

Abstract
Previous reports have shown that the human immunodeficiency virus (HIV) regulatory protein Tat has both pro-oxidant and pro-inflammatory properties, suggesting that Tat might contribute to the neurological complications of HIV. However, the intracellular mechanisms whereby Tat triggers free radical production and inflammation, and the relationship between Tat-induced free radicals and inflammatory reactions, are still subject to debate. The present study was undertaken to evaluate the specific effects of Tat on NADPH oxidase in microglia and macrophages, and to determine the specific role of NADPH oxidase in Tat-induced cytokine/chemokine release and neurotoxicity. Application of Tat to microglia or macrophages caused dose-and time-dependent increases in superoxide formation that were prevented by both pharmacologic NADPH oxidase inhibitors and by specific decoy peptides (gp91ds). Furthermore, inhibition of NADPH oxidase attenuated Tat-induced release of interleukin-6 (IL-6), tumor necrosis factor alpha (TNFα), and monocyte chemoattractant protein 1 (MCP-1), and decreased microglial-mediated neurotoxicity. Finally, macrophages derived from NADPH oxidase-deficient mice displayed reduced superoxide production, released lower levels of cytokines/chemokines, and induced less neurotoxicity in response to Tat compared to wild-type macrophages. Together, these data describe a specific and biologically significant signaling component of the macrophage/microglial response to Tat, and suggest the neuropathology associated with HIV infection might originate in part with Tat-induced activation of NADPH oxidase. Antioxid. Redox Signal. 11, 193–204.
Introduction
Over 1 million people in the United States may be infected with human immunodeficiency virus (HIV), and of those, 10–20% could eventually suffer from HIV dementia (HIVD) (19). The essential features of HIVD are progressive cognitive impairments accompanied by motor dysfunction, speech deficits, and behavioral changes (28). HIVD generally occurs in the context of advanced immunosuppression, but can present as the initial clinical manifestation of AIDS, particularly in pediatric cases, where it is associated with delayed development of motor and cognitive milestones (16). Despite these clinically significant changes in neurological function in HIVD, neurons themselves are not infected by HIV. Macrophages, including brain resident microglia and invading peripheral cells, are the principal brain-resident cells able to support and propagate productive HIV infection. Furthermore, extensive microglial/macrophage activation is a characteristic feature of HIVD brains, and has been shown to correlate with the clinical manifestations of HIVD (18, 20, 43). While the factor(s) responsible for widespread macrophage activation have not been clearly identified, observations that mRNA for the HIV viral regulatory protein Tat is elevated in patients with HIV dementia (45, 47), and that Tat is actively secreted by infected cells (15, 42), suggest a possible role for this protein in the progression of HIVD. In support of this hypothesis, Tat has been shown to be potently pro-inflammatory in macrophages (3, 7, 30, 39), and to correlate positively with human immunodeficiency virus (HIV)- and simian/human immunodeficiency virus (SHIV)-induced encephalitis (22). Taken together, these data strongly suggest that HIV-Tat may be an important mediator of potentially toxic macrophage reactions in the brain.
Microglia and macrophages are phagocytic immunocompetent cells that participate in both nonspecific innate immunity and also adaptive, humoral immune reactions, and thus can be potent sources of cytokines, reactive oxygen and nitrogen intermediates, and excitotoxins (6, 32, 41). Thus, these cells are important players in local immune responses, but the exact mechanisms underlying the phenotypical and functional transformation of these cells are still unclear. Interestingly, published studies have introduced the idea that the oxidative burst might direct intracellular inflammatory signal transduction in microglia/macrophages (13, 34, 37). Oxidative bust activity is carried out by NADPH oxidase, a superoxide-producing enzyme system consisting of membrane (gp91phox and p22phox) and cytosolic (p47phox, p67phox, and p40phox) components (10). Upon activation, the cytosolic component p47phox is phosphorylated, causing the cytosolic complex to translocate to the membrane. Once the entire complex is assembled at the membrane, p67phox interacts via its activation domain with the membrane-bound subunits, causing gp91phox to drive the transfer of electrons from NADPH to oxygen (44). Activation of NADPH oxidase and increased oxidative burst activity is a characteristic feature of macrophage activation both in vitro and in vivo, and experimental evidence suggests that free radical production in brain-resident immune cells may drive intracellular inflammatory signaling and the promulgation of the inflammatory cascade in the brain, in addition to contributing to local concentrations of free radicals. For instance, the activity of NADPH oxidase has been specifically implicated in activation of nuclear factor kappa B (NF-κB) and synthesis of tumor necrosis factor alpha (TNFα) (24), which are proposed to be important mediators of HIV-related neuronal damage (46). Likewise, NADPH-driven oxidative burst activity has also been shown to be critical for microglial release of glutamate (2). Finally, the activation of NADPH oxidase in microglia has been shown to participate in microglial-mediated neurotoxicity in experimental models of both Alzheimer's disease (23) and Parkinson's disease (17). Thus, NADPH oxidase may be an important regulator of immune-mediated and/or oxidative brain injury, but this enzyme system has not been well studied in terms of HIV and neuroAIDS.
To thus determine if NADPH oxidase plays an important role in Tat-induced neurotoxic inflammatory signaling in brain macrophages, the effects of recombinant Tat 1–72 on NADPH oxidase activity in microglial cell lines, primary rodent microglia, and primary macrophages was documented. Additionally, the role of NADPH oxidase in cytokine/chemokine release was determined through use of the NADPH oxidase inhibitors diphenylene iodonium (DPI) and apocynin. The broad-spectrum flavo-cytochome oxidase inhibitor DPI is commonly used to inhibit macrophage NADPH oxidase, although it may also affect cytochrome oxidases at higher doses (5–10 μm) (9). Thus, the more specific but less potent inhibitor apocynin, which acts by preventing the assembly of the NADPH oxidase subunits (40), was also employed. Finally, the role of NADPH oxidase in Tat-induced cytokine/chemokine release and neurotoxicity was confirmed through the study of macrophages derived from either wild-type or gp91phox-deficient mice, and through use of a specific decoy peptide that prevents the assembly of NADPH oxidase. This peptide inhibitor, gp91ds, is a fusion peptide made from a 9-mer, CSTRIRRQL, that inhibits assembly of NADPH oxidase by mimicking the gp91phox docking site for the cytoplasmic p47phox subunit, conjugated to an additional 9-mer sequence from the basic domain of the HIV Tat protein that drives membrane permeability. This peptide has been used extensively to selectively block NADPH oxidase in both in vitro and in vivo systems (33, 38, 48).
Materials and Methods
NADPH oxidase inhibitors
DPI and apocynin were purchased from Sigma-Aldrich (St. Louis, MO), were solubilized at 10 mM stocks in DMSO and stored at −20°C. The NADPH oxidase docking sequence peptide (gp91ds–(H)RKKRRQRRR-CSTRIRRQL(NH3)) and the control, scrambled peptide ((H)RKKRRQRRR-CLRITRQSR(NH3)) were custom-synthesized by Bachem Americas, Inc. (Torrence, CA). Peptides were aliquoted into 5 mM stock solutions in 150 mM NaCl acidified with 0.01 N acetic acid and stored at −20°C. Microglia were incubated with gp91ds or scrambled peptide (10–50 μM) or vehicle for 1 h in serum-free medium before addition of Tat.
