

Abstract
Methylmercury is a potent neurotoxin that causes severe neurological disorders in fetuses and young children. Recent studies indicated that MeHg could alter levels of immune mediators produced by cells of the central nervous system. Results from this study indicated that MeHg could greatly induce IL-6 release from primary mouse glial cultures. This property was not shared by other cytotoxic heavy metals, such as CdCl2 or HgCl2. MeHg was known to induce cytosolic phospholipase A2 (PLA2) activation and expression, and this enzyme was required for IL-6 induction in some experimental systems. Further experiments using structurally distinct pharmacological agents were performed to test the hypothesis that MeHg induced PLA2 activation was necessary for MeHg induced IL-6 release. Results indicated that AACOCF3 (≥ 10 μM), MAFP (≥ 0.625 μM) and BEL (≥ 0.625 μM) significantly reduced MeHg induced IL-6 release in glia. However, these PLA2 inhibitors did not block MeHg induced GSH depletion. These results suggested that PLA2 activation was required for MeHg to induce glial IL-6 release.
Keywords: IL-6, methylmercury, phospholipase A2
Methylmercury (MeHg) is an environmental toxin that exerts its toxic effects in various organ systems [6]. Severe neurological disorders occur in victims of MeHg poisoning, with the most severe episodes being in fetuses and young children. Within the CNS, neurons in the cerebellum and occipital lobe are severely affected [6, 7]. Glia are also susceptible to MeHg toxicity, which results in their failure to perform normally and may contribute to neuronal death [1]. Other than cells in the CNS, MeHg is also toxic to other cell types including those in the immune system. For example, toxic effects of this compound on B lymphocytes [21] and T lymphocytes [22] were reported.
Glia in the CNS release a number of cytokines that are important for proper CNS development [17] and function [9]. Insults to the CNS can lead to altered patterns of cytokine release by glia, which can be beneficial or detrimental to the CNS against the specific insult [25]. Other than direct effects on immune cells, MeHg also alters immune mediators secreted by cells of the nervous system [10]. Our previous studies indicate that MeHg causes IL-6 release in various glial cell lines across different species [4, 13]. Interestingly, MeHg does not cause indiscriminative release of cytokines. For instance, while MeHg causes IL-6 release into culture medium of human glioma cells, no TNF-α or IL-1β release can be observed in the same culture medium [4]. Whether this IL-6 induction is unique to MeHg or it is a property shared by HgCl2 is not known. In addition, whether IL-6 induction is a general property of cytotoxic heavy metals remains to be determined.
MeHg has multiple cellular targets and interferes with normal cellular functions. For example, it can cause significant depletion of glutathione (GSH), an important cellular antioxidant [11]. It is not clear which signaling system is responsible for the IL-6 induction caused by MeHg. This agent is known to alter the activities of phospholipases. MeHg can enhance cytosolic phospholipase A2 (PLA2) activity in cultured neurons [19, 26], astrocytes [20] and vascular endothelial cells [16]. PLA2 can hydrolyze the sn-2 ester bond in phospholipids. This causes release of free fatty acids (e.g., arachidonic acid) and lysophospholipids, a process involved in cell injury and death caused by various cytotoxic agents [8]. Interestingly, there is evidence that PLA2 activity has an important role in IL-6 induction. For example, IL-6 production induced by LPS in the U937 cells is dependent on PLA2. Inhibition of this enzyme by AACOCF3, an analog of arachidonic acid, can reduce IL-6 secretion [27]. This raises the possibility that activation of PLA2 by MeHg is an important step leading to glial IL-6 production. This hypothesis was tested in this study.
Mixed mouse cerebral glia derived from 1–2 days old C57BL/6 mice were prepared as described previously [5]. Cells were plated in 75 cm2 flasks, expanded once into 150 cm2 flasks before plating into multi-well culture plates for experiments. As previously reported, astrocytes constituted the majority of cells in these cultures because more than 90% of culture surface was covered by cells positive for glial fibrillary acidic protein (GFAP) staining [5]. The growth medium was composed of Dulbecco’s Modified Eagle’s Medium/F12 (DMEM/F12 medium) supplemented with 5% newborn calf serum and 2.5 mM glutamine. To prepare for experiments, cells were plated into culture plates in this growth medium at 140,000 cells/well in 24-well plates with 700 μl medium per well (IL-6 measurement and viability assay) or 20,000 cells/well in 96-well plates with 100 μl medium per well (GSH measurement). Testing agent was added to each well two hours after plating. Following overnight incubation, the medium was switched to DMEM/F12 supplemented with 1% newborn calf serum and 2.5 mM glutamine for MeHg treatment.
