 0.78 branch points/neuron, n=18; p<0.01; Figure 4A, C). Intriguingly, TrkB-Fc did not prevent the increase in axonal length (wt MECP2 + TrkB-Fc = 246 ± 42.10μm, n=5 vs. wt MECP2 = 253.6 ± 10.79μm, n=5; p>0.05) or the number of axonal branch points (wt MECP2 + TrkB-Fc = 4.6 ± 0.51 branch points/neuron, n=5 vs.wt MECP2 = 4.2 ± 0.37 branch points/neuron, n=5; p>0.05) caused by wt MECP2 overexpression (Fig. 4A, B). These results suggest that enhanced BDNF signaling is responsible for the enhanced dendritic branching caused by wt MECP2 overexpression, while other factors participate in MECP2-induced axonal overgrowth.
Figure 4. The BDNF Scavenger TrkB-Fc Prevented the Increase in Dendritic Branching Induced by Wildtype MECP2 Overexpression, without Affecting Axonal Growth.
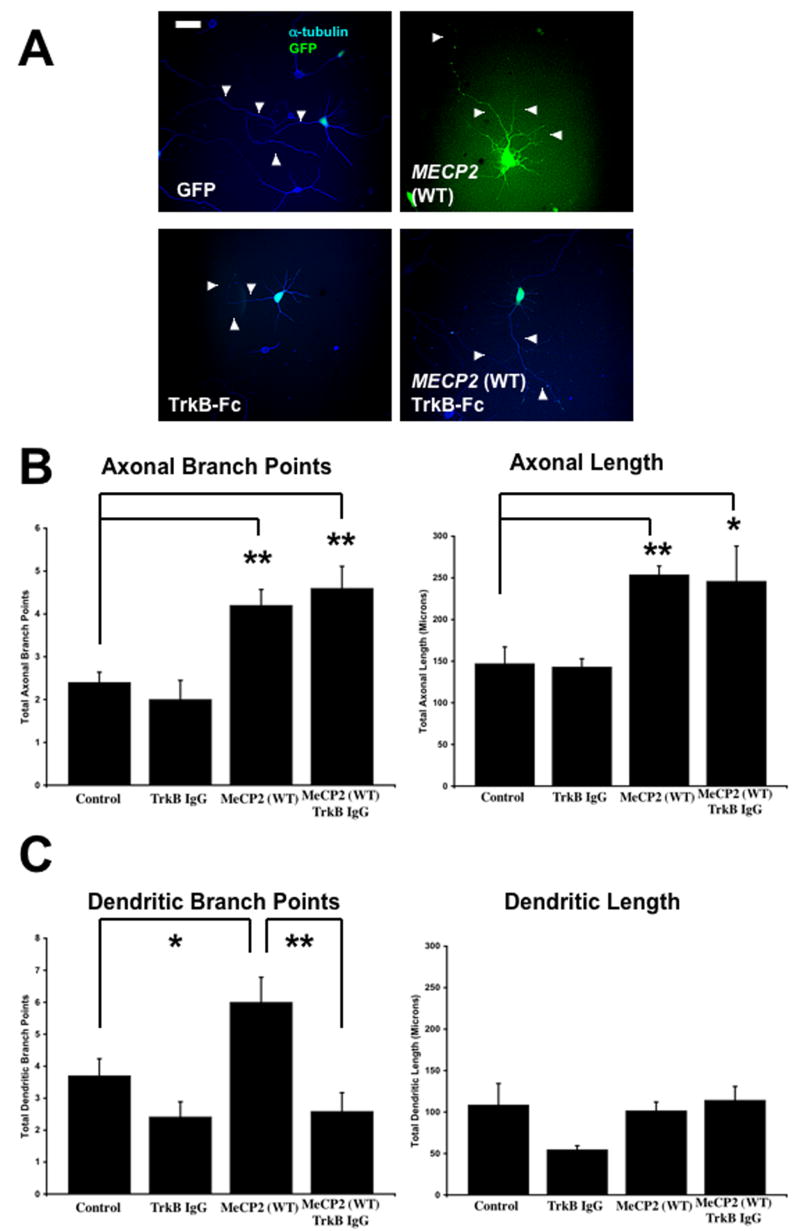
A. Representative examples of hippocampal neurons co-transfected with a control GFP plasmid and a plasmid to overexpress wildtype MECP2 (scale bar = 10μm). For quantitative morphological analysis, the neurons were co-transfected with eGFP (green) or stained with anti-α-tubulin antibodies (blue). B. Population data on axonal length and branch points. C. Population data on dendritic length and branch points.
6. Bdnf Overexpression Fully Rescued Dendritic Growth in Mecp2 Knockdown Neurons, while its Effect in Cells Expressing Rett-Associated Mutant MECP2 was Only Partial
Considering the potent effects of BDNF in dendritic and axonal growth (McAllister et al., 1999; Vicario-Abejon et al., 2002), we next tested whether Bdnf overexpression could rescue the dendritic atrophy caused by shRNA-mediated knockdown of endogenous Mecp2. Indeed, Bdnf overexpression significantly increased dendritic length in neurons where Mecp2 was knockdown with shRNA (Mecp2 shRNA + BDNF-GFP = 275.5 ± 34.81μm, n=6 vs. control shRNA = 105.2 ± 7.54μm, n=5; p<0.001; Fig. 5A, C). In fact, the effect of Bdnf overexpression on dendritic length was significantly larger in Mecp2 knockdown neurons (162% increase), than in control GFP neurons (147% increase; p>0.05). Furthermore, Bdnf overexpression significantly increased axonal length and branching after shRNA-mediated Mecp2 knockdown, despite the lack of effect of Mecp2 knockdown in these axonal features (Fig. 5A, B). Axonal length was 202.1 ± 14.33μm in neurons co-transfected with BDNF-GFP and Mecp2 shRNA (n=5), compared to 139.7 ± 18.81μm in shRNA control cells (n=5; p=0.03). Neurons co-transfected with BDNF-GFP and Mecp2 shRNA had an average of 4.2 ± 0.37 branch points/neuron (n=5), compared to 2.6 ± 0.51 branch points/neuron in shRNA control cells (n=5 p=0.04).
Figure 5. Bdnf Overexpression Rescued Dendritic Atrophy Caused by Mecp2 Knockdown, Increasing Dendritic Length as much as in BDNF-Transfected Neurons.