Preparation of Tat
Recombinant Tat 1-72 was produced and purified as described previously (5). Briefly, the Tat gene encoding the first 72 amino acids of HIV-1BRU (obtained from Dr. Richard Graynor, through the NIH AIDS repository) was inserted into an Eschrichia coli vector Pin Point Xa-2 (Promega, Madison, WI). Biotinylated Tat was purified on a column of soft release avidin resin, cleaved from the fusion protein using factor Xa, eluted from the column, and desalted with a PD10 column. The protein concentrations were determined, and Tat was aliquoted, lyophilizedm and stored at −80°C.
Cell culture and treatment
N9 murine microglial cells (kindly provided by Dr. Paola Ricciardi-Castagnoli, Consiglio Nazionale Delle Ricerche, Milano, Italy) were maintained in Iscove's Modified Dulbecco's Medium (IMDM; Gibco BRL, Gaithersburg, MD) supplemented with 5% heat-inactivated FBS (Sigma, St. Louis MO) and 25 μM β-mercaptoethanol. All experiments were conducted on cells that had been passaged no >20 times, plated at 70–80% confluence in phenol red-free, serum-free medium lacking β-mercaptoethanol.
Primary microglia were isolated mixed glial cultures derived from neonatal Sprague-Dawley rat pups, as described previously (4). Briefly, mixed glial cultures were generated from the brain tissues (cerebral cortices without meningies) and maintained in Modified Eagle Medium (Gibco BRL) with 10% fetal bovine serum. After 7–10 days in vitro, microglia were isolated from confluent mixed glial cultures by differential panning (45 min at 200 rpm on an orbital skaker). Cells were pelleted and subcultured at 5 × 105 cells/ml, and used within 24 h after subculturing. Such cultures generally contain only microglial cells with very few type I or II astrocytes (less that 8% of total cells).
Primary macrophages were isolated from adult wild-type and gp91phox-deficient mice by peritoneal lavage. Briefly, mice were killed by cervical dislocation under isoflurane anesthesia, and the peritoneal exudate harvested by lavage of the peritoneal cavities with 8 ml of sterile phosphate-buffered saline (PBS). Lavage fluids were centrifuged at 200 g for 5 min at 4°C, and the pellet was washed in 2 ml of IMDM. Total viable cell number was determined, and the peritoneal cells were cultured in IMDM (5 × 106 cells/ml for NADPH oxidase assays and 5 × 105 cells/ml for assays of cytokine/neurotoxin release) for 1 h at 37°C. The medium was then changed to remove nonadherent cells, and cells were treated immediately for NADPH oxidase assays or for assays of cytokine/neurotoxin release.
Analyses of superoxide production
The NADPH-dependent production of superoxide was measured by documenting lucigenin luminescence. Briefly, following treatment, cells were lysed by three freeze-thaw cycles and then brought up in protease inhibitor-containing PBS at 4°C. Samples (10–25 μg total protein) were incubated with 5 μM lucigenin and 100 μM NADPH. NADPH oxidase activity was measured immediately using a luminescence plate reader at 37°C. To verify the specific role of NADPH oxidase in the measured luminescence, background levels of luminescence for each sample was generated by measuring luminescence in the presence of 1 μM DPI.
Analyses of p47phox translocation
To verify p47translocation to the microglial cell membrane, N9 cells were treated with 100 nM Tat or 10 μM PMA for 1 h, after which the cells were washed, and homogenized in a solution containing 0.32 M sucrose; 5 mM Tris-HCI, pH 7.5; and protease inhibitors. At this point, the samples were normalized for protein content, and an aliquot was removed from each sample, denatured in SDS, and used to represent the total amount of p47. The remaining homogenate was then centrifuged at 1000 g for 5 min to remove nuclei. The supernatant was centrifuged at 12,500 g for 15 min to remove the crude mitochondrial pellet, and the resultant supernatant was centrifuged at 100,000 g for 45 min to isolate the crude membrane pellet. The membranes were homogenized in phosphate buffer (0.05 M potassium phosphate buffer, pH 7.4), denatured in SDS, and proteins from both total and membrane fractions were electrophoretically separated in polyacrylamide gels and blotted onto nitrocellulose. Blots were processed using a primary antibody directed against murine p47phox (Millipore, Billerica, MA), followed by horseradish peroxidase-conjugated secondary antibody, and visualized using a chemiluminescence system (Amersham Biosciences, Piscataway, NJ).
ELISAs
Levels of IL-6, MCP-1, and TNFα in cell culture medium were measured by ELISA as described previously (3). In each case, a dose-response and time-course curve was generated, and the threshold sensitivity was typically 1 pg/ml.
Neurotoxicity assays
Macrophage- and microglia-mediated neuronal injury was assessed in cultured primary hippocampal neurons exposed to conditioned medium. Primary hippocampal neurons were isolated and plated at a density of 1 × 105 cells/ml in 30 mm dishes in 1.5 ml of Neurobasal medium (Gibco BRL, Gaithersburg, MD) supplemented with B27 supplements, as described previously (11). After 6 days in vitro, 500 μl of the medium was removed, and replaced with 500 μl of either fresh Neurobasal/B27, unconditioned medium, or medium conditioned by macrophages or microglia (see next paragraph below). Neuronal survival was assessed via repeated measures of neuronal morphology as described previously (11). Briefly, viable neurons were counted in premarked microscope fields (five distinct fields per dish) before treatment and 24 h after treatment, with the viability of neurons assessed by morphological criteria. Neurons with intact neurites of uniform diameter and a soma with a smooth appearance were considered viable, while neurons with fragmented neurites and vacuolated and/or swollen soma were considered nonviable, as were neurons that detached from the dish over the course of treatment. The number of viable neurons both before and after treatment was determined, and survival was expressed as the percentage of total neurons present before treatment that remained viable after 24 h.
To generate conditioned medium, peritoneal macrophages isolated from either wild-type or gp91phox knock out adult male mice were cultured and treated with either vehicle (Hank's balanced salt solution, HBSS), Tat (100 nM), or combined LPS and IFNγ (100 ng/ml and 100 U/ml, respectively) and allowed to condition serum-free IMDM for 24 h. The medium was then removed, filtered to remove any cells, and applied immediately to cultured hippocampal neurons. Alternatively, primary microglia were isolated from mixed glial cultures, plated in serum-free DMEM, and treated with either gp91ds or scrambled peptides (50 μM) for 60 min, after which 100 nM Tat was applied. After a 3-h exposure, the cells were washed to remove all traces of Tat and/or the peptides, and allowed to condition serum-free medium for an additional 24 h. The medium was then removed, filtered to remove any detached cells, and applied immediately to cultured hippocampal neurons
Statistical analyses
All data were analyzed using one-way or two-way ANOVA, followed by Bonferroni's post-hoc analysis, respectively, to determine statistical significance; p values < 0.05 were designated as statistically significant, and are indicated in the text as *, **, or *** corresponding to p values < 0.05, < 0.01, or < 0.001, respectively.
