

Abstract
Existing treatments for major depressive disorder (MDD) usually take weeks to months to achieve their antidepressant effects, and a significant number of patients do not have adequate improvement even after months of treatment. In addition, increased risk of suicide attempts is a major public health concern during the first month of standard antidepressant therapy. Thus, improved therapeutics that can exert their antidepressant effects within hours or a few days of their administration are urgently needed, as is a better understanding of the presumed mechanisms associated with these rapid antidepressant effects. In this context, the N-methyl-D-aspartate (NMDA) antagonist ketamine has consistently shown antidepressant effects within a few hours of its administration. This makes it a valuable research tool to identify biomarkers of response in order to develop the next generation of fast-acting antidepressants. In this review, we describe clinical, electrophysiological, biochemical, and imaging correlates as relevant targets in the study of the antidepressant response associated with ketamine, and their implications for the development of novel, fast-acting antidepressants. We also review evidence that alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) to NMDA throughput may represent a convergent mechanism for the rapid antidepressant actions of ketamine. Overall, understanding the molecular basis of this work will likely lead to the ultimate development of improved therapeutics for MDD.
Keywords: antidepressant, depression, glutamate, ketamine, rapid onset
1. Introduction
The need for new, improved antidepressants
Major depressive disorder (MDD) is a serious, recurrent, heterogeneous, and disabling psychiatric illness that affects millions of individuals worldwide, and has a major negative impact on public health and productivity (Baune, Adrian, & Jacobi, 2007; Kessler et al., 2006). More than half a century ago, it was observed that the antidepressant imipramine was clinically effective in treating MDD. Since then, a great many other antidepressants have been developed; these are largely similar to imipramine in their mechanism of action, but are more selective. For instance, selective serotonin reuptake inhibitors (SSRIs) are safer and better tolerated than tricyclic antidepressants, but they have not been equivocally shown to offer advantages in terms of efficacy or speed of onset of action. Most surprisingly, recent studies clearly show that antidepressants are of only limited effectiveness for many patients. For example, in the largest open-label study (STAR*D) evaluating standard therapies for MDD, less than one-third of patients receiving standard antidepressants experienced remission after up to four months of treatment (Thase et al., 2005; Trivedi et al., 2006). Furthermore, remission in half of the patients often required six months of treatment and two antidepressant trials (Judd et al., 2002).
In addition to the issue of efficacy, the delayed onset of therapeutic effects associated with standard antidepressants is still a major challenge in the treatment of MDD. This is a critical public health problem, because high rates of mortality and morbidity are present during this latency period, and these are associated with a worse long-term course (Machado-Vieira, Salvadore, Luckenbaugh, Manji, & Zarate, 2008). Furthermore, despite the relatively large number of antidepressants available (Rush et al., 2006; Trivedi et al., 2006), our limited understanding of both the pathophysiology of MDD and the mechanisms of action involved in the therapeutic effects of antidepressants have been significant impediments to developing improved treatments. The key mechanism of action in the effects of standard antidepressants is thought to be their ability to increase monoamines in the synapses. Given that the clinical efficacy of these traditional agents is typically observed only after weeks of treatment, recent theories have suggested that monoamines are only primary effectors, modulating downstream signaling pathways responsible for their therapeutic efficacy (Manji & Lenox, 2000; Payne, Quiroz, Zarate, & Manji, 2002).
In recent years, it has become increasingly clear that there is an urgent need to develop pharmacological treatments for MDD that exert rapid and sustained antidepressant effects within hours or even a few days. In this context, the finding that the N-methyl-D-aspartate (NMDA) antagonist ketamine induces a rapid antidepressant response within hours has led to exciting new research into cellular mechanisms that affect rapid antidepressant action. Relatedly, accumulating evidence suggests that alterations in the regulation of glutamatergic neurotransmission contribute to the pathophysiology of MDD, as well as to the mechanism of existing antidepressants. This supporting evidence comes from: 1) findings of glutamatergic abnormalities in patients with MDD; 2) effects of existing antidepressants and mood stabilizing medications on the glutamatergic system; 3) preclinical evidence suggesting that drugs targeting various components of glutamate neurotransmission possess antidepressant and anxiolytic properties; and 4) recent studies demonstrating the effectiveness of glutamate-modulating agents in the treatment of mood disorders.
Thus, it appears that agents targeting the glutamatergic system may be key to developing a new generation of improved treatments for this devastating illness. In this article, we describe recent advances in this area with a special focus on the role of ketamine as a proof of concept agent for the development of new, improved treatments for MDD (Sanacora, Zarate, Krystal, & Manji, 2008).
General overview of the glutamatergic system
Glutamate is a critical excitatory neurotransmitter in the human brain system and is known to play a major role in cellular plasticity and cellular resilience (Sanacora et al., 2008). Diverse clinical studies have supported a critical role for the glutamatergic system in the pathophysiology of MDD, and it is believed to be a key target in mood regulation (reviewed in (Maeng & Zarate, 2007; Sanacora et al., 2008; Zarate, Quiroz, Payne, & Manji, 2002)).
Glutamate acts pre- and postsynaptically through the activation of diverse receptors characterized by structural characteristics. Ionotropic glutamate receptors—NMDA, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate (KA)—are ion channels that open the channel pore and allow ionic influx to the cell when activated. This influx regulates the neuronal surface’s polarization, thus activating intracellular signaling cascades. In contrast, the metabotropic receptors (mGluRs) are G-protein–coupled receptors that directly modulate the second messenger pathways. The mGluRs in group I, including mGluR1 and mGluR5, stimulate the hydrolyzation of phosphoinositide phospholipids in the cell’s plasma membrane. The receptors in groups II (including mGluR2 and mGluR3) and III (including mGluRs 4, 6, 7, and 8), with some exceptions, prevent the formation of cyclic adenosine monophosphate (cAMP) by activating a G protein that inhibits the enzyme adenylyl cyclase, which forms cAMP from adenosine triphosphate (ATP). This review focuses on the ionotropic NMDA and AMPA receptors, which are believed to be directly involved in the antidepressant actions of ketamine.
NMDA and AMPA receptors
The NMDA receptor channel includes the combination of NR1, NR2 (NR2A–NR2D), and NR3 (NR3A and NR3B) subunits. Glutamate’s binding site is believed to be in the NR2 subunit, while the NR1 subunit is the binding site for the glutamate co-agonist glycine. Two sites have been identified inside the ion channel: the “s” site, and the phencyclidine (PCP) site; the latter is ketamine’s binding site. The AMPA receptors mediate the rapid, desensitizing excitation at most of the synapses, and are responsible for the early synaptic response to glutamate. When activated, AMPA receptors open the pore, thus allowing the influx of sodium and the consequent neuronal membrane’s depolarization. The AMPA receptors comprise four subunits (GluR1–GluR4). At mature synapses, AMPA receptors are frequently c-oexpressed with NMDA receptors, where in concert they contribute to the synaptic plasticity processes involved in learning, memory, and neuroprotection (Malinow & Malenka, 2002).
2. Evidence for the involvement of the glutamatergic system in the pathophysiology of MDD: findings from human studies
Changes in the glutamatergic system have been described in the central nervous system (CNS) (CSF and brain tissue) as well as the periphery in subjects with MDD (Sanacora et al., 2008). It is not clear whether these peripheral changes in glutamate levels are a central dysfunction or represent a secondary finding regulated by other variables, including use of medication and peripheral sources of glutamate (Altamura et al., 1993; Frye, Tsai, Huggins, Coyle, & Post, 2007; Kim, Schmid-Burgk, Claus, & Kornhuber, 1982; Mauri et al., 1998; Mitani et al., 2006); central glutamatergic measurements thus appear to be more reliable. However, the rapid turnover between glutamate and glutamine, as well as the current lack of replication and contradictory findings in this area have limited our understanding of the potential role of glutamatergic biomarkers in the pathophysiology of MDD (Altamura, Maes, Dai, & Meltzer, 1995; Cortese & Phan, 2005; Francis, 2003; Levine et al., 2000; Maes, Verkerk, Vandoolaeghe, Lin, & Scharpe, 1998).