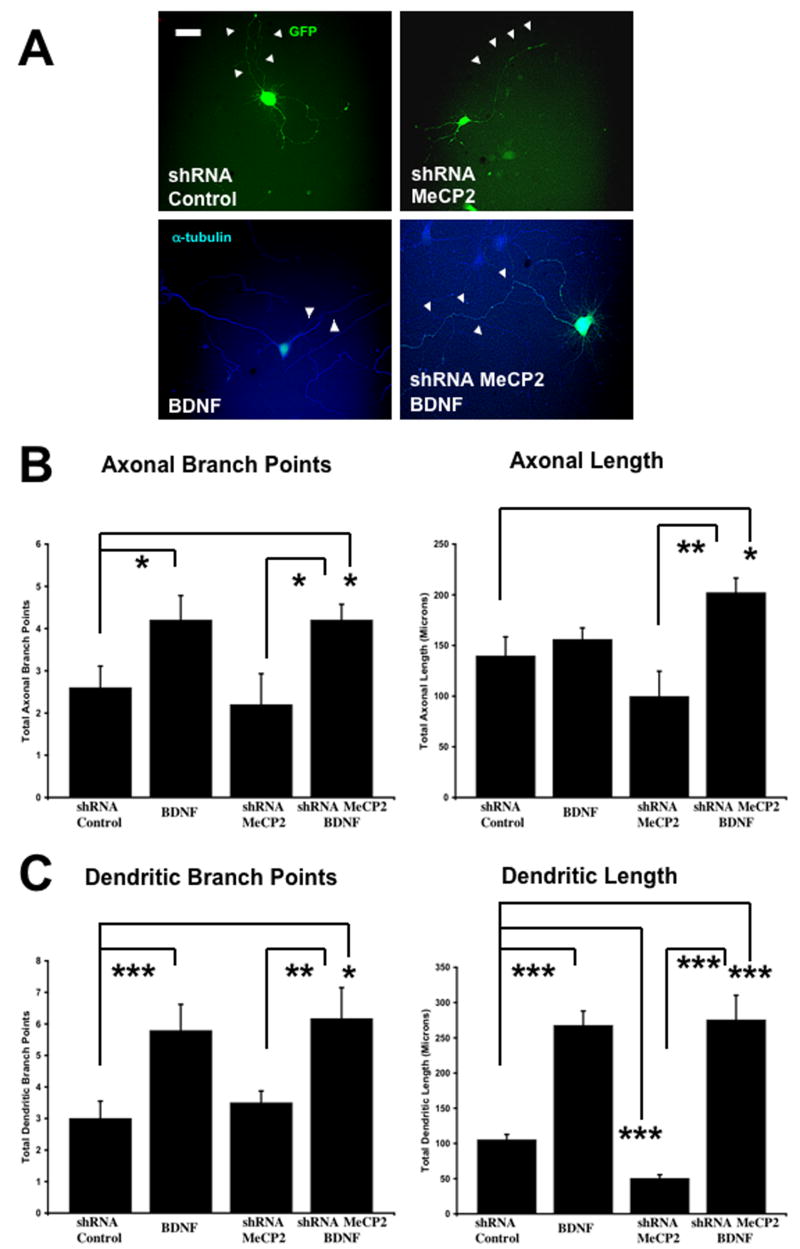
A. Representative examples of neurons transfected with an shRNA control plasmid, an shRNA plasmid to knockdown endogenous Mecp2, and a plasmid to overexpress Bdnf (scale bar = 10μm). For quantitative morphological analyses, neurons were co-transfected with eGFP (green) or stained with anti-α-tubulin antibodies (blue). B. Population data on axonal length and branch points. C. Population data on dendritic length and branch points.
Lastly, we tested if Bdnf overexpression could rescue the dendritic atrophy caused by overexpression of RTT-associated MECP2 mutations. Indeed, Bdnf overexpression prevented the decrease in dendritic length caused by the R106W MECP2 mutation: dendritic length was 127.8 ± 14.77μm in neurons co-transfected with BDNF-GFP and R106W (n=11), compared to 43.26 ± 4.25μm in neurons expressing R106W alone (n=20; p<0.05). Bdnf overexpression also prevented the dendritic atrophy caused by the T158M mutation (T158M + BDNF-GFP = 99.17 ± 16.62μm, n = 21 vs. T158M = 51.69 ± 7.7μm, n=20; p<0.05) (Fig. 6A, C). However, Bdnf overexpression did not prevent the reduction in dendritic branch points resulting from the expression of the R106W MECP2 mutation (R106W + BDNF-GFP = 2.46±0.91 branch points/neuron, n=11 vs. R106W = 2.1 ± 0.34 branch points/neuron, n=20; p>0.05) (Fig. 6A, C). Furthermore, Bdnf overexpression did not prevent the reduction in dendritic branch points caused by the T158M mutation (T158M + BDNF-GFP = 2.46 ± 0.43 branch points/neuron, n=21 vs. T158M = 2 ± 0.37 branch points/neuron, n=20; p>0.05; Figure 6A, C). It's important to note that the effects of Bdnf overexpression in neurons expressing RTT-associated MECP2 mutations were only partial, i.e. dendritic length was comparable to that of neurons expressing only GFP, but not to that of neurons expressing BDNF-GFP (compare GFP alone vs. BDNF-GFP in Fig. 5). Lastly, Bdnf was only able to rescue dendritic impairments, because axonal atrophy was not prevented by co-transfection of MECP2 mutations and Bdnf (Fig. 6A, C).
Figure 6. Bdnf Overexpression Partially Rescued Dendritic Atrophy in Neurons Expressing RTT-Associated MECP2 Mutations, Increasing Dendrite Length Only to Control Levels.