Results
NADPH oxidase mediates Tat-induced superoxide release in microglia and macrophages
Initial experiments in this investigation were designed to evaluate the specific role of NADPH oxidase in HIV-Tat-induced superoxide production in microglia and macrophages. Previous reports using a variety of techniques have shown that Tat can induce or augment superoxide production in cultured cells (39). Furthermore, reports from our lab have used immunoabsorbed preparations of Tat to document that the effects of Tat on superoxide production were specific to the protein and not based on endotoxin contaminants present in the recombinant preparation (3). NADPH oxidase activity in Tat-treated microglia was measured by quantifying the NADPH-dependent, superoxide driven oxidation of lucigenin, as described in Methods. To determine the time course of Tat-induced NADPH oxidase activity, N9 microglia were treated with 100 nM Tat 1-72 or 10 μM PMA for 15 min up to 24 h, after which cells were collected and analyzed. Data show that while lucigenin oxidation induced by unstimulated cells was generally very stable over 24 h in serum-free medium (Fig. 1A), application of Tat caused significant increases in luminescence as early as 1 h after application (Fig. 1A). Tat-induced increases in luminescence were sustained for up to 8 h after Tat application, returning to baseline levels after 24 h (Fig. 1A). PMA was used as a positive control, and PMA-induced luminescence was increased significantly over control after 15 min, and remained increased over the duration of the 24 h experiment (Fig. 1A). Data obtained using lucigenin were confirmed using MCLA (2-methyl-6-(p-methoxyphenyl)-3,7-dihydroimidazo(1,2-alpha)pyrazin-3-one) in place of lucigenin, and also by documenting superoxide generation using flow-cytometric analyses of DHE oxidation. Both assays confirmed the ability of Tat and PMA to induce significant increases in superoxide radical (data not shown). Tat-induced lucigenin oxidation in N9 cells was also dose-dependent, with concentrations as low as 50 nM causing significant elevations in luminescence 4 h following Tat application (Fig. 1B). To confirm the role of NADPH oxidase in Tat-induced lucigenin oxidation, the effects of the NADPH oxidase inhibitors DPI and apocynin were tested against Tat-induced lucigenin oxidation in N9 cells. Data show that exposure to 1 μM DPI or 250 μM apocynin completely prevented Tat-induced increases in luminescence (Fig. 1B), while analyses of trypan blue exclusion indicated that these inhibitors were not toxic to N9 cells at the doses employed (data not shown). Finally, the translocation of p47phox to the plasma membrane, a necessary step for NADPH oxidase activity, was confirmed in N9 cells treated with 100 nM Tat or 10 μM PMA for 1 h. The localization of p47phox in the plasma membrane was determined by Western blot analysis as described in Methods. Visual evaluation of blots indicates that both Tat and PMA induce marked increases in p47phox levels in plasma membrane fractions, while total levels of p47 were unchanged by treatment (Fig. 1C). Densitometric evaluations of blots revealed average (mean ± SEM) band intensities presented as percent control as follows: membrane pool: Control = 100 ± 16.13, Tat = 182.63 ± 16.7, PMA = 200.1 ± 11.82; total lysate: Control = 100 ± 6.9, Tat = 83.63 ± 6.8, PMA = 93.9 ± 7.02.
FIG. 1.
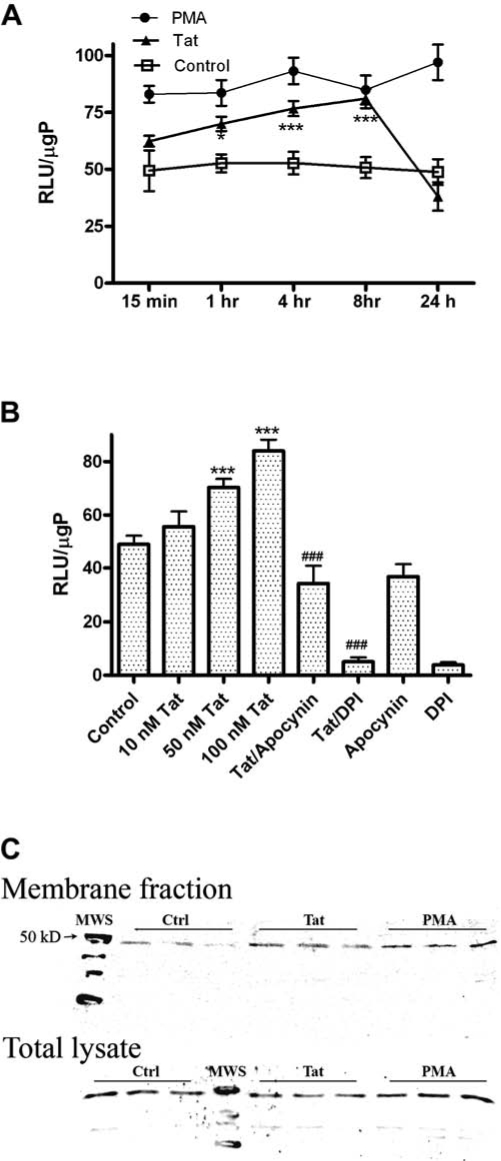
Dose-and time-dependent effects of Tat NADPH oxidase activity in N9 cells. N9 microglial cells were grown as described in Methods, and were treated with Tat or PMA for measures of NADPH oxidase activity. (A) N9 microglia were treated with either vehicle control (HBSS, open squares), 100 nM Tat (closed triangles), or 10 μM PMA (closed circles) for increasing periods of time. Cells were then harvested, lysed, and NADPH oxidase activity in the homogenates was analyzed by quantifying the light produced by NADPH-dependent oxidation of lucigenin, as described in Methods. Data were obtained from three separate experiments with 5–6 samples per group in each experiment, and were analyzed by two-way ANOVA. * and *** indicate statistically significant increases (p < 0.05 and p < 0.001, respectively) in NADPH oxidase activity in Tat-treated cells as compared to vehicle-treated (control) microglia. (B) N9 microglia treated with increasing doses of Tat for 4 h. Additional cells were treated with 1 μM DPI or 250 μM apocynin, both inhibitors of NADPH oxidase, in the presence or absence of 100 nM Tat for 4 h. Cells were then harvested, lysed, and NADPH oxidase activity in the homogenates was analyzed as described in Methods. Data were obtained from two separate experiments with 6 samples per group in each experiment, and were analyzed by one-way ANOVA. *** indicates the statistically significant increases (p < 0.001) in NADPH oxidase activity cells treated with 50 and 100 nM Tat as compared to vehicle-treated (control) microglia, while ### indicates the statistically significant decrease (p < 0.001, respectively) in NADPH oxidase activity when cells are treated with 100 nM Tat in the presence of DPI or apocynin. (C) N9 cells were treated with vehicle (Ctrl), 100 nM Tat, or 10 μM PMA for 60 min. Cells were then lysed, normalized for protein content, and the total and plasma membrane fractions were isolated from homogenates, as described in Methods. The proteins were separated on 12% polyacrylimide gels and blotted with anti-p47phox to visualize the differential presence of 47phox in both total and plasma membrane fractions. Data are representative of three separate experiments, with n = 3 for each treatment on each blot.
To ensure that Tat had similar effects in primary microglia cells as in N9 cells (and hence were not an artifact of cell immortalization), the effects of Tat on NADPH oxidase activity in primary microglia were determined. Primary microglia were isolated from mixed glial cultures as described in Methods, and exposed to increasing doses of Tat or 10 μM PMA for 4 h, after which NADPH oxidase activity was measured using lucigenin as described in Methods. Interestingly, basal levels of NADPH oxidase in unstimulated cells were approximately threefold higher in primary microglia than in N9 cells (131.6 ± 27.57 compared to 52.69 ± 4.99 RLU/μgP in N9 cells). However, as was observed in N9 cells, Tat caused significant, dose-dependent increases in NADPH oxidase activity in primary microglia, that were again completely prevented by DPI (Fig. 2).