Imaging, post-mortem, and genetic studies
Brain glutamate levels have been measured in vivo using proton magnetic resonance spectroscopy (1H-MRS), which quantifies the peak of glutamate resonances, comprising glutamate, glutamine, and gamma-aminobutyric acid (GABA) components. This lack of specificity for glutamate, as well as the few studies able to measure this peak in areas other than the occipital cortex (which has a limited role in behavioral regulation), supports the need for new studies or tools to further evaluate brain glutamatergic system regulation in MDD. Overall, data indicating elevated glutamate levels in the occipital cortex and reduced levels in the anterior cingulate cortex are the most relevant findings in this area (Hasler et al., 2007; Sanacora et al., 2004). Notably, modern 13C-MRS studies able to measure glutamate/glutamine cycling rate may soon provide detailed data about glutamatergic function changes associated with MDD (de Graaf, Mason, Patel, Behar, & Rothman, 2003; Sanacora et al., 2008).
Similarly, post-mortem and genetic studies support the role of glutamatergic system dysfunction in MDD. For instance, increased levels of glutamate and decreased levels of GluR2 and GluR3 receptor levels have been found in the prefrontal cortex of individuals with MDD (Beneyto & Meador-Woodruff, 2006; Hashimoto, Sawa, & Iyo, 2007; Scarr, Pavey, Sundram, MacKinnon, & Dean, 2003). Reduced NMDA receptor binding, NR1 subunit expression, excitatory amino-acid transporters 1 and 2 of the glia, and glutamine synthetase have also been found in the temporal and two frontal brain regions of subjects with MDD (Choudary et al., 2005; Nudmamud-Thanoi & Reynolds, 2004). A similar decrease in the expression of SAP102 (a synapse-associated protein that primarily interacts with the NR2B subunit) has been observed in the striatum of subjects with MDD (Kristiansen & Meador-Woodruff, 2005).
3. The role of NMDA and AMPA receptor modulators as MDD therapeutics
Emerging data suggest that glutamate plays a critical role in both the acute and long-term action of antidepressants. Both in vitro and human studies show that new glutamate modulating agents as well as traditional antidepressants (Pittaluga et al., 2007; Sernagor, Kuhn, Vyklicky, & Mayer, 1989) directly or indirectly target the glutamatergic system; furthermore, recent investigations specifically indicate the antidepressant efficacy of glutamate modulating agents in MDD. The glutamatergic modulators being studied have been shown to either target glutamate receptors (NMDA, AMPA, and metabotropic) directly, or to target glutamate before its release into the extracellular space (reviewed in (Maeng & Zarate, 2007; Sanacora et al., 2008; Zarate et al., 2002)).
With regards to MDD, the role of the glutamatergic system has been studied for decades (Skolnick, 1999; Skolnick, Legutko, Li, & Bymaster, 2001) mainly in the form of preclinical studies. Early reports described the action of antidepressants on glutamatergic receptors and the antidepressant-like effects of NMDA antagonists in animal models. Indeed, diverse animal studies have demonstrated a role for glutamatergic agents in the treatment of depressive-like states (Manji et al., 2003). In this context, stress and neuroplasticity have also been shown to play a role in MDD, and these are also critically regulated by the glutamatergic system (Pittenger & Duman, 2008). However, for unclear reasons, work on glutamate in mood disorders remained at a lull until recently, when a series of preclinical and clinical studies “re-discovered” the importance of glutamate in mood disorders; it is now a very active area of research.
Several other glutamatergic modulators have been tested in both human and animal studies of treatment-resistant MDD and found to be of considerable utility. These promising agents include NMDA antagonists, AMPA potentiators, inhibitors of glutamate-release agents, and enhancers of glutamate transporters. For instance, NMDA receptor antagonists have antidepressant-like effects in diverse paradigms (Maeng & Zarate, 2007; Moryl, Danysz, & Quack, 1993; Papp & Moryl, 1994, 1996; Przegalinski, Tatarczynska, Deren-Wesolek, & Chojnacka-Wojcik, 1997; Trullas & Skolnick, 1990) (reviewed in (Zarate et al., 2003; Zarate et al., 2002)). Studies have noted that dizocilpine (MK-801), a channel blocker, and CGP 37849, an NMDA receptor antagonist, have antidepressant-like effects when administered alone or in combination with standard antidepressants (Maj, Rogoz, Skuza, & Sowinska, 1992; Meloni et al., 1993; Padovan & Guimaraes, 2004; Papp & Moryl, 1993; Skolnick, Miller, Young, Boje, & Trullas, 1992; Trullas & Skolnick, 1990).
Notably, Preskorn and colleagues recently investigated the antidepressant efficacy of the NR2B subunit-selective antagonist CP-101,606 (Preskorn et al., 2008). NMDA receptors containing the NR2B subunit are localized primarily in the forebrain, including the hippocampus, which is implicated in the pathophysiology of MDD (Campbell & MacQueen, 2004). Unlike ketamine, which blocks the receptor-gated ion channel, CP-101,606 inhibits NMDA receptors by an allosteric mechanism (Mott et al., 1998). The study evaluated a group of patients who did not respond to the SSRI paroxetine, and who were subsequently randomized to receive a single, double-blind, IV infusion of either CP-101,606 (1.5 hours at a dose of 0.75mg/kg) or placebo adjunctive to paroxetine 40mg/day. An initial higher dose during an eight-hour infusion had been previously tested and found to induce dissociative symptoms in four of seven patients. In this study, CP-101,606 was superior to placebo on the primary outcome measure (Montgomery-Asberg Depression Rating Scale (MADRS) score at Day 5). Sixty percent of these treatment-resistant patients with MDD using the active compound met criteria for response (50% improvement), and 33% were in remission at Day 5. It is important to note that among responders at Day 5, more than half maintained a sustained response after 12 days, and 32% maintained a response one month after infusion. Dissociative side effects were present in four of the first seven subjects randomized and, subsequently, the study medication dose was significantly lowered. Six out of 15 (40%) patients in the CP-101,606 group experienced dissociative side effects, compared with two of 15 (13%) in the placebo group. An 8.4-point greater reduction in MADRS scores was observed in patients receiving CP-101,606 than those receiving placebo. This study did have several limitations, including that the study compound was not tested as monotherapy, and that paroxetine also affects the glutamate system (Zarate & Manji, 2008). In addition, because the dissociative side effects of the study compound were quite apparent and required dose modification (both total dose and duration of the infusion), blinding in the study may have been compromised. Nevertheless, these results are quite promising and warrant further exploration in a larger study.
This article cannot review the role of all these different agents in mood disorders; the interested reader is referred elsewhere for more information (see (Sanacora et al., 2008)). Instead, this paper will focus on one agent—the NMDA antagonist ketamine—as an example of how our understanding of one agent can lead to novel insights into the pathophysiology of mood disorders, as well as the development of biomarkers and improved therapeutics.
4. Ketamine studies in MDD: a valuable proof of concept tool for the development of improved therapeutics
Ketamine: overview
Ketamine (dl2-(o-chlorophenyl)-2-(methylamino) cyclohexanone hydrochloride) is a non-competitive NMDA antagonist (Harrison & Simmonds, 1985) and a derivative of PCP. The original name for ketamine was CI581, and its discovery is credited to Dr. Calvin Stevens (Wayne State University) who isolated the compound in 1961. Ketamine is water- and lipid-soluble, and is easily administered through diverse routes with ample distribution in the body. Ketamine metabolism is regulated by hepatic microsomal enzymes. It causes bronchodilation and stimulation of the sympathetic nervous system and cardiovascular system. In clinics, ketamine, and particularly S(+)-ketamine, is used for premedication, sedation, and induction and maintenance of general anesthesia; indeed, ketamine has been widely used as a standard anesthetic in children and adults at doses higher than 2mg/kg (Green et al., 1998). Ketamine is a suitable anesthetic agent for trauma victims, septic shock, patients with pulmonary diseases, and hypovolemia (Sinner & Graf, 2008). Metabolites are excreted renally (90%) and fecally (5%), with 4% of an administered dose excreted unchanged in urine. Anesthesia lasts five to 10 minutes for IV administration and 12-25 minutes for IM administration.