For IL-6 measurement, culture medium from each well was collected after overnight (>18 hrs) MeHg treatment, stored at −70°C for later IL-6 ELISA analysis. Cell viability was determined immediately by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay [12, 13]. The OD of each well was measured by a plate reader (Spectra Max 190, Molecular Devices, Sunnyvale, CA) with a filter setting at 570 nm (reference filter setting was 630 nm). Our previous studies indicated that the MTT viability assay agreed well with results from the trypan blue exclusion viability assay [13]. Mouse IL-6 ELISA kits were obtained from eBioscience (San Diego, CA). The assay was set up in duplicates, and a standard curve was run in parallel, per the manufacturer’s instructional manual.
A set of experiments was conducted to measure cellular GSH levels according to the protocol described previously [5]. After MeHg treatment, cultures were incubated with 40 μM monochlorobimane (prepared in Hank’s Balanced Salt Solution) at 37°C for 20 min. The fluorescence of each well was detected by a fluorescence plate reader (Spectra Max Gemini XS, Molecular Devices, Sunnyvale, CA) with the following settings: excitation 390 nm, emission 485 nm.
PLA2 inhibitors including arachidonyl trifluoromethyl ketone (AACOCF3), bromoenol lactone (BEL) and methyl arachidonyl fluorophosphonate (MAFP) were from Cayman Chemical (Ann Arbor, MI). MeHgCl, HgCl2, CdCl2, monochlorobimane and other general biochemical reagents were from Sigma (St. Louis, MO) unless otherwise stated. Results were pooled from 12 replicate samples derived from 4 independent experiments (IL-6 ELISA and MTT assay) or 12 replicate samples derived from 3 independent experiments (GSH assay) and expressed as mean ± SEM. Statistical analyses were performed by one-way or two-way ANOVA. The Bonferroni test was used for post-hoc analysis, and p< 0.05 was considered as significant difference.
As a follow-up of our previous experiments using glial cell lines [4, 13], the first set of experiments determined the effect of MeHg on IL-6 secretion in primary mouse glia and correlated this to its cytotoxicity. Mouse glia were treated with various concentrations of MeHg overnight (>18 hrs), and then IL-6 production and cell viability in each well were determined. Results indicated that MeHg at 2.5, 5, or 10 μM reduced the viability from 100% (control) to ~100%, ~88%, or ~38%, respectively (Fig. 1A, open circles, left-side axis). MeHg at these concentrations caused IL-6 secretion from ~14 (control) to ~40, ~185 or ~227 pg/ml, respectively (Fig. 1A, solid circles, right-side axis). Subsequent experiments indicated that prolonged MeHg incubation beyond one day did not further increase IL-6 release (not shown). Since these glial cultures did contain a small number of microglia, experiments were performed to determine the contribution of microglia to the observed IL-6 release. Results indicated that treatment of purified microglia (280,000 cells/well) overnight with 5 μM MeHg caused IL-6 release from ~5 pg/ml (control) to ~18 pg/well. In contrast, mixed glia at 140,000 cells/well could generate ~185 pg/ml IL-6 under similar experimental conditions (Fig. 1A). We thus concluded that, even though microglia could generate IL-6 after MeHg treatment, they only had a minor contribution to the IL-6 release we detected in this mixed glial culture.
Fig. 1. Effect of MeHg, CdCl2and HgCl2on IL-6 release and cellular viability in primary mouse glia.