FIG. 2.

Effects of Tat on NADPH oxidase activity in primary microglia. Primary rodent microglia were isolated from neonatal animals and grown as described in Methods. Cells were treated with increasing doses of Tat for 4 h, and additional cells were treated with Tat in the presence of 1 μM DPI as a negative control, while 10 μM PMA served as a positive control. Cells were then harvested, lysed, and NADPH oxidase activity in the homogenates was analyzed as described in Methods. Data were obtained from three separate experiments with 4 samples per group in each experiment, and were analyzed by one-way ANOVA. * and *** indicate statistically significant increases (p < 0.05 and p < 0.001, respectively) in NADPH oxidase activity in cells treated with 50 nM and 100 nM Tat as compared to vehicle-treated (control) microglia, while ### indicates the statistically significant decrease (p < 0.001) in NADPH oxidase activity in cells treated with 100 nM Tat in the presence of 1 μM DPI.
As a final control to confirm that Tat-induced lucigenin oxidation was based on NADPH oxidase activity, experiments were undertaken using primary peritoneal macrophages isolated from wild-type (WT) mice and from mice deficient in the catalytic gp91phox subunit of NADPH oxidase (NOX KO). Macrophages from 6 individual WT and 6 individual NOX KO mice were cultured as described in Methods and treated with increasing doses of Tat or 10 μM PMA for 60 min, after which NADPH oxidase activity was measured as described in Methods. Tat caused significant, dose-dependent increases in NADPH oxidase activity in peritoneal macrophages isolated from WT mice (Fig. 3). While detectable luminescence was elicited by NOX KO peritoneal macrophages, levels of lucigenin oxidation were approximately sixfold higher (344.5 ± 36.5 compared to 60.4 ± 10.0 RLU/1,500,000 unstimulated cells) in cells derived from WT mice compared to cells derived from NOX KO mice (Fig. 3).
FIG. 3.

Effects of Tat on NADPH oxidase activity in primary macrophages isolated from wild-type and gp91phox deficient mice. Primary rodent peritoneal macrophages were isolated from adult wild-type (WT) and p91phox knock-out (NOX KO) mice, as described in Methods. Cells were seeded into 96-well plates at a density of 5 × 106 cells/ml and treated with increasing doses of Tat for 60 min. Additional cells were treated with 10 μM PMA. Cells were then harvested, lysed, and NADPH oxidase activity was then analyzed as described in Methods. Data were obtained from two separate experiments with three samples per group in each experiment, and were analyzed by two-way ANOVA. *** indicates the statistically significant increases (p < 0.001) in NADPH oxidase activity in WT cells treated with all doses of Tat and PMA as compared to vehicle-treated (control) macrophages, while ### indicates the statistically significant decrease (p < 0.001) in NADPH oxidase activity in similarly treated NOX KO cells.
Inhibition of NADPH oxidase decreases Tat-induced cytokine/chemokine release
To determine the role of NADPH oxidase activity in Tat-induced cytokine production, N9 cells were treated with increasing concentrations of Tat for 24 h, and the cell culture medium was tested for TNF-α, IL-6, and MCP-1 using ELISA. Tat caused significant, time-dependent increases in the secretion of all three inflammatory signals (Fig. 4A, B, and C). However, Tat-induced release of TNFα, IL-6, and MCP-1 was significantly reduced in N9 cells that had been co-treated with apocynin or DPI (Fig. 4A, B, and C). To again verify that Tat induced the same response in primary cells as N9 cells, the effects of Tat and apocynin on cytokine/chemokine release from primary microglia was determined. Primary microglial cultures were isolated and purified as described in Methods, and then exposed 100 nM Tat in the presence or absence of 250 μM apocynin for 24 h. The concentration of inflammatory mediators in the cell culture medium was then determined as described in Methods. As was observed in N9 cells, primary microglia exposed to Tat released significant quantities of TNF-α, IL-6, and MCP-1, and this release of inflammatory mediators was significantly attenuated by apocynin (Fig. 5).
FIG. 4.

Effects of NADPH oxidase inhibition on Tat-induced cytokine and chemokine release from cultured microglial cell lines. Cultured N9 cells were grown as described in Methods. Cells were treated with Tat (10–100 nM) in the presence or absence of 250 μM apocynin or 1 μM DPI for 24 h, after which the medium was harvested, centrifuged, and analyzed for levels of TNFα (A), IL-6 (B), and MCP-1 (C). Data were obtained from three separate experiments with 6–8 samples per group in each experiment, and were analyzed by two-way ANOVA; * and *** indicate statistically significant increases (p > 0.05 and p > 0.001, respectively) in cytokine/chemokine release from Tat-treated cells compared to control treated cells. ### indicates the statistically significant attenuation (p > 0.001) of cytokine/chemokine release observed when N9 cells were treated with Tat in the presence of apocynin or DPI.
FIG. 5.

Effects of NADPH oxidase inhibition on Tat-induced cytokine and chemokine release from primary microglia. Primary rodent microglia were isolated from neonatal rat pups, as described in Methods. Cells were treated with 100 nM Tat in the presence or absence of 250 μM apocynin for 24 h, after which the medium was harvested, centrifuged, and analyzed for levels of TNFα (A), IL-6 (B), and MCP-1 (C), as described in Methods. Data were obtained from three separate experiments with 4–6 samples per group in each experiment, and were analyzed by one-way ANOVA. * and *** indicate statistically significant increases (p < 0.05 and p < 0.001, respectively) in cytokine/chemokine release from cells treated with Tat as compared to vehicle-treated (control) microglia, while # and ### indicate the statistically significant decrease (p < 0.05 and p < 0.001, respectively) in cytokine/chemokine release from primary microglia treated with 100 nM Tat in the presence of apocynin.
Primary macrophages isolated from WT and NOX KO mice were used to confirm the role of NADPH oxidase in Tat-induced cytokine and chemokine release. Primary macrophages were isolated and cultured as described in Methods, and then exposed to 100 nM Tat for 24 h. The release of TNF-α, IL-6, and MCP-1 in the cell culture medium was then determined as described in Methods. As was observed in N9 and primary microglia, 100 nM Tat caused the secretion of significant quantities of TNF-α, IL-6, and MCP-1 into the medium of cultured WT macrophages (Fig. 6), but this release of inflammatory mediators was significantly decreased in the medium of cells isolated from NOX KO mice (Fig. 6).
FIG. 6.

Effects of Tat on cytokine/chemokine release from primary macrophages isolated from wild-type and gp91phox deficient mice. Primary macrophages were isolated from adult wild-type (WT) and p91phox knock-out (NOX KO) mice as described in Methods. Cells were plated and treated with 100 nM Tat for 24 h, after which the medium was harvested, centrifuged, and analyzed for levels of TNFα (A), IL-6 (B), and MCP-1 (C), as described in Methods. Data were obtained from three separate experiments with 4 samples per group in each experiment, and were analyzed by two-way ANOVA. *** indicate statistically significant increases (p < 0.001) in cytokine/chemokine release from cells treated with Tat as compared to vehicle-treated (control) macrophages. # and ### indicate statistically significant decreases (p < 0.05 and p < 0.001, respectively) in cytokine/chemokine release from Tat treated NOX KO cells as compared to Tat-treated WT cells.