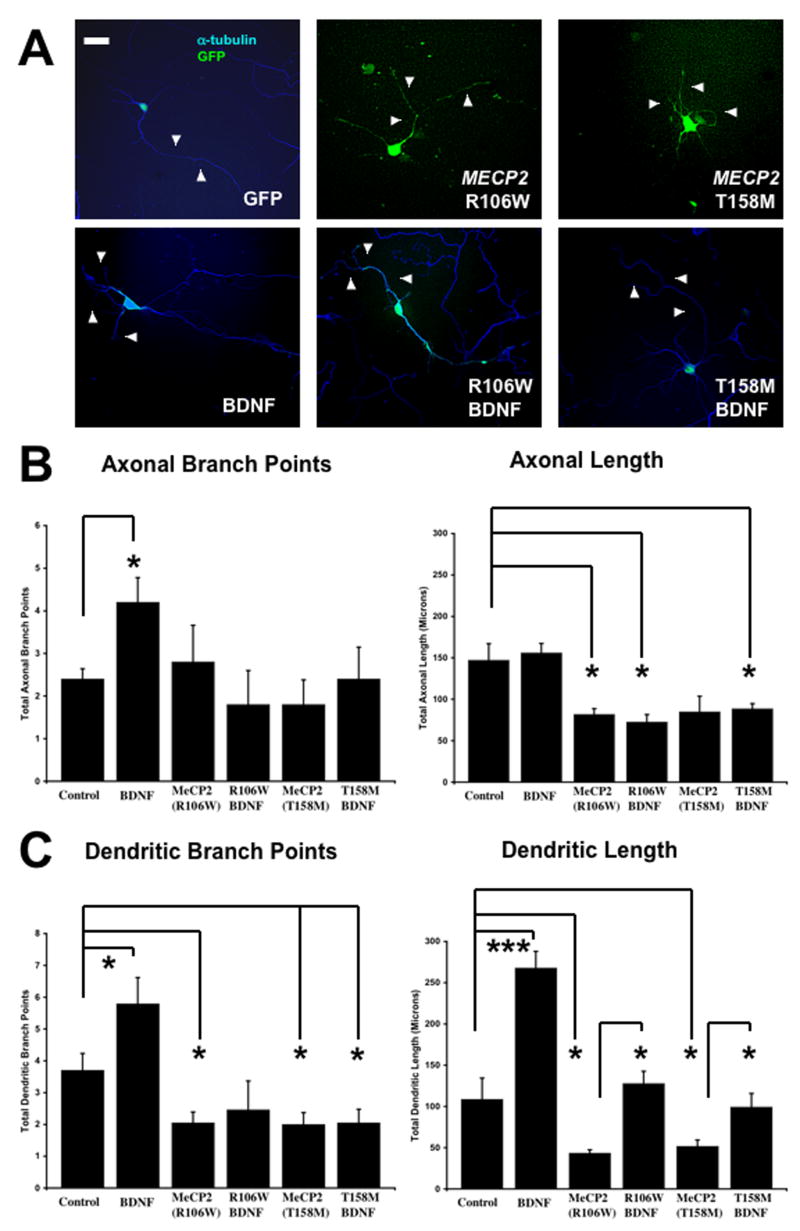
A. Representative examples of neurons transfected with a control GFP plasmid, plasmids to overexpress to different missense MECP2 mutations (R106W or T158M) commonly found in Rett syndrome patients, and a plasmid to overexpress Bdnf (scale bar = 10μm). For quantitative morphological analyses, neurons were co-transfected with eGFP (green) or stained with anti-α-tubulin antibodies (blue). B. Population data on axonal length and branch points. C. Population data on dendritic length and branch points.
Discussion
Here, we presented several new observations regarding the role of MeCP2 in the terminal differentiation of hippocampal neurons, specifically the growth and branching of their dendrites and axons. First, endogenous Mecp2 knockdown reduced dendritic length without affecting axonal morphology. Second, overexpression of wildtype human MECP2 increased dendritic branching, as well as axonal length and branching. Third, overexpression of missense MECP2 mutations common in Rett syndrome reduced dendritic length and branching, as well as axonal length. It is important to note that the endogenous Mecp2 gene was not manipulated in these wildtype or mutant MECP2 overexpression experiments. Fourth, either endogenous Mecp2 knockdown or overexpression of wildtype MECP2 – but not of Rett-associated MECP2 mutations – increased intracellular BDNF levels in hippocampal neurons. Fifth, the BDNF scavenger TrkB-Fc prevented the increase in dendritic branching caused by wildtype human MECP2 overexpression. Lastly, Bdnf overexpression fully rescued dendritic growth in Mecp2 knockdown neurons, while its effect in cells expressing Rett-associated mutant MECP2 was only partial. Taken together, these results demonstrate that MeCP2 plays varied roles in dendritic and axonal development during neuronal terminal differentiation, and that some of these effects are mediated by autocrine actions of BDNF.
The expression of MECP2/Mecp2 in humans and rodents increases during neuronal development and maturation (Akbarian et al., 2001; Cohen et al., 2003, Jung, 2003 #21226; LaSalle et al., 2001; Shahbazian et al., 2002; Zoghbi, 2003). Consistently, the development of axons and dendrites has been shown to be affected by MeCP2 levels (Cassel et al., 2004; Kaufmann et al., 2005; Kishi and Macklis, 2004; Matarazzo et al., 2004; Matarazzo and Ronnett, 2004; Mullaney et al., 2004). Furthermore, cultured hippocampal neurons from Mecp2 null mice formed fewer excitatory autapses than wildtype neurons, while neurons from MECP2 overexpressing mice (∼twofold the endogenous levels) showed a higher density of excitatory autapses than wildtype neurons after 2 weeks in vitro (Chao et al., 2007). Together with the present results, these observations suggest that overexpression of wildtype MECP2 enhances synapse formation because it promotes dendritic growth during early neuronal development. In addition, this interpretation predicts that endogenous Mecp2 knockdown, or the expression of Rett-associated MECP2 mutations will result in reduced dendritic spine density, an issue investigated in the companion manuscript (Chapleau et al., manuscript NBD-08-380 under revision after first review). It is important to note that the only published study of the consequences of mutant MECP2 expression on dendritic and axonal morphology described increased dendritic and axonal branching (not length) in embryonic day-15 cortical neurons overexpressing MeCP2293, a truncated protein caused by an arginine to nonsense mutation in RTT individuals (Jugloff et al., 2005). This discrepancy with our observations (reduced dendritic length and branching, as well as axonal length after mutant MECP2 overexpression) may result from different brain regions (cortex vs. hippocampus), developmental ages (E15 vs. E18), when were neurons transfected (5 days in vitro vs. 30min after dissociation), the duration of mutant MECP2 overexpression (24hs vs. 96hs), or the method of transfection (lipofection vs. electroporation).