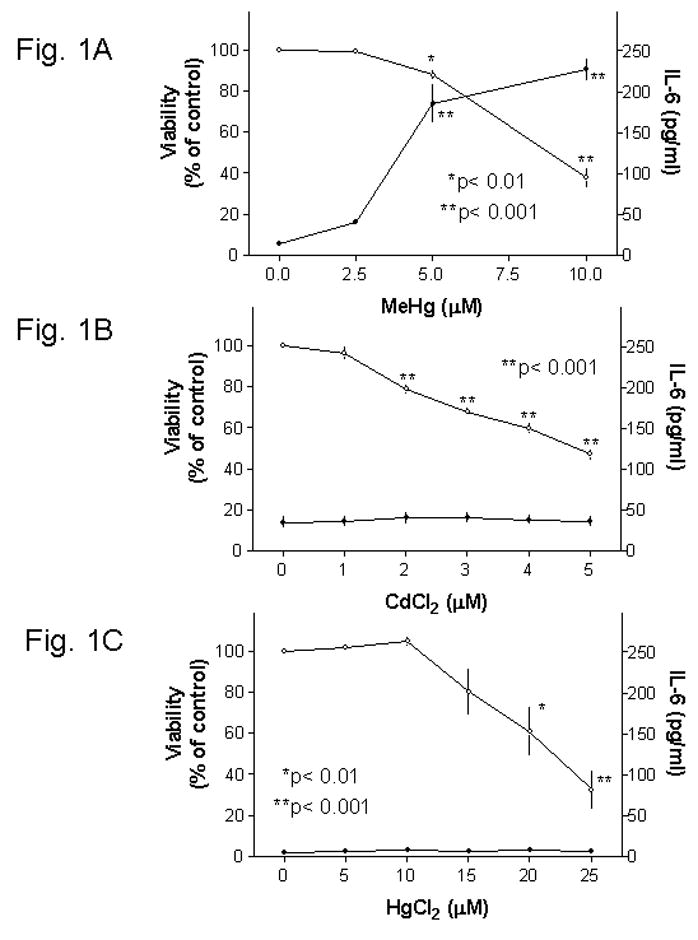
Mouse glia were treated with MeHg, CdCl2 or HgCl2 overnight (>18 hrs), and then the medium in each well was collected for IL-6 ELISA and cellular viability was determined by the MTT viability assay. Results indicated that MeHg (Fig. 1A) caused concentration-dependent decrease of viability (open circles, left-side axis) and concurrent increase of IL-6 release (solid circles, right-side axis). Both CdCl2 (Fig. 1B) and HgCl2 (Fig. 1C) caused concentration-dependent decrease of viability (open circles, left-side axis). However, these two agents did not induce IL-6 release (solid circles, right-side axis).
The following set of experiments indicated that CdCl2 did not induce IL-6 production (Fig. 1B, solid circles) despite its cytotoxic effect (Fig. 1B, open circles). In addition, HgCl2, another toxic mercury compound, did not induce IL-6 release (Fig. 1C). These results suggested that cytotoxicity exerted by a toxic metal is not a sufficient cause for IL-6 production, and that the induction of IL-6 appeared to be an unique property of MeHg.
Subsequent experiments indicated that even though MeHg caused IL-6 production and cytotoxicity simultaneously, very cytotoxic concentrations of MeHg (e.g., those that reduced the viability to ~25% or below) induced very little IL-6 (data not shown). A moderate cytotoxic concentration (5 μM) of MeHg (see Fig. 1A, open circles) was used for the following sets of experiments.
Shanker et al. [20] reported that 5 μM MeHg caused a fast (within 10 min) and significant increase (~300% of control) of PLA2 activity in primary astrocytes. Furthermore, AACOCF3, a specific inhibitor for this enzyme, completely blocked the activation. To test whether activation of PLA2 was required for MeHg induced IL-6 production, cells were pretreated with AACOCF3 overnight, followed by 5 μM MeHg (without the inhibitor) for one day, and then the IL-6 production and viability were determined. IL-6 release caused by 5 μM MeHg was used as 100% for comparison in this set of experiments. Results indicated that inhibition of PLA2 by this agent led to a concentration dependent decrease of IL-6 secretion (Fig. 2A, solid circles). The IL-6 secretion dropped from 100% (defined by the IL-6 levels induced by 5 μM MeHg) to ~79%, ~59% or ~15% as a result of 10, 20 or 40 μM AACOCF3 pretreatment, respectively. AACOCF3 at 10–20 μM appeared to increase cell viability in MeHg treated cultures, but the difference was not statistically significant (Fig. 2A, open circles).