Ketamine’s primary mechanism of action is blocking the NMDA receptor at the PCP site within the ionotropic channel. It has a high affinity for the NMDA receptor, with slow open-channel blocking/unblocking kinetics, and a specific type of channel closure (called “trapping block”). Simultaneously, it induces a substantial presynaptic release of glutamate by increasing the firing rate of glutamatergic neurons after activating GABAergic inputs (Moghaddam, Adams, Verma, & Daly, 1997). As mentioned previously, many animal models have noted ketamine’s significant antidepressant and anxiolytic effects (Aguado, San Antonio, Perez, del Valle, & Gomez, 1994; Garcia et al., 2008; Maeng et al., 2008; Mickley et al., 1998; Silvestre, Nadal, Pallares, & Ferre, 1997; Zarate et al., 2003), and it has been suggested that some of the properties described above are critical to these effects.
Clinical studies
One initial study found improvement in treatment-resistant depressive symptoms within 72 hours after ketamine infusion in seven subjects with MDD (Berman et al., 2000). More recently, a fast (within two hours), robust, and relatively sustained antidepressant effect (lasting one to two weeks) was found after infusion of ketamine in patients with treatment-resistant MDD (Zarate et al., 2006). In this randomized, double-blind placebo-controlled crossover study, a single intravenous subanesthetic dose of ketamine (0.5 mg/kg for 40 minutes) induced this significant, rapid, and sustained effect. More than 70% of patients met the criteria for response (50% improvement) at 24 hours after infusion, and 35% showed a sustained response after one week. Two patients maintained their response for at least two weeks in the ketamine group. In contrast, no patient presented sustained response after one day following infusion in the placebo group. Mild perceptual disturbances were observed in most patients only in the first hour after infusion; no serious adverse events occurred.
Patients were rated 60 minutes prior to infusion and at 40, 80, 110, and 230 minutes, as well as one, two, three, and seven days after the single intravenous dose. Significant improvement in the Hamilton Depression Rating Scale (HAM-D) with ketamine over placebo was continuously observed from 110 minutes through seven days. The effect size for the drug difference was very large (d = 1.46; 95% CI, 0.91–2.01) after 24 hours, and moderate to large (d = 0.68; 95% CI, 0.13–1.23) after one week. Similar improvements were noted using the Beck Depression Inventory and Visual Analogue Scale depression scores. The core symptoms of MDD were significantly attenuated or, in many cases, completed remitted in the first few hours following ketamine infusion. A linear mixed model with HAM-D showed significant main effects for drug, time, and an interaction between drug and time (Zarate et al., 2006).
Notably, the response rates obtained with ketamine after 24 hours (71%) are comparable to those reported after six to eight weeks of treatment with standard monoaminergic-based antidepressants (65%) (Entsuah, Huang, & Thase, 2001; Thase et al., 2005), highlighting the relevance of these findings. It is important to mention that the antidepressant efficacy of ketamine has been noted much beyond its short half-life (Zarate et al., 2006). There has never been a report of any other somatic or pharmacological intervention that consistently and reproducibly results in such a dramatically rapid and prolonged response—well beyond the half-life of the drug—with a single administration. Furthermore, the findings of a rapid antidepressant response to ketamine in patients with treatment-resistant MDD have now been replicated in a different cohort involving 26 subjects (Phelps et al., 2008). The magnitude and time-frame of response to ketamine in this study was similar to our previous finding.
Subsequent reports of ketamine’s antidepressant effects concur with the studies described above. For instance, similar effects were observed with ketamine in depressed patients who were in pre- and post-operative states (Goforth & Holsinger, 2007; Kudoh, Takahira, Katagai, & Takazawa, 2002), in patients with MDD and concurrent pain syndrome (Correll & Futter, 2006), as well as during a course of ECT (Ostroff, Gonzales, & Sanacora, 2005), thus reinforcing its relevance as a therapeutic tool to achieve rapid antidepressant response in MDD. In addition, case reports of ketamine’s rapid antidepressant efficacy in treatment-resistant MDD comorbid with alcohol dependence and pain syndrome have also been described (Liebrenz, Stohler, & Borgeat, 2007).
Despite ketamine’s safety profile and lack of physical dependence (Britt & McCance-Katz, 2005; Green et al., 1998), one issue of particular import is ketamine’s sedative and psychotomimetic side effects, which will probably continue to limit its clinical use in larger samples. Relatedly, an increased propensity to psychotomimetic effects and tolerance to ketamine’s antidepressant effects might occur after repeated doses. For instance, a recent case report noted that a second infusion of ketamine showed limited effect one month after an initial infusion (Liebrenz et al., 2007). Similarly, repeated exposure to ketamine increases the risk of severe psychosis, dissociative episodes, and severe emotional distress or euphoria in patients and healthy subjects (Dillon, Copeland, & Jansen, 2003). Misuse and abuse of therapeutically relevant agents in psychiatry is not a new phenomenon. Previous reports of misuse of agents such as benzodiazepines, anti-cholinergic drugs, and stimulants were common.
Despite ketamine’s sedative and psychotomimetic side effects, it does induce a consistently reproducible antidepressant effect within a short period of time. Thus, its true worth may be as a research tool. Currently, our laboratory is using ketamine to develop biomarkers of response in order to develop the next generation of antidepressants. More specifically, we are examining clinical (e.g, family history), electrophysiological (magnetoencephalographic (MEG) and anterior cingulate cortex (ACC) activity), biochemical, and imaging correlates of antidepressant response to ketamine. Such work is important because identifying the biological correlates associated with ketamine’s rapid antidepressant effects may help identify valuable, specific biomarkers and novel targets for the development of new compounds that can similarly produce rapid antidepressant effects, but without ketamine’s side effects. Below, we describe some of the clinical, electrophysiological, and biochemical correlates of antidepressant response to ketamine.
Family history of alcoholism
In the past few years there has been increased interest in the joint study of the pathophysiology of MDD and risk of alcoholism, as the glutamate system appears to play a major role in both conditions. One recent study found that alcohol-dependent individuals had marked reductions to the subjective intoxicating effects of ketamine compared to healthy controls (Krystal et al., 2003). The same group subsequently found that healthy individuals with a positive family history of alcohol dependence had fewer perceptual alterations and lower dysphoric mood after receiving ketamine than those without a family history (Petrakis et al., 2004). We recently extended these findings to assess antidepressant response in patients with treatment-resistant MDD and found that subjects with MDD and a family history of alcohol dependence showed significantly greater improvement in depression rating scales scores after ketamine infusion compared to those who had no family history of alcohol dependence (Phelps et al., 2008).
The precise reasons underlying the better response of patients with treatment-resistant MDD with a positive family history of alcohol dependence is essentially unknown. However, emerging data suggest that genetically determined alterations in NMDA receptor subunits may be associated with alcohol dependence. For instance, a recent study found evidence of an association between NR2A and alcohol dependence (Schumann et al., 2008), and alcohol has been shown to regulate NR2A expression in brain regions implicated in addiction-related neurobiological processes, including the amygdala and hippocampus (Boyce-Rustav & Holmes, 2006). Thus, it is possible that genetic variations in NMDA subunits, particularly NR2A, increase vulnerability to alcohol dependence by altering the sensitivity of the NMDA complex. Because ketamine acts as a partial NR2A antagonist (Petrenko et al., 2004), it is possible that this difference in NR2A sensitivity may impart a greater response to ketamine’s antidepressant effects.