The altered molecular pathway(s) that contribute to the pathogenesis of Rett syndrome (RTT) remain unknown and under exhaustive investigation. Numerous gene expression profiles have been conducted on samples from Rett patients or from Mecp2-deficient mouse models (Ballestar et al., 2005; Chahrour et al., 2008; Colantuoni et al., 2001; Delgado et al., 2006; Deng et al., 2007; McGill et al., 2006; Nuber et al., 2005; Tudor et al., 2002), with each report arriving to different conclusions regarding deregulated protein(s) in RTT through either direct or indirect interactions with MeCP2. One prominent and quite consistent gene target of MeCP2 transcriptional regulation is Bdnf (Caballero and Hendrich, 2005; Chahrour et al., 2008; Chen et al., 2003; Klose and Bird, 2003; Martinowich et al., 2003; Wade, 2004). Chen et al. (Chen et al., 2003) first demonstrated that MeCP2 represses Bdnf transcription by directly binding to mouse Bdnf promoter IV, which is activated by neuronal activity and Ca2+ influx through voltage-gated Ca2+ channels (Tao et al., 1998). In the absence of neuronal activity (i.e. in the presence of TTX), exon IV mRNA transcripts were twofold higher in cultured cortical neurons from Mecp2 null mice than in neurons from wildtype mice (Chen et al., 2003). However, it was later found that BDNF protein levels measured by ELISA were found to be lower in brain samples from Mecp2 null mice compared to wildtype mice at 6-8 weeks of age (Chang et al., 2006). Given that Bdnf mRNA and protein levels are tightly regulated by neuronal activity, the reduced firing frequency observed in cortical neurons from Mecp2 null mice (Dani et al., 2005) may be the cause of impaired activity-dependent BDNF protein synthesis, as estimated by BDNF protein ELISA measurements (Chang et al., 2006; Ogier et al., 2007; Wang et al., 2006). In addition, MECP2 overexpression in cultured cortical neurons resulted in elevated Bdnf mRNA levels, likely via a homeostatic regulatory loop that includes the CREB-induced microRNA132, which negatively regulates MeCP2 levels (Klein et al., 2007). Thus, the functional significance and the specific mechanism(s) of the interaction between BDNF and MeCP2 remain unknown and hotly debated (Cohen et al., 2008; Sun and Wu, 2006). At the clinical level, the role of neurotrophins such as BDNF in Rett syndrome is also unclear. Initial studies described lower levels of NGF in blood serum or cerebral spinal fluid of RTT patients compared to non-RTT individuals, but no differences in BDNF levels (Lappalainen et al., 1996; Riikonen, 2003; Riikonen and Vanhala, 1999; Vanhala et al., 1998). However, a recent study reports that BDNF mRNA levels are reduced in brain samples from RTT patients (Abuhatzira et al., 2007).
Considering the powerful actions of BDNF on dendritic, axonal and, eventually, synaptic development, (McAllister et al., 1999; Vicario-Abejon et al., 2002), and the observations that increasing BDNF protein levels reverted some crucial RTT-like phenotypes in Mecp2 knockout mice (Chang et al., 2006; Ogier et al., 2007), a “BDNF hypothesis of Rett syndrome” has been proposed (Amaral et al., 2007). Consistent with this hypothesis, the membrane impermeable TrkB-Fc fusion protein, which scavenges extracellular BDNF (Shelton et al., 1995), prevented the increase in dendritic complexity caused by overexpression of wildtype MECP2 in hippocampal neurons. It remains to be tested whether a similar manipulation of extracellular BDNF levels prevents the effect of MECP2 overexpression reported in cortical dendrites (Jugloff et al., 2005) and hippocampal excitatory autapses (Chao et al., 2007). Despite the current debate regarding the functional interaction between BDNF and MeCP2 and their role in Rett syndrome (Cohen et al., 2008; Sun and Wu, 2006), our current knowledge indicates that MeCP2 acts both directly and indirectly to control the expression of Bdnf. Indeed, our present results show that both knockdown of endogenous Mecp2 as well as overexpression of wildtype MECP2 increased intracellular BDNF protein levels. Whether these elevated intracellular BDNF levels are accompanied by enhanced activity-dependent release and autocrine/paracrine signaling remains to be determined. For example, neurons of the nodose ganglion from Mecp2-null mice show lower total BDNF levels than wildtype neurons, but are able to release it at wildtype levels (Wang et al., 2006). Intriguingly, overexpression of RTT-associated missense mutations did not affect intracellular BDNF protein levels, which may or may not be released appropriately by neuronal activity. Overall, our observations support the view that the developmental increase in MECP2/Mecp2 expression controls BDNF protein levels, which in turn participate in the growth and maturation of dendritic architecture, and that Rett-associated MECP2 mutations impair neuronal terminal differentiation by deregulating BDNF levels and/or its activity-dependent release.
The observations that: (1) Bdnf overexpression rescued the hypoactivity in wheel running exhibited by Mecp2 knockout mouse, and the low frequency of action potential firing observed in their cortical neurons (Chang et al., 2006); and (2) treatment of Mecp2 null mice with AMPAkines (which increases BDNF mRNA and protein levels) rescued the irregular respiratory patterns exhibited by Mecp2 null mice (Ogier et al., 2007), prompt us to test whether Bdnf overexpression could rescue the stunted dendritic growth caused by either endogenous Mecp2 knockdown or overexpression of mutant MECP2. Indeed, Bdnf was able to increase dendritic length in neurons after Mecp2 knockdown, whose dendrites reached average lengths comparable to neurons transfected with Bdnf alone. Intriguingly, the effect in neurons expressing Rett-associated MECP2 mutations was only partial, because it increased dendritic length only to control levels observed in GFP-expressing neurons, without the enhanced overgrowth produced by Bdnf overexpression. Despite this partial effect, these results demonstrate that BDNF is able to revert the dendritic atrophy caused by Rett-associated MECP2 mutations.
In addition to Bdnf, the search for gene targets of MeCP2 has yielded a few intriguing candidates (Bienvenu and Chelly, 2006). For example, the genes encoding CRH, corticotropin-releasing hormone (McGill et al., 2006), FXYD1 – a transmembrane modulator of the Na+/K+-ATPase (Deng et al., 2007) and the inhibitors of differentiation ID1-4 (Peddada et al., 2006) are all upregulated in Mecp2 null mice and in Rett patients, in addition to participating in neuronal and dendritic development. Recently, a more targeted study identified 2,582 genes symmetrically misregulated in the hypothalamus of Mecp2 null and MECP2 overexpressing mice, suggesting that they represent primary targets (Chahrour et al., 2008). Unexpectedly, 85% of them seem to be activated by MeCP2 because they were up-regulated in MECP2 overexpressing and down-regulated in Mecp2 null mice (Chahrour et al., 2008). The contribution of any of these genes in our results, as well as any potential interactions with Bdnf transcription and translation remains unknown.