Fig. 2. Inhibition of MeHg induced IL-6 release by PLA2inhibitors.
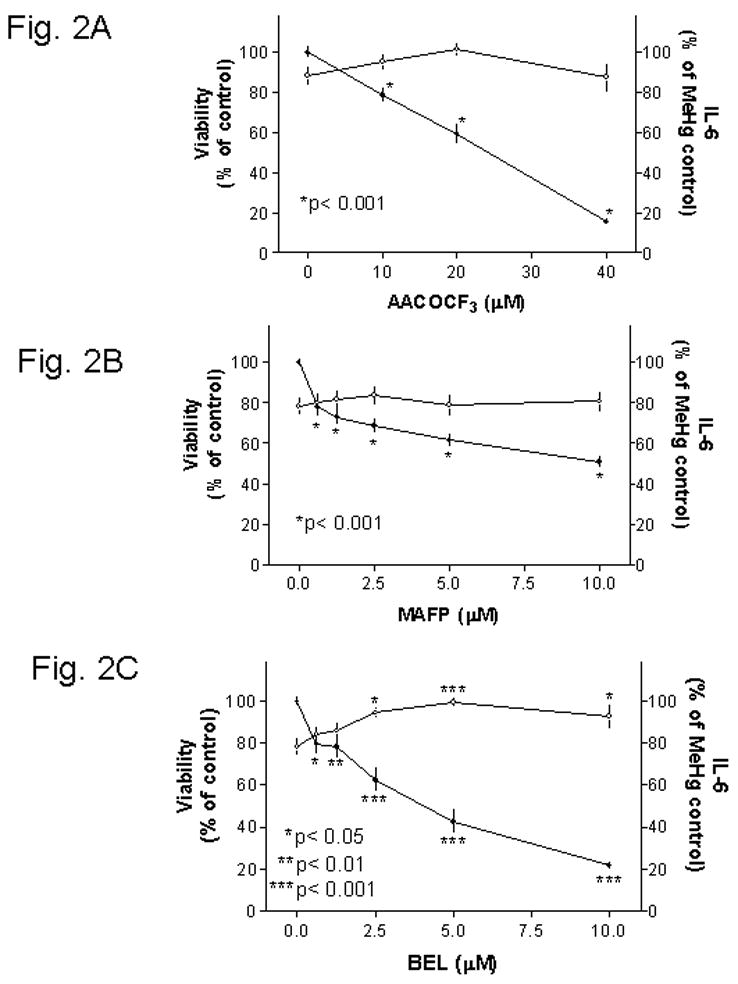
Mouse glia were treated with various concentrations of AACOCF3 (Fig. 2A), MAFP (Fig. 2B) or BEL (Fig. 2C) two hours after plating. After overnight incubation, cells were treated with 5 μM MeHg (without inhibitors) overnight, and then the viability (open circles, left-side axis) and IL-6 levels in the medium (solid circles, right-side axis) were determined. IL-6 release caused by 5 μM MeHg was used as 100% in this set of analyses. Results indicated that all three PLA2 inhibitors decreased MeHg induced IL-6 release. Among them, BEL was the only one that could significantly prevent MeHg cytotoxicity.
In addition to AACOCF3, PLA2 can be inhibited by MAFP (structurally similar to AACOCF3) [2, 15] and BEL (structurally distinct from those two above) [14]. MAFP at 0.625 μM or higher concentrations significantly reduced MeHg induced IL-6 release (Fig. 2B, solid circles). With 10 μM MAFP, the IL-6 release was ~51% of that caused by 5 μM MeHg. MAFP at this concentration range did not affect cytotoxicity caused by MeHg (Fig. 2B, open circles).
The other PLA2 inhibitor, BEL, also led to a concentration dependent inhibition of MeHg induced IL-6 release (Fig. 2C, solid circles). Significant inhibition could be observed at 0.625 μM BEL. At 10 μM BEL, the IL-6 release was ~22% of that caused by MeHg. Concurrent viability assay indicated that this agent could prevent MeHg induced cytotoxicity (Fig. 2C, open circles).