ACC activity
In a study using conventional antidepressants, Mayberg and colleagues (1997) were the first to show that higher rostral ACC (BA 24a/b) metabolism differentiated treatment responders from non-responders at six weeks (Mayberg et al., 1997). A recent functional MRI study found that faster improvement of depressive symptoms with fluoxetine was predicted by more positive functional activation of the ACC during the face emotional task (Chen et al., 2007). Similarly, patients with better antidepressant response to nortriptyline also had hyperactivity (higher theta activity) in the rostral ACC (Brodmann’s area 24/32), thus supporting the role of ACC activation as a predictor of antidepressant response (Pizzagalli et al., 2001). Other antidepressants, as well as sleep deprivation, have also been shown to produce similar effects (Saxena et al., 2003; Wu al., 1999).
Interestingly, the antidepressant effects of ECT also appear to be correlated with increased metabolism in the left subgenual ACC (McCormick et al., 2007). Likewise, high pretreatment ACC activity predicted better response to repetitive transcranial magnetic stimulation (rTMS) (Langguth et al., 2007). A consistent association between increased subgenual cingulate (Cg25) activity and acute sadness has also been noted using positron emission tomography (PET); in this study, application of chronic deep brain stimulation (DBS) close to the subgenual cingulate region (Brodmann’s area 25) reduced the elevated activity in this area, and this reduction was associated with a significant and sustained remission of depressive symptoms in four out of six patients (Mayberg et al., 2005).
One recent study tested the hypothesis that MEG recordings could provide a neurophysiologic biomarker associated with ketamine’s rapid antidepressant effects. It was found that increased pretreatment rostral ACC activity in MDD patients was indeed positively correlated with rapid antidepressant response to ketamine infusion (Salvadore et al., 2008). The ACC activity in response to rapid exposure to fearful faces was measured in eleven drug-free MDD subjects matched with healthy controls. In this same study, it was also observed that amygdala response to fearful faces was negatively correlated with antidepressant response to ketamine 230 minutes after infusion (Salvadore et al., 2008). Interestingly, Deakin and colleagues (2008) also observed a direct regulation of orbitofrontal and subgenual ACC blood oxygenation level dependent (BOLD) activity by ketamine in healthy individuals. In addition, they observed three neuroanatomical systems potentially related to ketamine’s side effects, especially psychosis: (1) deactivations in the ventral anterior limbic cortex; (2) activation in the mid-posterior cingulate; and (3) activations in the temporal lobe hippocampus/parahippocampal cortex and superior, middle, and inferior temporal cortices (Deakin et al., 2008).
Brain-derived neurotrophic factor (BDNF)
BDNF is the most studied neurotrophin in the pathophysiology of MDD. One model suggested that the pathophysiology of mood disorders was associated with decreased neurotrophin levels during mood episodes; treatment with antidepressants and mood stabilizers is associated with clinical improvement. Because of this association, we investigated whether changes in BDNF levels were associated with the initial antidepressant effects of ketamine, but found that these antidepressant effects were not mediated by BDNF. Plasma BDNF levels showed no changes from baseline after ketamine infusion and up to 230 minutes post-infusion (a time point when antidepressant effects and response were manifest) using an anti-BDNF sandwich-ELISA kit. Given that BDNF levels were evaluated only during the first day of treatment, it might be also important to study its role in ketamine’s sustained antidepressant effects. The study did, however, confirm ketamine’s efficacy as a fast-acting antidepressant (Machado-Vieira et al., in press).
Intriguingly, both ketamine and standard monoaminergic antidepressants seem to act directly at the NMDA and AMPA receptors (Du et al., 2004; Du et al., 2007; Maeng et al., 2008); however, it appears that ketamine’s glutamatergic targets may rapidly activate early neuroplastic changes and synaptic potentiation through AMPA receptors, but not be related to BDNF effects. In contrast, traditional antidepressants need to activate long neurotrophic signaling cascades, which may be the reason for their delayed effects.
5. Enhancing AMPA to NMDA throughput as a convergent mechanism for the rapid antidepressant actions of ketamine
In order to develop the next generation of therapeutics that work more rapidly than ketamine and are better tolerated, it becomes crucial to understand the molecular and cellular basis for ketamine’s antidepressant effects. With this goal in mind, our laboratory conducted a series of preclinical tests with ketamine in animal models of MDD. Preclinical evidence indicates a relevant interplay between AMPA and NMDA receptors in the rapid antidepressant effects of ketamine (Maeng & Zarate, 2007; Maeng et al., 2008). Indeed, ketamine decreased immobility time in the forced swim test—an animal model of depression—and this effect was abolished when the AMPA antagonist NBQX was given prior to its infusion. Conversely, AMPA inhibition with NBQX did not regulate imipramine’s antidepressant-like effects (Maeng et al., 2008), thus reinforcing the selectivity of this effect to ketamine and maybe other glutamatergic modulators. More recently, studies from our laboratory found that Ro 25-6981, a selective NR2B subunit antagonist, had antidepressant-like properties in rodents. As with ketamine, these effects appear to be largely mediated through AMPA receptors (Maeng et al., 2008).
This issue is particularly interesting because synaptic potentiation is thought to be involved in ketamine’s acute antidepressant effects (Salvadore et al., 2008). As noted previously, ketamine may produce AMPA-mediated synaptic potentiation rapidly, whereas traditional antidepressants do so in a delayed manner through a cascade of intracellular signaling changes (Sanacora et al., 2008), thus potentially explaining the differential time of onset for antidepressant effects. Sleep deprivation, which is also known to induce rapid antidepressant effects, similarly enhances AMPA-mediated synaptic plasticity (Faraguna, Vyazovskiy, Nelson, Tononi, & Cirelli, 2008). Furthermore, ketamine appears to potentiate synaptic efficacy in the amygdala–accumbens pathway (Kessal, Chessel, Spennato, & Garcia, 2005). Notably, synaptic potentiation is known to involve AMPA trafficking, which enhances AMPA throughput. Therefore, it is possible that enhanced glutamatergic throughput of AMPA relative to NMDA receptors after ketamine treatment may result in increased synaptic potentiation and activation of early neuroplastic genes (possibly not related to increased BDNF levels). The consequent increase in glutamate release then preferentially favors AMPA receptors over NMDA receptors because the latter are blocked by ketamine; thus, the net effect of ketamine’s antidepressant effect on a cellular level is increased glutamatergic throughput.
Similar to NMDA antagonists, the antidepressant-like effects of the mGluR2/3 (group II mGluRs) antagonists can be prevented by co-administration of an AMPA receptor antagonist, suggesting that increased synaptic AMPA receptor activity may be a common pathway by which glutamatergic-mediated antidepressant effects originate. Specifically, the mGlu2/3 receptor antagonist (LY341495) appears to shorten the time required for standard antidepressants to exert their full effects (Matrisciano et al., 2005). Moreover, another recent study found that the AMPA antagonist 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f ]quinoxaline-7-sulfonamide blocked the antidepressant-like activity of a similar group II mGluR antagonist (MGS-0039) in the tail suspension test, another animal model of depression (Karasawa, Shimazaki, Kawashima, & Chaki, 2005). Finally, Chourbaji and colleagues (2008) recently found that mice with a deletion of the main AMPA receptor subunit GluR1 appeared to be a good animal model for depression, thus reinforcing the role of increased AMPA activity or decreased NMDA function as relevant paradigms for MDD (Chourbaji et al., 2008).