NADPH oxidase inhibition decreases Tat-induced neurotoxicity
Microglial activation has been repeatedly documented in the context of brain injury in vivo, with many investigators proposing a critical role for microglia and macrophages in promoting neuronal injury (reviewed in (32)). To determine if Tat elicits a neurotoxic phenotype in macrophage cells, and if NADPH oxidase participates in Tat-induced, macrophage-mediated neurotoxicity, experiments were designed to measure neuronal injury, using a conditioned medium-based assay as described in Methods. For these experiments, primary hippocampal neurons were isolated and plated as described in Methods, and after 6 days in vitro, exposed to either Neurobasal/B27 medium, fresh (unconditioned) IMDM, or IMDM conditioned by WT or NOX KO peritoneal macrophages cells treated with vehicle, Tat, or LPS/IFN. Examination of neuronal morphology 24 h after the application of the conditioned medium indicated that IMDM collected from untreated WT or NOX KO macrophages resulted in similar morphological changes to cultured neurons (Fig. 7A). However, medium from WT macrophages treated with either Tat or LPS/IFN appeared to cause marked neurotoxicity (Fig. 7A), while medium collected from Tat-or LPS/IFN-treated NOX KO macrophages did not (Fig. 7A). Blinded cell counts based on repeated measures of morphology were conducted to quantify neuronal survival as described in Methods. Data confirm that medium conditioned by vehicle-treated cells (either WT or NOX KO) did not induce toxicity over that caused by unconditioned IMDM (Fig. 7B). Medium conditioned by WT cells exposed to either Tat or LPS/IFN induced significant declines in neuronal survival (Fig. 7B). However, no toxicity was induced by medium collected from Tat-treated in NOX KO macrophages (Fig. 7B). Interestingly, medium conditioned by NOX KO cells treated with LPS/IFN did cause modest but significant decreases in neuronal survival (Fig. 7B), although this effect was still significantly different than that induced by WT cells exposed to LPS/IFN (Fig. 7B). This data suggests that the combination of LPS and IFN is able to act at least in part independently of NADPH oxidase in inducing a neurotoxic phenotype in macrophages.
FIG. 7.

Effects of NADPH oxidase knock-out on macrophage-mediated neurotoxicity. Primary hippocampal neurons were plated as described in Methods, and after 6 days in vitro, exposed to either Neurobasal/B27 medium, fresh (unconditioned) IMDM, or IMDM conditioned by WT or NOX KO peritoneal macrophages cells treated with either vehicle, Tat, or combined LPS and IFN (100 ng/ml and 100 U/ml, respectively). Neuronal injury in primary hippocampal neurons exposed to medium conditioned by macrophages was determined as described in Methods. (A) Representative images of primary hippocampal neurons taken either immediately before (Ini) or 24 h after (Fin) exposure to IMDM conditioned by either wild type (WT) or pg91phox knockout (KO) macrophages treated for 24 h with vehicle (HBSS), 100 nM Tat, or LPS/IFN. Arrows indicate specific neurons that were healthy at the onset of exposure but did not survive exposure to conditioned medium. (B) Blinded cell counts based on repeated measures of morphology were conducted to quantify neuronal survival, as described in Methods, and neuronal survival data are expressed in relation to the percentage of survival observed in cells exposed only to Neurobasal/B27 medium for 24 h (% ctrl). Data were obtained from two separate experiments with 3–4 samples per group in each experiment, and were analyzed by two-way ANOVA. *** indicates the statistically significant decrease (p < 0.001) in neuronal survival following exposure to medium from WT macrophages treated with Tat or combined LPS/IFN, while * indicates the statistically significant decrease (p < 0.05) in neuronal survival following exposure to medium from NOX KO macrophages treated with combined LPS/IFN compared to medium from vehicle-treated (control) macrophages. # and ### indicate statistically significant increases (p < 0.05 and p < 0.001, respectively) in survival in neurons exposed to medium conditioned by NOX KO cells as compared to neurons exposed to medium from identically treated WT cells.
Decoy gp91ds peptides prevent NADPH oxidase activity, cytokine release, and neurotoxicity in primary microglia
As a final control to compensate for both the potential nonspecific actions of the pharmacological NADPH oxidase inhibitors and the potential compensatory mechanisms that might affect the gp91 KO cells, experiments were conducted in primary microglia treated with a specific decoy peptide that prevents the assembly of NADPH oxidase. This peptide inhibitor, gp91ds, is a fusion peptide that inhibits assembly of NADPH oxidase by mimicking the gp91phox docking site for the cytoplasmic p47phox subunit (33, 38, 48). Thus, to completely document the role of NADPH oxidase in Tat-induced microglial reactivity, the effects of Tgp91ds on NADPH oxidase activity, cytokine release, and neurotoxicity were determined. Primary microglia were isolated from mixed glial cultures as described in Methods, and incubated with gp91ds or scrambled peptide (10 and 50 μM) or vehicle (150 mM NaCl with 0.01 N acetic acid) for 1 h in serum-free medium before addition of 100 nM Tat. After 4 h the cells were harvested, and NADPH oxidase activity was measured as described in Methods. Data show that Tat-induced increases in NADPH oxidase activity in primary microglia were dose-dependently decreased by gp91ds, but not by the scrambled control peptide (Fig. 8A). To again verify the role of NADPH oxidase in Tat-induced cytokine release, the effects of gp91de and scrambled peptide on TNF release from primary microglia were determined. Primary microglial cultures were isolated and purified as described in Methods, pretreated with 50 μM gp91ds or scrambled peptide for 60 min, and then exposed 100 nM Tat for 24 h. The concentration of TNF in the cell culture medium was then determined as described in Methods. Data show that Tat-induced increases in TNF release from primary microglia were significantly decreased by gp91ds but not by the scrambled control peptide (Fig. 8B). Likewise, gp91ds, but not the vehicle or the scrambled peptide, inhibited Tat-induced release of IL-6 and MCP-1 (data not shown).
FIG. 8.