Given the observations that PLA2 inhibitors prevented MeHg induced IL-6 release, experiments were performed to determine whether these agents could block other aspects of MeHg cytotoxicity. The effect of these inhibitors on MeHg induced GSH depletion was analyzed in this set of experiments. The concentrations used in this set of experiments were: 20 μM AACOCF3, 10 μM MAFP and 5 μM BEL. These were concentrations that could significantly decrease IL-6 release caused by 5 μM MeHg (Fig. 2). Experiments were thus performed to determine whether they could prevent GSH depletion caused by 5 μM MeHg.
MeHg treatment on mouse glia led to a concentration dependent decrease of GSH levels (Fig. 3A). With 5 μM MeHg, the GSH levels was significantly (p< 0.001) decreased to ~78% of control (Fig. 3A and comparison between 1st set of columns in Fig. 3B). Pretreatment of cells with AACOCF3 led to an increase of cellular GSH levels to ~113% of control, which was significantly (p< 0.001) depressed after cells were treated with MeHg (Fig. 3B, comparison between 2nd set of columns). Pretreatment of cells with MAFP did not alter cellular GSH levels, and MeHg significantly (p<0.001) caused GSH depletion (Fig. 3B, comparison between 3rd set of columns). Pretreatment of cells with BEL increased cellular GSH levels to ~134% of control, and this was significantly (p< 0.001) depressed after cells were treated with MeHg (Fig. 3B, comparison between 4th set of columns). These results indicated that even though these agents could prevent IL-6 release induced by MeHg, they could not prevent the GSH depletion caused by MeHg. However, it should be noted that both AACOCF3 and BEL could significantly increase cellular GSH levels (to ~113% and ~134% of control, respectively, Fig. 3B). This property might partially account for their abilities to prevent MeHg induced cell death (see Fig. 2A and 2C, respectively). See below for further discussion of this subject.
Fig. 3. Effect of PLA2inhibitors on MeHg induced GSH depletion.
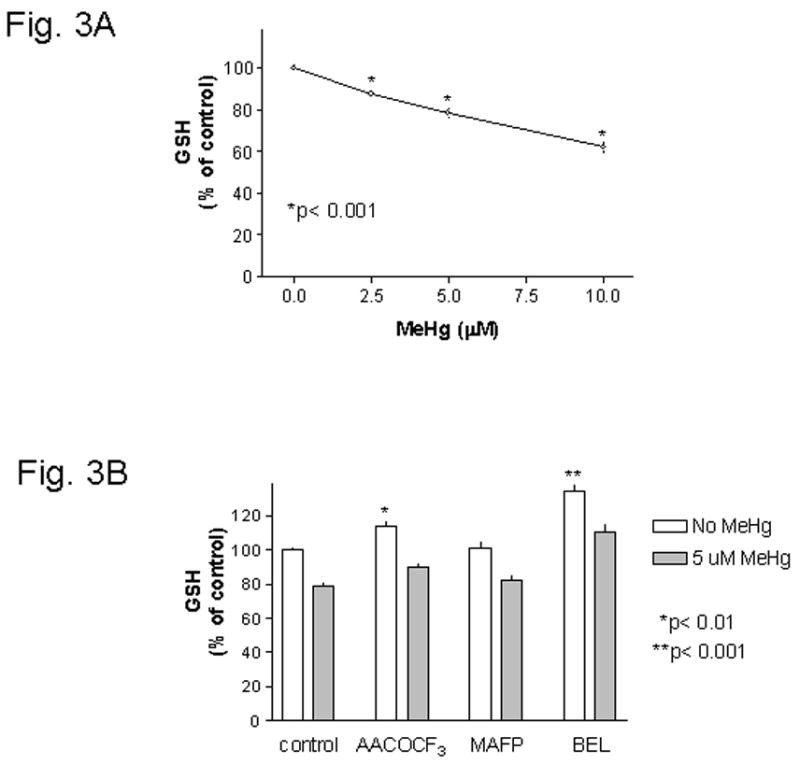
Fig. 3A : Concentration dependent depletion of cellular GSH by MeHg. Mouse glia were treated with various concentrations of MeHg for 4 hours, and then the cellular GSH levels were determined. MeHg at 2.5 μM or above caused significant GSH depletion. Fig. 3B : PLA2 inhibitors did not prevent MeHg induced GSH depletion. Mouse glia were treated with 20 μM AACOCF3, 10 μM MAFP or 5 μM BEL two hours after plating. After overnight incubation, cells were treated with 5 μM MeHg (without inhibitors) for 4 hours, and then cellular GSH was determined. Two-way ANOVA analysis indicated that both MeHg and PLA2 inhibitors had significant effects on GSH levels. There was no interactions between these two treatments. Post-test Bonferroni analyses between each set of columns indicated that MeHg always caused significant (p< 0.001) GSH depletion regardless of whether cells were untreated or were treated with PLA2 inhibitors. Further analysis indicated that both AACOCF3 (p< 0.01) and BEL (p< 0.001) could significantly raise cellular GSH levels above control.