It is important to note that downregulation in NMDA receptor function has also been described as an important downstream target for standard monoaminergic antidepressants. The period of latency necessary to achieve antidepressant effects with these agents may represent the time necessary to exert direct modulatory effects at the NMDA receptor-channel complex. At the same time, inducing direct regulation of the NMDA receptors should result in more rapid antidepressant effects.
There is also evidence that AMPA receptors appear to respond to chronic treatment with standard antidepressants (Paul & Skolnick, 2003), but the mechanisms involved seem be unrelated to those associated with ketamine’s rapid onset of antidepressant effects. Indeed, chronic treatment with standard antidepressants has also been shown to enhance AMPA receptor surface levels (Du et al., 2004; Du et al., 2007). Taken together, these findings suggest that AMPA receptor amplification reduces NMDA receptor function through activation of plasticity pathways. AMPA-mediated synaptic potentiation is involved in the early antidepressant effects of ketamine, while AMPA-activating neurotrophic intracellular signaling cascades modulate long-term antidepressant effects involving monoaminergic regulation.
6. Conclusion and perspectives
The fact that monoaminergic antidepressants take weeks to achieve their full effect leaves patients receiving these medications particularly vulnerable to impairment in global functioning and at high risk of self-harm. The long and risky period of latency in MDD, as well as the persistent residual symptoms, low rates of remission, and frequent relapses are challenges that need to be better addressed by the next generation of improved therapeutics.
Several glutamatergic compounds are being tested in “proof-of-concept” studies in patients with severe mood disorders (Sanacora et al., 2008). As this article has explored, using ketamine as a proof of concept tool to 1) develop biomarkers of response to further our understanding of what bio-signatures are necessary, and 2) determine what aspects of glutamatergic receptor/subunit modulation are relevant to the next generation of improved therapeutics for the treatment of MDD is likely to enhance drug discovery. Specifically, using ketamine as a starting point to explore improved therapeutics for MDD can stimulate the continuous development and testing of new fast-acting antidepressants displaying increased selectivity. For instance, initial results with specific drugs that have fewer side effects than ketamine, such as NR2B antagonists, are promising. It will also be important to determine whether antagonizing other NMDA subunits would lead to antidepressant effects. Evidence is mounting that upregulation and/or overstimulation of the NR2A receptor subtype of the NMDA receptor plays a pivotal role in the etiology of MDD (Boyce-Rustav & Holmes, 2006; Sanacora et al., 2008); for instance, it was recently reported that NR2A knockout mice exhibit a highly robust anxiolytic- and antidepressant-like phenotype. For that reason, it will be important to test selective NR2A antagonists and mixed NR2A and NR2B antagonists in proof-of-concept studies in MDD to determine their relevance to therapeutic antidepressant effects.
Given the urgency associated with our need to develop new treatments for MDD that are more effective and work more rapidly than existing ones, it is heartening that so many further studies on the potential mechanisms involved in the faster antidepressant actions of new glutamatergic modulators are underway. We believe this represents a major and important step forward in the development of the next generation of antidepressants. As the search for effective treatments in MDD continues, it is crucial to change the way we understand and conduct drug development. As with other areas of medicine, our gradual understanding of the pathophysiology of MDD and the mechanism of action of antidepressants suggests that an antidepressant response occurring within hours is now an obtainable goal. Despite ketamine’s sedative and psychotomimetic adverse effects, overall it has been shown to induce a consistently reproducible antidepressant effect within a short period of time. Thus, its true worth may be as a research tool in our search for similar agents that can induce sustained effects after repeated doses. The work with ketamine described above provides a direct example that rapid response is possible. Most significantly, pharmacological treatments that exert a rapid and sustained antidepressant effect within hours or even a few days could significantly impact care for our patients as well as public health worldwide.
Figure 1. Potential targets for ketamine and similar agents induce rapid and sustained antidepressant effects.
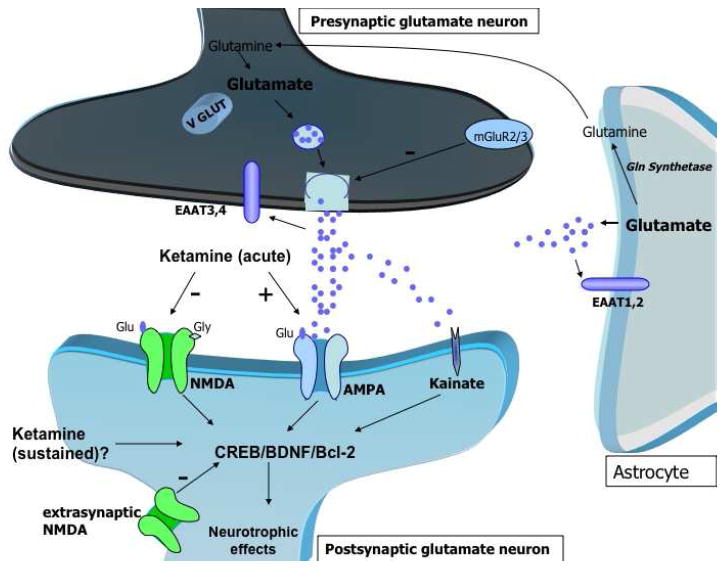
Glutamate is packaged into presynaptic vesicles by the vesicular glutamate transporters (VGLUTs), which critically modulate glutamate concentration in the synaptic vesicles and its consequent release in the synaptic cleft. Also, presynaptic group II mGluR modulation controls glutamate accumulation in the synaptic vesicles. Glutamate clearance from the extracellular space occurs through the high-affinity EAATs presented in the glia and presynaptic neuron. The EAATs play a key role in maintaining adequate neuronal function by reducing potentially toxic extracellular glutamate levels. Notably, ketamine’s rapid antidepressant effects have been shown to be modulated by AMPA relative to NMDA throughput. Excessive glutamate also stimulates the extrasynaptic NMDA receptors, which antagonizes the activation of neurotrophic cascades. The potential sustained (sub-acute) antidepressant effects of ketamine are hypothesized to be mediated by increases in CREB and BDNF expression, as well as the anti-apoptotic protein Bcl-2.
AMPA (alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid); NMDA (N-methyl-D-aspartate); VGLUTs (vesicular Glu transporters); metabotropic Glu receptors (mGluRs); EAAT (excitatory amino-acid transporters); BDNF (brain-derived neurotrophic factor); CREB (cAMP response element binding); B-cell lymphoma 2 (Bcl-2)
Acknowledgments
Funding for this work was supported by the Intramural Research Program of the National Institute of Mental Health (NIMH) and a NARSAD Award (CAZ).