Effects of gp91ds and scrambled peptides on Tat-induced microglial reactivity in primary microglia. Primary rodent microglia were isolated from neonatal animals and grown as described in Methods. (A) For analyses of NAPDH oxidase, cells were pretreated for 60 min with vehicle (150 mM NaCl with 0.01 N acetic acid) or increasing doses of gp91ds or the scrambled control peptide, and then treated with 100 nM Tat for 4 h. Cells were then harvested, lysed, and NADPH oxidase activity in the homogenates was analyzed as described in Methods. Data were obtained from two separate experiments with 4 samples per group in each experiment and were analyzed by ANOVA. *** indicates statistically significant increases (p < 0.001) in NADPH oxidase activity in cells treated Tat as compared to vehicle-treated (control) microglia, while ## and ### indicates the statistically significant decreases (p < 0.01 and p < 0.001, respectively) in NADPH oxidase activity in cells treated with Tat in the presence of 10 and 50 μM of gp91ds. (B) For analyses of cytokine release, primary microglia were treated with 100 nM Tat in the presence or absence of 50 μM of gp91ds or scrambled peptide (or vehicle) for 24 h, after which the medium was harvested, centrifuged, and analyzed for levels of TNFα as described in Methods. Data were obtained from two separate experiments with 4 samples per group in each experiment and were analyzed by ANOVA. *** indicates the statistically significant increase (p < 0.001) in TNFα release from cells treated with Tat as compared to control microglia, while ### indicates the statistically significant decrease (p < 0.001) in cytokine release from primary microglia treated with Tat in the presence of gp91ds. (C) For neurotoxicity assays, primary hippocampal neurons were plated as described in Methods, and after 6 days in vitro, exposed to either Neurobasal/B27 medium, fresh (unconditioned) DMEM, or DMEM conditioned by primary microglia that had been previously treated with Tat in the absence or presence of gp91ds and the scrambled control peptide. For these experiments, microglia were exposed to gp91ds or scrambled peptides (50 μM) for 60 min, after which 100 nM Tat was applied. After a 3-h exposure, the cells were washed to remove all traces of Tat and/or the peptides, and allowed to condition the serum-free medium for an additional 24 h. Blinded cell counts based on repeated measures of morphology were conducted to quantify neuronal survival as described in Methods, and neuronal survival data are expressed in relation to the percentage of survival observed in cells exposed only to Neurobasal/B27 medium for 24 h (% ctrl). Data were obtained from two separate experiments with 2–5 samples per group in each experiment and were analyzed by two-way ANOVA. *** indicates the statistically significant decrease (p < 0.001) in neuronal survival following exposure to medium from microglia treated with Tat, while ### indicates the statistically significant prevention (p < 0.001) of cytotoxicity in neurons exposed to medium conditioned primary microglia treated with Tat in the presence of gp91ds.
Finally, to both verify the ability of primary microglia to induce neurotoxicity and to determine if NADPH oxidase modulates microglial-mediated neurotoxicity, experiments were designed to measure microglial-mediated neuronal injury, using the conditioned medium-based assay as described in Methods. Primary hippocampal neurons were isolated and plated as described in Methods, and after 6 days in vitro, exposed to either Neurobasal/B27 medium, fresh (unconditioned) DMEM, or DMEM conditioned by primary microglia that had been previously treated with Tat in the absence or presence of gp91ds and the scrambled control peptide. For these experiments, microglia were exposed to gp91ds or scrambled peptides (50 μM) for 60 min, after which 100 nM Tat was applied. After a 3-h exposure, the cells were washed to remove all traces of Tat and/or the peptides, and allowed to condition the serum-free medium for an additional 24 h. Blinded cell counts based on repeated measures (before the addition of the conditioned medium and again 24 h later) of morphology were conducted to quantify neuronal survival, as described in Methods. Data show that medium conditioned by microglia exposed just to gp91ds, the scrambled peptide, or their vehicle did not induce toxicity over that caused by unconditioned DMEM (Fig. 8C). However, as was observed using macrophages, medium conditioned by microglia exposed to Tat induced significant declines in neuronal survival (Fig. 8C). However, Tat-induced microglia-mediated neurotoxicity was prevented by gp91ds, but not the scrambled peptide (Fig. 8C).
Discussion
Data in this article document the specific and biologically significant role that NADPH oxidase plays in mediating the neurotoxic inflammatory effects of HIV-Tat on cultured microglia and macrophages. Specifically, data show that Tat causes dose-and time-dependent increase in NADPH oxidase activity in cultured microglial N9 cell lines, in primary neonatal rat microglia, and in primary adult mouse macrophages. Additionally, while Tat is well known to elicit the release of neuroactive cytokines from cultured monocytic cells (3), data in this manuscript demonstrates that Tat-induced cytokine and chemokine release depends in large part on NADPH oxidase activity. Finally, genetic deletion of the NADPH oxidase subunit gp91phox nearly completely prevents macrophage-mediated, Tat-induced neurotoxicity. Collectively, these data support a specific and central role for NADPH oxidase in directing neurotoxic inflammatory signaling in microglia/macrophages in response to the HIV protein Tat. Thus, this study suggests that the development of specific NOX inhibitors might be considered to better manage HIV-related neuroinflammation in clinical settings.
Epidemiological studies suggest that nearly one-fifth of patients with AIDS could develop indications of neuroAIDS, including HIVD (19). In addition to the toll exacted on the individual patient, the development of dementia in AIDS patients has important public health implications in that the development of HIVD can decrease job performance and limit self-care dependence (21). Thus, there is considerable impetus to resolve the viral and/or host mechanisms that drive the progression of HIVD. In this regard, the viral regulatory protein Tat has repeatedly been implicated in the pathogenesis of neuroAIDS (reviewed in (25)). For example, chronic Tat expression in intact brain causes significant alterations in histological markers of microglial inflammation, synaptic density, and impairments in behavioral performance (14). Collectively, these data support the hypothesis that Tat per se could contribute to the pathogenesis of neuroAIDS and HIVD, and encourage study into the specific cellular mechanisms of Tat-mediated brain dysfunction.
Microglia and macrophages could drive neuroAIDS via variety of mechanisms, including by serving as viral reservoirs and sites of HIV-1 replication as well as acting as sources of neurotoxic cytokines and pro-oxidants (reviewed in (26)). Data presented in this manuscript are in agreement with previous reports that document that Tat can specifically and significantly increase macrophage/microglia reactivity and macrophage-mediated neurotoxicity (30). Furthermore, these data are especially significant in light of observations that the presence of activated macrophages correlates with the incidence and severity of HIVD (20, 43). Tat can interact with many cell surface proteins, including integrins, chemokine receptors, and glutamate receptors (reviewed in (25)), but the mechanisms whereby Tat initiates the phenotypical and functional transformation of microglia/macrophages are still unresolved. As the identification of specific triggers of Tat-induced, microglia-mediated neuronal injury would present valuable targets for pharmaceutical development, this study was undertaken to determine if the cell surface enzyme complex NADPH oxidase might participate in the transduction of Tat-induced microglial/macrophage activation. NADPH oxidase is a superoxide-producing enzyme system, and activation of NADPH oxidase is a characteristic feature of microglial activation both in vitro and in vivo. While intended to kill invading pathogens, ROS generated by activated microglia could directly contribute to brain injury by inducing lipid peroxidation, DNA fragmentation and protein oxidation in surrounding cells—a phenomena called “bystander lysis” (12, 29). However, experimental evidence presented in this manuscript also suggests that free radical production in immune cells is also critical to intracellular signal transduction and the promulgation of the inflammatory cascade in the brain. For example, reports from our labs and others have documented the important role that p42/44 MAPK plays in the effects of Tat in microglia/macrophages (3, 8), while complementary studies in endothelial cells have shown that Tat-induced p42/44 MAPK activation is NADPH oxidase dependent (36). Furthermore, these data are in agreement with previous reports documenting that NADPH-driven oxidant production participates in the induction of other important inflammatory signaling pathways, particularly NF-κB (24). In addition to activation and nuclear translocation, the actual composition of NF-κB is responsive to changes in the intracellular oxidative environment (49), and the relative expression of p50 homodimers, which do not posses transactivation domains, could be an important mechanism to control inflammatory signaling. However, it seems likely that inflammatory signaling in microglia is not completely redox driven, as NADPH oxidase inhibition (either genetic or pharmacologic) only partially inhibited Tat-induced cytokine release. Thus, there may exist several overlapping and/or distinct intracellular pathways that can all lead to functional microglial activation. Indeed, investigation of the interactions of redox- and nonredox-based signaling pathways could lead to much better pharmacological control over microglial reactivity. Thus, ongoing studies are examining the role of NADPH oxidase in the regulation of important intracellular signaling pathways, including MAPK, NF-κB, and SOCS, in microglial reactivity.