Verity et al., reported that MeHg increase PLA2 activities in cultured cerebellar granule neurons [26]. The finding was confirmed by Shanker et al., who reported that MeHg, in addition to activating PLA2 (within minutes), also increased PLA2 protein expression (within hours) in primary astrocytes [20] and primary neurons [19]. Activation of PLA2 is involved in damaging effects to cells caused by a variety of cytotoxic agents [8]. Pharmacological inhibition of this enzyme can reduce the formation MeHg-induced reactive oxygen species, thus may help to reduce MeHg induced cytotoxicity [18].
Results from this study further indicate that PLA2 activation by MeHg is required for its ability to induce IL-6 release. Activation of PLA2 can cause generation of arachidonic acid and lysophosphatidyl choline, both of which are potent stimulators of IL-6 release [3, 24]. While PLA2 inhibitors specifically prevented IL-6 released caused by MeHg, they did not prevent every aspect of MeHg cytotoxicity. For example, they did not prevent GSH depletion caused by MeHg (Fig. 3B).
Whether inhibition of PLA2 could promote cell survival in MeHg treated cultures was a subject of investigation in previous reports. Verity et al. reported that 10 μM MeHg induced PLA2 activation in cultured cerebellar granule neurons could be inhibited 50% by 100 μM mecaprine. However, this did not lead to a reduced cytotoxicity [26]. In contrast, Mazerik et al. reported that 1 μM AACOCF3 pretreatment led to significant less cytotoxicity caused by 10 μM MeHg in bovine pulmonary artery endothelial cells [16]. The discrepancy between these findings may be caused by different cell type used, different experimental protocols (e.g., pretreatment duration) and how cytotoxic MeHg exhibited in those particular experimental conditions.
Results from this study using a moderate cytotoxic concentration (5 μM) of MeHg and an overnight pretreatment paradigm indicated that inhibition of PLA2 by 20 μM AACOCF3 could slightly raise cell viability in MeHg treated cultures even though the difference was not statistically significant (Fig. 2A). On the other hand, 10 μM MAFP had no effect on MeHg induced cytotoxicity (Fig. 2B). In contrast, 5 μM BEL significantly prevented MeHg induced cytotoxicity (Fig. 2C). Further studies indicated that the ability of these agents to raise cellular GSH levels (Fig. 3B) correlated well with their ability to prevent MeHg induced cytotoxicity. As a result, the agent that could best raise GSH levels (i.e., BEL) was the most effective in preventing MeHg induced cell death. These results were consistent with an earlier report that indicated cells with higher GSH levels were more resistant to mercury cytotoxicity [23].
In conclusion, MeHg causes IL-6 release in primary mouse glia. Even though cytotoxicity is accompanied with IL-6 release, cytotoxicity itself caused by heavy metals is not sufficient for IL-6 induction. MeHg induced PLA2 activation is necessary for IL-6 induction. While the detailed downstream events necessary for IL-6 induction remains to be elucidated, results from others suggest that both arachidonic acid and lysophosphatidyl choline released by PLA2 are potent stimulator for IL-6 production [3, 24]. Built on those earlier studies describing the ability of MeHg to increase PLA2 activity and expression in glia [20], the current study extends those findings and establishes that an increase of IL-6 release is a cellular consequence of glial PLA2 activation. Whether this IL-6 release by glia attenuates or aggravates MeHg neurotoxicity in animals remains to be determined.