DNA methylation is a common mechanism of silencing genes during cellular differentiation, and MeCP2 may play a critical role in neuronal terminal differentiation by reading this epigenetic code. Indeed, gene methylation is required for NGF to induce neurite outgrowth in PC12 cells (Persengiev and Kilpatrick, 1996), a signature of their differentiation into neurons. Consistently, the expression of the inhibitors of differentiation genes (ID1, ID2 and ID3) is reduced during NGF-induced PC12 differentiation, an effect sensitive to inhibition of DNA methyltransferase activity (Persengiev and Kilpatrick, 1997). Intriguingly, all four ID genes are targets of MeCP2 transcriptional repression, demonstrated by increased mRNA and protein levels in brain samples from Mecp2 null mice and RTT individuals (Peddada et al., 2006). The observation that Mecp2 null mice express lower levels of Neurod1, a gene target of ID proteins that is critical for neuronal development (Andres-Barquin et al., 2000; Chae et al., 2004), strongly suggest that Mecp2-deficient neurons are arrested in an immature stage of differentiation by the inability to properly read the epigenetic code represented by DNA methylation of gene promoters (Shahbazian and Zoghbi, 2002). In summary, our results are consistent with a role of MeCP2 in early neuronal development and differentiation, providing novel evidence supporting the involvement of BDNF in the changes caused by MeCP2 deregulation leading to prominent features of the neuropathology of Rett syndrome.
Supplementary Material
A. Western immunoblots of PC-12 cells transfected with human wt MECP2-expressing plasmids, or different shRNA interfering sequence plasmids to knockdown endogenous Mecp2. Sequence #2 produced a ∼65% reduction in MeCP2 protein levels. B. Primary hippocampal neurons were co-transfected with shRNA sequence #2 and eGFP, and immunostained with anti-MeCP2 antibodies (red) to determine MeCP2 protein levels by quantitative immunofluorescence. Inset shows the same field of view in the green fluorescence channel to identify the GFP transfected neuron. Note that the MeCP2 positive (red) neuron on the right, which can be identified as GFP positive (green) in the inset, is dimmer than the one on the left due to the expression of the MeCP2 shRNA plasmid #2. C. Primary hippocampal neurons were transfected with expression plasmids to overexpress wildtype MECP2 or the T158M mutation and immunostained with anti-MeCP2 antibodies (red) to determine MeCP2 protein levels by quantitative immunofluorescence. Green fluorescence represent neurons transfected with eGFP-tagged MeCP2, and red immunofluorescence shows MeCP2 positive neurons. As in B., the inset shows the green channel. D. Population data from the quantitative fluorescence estimation of MeCP2 levels in hippocampal neurons. Mecp2 knockdown caused a ∼55% reduction in MeCP2 immunofluorescence, while transfection with expression plasmids resulted in a twofold increase in MeCP2 immunofluorescence, all compared to untransfected neurons within the same fields of view.
A. Representative image of a 4 div-hippocampal neuron transfected with eGFP. B. Tracing of the same neuron to determine the length and branch points of axons and dendrites. After 4 div, neurons have a lone axon and several dendrites that are highly branched. For quantitative morphological analyses, neurons were co-transfected with eGFP or stained with anti-α-tubulin antibodies (scale bar = 10μm).
A. Representative examples of neurons transfected with a control GFP plasmid and a plasmid to overexpress Bdnf (scale bar = 10μm). Neurons were also exposed to the fusion protein TrkB-Fc to sequester extracellular BDNF released from Bdnf-overexpressing neurons. For quantitative morphological analyses, the neurons were co-transfected with eGFP (green) or stained with anti-α-tubulin antibodies (blue). B. Population data on axonal length and branch points. C. Population data on dendritic length and branch points.
Acknowledgments
Supported by NIH grants NS40593 and NS057780, IRSF and the Civitan International Foundation. We also thank the assistance of the UAB Intellectual and Developmental Disabilities Research Center (P30-HD38985) and the UAB Neuroscience Cores (P30-NS47466, P30-NS57098). Human tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD. We thank Dr. Carolyn Schanen (Nemours Biomedical Research, Alfred I. duPont Hospital for Children, Wilmington, DE, USA), and Mr. J. Matthew Rutherford for discussions and comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abuhatzira L, Makedonski K, Kaufman Y, Razin A, Shemer R. MeCP2 deficiency in the brain decreases BDNF levels by REST/CoREST-mediated repression and increases TRKB production. Epigenetics. 2007;2:214–22. doi: 10.4161/epi.2.4.5212. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Chen RZ, Gribnau J, Rasmussen TP, Fong H, Jaenisch R, Jones EG. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis. 2001;8:784–91. doi: 10.1006/nbdi.2001.0420. [DOI] [PubMed] [Google Scholar]
- Amaral MD, Chapleau CA, Pozzo-Miller L. Transient receptor potential channels as novel effectors of brain-derived neurotrophic factor signaling: Potential implications for Rett syndrome. Pharmacol Ther. 2007;113:394–409. doi: 10.1016/j.pharmthera.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral MD, Pozzo-Miller L. TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J Neurosci. 2007;27:5179–89. doi: 10.1523/JNEUROSCI.5499-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Andres-Barquin PJ, Hernandez MC, Israel MA. Id genes in nervous system development. Histol Histopathol. 2000;15:603–18. doi: 10.14670/HH-15.603. [DOI] [PubMed] [Google Scholar]
- Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54:195–201. doi: 10.1097/00005072-199503000-00006. [DOI] [PubMed] [Google Scholar]
- Armstrong DD. Neuropathology of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:72–6. doi: 10.1002/mrdd.10027. [DOI] [PubMed] [Google Scholar]
- Armstrong DD, Dunn JK, Schultz RJ, Herbert DA, Glaze DG, Motil KJ. Organ growth in Rett syndrome: a postmortem examination analysis. Pediatr Neurol. 1999;20:125–9. doi: 10.1016/s0887-8994(98)00124-6. [DOI] [PubMed] [Google Scholar]