Abbreviations
- ACC
anterior cingulated cortex
- AMPA
alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- MDD
major depressive disorder
- NMDA
N-methyl-D-aspartate
Footnotes
Financial Disclosure: Ioline Henter provided outstanding editorial assistance. A patent application for the use of ketamine in depression has been submitted listing Dr. Zarate among the inventors. Dr. Zarate has assigned his rights on the patent to the US government.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguado L, San Antonio A, Perez L, del Valle R, Gomez J. Effects of the NMDA receptor antagonist ketamine on flavor memory: conditioned aversion, latent inhibition, and habituation of neophobia. Behav Neural Biol. 1994;61(3):271–281. doi: 10.1016/s0163-1047(05)80010-x. [DOI] [PubMed] [Google Scholar]
- Altamura CA, Maes M, Dai J, Meltzer HY. Plasma concentrations of excitatory amino acids, serine, glycine, taurine and histidine in major depression. Eur Neuropsychopharmacol. 1995;5(Suppl):71–75. doi: 10.1016/0924-977x(95)00033-l. [DOI] [PubMed] [Google Scholar]
- Altamura CA, Mauri MC, Ferrara A, Moro AR, D’Andrea G, Zamberlan F. Plasma and platelet excitatory amino acids in psychiatric disorders. Am J Psychiatry. 1993;150(11):1731–1733. doi: 10.1176/ajp.150.11.1731. [DOI] [PubMed] [Google Scholar]
- Baune BT, Adrian I, Jacobi F. Medical disorders affect health outcome and general functioning depending on comorbid major depression in the general population. J Psychosom Res. 2007;62(2):109–118. doi: 10.1016/j.jpsychores.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse. 2006;60(8):585–598. doi: 10.1002/syn.20329. [DOI] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustav JM, Holmes A. Ethanol-related behaviors in mice lacking the NMDA receptor NR2A subunit. Psychopharmacology (Berl) 2006;187:455–466. doi: 10.1007/s00213-006-0448-6. [DOI] [PubMed] [Google Scholar]
- Britt GC, McCance-Katz EF. A brief overview of the clinical pharmacology of “club drugs”. Subst Use Misuse. 2005;40(910):1189–1201. doi: 10.1081/JA-200066730. [DOI] [PubMed] [Google Scholar]
- Campbell S, MacQueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatr Neurosci. 2004;29:417–426. [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Ridler K, Suckling J, Williams S, Fu CH, Merlo-Pich E, et al. Brain imaging correlates of depressive symptom severity and predictors of symptom improvement after antidepressant treatment. Biol Psychiatry. 2007;62:407–414. doi: 10.1016/j.biopsych.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci U S A. 2005;102(43):15653–15658. doi: 10.1073/pnas.0507901102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourbaji S, Vogt MA, Fumagalli F, Sohr R, Frasca A, Brandwein C, et al. AMPA receptor subunit 1 (GluR-A) knockout mice model the glutamate hypothesis of depression. FASEB J. 2008;22:3129–3134. doi: 10.1096/fj.08-106450. [DOI] [PubMed] [Google Scholar]
- Correll GE, Futter GE. Two case studies of patients with major depressive disorder given low-dose (subanesthetic) ketamine infusions. Pain Med. 2006;7(1):92–95. doi: 10.1111/j.1526-4637.2006.00101.x. [DOI] [PubMed] [Google Scholar]
- Cortese BM, Phan KL. The role of glutamate in anxiety and related disorders. CNS Spectr. 2005;10(10):820–830. doi: 10.1017/s1092852900010427. [DOI] [PubMed] [Google Scholar]
- de Graaf RA, Mason GF, Patel AB, Behar KL, Rothman DL. In vivo 1H-[13C]-NMR spectroscopy of cerebral metabolism. NMR Biomed. 2003;16(67):339–357. doi: 10.1002/nbm.847. [DOI] [PubMed] [Google Scholar]
- Deakin JF, Lees J, McKie S, Hallak JE, Williams SR, Dursun SM. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65:154–164. doi: 10.1001/archgenpsychiatry.2007.37. [DOI] [PubMed] [Google Scholar]
- Dillon P, Copeland J, Jansen K. Patterns of use and harms associated with non-medical ketamine use. Drug Alcohol Depend. 2003;69:23–28. doi: 10.1016/s0376-8716(02)00243-0. [DOI] [PubMed] [Google Scholar]
- Du J, Gray NA, Falke CA, Chen W, Yuan P, Szabo ST, et al. Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J Neurosci. 2004;24(29):6578–6589. doi: 10.1523/JNEUROSCI.1258-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Suzuki K, Wei Y, Wang Y, Blumenthal R, Chen Z, et al. The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: relationship to clinical effects in mood disorders. Neuropsychopharmacology. 2007;32(4):793–802. doi: 10.1038/sj.npp.1301178. [DOI] [PubMed] [Google Scholar]
- Entsuah AR, Huang H, Thase ME. Response and remission rates in different subpopulations with major depressive disorder administered venlafaxine, selective serotonin reuptake inhibitors, or placebo. J Clin Psychiatry. 2001;62(11):869–877. doi: 10.4088/jcp.v62n1106. [DOI] [PubMed] [Google Scholar]
- Faraguna U, Vyazovskiy VV, Nelson AB, Tononi G, Cirelli C. A causal role for brain-derived neurotrophic factor in the homeostatic regulation of sleep. J Neurosci. 2008;28:4088–4095. doi: 10.1523/JNEUROSCI.5510-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis PT. Glutamatergic systems in Alzheimer’s disease. Int J Geriatr Psychiatry. 2003;18(Suppl 1):S15–21. doi: 10.1002/gps.934. [DOI] [PubMed] [Google Scholar]
- Frye MA, Tsai GE, Huggins T, Coyle JT, Post RM. Low cerebrospinal fluid glutamate and glycine in refractory affective disorder. Biol Psychiatry. 2007;61(2):162–166. doi: 10.1016/j.biopsych.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Garcia LS, Comim CM, Valvassori SS, Reus GZ, Barbosa LM, Andreazza AC, et al. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):140–144. doi: 10.1016/j.pnpbp.2007.07.027. [DOI] [PubMed] [Google Scholar]
- Goforth HW, Holsinger T. Rapid relief of severe major depressive disorder by use of preoperative ketamine and electroconvulsive therapy. J Ect. 2007;23(1):23–25. doi: 10.1097/01.yct.0000263257.44539.23. [DOI] [PubMed] [Google Scholar]
- Green SM, Rothrock SG, Lynch EL, Ho M, Harris T, Hestdalen R, et al. Intramuscular ketamine for pediatric sedation in the emergency department: safety profile in 1,022 cases. Ann Emerg Med. 1998;31(6):688–697. doi: 10.1016/s0196-0644(98)70226-4. [DOI] [PubMed] [Google Scholar]
- Harrison NL, Simmonds MA. Quantitative studies on some antagonists of N-methyl D-aspartate in slices of rat cerebral cortex. Br J Pharmacol. 1985;84(2):381–391. doi: 10.1111/j.1476-5381.1985.tb12922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Sawa A, Iyo M. Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry. 2007;62(11):1310–1316. doi: 10.1016/j.biopsych.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 2007;64:193–200. doi: 10.1001/archpsyc.64.2.193. [DOI] [PubMed] [Google Scholar]
- Judd LL, Akiskal HS, Schettler PJ, Endicott J, Maser J, Solomon DA, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59(6):530–537. doi: 10.1001/archpsyc.59.6.530. [DOI] [PubMed] [Google Scholar]
- Karasawa J, Shimazaki T, Kawashima N, Chaki S. AMPA receptor stimulation mediates the antidepressant-like effect of a group II metabotropic glutamate receptor antagonist. Brain Res. 2005;1042(1):92–98. doi: 10.1016/j.brainres.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Kessal K, Chessel A, Spennato G, Garcia R. Ketamine and amphetamine both enhance synaptic transmission in the amygdala-nucleus accumbens pathway but with different time-courses. Synapse. 2005;57(1):61–65. doi: 10.1002/syn.20154. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Akiskal HS, Ames M, Birnbaum H, Greenberg P, Hirschfeld RM, et al. Prevalence and effects of mood disorders on work performance in a nationally representative sample of U.S. workers. Am J Psychiatry. 2006;163(9):1561–1568. doi: 10.1176/appi.ajp.163.9.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Schmid-Burgk W, Claus D, Kornhuber HH. Increased serum glutamate in depressed patients. Arch Psychiatr Nervenkr. 1982;232(4):299–304. doi: 10.1007/BF00345492. [DOI] [PubMed] [Google Scholar]