It is important to note that while activated microglia and macrophages have been repeatedly documented in association with brain injury, exact cause and effect relationships have not been fully established. Thus, the data in this manuscript may be telling in that it is clearly shown that medium from cultured macrophages or microglia can induce significant toxicity in hippocampal neurons, and that this neuronal injury is dependent in large part on NADPH oxidase. There are several mechanisms whereby an altered redox environment in microglia could evoke neuronal injury in these model systems. The release of cytokines and chemokines, which data show are dependent on NADPH oxidase, are thought to be important regulators of neuronal function. For example, the production of TNF by Tat-treated microglia has been shown to be important to neurotoxicity (5). Likewise, MCP-1, a major chemokine involved in macrophage recruitment and microglial activation, is an extremely reliable biomarker for cognitive decline with HIV or simian immunodeficiency viral models of neuroAIDS (27). An additional attractive theory involves the regulation of extracellular glutamate and the induction of excitotoxicity. Microglia express specific glutamate transporters (31), and more importantly, these cells can release significant concentrations of glutamate via a membrane associated cystine-glutamate anti-porter (Xc) whose expression is significantly upregulated in activated cells (35). Indeed, as upregulation of the Xc antiporter in immunocompetent cells is thought to be a self-protective mechanism to increase intracellular cysteine and glutathione so that the cells are not damaged by their own oxidative burst, it can be proposed that any stimulus that increases microglial NADPH oxidase activity will also increase glutamate release. In support of this potential scenario, a series of elegant reports have indeed shown that microglial activation in general, and NADPH oxidase in particular, is associated with the release of neurotoxic levels of glutamate (1).
Acknowledgments
The authors are grateful to Dr. P. Ricciardi–Castagnoli, of Consiglio Nazionale Delle Ricerche, Milano, Italy for the N9 cell lines. Additional gratitude goes to Drs. Patrick Pagano and Le Zheng for assistance and advice regarding the gp91ds peptide. This work was supported by grants from the National Institutes of Health (NS46267, DA19398, and RR15592).
Abbreviations
DPI, diphenylene iodonium; DMEM, Dulbecco's minimum essential medium; FBS, fetal bovine serum; gp91ds, a decoy peptide based on the docking sequence of gp91phox that binds p47phox; HBSS, Hank's balanced salt solution; HIVD, HIV dementia; IMDM, Iscove's Modified Dulbecco's Medium, NOX KO, NADPH oxidase catalytic gp91phox subunit knock out; PBS, phosphate-buffered saline; PMSF, phenylmethylsulfonyl fluoride; RLU, relative light unit; WT, wild type.
Disclosure Statement
No competing financial interests exist.
References
- 1.Barger SW. Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- 2.Barger SW. Goodwin ME. Porter MM. Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruce–Keller AJ. Barger SW. Moss NI. Pham JT. Keller JN. Nath A. Pro-inflammatory and pro-oxidant properties of the HIV protein Tat in a microglial cell line: attenuation by 17beta-estradiol. J Neurochem. 2001;78:1315–1324. doi: 10.1046/j.1471-4159.2001.00511.x. [DOI] [PubMed] [Google Scholar]
- 4.Bruce–Keller AJ. Geddes JW. Knapp PE. McFall RW. Keller JN. Holtsberg FW. Steiner SM. Partharasy S. Mattson MP. Anti-death properties of TNF against metabolic poisoning: mitochondrial stabilization by MnSOD. J Neuroimmunol. 1999;93:53–71. doi: 10.1016/s0165-5728(98)00190-8. [DOI] [PubMed] [Google Scholar]
- 5.Chen P. Mayne M. Power C. Nath A. The Tat protein of HIV-1 induces tumor necrosis factor-alpha production. J Biol Chem. 1997;272:22385–22388. doi: 10.1074/jbc.272.36.22385. [DOI] [PubMed] [Google Scholar]
- 6.Colton AC. Gilbert DL. Production of superoxide by a CNS macrophage, the microglia. FEBS Lett. 1987;223:284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- 7.D'Aversa TG. Eugenin EA. Berman JW. NeuroAIDS: contributions of the human immunodeficiency virus-1 proteins Tat and gp120 as well as CD40 to microglial activation. J Neurosci Res. 2005;81:436–446. doi: 10.1002/jnr.20486. [DOI] [PubMed] [Google Scholar]
- 8.D'Aversa TG. Yu KO. Berman JW. Expression of chemokines by human fetal microglia after treatment with the human immunodeficiency virus type 1 protein Tat. J Neurovirol. 2004;10:86–97. doi: 10.1080/13550280490279807. [DOI] [PubMed] [Google Scholar]
- 9.Day BJ. Kariya C. A novel class of cytochrome P450 reductase redox cyclers: Cationic manganoporphyrins. Tox Sci. 2005;85:713–719. doi: 10.1093/toxsci/kfi108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeLeo FR. Quinn MT. Assembly of the phagocyte NADPH oxidase: Molecular interaction of oxidase proteins. J Leukoc Biol. 1996;60:677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- 11.Ding Q. Dimayuga E. Markesbery WR. Keller JN. Proteasome inhibition induces reversible impairments in protein synthesis. FASEB J. 2006;20:1055–1063. doi: 10.1096/fj.05-5495com. [DOI] [PubMed] [Google Scholar]
- 12.Dringen R. Oxidative and antioxidative potential of brain microglial cells. Antioxid Redox Signal. 2005;7:1223–1233. doi: 10.1089/ars.2005.7.1223. [DOI] [PubMed] [Google Scholar]
- 13.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 14.El–Hage N. Wu G. Wang J. Ambati J. Knapp PE. Reed JL. Bruce–Keller AJ. Hauser KF. HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia. 2006;53:132–146. doi: 10.1002/glia.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ensoli B. Buonaguro L. Barillari G. Fiorelli V. Gendelman R. Morgan R. Wingfield P. Gallo RC. Release, uptake, and effects of extracellular human immunodeficiency virus type-1 Tat protein on cell growth and viral replication. J Virol. 1993;67:277–287. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epstein LG. Sharer LR. Oleske JM. Connor EM. Goudsmit J. Bagdon L. Robert–Guroff M. Koenigsberger MR. Neurological manifestations of HIV infection in children with AIDS. Pediatrics. 1986;78:678–687. [PubMed] [Google Scholar]
- 17.Gao HM. Liu B. Zhang W. Hong JS. Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson's disease. FASEB J. 2003;13:1954–1956. doi: 10.1096/fj.03-0109fje. [DOI] [PubMed] [Google Scholar]