Acknowledgments
This work was partially supported by funds from Research to Prevent Blindness. Technical support provided by a P30 facility funded by the NIH (NS047546) was greatly appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aschner M, Allen JW, Kimelberg HK, LoPachin RM, Streit WJ. Glial cells in neurotoxicity development. Annu Rev Pharmacol Toxicol. 1999;39:151–173. doi: 10.1146/annurev.pharmtox.39.1.151. [DOI] [PubMed] [Google Scholar]
- 2.Balsinde J, Dennis EA. Distinct roles in signal transduction for each of the phospholipase A2 enzymes present in P388D1 macrophages. J Biol Chem. 1996;271:6758–6765. doi: 10.1074/jbc.271.12.6758. [DOI] [PubMed] [Google Scholar]
- 3.Bordin L, Priante G, Musacchio E, Giunco S, Tibaldi E, Clari G, Baggio B. Arachidonic acid-induced IL-6 expression is mediated by PKC alpha activation in osteoblastic cells. Biochemistry. 2003;42:4485–4491. doi: 10.1021/bi026842n. [DOI] [PubMed] [Google Scholar]
- 4.Chang JY. Methylmercury causes glial IL-6 release. Neurosci Lett. 2007;416:217–220. doi: 10.1016/j.neulet.2007.01.076. [DOI] [PubMed] [Google Scholar]
- 5.Chang JY, Tsai PF. Prevention of methylmercury-induced mitochondrial depolarization, glutathione depletion and cell death by 15-deoxy-delta-12,14-prostaglandin J(2) Neurotoxicology. 2008;29:1054–1061. doi: 10.1016/j.neuro.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clarkson TW, Magos L, Myers GJ. The toxicology of mercury--current exposures and clinical manifestations. N Engl J Med. 2003;349:1731–1737. doi: 10.1056/NEJMra022471. [DOI] [PubMed] [Google Scholar]
- 7.Costa LG, Aschner M, Vitalone A, Syversen T, Soldin OP. Developmental neuropathology of environmental agents. Annu Rev Pharmacol Toxicol. 2004;44:87–110. doi: 10.1146/annurev.pharmtox.44.101802.121424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cummings BS, McHowat J, Schnellmann RG. Phospholipase A(2)s in cell injury and death. J Pharmacol Exp Ther. 2000;294:793–799. [PubMed] [Google Scholar]
- 9.Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 10.Eskes C, Honegger P, Juillerat-Jeanneret L, Monnet-Tschudi F. Microglial reaction induced by noncytotoxic methylmercury treatment leads to neuroprotection via interactions with astrocytes and IL-6 release. Glia. 2002;37:43–52. doi: 10.1002/glia.10019. [DOI] [PubMed] [Google Scholar]
- 11.Fonnum F, Lock EA. The contributions of excitotoxicity, glutathione depletion and DNA repair in chemically induced injury to neurones: exemplified with toxic effects on cerebellar granule cells. J Neurochem. 2004;88:513–531. doi: 10.1046/j.1471-4159.2003.02211.x. [DOI] [PubMed] [Google Scholar]
- 12.Garg TK, Chang JY. 15-deoxy-delta 12, 14-Prostaglandin J2 prevents reactive oxygen species generation and mitochondrial membrane depolarization induced by oxidative stress. BMC Pharmacol. 2004;4:6. doi: 10.1186/1471-2210-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garg TK, Chang JY. Methylmercury causes oxidative stress and cytotoxicity in microglia: Attenuation by 15-deoxy-delta 12, 14-Prostaglandin J2. J Neuroimmunol. 2006;171:17–28. doi: 10.1016/j.jneuroim.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 14.Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2. Mechanism-based discrimination between calcium-dependent and -independent phospholipases A2. J Biol Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- 15.Lio YC, Reynolds LJ, Balsinde J, Dennis EA. Irreversible inhibition of Ca(2+)-independent phospholipase A2 by methyl arachidonyl fluorophosphonate. Biochim Biophys Acta. 1996;1302:55–60. doi: 10.1016/0005-2760(96)00002-1. [DOI] [PubMed] [Google Scholar]