- Ballestar E, Ropero S, Alaminos M, Armstrong J, Setien F, Agrelo R, Fraga MF, Herranz M, Avila S, Pineda M, Monros E, Esteller M. The impact of MECP2 mutations in the expression patterns of Rett syndrome patients. Hum Genet. 2005;116:91–104. doi: 10.1007/s00439-004-1200-0. [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K. Culturing nerve cells. MIT Press; Boston: 1998. [Google Scholar]
- Bienvenu T, Carrie A, de Roux N, Vinet MC, Jonveaux P, Couvert P, Villard L, Arzimanoglou A, Beldjord C, Fontes M, Tardieu M, Chelly J. MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum Mol Genet. 2000;9:1377–84. doi: 10.1093/hmg/9.9.1377. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7:415–26. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- Caballero IM, Hendrich B. MeCP2 in neurons: closing in on the causes of Rett syndrome. Hum Mol Genet. 2005;14(Spec No 1):R19–26. doi: 10.1093/hmg/ddi102. [DOI] [PubMed] [Google Scholar]
- Cassel S, Revel MO, Kelche C, Zwiller J. Expression of the methyl-CpG-binding protein MeCP2 in rat brain. An ontogenetic study. Neurobiol Dis. 2004;15:206–11. doi: 10.1016/j.nbd.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Chae JH, Stein GH, Lee JE. NeuroD: the predicted and the surprising. Mol Cells. 2004;18:271–88. [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science. 2008;320:1224–9. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–8. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chechlacz M, Gleeson JG. Is mental retardation a defect of synapse structure and function? Pediatr Neurol. 2003;29:11–7. doi: 10.1016/s0887-8994(03)00152-8. [DOI] [PubMed] [Google Scholar]
- Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long- term potentiation. J Neurosci. 1999;19:7983–90. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–9. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Cline H, Haas K. The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: a review of the synaptotrophic hypothesis. J Physiol. 2008;586:1509–17. doi: 10.1113/jphysiol.2007.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen DR, Matarazzo V, Palmer AM, Tu Y, Jeon OH, Pevsner J, Ronnett GV. Expression of MeCP2 in olfactory receptor neurons is developmentally regulated and occurs before synaptogenesis. Mol Cell Neurosci. 2003;22:417–29. doi: 10.1016/s1044-7431(03)00026-5. [DOI] [PubMed] [Google Scholar]
- Cohen S, Zhou Z, Greenberg ME. Medicine. Activating a repressor. Science. 2008;320:1172–3. doi: 10.1126/science.1159146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Jeon OH, Hyder K, Chenchik A, Khimani AH, Narayanan V, Hoffman EP, Kaufmann WE, Naidu S, Pevsner J. Gene expression profiling in postmortem Rett Syndrome brain: differential gene expression and patient classification. Neurobiol Dis. 2001;8:847–65. doi: 10.1006/nbdi.2001.0428. [DOI] [PubMed] [Google Scholar]
- Craig AM, Banker G. Neuronal polarity. Annu Rev Neurosci. 1994;17:267–310. doi: 10.1146/annurev.ne.17.030194.001411. [DOI] [PubMed] [Google Scholar]
- Cusack SM, Rohn TT, Medeck RJ, Irwin KM, Brown RJ, Mercer LM, Oxford JT. Suppression of MeCP2beta expression inhibits neurite extension in PC12 cells. Exp Cell Res. 2004;299:442–53. doi: 10.1016/j.yexcr.2004.05.035. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–5. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado IJ, Kim DS, Thatcher KN, Lasalle JM, Van den Veyver IB. Expression profiling of clonal lymphocyte cell cultures from Rett syndrome patients. BMC Med Genet. 2006;7:61. doi: 10.1186/1471-2350-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng V, Matagne V, Banine F, Frerking M, Ohliger P, Budden S, Pevsner J, Dissen GA, Sherman LS, Ojeda SR. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum Mol Genet. 2007;16:640–50. doi: 10.1093/hmg/ddm007. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–68. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdfelder E, Faul F, Buchner A. GPOWER: A general power analysis program. Behavior Research Methods. 1996;28:1–11. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–9. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Gresch O, Engel FB, Nesic D, Tran TT, England HM, Hickman ES, Korner I, Gan L, Chen S, Castro-Obregon S, Hammermann R, Wolf J, Muller-Hartmann H, Nix M, Siebenkotten G, Kraus G, Lun K. New non-viral method for gene transfer into primary cells. Methods. 2004;33:151–63. doi: 10.1016/j.ymeth.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:61–5. doi: 10.1002/mrdd.10020. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Goutieres F, Hanefeld F, Rett A, Wilson J. Rett syndrome: criteria for inclusion and exclusion. Brain Dev. 1985;7:372–3. doi: 10.1016/s0387-7604(85)80048-6. [DOI] [PubMed] [Google Scholar]
- Jellinger K, Armstrong D, Zoghbi HY, Percy AK. Neuropathology of Rett syndrome. Acta Neuropathol (Berl) 1988;76:142–58. doi: 10.1007/BF00688098. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Jeon OH, Pevsner J, Blue ME, Naidu S. Neurobiology of Rett syndrome: a genetic disorder of synapse development. Brain Dev. 2001;23 1:S206–13. doi: 10.1016/s0387-7604(01)00351-5. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Mullaney B, Blue ME. Neurobiology of Rett syndrome. J Child Neurol. 2003;18:688–92. doi: 10.1177/08830738030180100501. [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–91. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Jugloff DG, Jung BP, Purushotham D, Logan R, Eubanks JH. Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol Dis. 2005;19:18–27. doi: 10.1016/j.nbd.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–64. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Johnston MV, Blue ME. MeCP2 expression and function during brain development: implications for Rett syndrome's pathogenesis and clinical evolution. Brain Dev. 2005;27 1:S77–S87. doi: 10.1016/j.braindev.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, MacDonald SM, Altamura CR. Dendritic cytoskeletal protein expression in mental retardation: an immunohistochemical study of the neocortex in Rett syndrome. Cereb Cortex. 2000;10:992–1004. doi: 10.1093/cercor/10.10.992. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–91. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Naidu S, Budden S. Abnormal expression of microtubule-associated protein 2 (MAP-2) in neocortex in Rett syndrome. Neuropediatrics. 1995;26:109–13. doi: 10.1055/s-2007-979738. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Worley PF, Taylor CV, Bremer M, Isakson PC. Cyclooxygenase-2 expression during rat neocortical development and in Rett syndrome. Brain Dev. 1997;19:25–34. doi: 10.1016/s0387-7604(96)00047-2. [DOI] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27:306–21. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci. 2007;10:1513–4. doi: 10.1038/nn2010. [DOI] [PubMed] [Google Scholar]