- 18.Glass J. Fedor H. Wesselingh SL. McArthur JC. Immunocytochemical quantification of human immunodeficiency virus in the brain; Correlation with dementia. Ann Neurol. 1995;38:755–762. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- 19.Grant I. Sacktor H. McArthur J. HIV neurocognitive disorders. In: Gendelman H.E., et al., editors. The Neurology of AIDS. Oxford University Press; London: 2005. pp. 357–373. [Google Scholar]
- 20.Gray F. Adle–Biassette H. Chretien F. Lorin de la Grandmaison G. Force G. Keohane C. Neuropathology and neurodegeneration in human immunodeficiency virus infection. Pathogenesis of HIV-induced lesions of the brain, correlations with HIV-associated disorders and modifications according to treatments. Clin Neuropathol. 2001;20:146–155. [PubMed] [Google Scholar]
- 21.Heaton RK. Velim RA. McCutchan JA. Gulevich SJ. Atkinson JH. Wallace MR. Godfrey HP. Kirson DA. Grant I. Neuropsychological impairment in human immunodeficiency virus-infection: Implications for employment. HNRC Group. HIV Neurobehavioral Research Center. Psychosomatic Med. 1994;56:8–17. doi: 10.1097/00006842-199401000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Hudson L. J L. Nath A. Jones M. Raghavan R. Narayan O. Male D. Everall I. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol. 2000;6:145–155. doi: 10.3109/13550280009013158. [DOI] [PubMed] [Google Scholar]
- 23.Jana A. Pahan K. Fibrillar amyloid-beta peptides kill human primary neurons via NADPH oxidase-mediated activation of neutral sphingomyelinase. Implications for Alzheimer's disease. J Biol Chem. 2004;279:51451–51459. doi: 10.1074/jbc.M404635200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaul N. Forman HJ. Activation of NF kappa B by the respiratory burst of macrophages. Free Radic Biol Med. 1996;21:401–405. doi: 10.1016/0891-5849(96)00178-5. [DOI] [PubMed] [Google Scholar]
- 25.King JE. Eugenin EA. Buckner CM. Berman JW. HIV tat and neurotoxicity. Microbes Infect. 2006;8:1347–1357. doi: 10.1016/j.micinf.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 26.Kramer–Hammerle S. Rothenaigner I. Wolff H. Bell J. Brack–Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 27.Mankowski JL. Queen SE. Clements JE. Zink MC. Cerebrospinal fluid markers that predict SIV CNS disease. J Neuroimmunol. 2004;157:66–70. doi: 10.1016/j.jneuroim.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 28.McArthur JC. Brew BJ. Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4:543–555. doi: 10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- 29.McGeer PL. McGeer EG. The role of the immune system in neurodegenerative disorders. Mov Disord. 1997;12:855–858. doi: 10.1002/mds.870120604. [DOI] [PubMed] [Google Scholar]
- 30.Minghetti L. Visentin S. Patrizio M. Franchini L. Ajmone–Cat MA. Levi G. Multiple actions of the human immunodeficiency virus type-1 Tat protein on microglial cell functions. Neurochem Res. 2004;29:965–978. doi: 10.1023/b:nere.0000021241.90133.89. [DOI] [PubMed] [Google Scholar]
- 31.Nakajima K. Tohyama Y. Kohsaka S. Kurihara T. Ability of rat microglia to uptake extracellular glutamate. Neurosci Lett. 2001;307:171–174. doi: 10.1016/s0304-3940(01)01943-7. [DOI] [PubMed] [Google Scholar]
- 32.Nelson P. Soma L. Lavi E. Microglia in diseases of the central nervous system. Ann Med. 2002;34:491–500. doi: 10.1080/078538902321117698. [DOI] [PubMed] [Google Scholar]
- 33.Park L. Anrather J. Girouard H. Zhou P. Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27:1908–1918. doi: 10.1038/sj.jcbfm.9600491. [DOI] [PubMed] [Google Scholar]
- 34.Pawate S. Shen Q. Fan F. Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res. 2004;77:540–551. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- 35.Piani D. Spranger M. Frei K. Schaffner A. Fontana A. Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. Eur J Immunol. 1992;22:2429–2436. doi: 10.1002/eji.1830220936. [DOI] [PubMed] [Google Scholar]
- 36.Pu H. Tian J. Andras IE. Hayashi K. Flora G. Hennig B. Toborek M. HIV-1 Tat protein-induced alterations of ZO-1 expression are mediated by redox-regulated ERK 1/2 activation. J Cereb Blood Flow Metab. 2005;25:1325–1335. doi: 10.1038/sj.jcbfm.9600125. [DOI] [PubMed] [Google Scholar]
- 37.Qin L. Liu Y. Wang T. Wei S. Block ML. Wilson B. Liu B. Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- 38.Rey FE. Cifuentes ME. Kiarash A. Quinn MT. Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 39.Sheng WS. Hu S. Hegg CC. Thayer SA. Peterson PK. Activation of human microglial cells by HIV-1 gp41 and Tat proteins. Clin Immunol. 2000;96:243–251. doi: 10.1006/clim.2000.4905. [DOI] [PubMed] [Google Scholar]
- 40.Stolk J. Hiltermann TJ. Dijkman JH. Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- 41.Streit WJ. Walter SA. Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 42.Tardieu M. Hery C. Peudenier S. Boespflug O. Montagnier L. Human immunodeficiency virus type 1-infected monocytic cells can destroy human neural cells after cell-to-cell adhesion. Ann Neurol. 1992;132:11–17. doi: 10.1002/ana.410320104. [DOI] [PubMed] [Google Scholar]
- 43.Tyor WR. Wesselingh W. Griffith SW. McArther JC. Griffen DE. Unifying hypothesis of the pathogenesis of HIV-associated dementia complex, vacuolar myelopathy, and sensory neuropathy. J AIDS Human Ret. 1995;9:379–388. [PubMed] [Google Scholar]
- 44.Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci. 2002;59:1428–1459. doi: 10.1007/s00018-002-8520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wesselingh SL. Power C. Glass JD. Tyor WR. McArthur J. Farber JM. Griffin JW. Griffin DE. Intracerebral cytokine messenger RNA expression in acquired immunodefiency syndrome dementia. Ann Neurol. 1993;33:576–582. doi: 10.1002/ana.410330604. [DOI] [PubMed] [Google Scholar]
- 46.Westendorp MO. Shatrov VA. Schulze–Osthoff K. Frank R. Kraft M. Los M. Krammer PH. Droge W. Lehmann V. HIV-1 Tat potentiates TNF-induced NF-kappa B activation and cytotoxicity by altering the cellular redox state. EMBO J. 1995;14:546–554. doi: 10.1002/j.1460-2075.1995.tb07030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiley CA. Baldwin M. Achim CL. Expression of HIV regulatory and structural mRNA in the central nervous system. AIDS. 1996;10:843–847. doi: 10.1097/00002030-199607000-00007. [DOI] [PubMed] [Google Scholar]
- 48.Zhang M. Kho AL. Anilkumar N. Chibber R. Pagano PJ. Shah AM. Cave AC. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation. 2006;113:1235–1243. doi: 10.1161/CIRCULATIONAHA.105.581397. [DOI] [PubMed] [Google Scholar]
- 49.Zhou LZ. Johnson AP. Rando TA. NF kappa B and AP-1 mediate transcriptional responses to oxidative stress in skeletal muscle cells. Free Radic Biol Med. 2001;31:1405–1416. doi: 10.1016/s0891-5849(01)00719-5. [DOI] [PubMed] [Google Scholar]