- 16.Mazerik JN, Hagele T, Sherwani S, Ciapala V, Butler S, Kuppusamy ML, Hunter M, Kuppusamy P, Marsh CB, Parinandi NL. Phospholipase A2 activation regulates cytotoxicity of methylmercury in vascular endothelial cells. Int J Toxicol. 2007;26:553–569. doi: 10.1080/10915810701707759. [DOI] [PubMed] [Google Scholar]
- 17.Nakashima K, Taga T. Mechanisms underlying cytokine-mediated cell-fate regulation in the nervous system. Mol Neurobiol. 2002;25:233–244. doi: 10.1385/MN:25:3:233. [DOI] [PubMed] [Google Scholar]
- 18.Shanker G, Aschner M. Methylmercury-induced reactive oxygen species formation in neonatal cerebral astrocytic cultures is attenuated by antioxidants. Brain Res Mol Brain Res. 2003;110:85–91. doi: 10.1016/s0169-328x(02)00642-3. [DOI] [PubMed] [Google Scholar]
- 19.Shanker G, Hampson RE, Aschner M. Methylmercury stimulates arachidonic acid release and cytosolic phospholipase A2 expression in primary neuronal cultures. Neurotoxicology. 2004;25:399–406. doi: 10.1016/j.neuro.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 20.Shanker G, Mutkus LA, Walker SJ, Aschner M. Methylmercury enhances arachidonic acid release and cytosolic phospholipase A2 expression in primary cultures of neonatal astrocytes. Brain Res Mol Brain Res. 2002;106:1–11. doi: 10.1016/s0169-328x(02)00403-5. [DOI] [PubMed] [Google Scholar]
- 21.Shenker BJ, Berthold P, Rooney C, Vitale L, DeBolt K, Shapiro IM. Immunotoxic effects of mercuric compounds on human lymphocytes and monocytes. III. Alterations in B-cell function and viability. Immunopharmacol Immunotoxicol. 1993;15:87–112. doi: 10.3109/08923979309066936. [DOI] [PubMed] [Google Scholar]
- 22.Shenker BJ, Guo TL, IO, Shapiro IM. Induction of apoptosis in human T-cells by methyl mercury: temporal relationship between mitochondrial dysfunction and loss of reductive reserve. Toxicol Appl Pharmacol. 1999;157:23–35. doi: 10.1006/taap.1999.8652. [DOI] [PubMed] [Google Scholar]
- 23.Shenker BJ, Mayro JS, Rooney C, Vitale L, Shapiro IM. Immunotoxic effects of mercuric compounds on human lymphocytes and monocytes. IV. Alterations in cellular glutathione content. Immunopharmacol Immunotoxicol. 1993;15:273–290. doi: 10.3109/08923979309025999. [DOI] [PubMed] [Google Scholar]
- 24.Spangelo BL, Jarvis WD. Lysophosphatidylcholine stimulates interleukin-6 release from rat anterior pituitary cells in vitro. Endocrinology. 1996;137:4419–4426. doi: 10.1210/endo.137.10.8828503. [DOI] [PubMed] [Google Scholar]
- 25.Stoll G, Jander S, Schroeter M. Cytokines in CNS disorders: neurotoxicity versus neuroprotection. J Neural Transm Suppl. 2000;59:81–89. doi: 10.1007/978-3-7091-6781-6_11. [DOI] [PubMed] [Google Scholar]
- 26.Verity MA, Sarafian T, Pacifici EH, Sevanian A. Phospholipase A2 stimulation by methyl mercury in neuron culture. J Neurochem. 1994;62:705–714. doi: 10.1046/j.1471-4159.1994.62020705.x. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Xue H, Xu Q, Zhang K, Hao X, Wang L, Yan G. p38 kinase/cytosolic phospholipase A2/cyclooxygenase-2 pathway: a new signaling cascade for lipopolysaccharide-induced interleukin-1beta and interleukin-6 release in differentiated U937 cells. Prostaglandins Other Lipid Mediat. 2008;86:61–67. doi: 10.1016/j.prostaglandins.2008.03.002. [DOI] [PubMed] [Google Scholar]