- Klose R, Bird A. Molecular biology. MeCP2 repression goes nonglobal. Science. 2003;302:793–5. doi: 10.1126/science.1091762. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell. 2005;19:667–78. doi: 10.1016/j.molcel.2005.07.021. [DOI] [PubMed] [Google Scholar]
- Kojima M, Takei N, Numakawa T, Ishikawa Y, Suzuki S, Matsumoto T, Katoh-Semba R, Nawa H, Hatanaka H. Biological characterization and optical imaging of brain-derived neurotrophic factor-green fluorescent protein suggest an activity- dependent local release of brain-derived neurotrophic factor in neurites of cultured hippocampal neurons. J Neurosci Res. 2001;64:1–10. doi: 10.1002/jnr.1080. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Bird A. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004;32:1818–23. doi: 10.1093/nar/gkh349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo S. Methyl-CpG-binding protein MeCP2 represses Sp1-activated transcription of the human leukosialin gene when the promoter is methylated. Mol Cell Biol. 1998;18:5492–9. doi: 10.1128/mcb.18.9.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo S, Nomura Y, Segawa M, Fujita N, Nakao M, Dragich J, Schanen C, Tamura M. Functional analyses of MeCP2 mutations associated with Rett syndrome using transient expression systems. Brain Dev. 2001;23 1:S165–73. doi: 10.1016/s0387-7604(01)00345-x. [DOI] [PubMed] [Google Scholar]
- Kudo S, Nomura Y, Segawa M, Fujita N, Nakao M, Schanen C, Tamura M. Heterogeneity in residual function of MeCP2 carrying missense mutations in the methyl CpG binding domain. J Med Genet. 2003;40:487–93. doi: 10.1136/jmg.40.7.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen R, Lindholm D, Riikonen R. Low levels of nerve growth factor in cerebrospinal fluid of children with Rett syndrome. J Child Neurol. 1996;11:296–300. doi: 10.1177/088307389601100407. [DOI] [PubMed] [Google Scholar]
- LaSalle JM. Paradoxical role of methyl-CpG-binding protein 2 in Rett syndrome. Curr Top Dev Biol. 2004;59:61–86. doi: 10.1016/S0070-2153(04)59003-8. [DOI] [PubMed] [Google Scholar]
- LaSalle JM, Goldstine J, Balmer D, Greco CM. Quantitative localization of heterogeneous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in normal and Rett syndrome brain by laser scanning cytometry. Hum Mol Genet. 2001;10:1729–40. doi: 10.1093/hmg/10.17.1729. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–3. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Matarazzo V, Cohen D, Palmer AM, Simpson PJ, Khokhar B, Pan SJ, Ronnett GV. The transcriptional repressor Mecp2 regulates terminal neuronal differentiation. Mol Cell Neurosci. 2004;27:44–58. doi: 10.1016/j.mcn.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Matarazzo V, Ronnett GV. Temporal and regional differences in the olfactory proteome as a consequence of MeCP2 deficiency. Proc Natl Acad Sci U S A. 2004;101:7763–8. doi: 10.1073/pnas.0307083101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–78. doi: 10.1016/s0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- McGill BE, Bundle SF, Yaylaoglu MB, Carson JP, Thaller C, Zoghbi HY. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2006;103:18267–72. doi: 10.1073/pnas.0608702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mnatzakanian GN, Lohi H, Munteanu I, Alfred SE, Yamada T, MacLeod PJ, Jones JR, Scherer SW, Schanen NC, Friez MJ, Vincent JB, Minassian BA. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat Genet. 2004;36:339–41. doi: 10.1038/ng1327. [DOI] [PubMed] [Google Scholar]
- Moore CD, Thacker EE, Larimore J, Gaston D, Underwood A, Kearns B, Patterson SI, Jackson T, Chapleau C, Pozzo-Miller L, Theibert A. The neuronal Arf GAP centaurin alpha1 modulates dendritic differentiation. J Cell Sci. 2007;120:2683–93. doi: 10.1242/jcs.006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullaney BC, Johnston MV, Blue ME. Developmental expression of methyl-CpG binding protein 2 is dynamically regulated in the rodent brain. Neuroscience. 2004;123:939–49. doi: 10.1016/j.neuroscience.2003.11.025. [DOI] [PubMed] [Google Scholar]
- Naidu S. Rett syndrome: a disorder affecting early brain growth. Ann Neurol. 1997;42:3–10. doi: 10.1002/ana.410420104. [DOI] [PubMed] [Google Scholar]
- Nan X, Bird A. The biological functions of the methyl-CpG-binding protein MeCP2 and its implication in Rett syndrome. Brain Dev. 2001;23 1:S32–7. doi: 10.1016/s0387-7604(01)00333-3. [DOI] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–81. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–9. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Nuber UA, Kriaucionis S, Roloff TC, Guy J, Selfridge J, Steinhoff C, Schulz R, Lipkowitz B, Ropers HH, Holmes MC, Bird A. Up-regulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum Mol Genet. 2005;14:2247–56. doi: 10.1093/hmg/ddi229. [DOI] [PubMed] [Google Scholar]
- Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci. 2007;27:10912–7. doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peddada S, Yasui DH, LaSalle JM. Inhibitors of differentiation (ID1, ID2, ID3 and ID4) genes are neuronal targets of MeCP2 that are elevated in Rett syndrome. Hum Mol Genet. 2006;15:2003–14. doi: 10.1093/hmg/ddl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percy AK, Lane JB. Rett syndrome: model of neurodevelopmental disorders. J Child Neurol. 2005;20:718–21. doi: 10.1177/08830738050200090301. [DOI] [PubMed] [Google Scholar]
- Persengiev SP, Kilpatrick DL. Nerve growth factor induced differentiation of neuronal cells requires gene methylation. Neuroreport. 1996;8:227–31. doi: 10.1097/00001756-199612200-00046. [DOI] [PubMed] [Google Scholar]