- Kristiansen LV, Meador-Woodruff JH. Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr Res. 2005;78:87–93. doi: 10.1016/j.schres.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Petrakis IL, Krupitsky E, Schutz C, Trevisan L, D’Souza DC. NMDA receptor antagonism and the ethanol intoxication signal: from alcoholism risk to pharmacotherapy. Ann N Y Acad Sci. 2003;1003:176–184. doi: 10.1196/annals.1300.010. [DOI] [PubMed] [Google Scholar]
- Kudoh A, Takahira Y, Katagai H, Takazawa T. Small-dose ketamine improves the postoperative state of depressed patients. Anesth Analg. 2002;95(1):114–118. doi: 10.1097/00000539-200207000-00020. table of contents. [DOI] [PubMed] [Google Scholar]
- Langguth B, Wiegand R, Kharraz A, Landgrebe M, Marienhagen J, Frick U, et al. Pre-treatment anterior cingulate activity as a predictor of antidepressant response to repetitive transcranial magnetic stimulation (rTMS) Neuro Endocrinol Lett. 2007;28:633–638. [PubMed] [Google Scholar]
- Levine J, Panchalingam K, Rapoport A, Gershon S, McClure RJ, Pettegrew JW. Increased cerebrospinal fluid glutamine levels in depressed patients. Biol Psychiatry. 2000;47(7):586–593. doi: 10.1016/s0006-3223(99)00284-x. [DOI] [PubMed] [Google Scholar]
- Liebrenz M, Stohler R, Borgeat A. Repeated intravenous ketamine therapy in a patient with treatment-resistant major depression. World J Biol Psychiatry. 2007:1–4. doi: 10.1080/15622970701420481. [DOI] [PubMed] [Google Scholar]
- Machado-Vieira R, Salvadore G, Luckenbaugh DA, Manji HK, Zarate CA., Jr Rapid onset of antidepressant action: a new paradigm in the research and treatment of major depressive disorder. J Clin Psychiatry. 2008;69(6):946–958. doi: 10.4088/jcp.v69n0610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Vieira R, Yuan P, Brutsche N, DiazGranados N, Luckenbaugh D, Manji HK, et al. Brain derived neurotrophic factor and initial antidepressant response to an NMDA antagonist. J Clin Psychiatry. doi: 10.4088/JCP.08m04659. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng S, Zarate CA., Jr The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467–474. doi: 10.1007/s11920-007-0063-1. [DOI] [PubMed] [Google Scholar]
- Maeng S, Zarate CA, Jr, Du J, Schloesser RJ, McCammon J, Chen G, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63(4):349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Maes M, Verkerk R, Vandoolaeghe E, Lin A, Scharpe S. Serum levels of excitatory amino acids, serine, glycine, histidine, threonine, taurine, alanine and arginine in treatment-resistant depression: modulation by treatment with antidepressants and prediction of clinical responsivity. Acta Psychiatr Scand. 1998;97(4):302–308. doi: 10.1111/j.1600-0447.1998.tb10004.x. [DOI] [PubMed] [Google Scholar]
- Maj J, Rogoz Z, Skuza G, Sowinska H. The effect of CGP 37849 and CGP 39551, competitive NMDA receptor antagonists, in the forced swimming test. Pol J Pharmacol Pharm. 1992;44(4):337–346. [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Manji HK, Lenox RH. Signaling: cellular insights into the pathophysiology of bipolar disorder. Biol Psychiatry. 2000;48(6):518–530. doi: 10.1016/s0006-3223(00)00929-x. [DOI] [PubMed] [Google Scholar]
- Manji HK, Quiroz JA, Sporn J, Payne JL, Denicoff K, G NA, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry. 2003;53(8):707–742. doi: 10.1016/s0006-3223(03)00117-3. [DOI] [PubMed] [Google Scholar]
- Matrisciano F, Scaccianoce S, Del Bianco P, Panaccione I, Canudas AM, Battaglia G, et al. Metabotropic glutamate receptors and neuroadaptation to antidepressants: imipramine-induced down-regulation of beta-adrenergic receptors in mice treated with metabotropic glutamate 2/3 receptor ligands. J Neurochem. 2005;93(5):1345–1352. doi: 10.1111/j.1471-4159.2005.03141.x. [DOI] [PubMed] [Google Scholar]
- Mauri MC, Ferrara A, Boscati L, Bravin S, Zamberlan F, Alecci M, et al. Plasma and platelet amino acid concentrations in patients affected by major depression and under fluvoxamine treatment. Neuropsychobiology. 1998;37(3):124–129. doi: 10.1159/000026491. [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Brannan SK, Mahurin RK, Jerabek PA, Brickman JS, Tekell JL, et al. Cingulate function in depression: a potential predictor of treatment response. Neuroreport. 1997;8:1057–1061. doi: 10.1097/00001756-199703030-00048. [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–660. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- McCormick LM, Boles Ponto LL, Pierson RK, Johnson HJ, Magnotta V, Brumm MC. Metabolic correlates of antidepressant and antipsychotic response in patients with psychotic depression undergoing electroconvulsive therapy. J ECT. 2007;23:265–273. doi: 10.1097/yct.0b013e318150d56d. [DOI] [PubMed] [Google Scholar]
- Meloni D, Gambarana C, De Montis MG, Dal Pra P, Taddei I, Tagliamonte A. Dizocilpine antagonizes the effect of chronic imipramine on learned helplessness in rats. Pharmacol Biochem Behav. 1993;46:423–426. doi: 10.1016/0091-3057(93)90374-3. [DOI] [PubMed] [Google Scholar]
- Mickley GA, Schaldach MA, Snyder KJ, Balogh SA, Len T, Neimanis K, et al. Ketamine blocks a conditioned taste aversion (CTA) in neonatal rats. Physiol Behav. 1998;64(3):381–390. doi: 10.1016/s0031-9384(98)00097-3. [DOI] [PubMed] [Google Scholar]
- Mitani H, Shirayama Y, Yamada T, Maeda K, Ashby CR, Jr, Kawahara R. Correlation between plasma levels of glutamate, alanine and serine with severity of depression. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1155–1158. doi: 10.1016/j.pnpbp.2006.03.036. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17(8):2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moryl E, Danysz W, Quack G. Potential antidepressive properties of amantadine, memantine and bifemelane. Pharmacol Toxicol. 1993;72(6):394–397. doi: 10.1111/j.1600-0773.1993.tb01351.x. [DOI] [PubMed] [Google Scholar]
- Mott DD, Doherty JJ, Zhang S, Washburn MS, Fendley MJ, Lyuboslavsky P, et al. Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat Neurosci. 1998;1:659–667. doi: 10.1038/3661. [DOI] [PubMed] [Google Scholar]
- Nudmamud-Thanoi S, Reynolds GP. The NR1 subunit of the glutamate/NMDA receptor in the superior temporal cortex in schizophrenia and affective disorders. Neurosci Lett. 2004;372(12):173–177. doi: 10.1016/j.neulet.2004.09.035. [DOI] [PubMed] [Google Scholar]
- Ostroff R, Gonzales M, Sanacora G. Antidepressant effect of ketamine during ECT. Am J Psychiatry. 2005;162(7):1385–1386. doi: 10.1176/appi.ajp.162.7.1385. [DOI] [PubMed] [Google Scholar]
- Padovan CM, Guimaraes FS. Antidepressant-like effects of NMDA-receptor antagonist injected into the dorsal hippocampus of rats. Pharmacol Biochem Behav. 2004;77(1):15–19. doi: 10.1016/j.pbb.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Papp M, Moryl E. New evidence for the antidepressant activity of MK-801, a non-competitive antagonist of NMDA receptors. Pol J Pharmacol. 1993;45(56):549–553. [PubMed] [Google Scholar]