- Persengiev SP, Kilpatrick DL. The DNA methyltransferase inhibitor 5-azacytidine specifically alters the expression of helix-loop-helix proteins Id1, Id2 and Id3 during neuronal differentiation. Neuroreport. 1997;8:2091–5. doi: 10.1097/00001756-199707070-00001. [DOI] [PubMed] [Google Scholar]
- Philippart M. Rett and Angelman's syndromes: models of arrested development. Pediatr Neurol. 2001;25:288–94. doi: 10.1016/s0887-8994(01)00288-0. [DOI] [PubMed] [Google Scholar]
- Riikonen R. Neurotrophic factors in the pathogenesis of Rett syndrome. J Child Neurol. 2003;18:693–7. doi: 10.1177/08830738030180101101. [DOI] [PubMed] [Google Scholar]
- Riikonen R, Vanhala R. Levels of cerebrospinal fluid nerve-growth factor differ in infantile autism and Rett syndrome. Dev Med Child Neurol. 1999;41:148–52. doi: 10.1017/s0012162299000328. [DOI] [PubMed] [Google Scholar]
- Schultz RJ, Glaze DG, Motil KJ, Armstrong DD, del Junco DJ, Hubbard CR, Percy AK. The pattern of growth failure in Rett syndrome. Am J Dis Child. 1993;147:633–7. doi: 10.1001/archpedi.1993.02160300039018. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–24. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Zoghbi HY. Rett syndrome and MeCP2: linking epigenetics and neuronal function. Am J Hum Genet. 2002;71:1259–72. doi: 10.1086/345360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Phillips HS, Carroll K, Spencer SD, Levinson AD. Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci. 1995;15:477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada A, Mason CA, Morrison ME. TrkB signaling modulates spine density and morphology independent of dendrite structure in cultured neonatal Purkinje cells. J Neurosci. 1998;18:8559–70. doi: 10.1523/JNEUROSCI.18-21-08559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YE, Wu H. The ups and downs of BDNF in Rett syndrome. Neuron. 2006;49:321–3. doi: 10.1016/j.neuron.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–26. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002;99:15536–41. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–58. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Zhang XL, Hartman K, Winterer J, Muller W, Stanton PK, Pozzo-Miller L. BDNF increases release probability and the size of a rapidly recycling vesicle pool within rat hippocampal excitatory synapses. J Physiol. 2006;574:787–803. doi: 10.1113/jphysiol.2006.111310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Veyver IB, Zoghbi HY. Mutations in the gene encoding methyl-CpG-binding protein 2 cause Rett syndrome. Brain Dev. 2001;23 1:S147–51. doi: 10.1016/s0387-7604(01)00376-x. [DOI] [PubMed] [Google Scholar]
- Vanhala R, Korhonen L, Mikelsaar M, Lindholm D, Riikonen R. Neurotrophic factors in cerebrospinal fluid and serum of patients with Rett syndrome. J Child Neurol. 1998;13:429–33. doi: 10.1177/088307389801300903. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejon C, Owens D, McKay R, Segal M. Role of neurotrophins in central synapse formation and stabilization. Nat Rev Neurosci. 2002;3:965–74. doi: 10.1038/nrn988. [DOI] [PubMed] [Google Scholar]
- Wade PA. Dynamic regulation of DNA methylation coupled transcriptional repression: BDNF regulation by MeCP2. Bioessays. 2004;26:217–20. doi: 10.1002/bies.20018. [DOI] [PubMed] [Google Scholar]
- Wan M, Lee SS, Zhang X, Houwink-Manville I, Song HR, Amir RE, Budden S, Naidu S, Pereira JL, Lo IF, Zoghbi HY, Schanen NC, Francke U. Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am J Hum Genet. 1999;65:1520–9. doi: 10.1086/302690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Chan SA, Ogier M, Hellard D, Wang Q, Smith C, Katz DM. Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J Neurosci. 2006;26:10911–5. doi: 10.1523/JNEUROSCI.1810-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–30. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Western immunoblots of PC-12 cells transfected with human wt MECP2-expressing plasmids, or different shRNA interfering sequence plasmids to knockdown endogenous Mecp2. Sequence #2 produced a ∼65% reduction in MeCP2 protein levels. B. Primary hippocampal neurons were co-transfected with shRNA sequence #2 and eGFP, and immunostained with anti-MeCP2 antibodies (red) to determine MeCP2 protein levels by quantitative immunofluorescence. Inset shows the same field of view in the green fluorescence channel to identify the GFP transfected neuron. Note that the MeCP2 positive (red) neuron on the right, which can be identified as GFP positive (green) in the inset, is dimmer than the one on the left due to the expression of the MeCP2 shRNA plasmid #2. C. Primary hippocampal neurons were transfected with expression plasmids to overexpress wildtype MECP2 or the T158M mutation and immunostained with anti-MeCP2 antibodies (red) to determine MeCP2 protein levels by quantitative immunofluorescence. Green fluorescence represent neurons transfected with eGFP-tagged MeCP2, and red immunofluorescence shows MeCP2 positive neurons. As in B., the inset shows the green channel. D. Population data from the quantitative fluorescence estimation of MeCP2 levels in hippocampal neurons. Mecp2 knockdown caused a ∼55% reduction in MeCP2 immunofluorescence, while transfection with expression plasmids resulted in a twofold increase in MeCP2 immunofluorescence, all compared to untransfected neurons within the same fields of view.
A. Representative image of a 4 div-hippocampal neuron transfected with eGFP. B. Tracing of the same neuron to determine the length and branch points of axons and dendrites. After 4 div, neurons have a lone axon and several dendrites that are highly branched. For quantitative morphological analyses, neurons were co-transfected with eGFP or stained with anti-α-tubulin antibodies (scale bar = 10μm).
A. Representative examples of neurons transfected with a control GFP plasmid and a plasmid to overexpress Bdnf (scale bar = 10μm). Neurons were also exposed to the fusion protein TrkB-Fc to sequester extracellular BDNF released from Bdnf-overexpressing neurons. For quantitative morphological analyses, the neurons were co-transfected with eGFP (green) or stained with anti-α-tubulin antibodies (blue). B. Population data on axonal length and branch points. C. Population data on dendritic length and branch points.