- Papp M, Moryl E. Antidepressant activity of non-competitive and competitive NMDA receptor antagonists in a chronic mild stress model of depression. Eur J Pharmacol. 1994;263(12):1–7. doi: 10.1016/0014-2999(94)90516-9. [DOI] [PubMed] [Google Scholar]
- Papp M, Moryl E. Antidepressant-like effects of 1-aminocyclopropanecarboxylic acid and D-cycloserine in an animal model of depression. Eur J Pharmacol. 1996;316(23):145–151. doi: 10.1016/s0014-2999(96)00675-9. [DOI] [PubMed] [Google Scholar]
- Paul IA, Skolnick P. Glutamate and depression: clinical and preclinical studies. Ann N Y Acad Sci. 2003;1003:250–272. doi: 10.1196/annals.1300.016. [DOI] [PubMed] [Google Scholar]
- Payne JL, Quiroz JA, Zarate CA, Jr, Manji HK. Timing is everything: does the robust upregulation of noradrenergically regulated plasticity genes underlie the rapid antidepressant effects of sleep deprivation? Biol Psychiatry. 2002;52(10):921–926. doi: 10.1016/s0006-3223(02)01676-1. [DOI] [PubMed] [Google Scholar]
- Petrakis IL, Limoncelli D, Gueorguieva R, Jatlow P, Boutros NN, Trevisan L, et al. Altered NMDA glutamate receptor antagonist response in individuals with a family vulnerability to alcoholism. Am J Psychiatry. 2004;161:1776–1782. doi: 10.1176/ajp.161.10.1776. [DOI] [PubMed] [Google Scholar]
- Petrenko AB, Yamakura T, Fujiwara N, Askalany AR, Baba H, Sakimura K. Reduced sensitivity to ketamine and pentobarbital in mice lacking the N-methyl-D-aspartate receptor GluRepsilon1 subunit. Anesth Analg. 2004;99:1136–1140. doi: 10.1213/01.ANE.0000131729.54986.30. [DOI] [PubMed] [Google Scholar]
- Phelps LE, Brutsche N, Moral JR, Luckenbaugh DA, Manji HK, Zarate CA., Jr Family History of Alcohol Dependence and Initial Antidepressant Response to an N-methyl-D-aspartate Antagonist. Biol Psychiatry. 2008 Nov 7; doi: 10.1016/j.biopsych.2008.09.029. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittaluga A, Raiteri L, Longordo F, Luccini E, Barbiero VS, Racagni G, et al. Antidepressant treatments and function of glutamate ionotropic receptors mediating amine release in hippocampus. Neuropharmacology. 2007;53(1):27–36. doi: 10.1016/j.neuropharm.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008;33(1):88–109. doi: 10.1038/sj.npp.1301574. [DOI] [PubMed] [Google Scholar]
- Pizzagalli D, Pascual-Marqui RD, Nitschke JB, Oakes TR, Larson CL, Abercrombie HC, et al. Anterior cingulate activity as a predictor of degree of treatment response in major depression: evidence from brain electrical tomography analysis. Am J Psychiatry. 2001;158:405–415. doi: 10.1176/appi.ajp.158.3.405. [DOI] [PubMed] [Google Scholar]
- Preskorn S, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28:631–637. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Przegalinski E, Tatarczynska E, Deren-Wesolek A, Chojnacka-Wojcik E. Antidepressant-like effects of a partial agonist at strychnine-insensitive glycine receptors and a competitive NMDA receptor antagonist. Neuropharmacology. 1997;36(1):31–37. doi: 10.1016/s0028-3908(96)00157-8. [DOI] [PubMed] [Google Scholar]
- Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- Salvadore G, Cornwell BR, Colon-Rosario V, Coppola R, Grillon C, Zarate CA, Jr, et al. Increased Anterior Cingulate Cortical Activity in Response to Fearful Faces: A Neurophysiological Biomarker that Predicts Rapid Antidepressant Response to Ketamine. Biol Psychiatry. 2008 Sept 24; doi: 10.1016/j.biopsych.2008.08.014. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, et al. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7(5):426–437. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Brody AL, Ho ML, Zohrabi N, Maidment KM, Baxter LR., Jr Differential brain metabolic predictors of response to paroxetine in obsessive-compulsive disorder versus major depression. Am J Psychiatry. 2003;160(3):522–532. doi: 10.1176/appi.ajp.160.3.522. [DOI] [PubMed] [Google Scholar]
- Scarr E, Pavey G, Sundram S, MacKinnon A, Dean B. Decreased hippocampal NMDA, but not kainate or AMPA receptors in bipolar disorder. Bipolar Disord. 2003;5(4):257–264. doi: 10.1034/j.1399-5618.2003.00024.x. [DOI] [PubMed] [Google Scholar]
- Schumann G, Johann M, Frank J, Preuss U, Dahmen N, Laucht M, et al. Systematic analysis of glutamatergic neurotransmission genes in alcohol dependence and adolescent risky drinking behavior. Arch Gen Psychiatry. 2008;65:826–838. doi: 10.1001/archpsyc.65.7.826. [DOI] [PubMed] [Google Scholar]
- Sernagor E, Kuhn D, Vyklicky L, Jr, Mayer ML. Open channel block of NMDA receptor responses evoked by tricyclic antidepressants. Neuron. 1989;2(3):1221–1227. doi: 10.1016/0896-6273(89)90306-1. [DOI] [PubMed] [Google Scholar]
- Silvestre JS, Nadal R, Pallares M, Ferre N. Acute effects of ketamine in the holeboard, the elevated-plus maze, and the social interaction test in Wistar rats. Depress Anxiety. 1997;5(1):29–33. [PubMed] [Google Scholar]
- Sinner B, Graf BM. Ketamine. Handb Exp Pharmacol. 2008;182:313–333. doi: 10.1007/978-3-540-74806-9_15. [DOI] [PubMed] [Google Scholar]
- Skolnick P. Antidepressants for the new millennium. Eur J Pharmacol. 1999;375(13):31–40. doi: 10.1016/s0014-2999(99)00330-1. [DOI] [PubMed] [Google Scholar]
- Skolnick P, Legutko B, Li X, Bymaster FP. Current perspectives on the development of non-biogenic amine-based antidepressants. Pharmacol Res. 2001;43(5):411–423. doi: 10.1006/phrs.2000.0806. [DOI] [PubMed] [Google Scholar]
- Skolnick P, Miller R, Young A, Boje K, Trullas R. Chronic treatment with 1-aminocyclopropanecarboxylic acid desensitizes behavioral responses to compounds acting at the N-methyl-D-aspartate receptor complex. Psychopharmacology (Berl) 1992;107(4):489–496. doi: 10.1007/BF02245261. [DOI] [PubMed] [Google Scholar]
- Thase ME, Haight BR, Richard N, Rockett CB, Mitton M, Modell JG, et al. Remission rates following antidepressant therapy with bupropion or selective serotonin reuptake inhibitors: a meta-analysis of original data from 7 randomized controlled trials. J Clin Psychiatry. 2005;66(8):974–981. doi: 10.4088/jcp.v66n0803. [DOI] [PubMed] [Google Scholar]
- Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163(1):28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- Trullas R, Skolnick P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur J Pharmacol. 1990;185(1):1–10. doi: 10.1016/0014-2999(90)90204-j. [DOI] [PubMed] [Google Scholar]
- Wu J, Buchsbaum MS, Gillin JC, Tang C, Cadwell S, Wiegand M, et al. Prediction of antidepressant effects of sleep deprivation by metabolic rates in the ventral anterior cingulate and medial prefrontal cortex. Am J Psychiatry. 1999;156(8):1149–1158. doi: 10.1176/ajp.156.8.1149. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Du J, Quiroz J, Gray NA, Denicoff K, Singh JB, et al. Regulation of cellular plasticity cascades in the pathophysiology and treatment of mood disorders: role of the glutamatergic system. Ann N Y Acad Sci. 2003;1003:273–291. doi: 10.1196/annals.1300.017. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Manji HK. The promising role of the glutamate system in mood disorders. In: Heresco-Levy U, Javitt D, editors. Glutamate in Neuropsychiatric Disorders. Kerala, India: Research Signpost; 2008. [Google Scholar]
- Zarate CA, Quiroz J, Payne J, Manji HK. Modulators of the glutamatergic system: implications for the development of improved therapeutics in mood disorders. Psychopharmacol Bull. 2002;36(4):35–83. [PubMed] [Google Scholar]
- Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]