

Abstract
Homologous genes originate from a common ancestor through vertical inheritance, duplication, or horizontal gene transfer. Entire homolog families spawned by a single ancestral gene can be identified across multiple genomes based on protein sequence similarity. The sequences, however, do not always reveal conclusively the history of large families. To study the evolution of complete gene repertoires, we propose here a mathematical framework that does not rely on resolved gene family histories. We show that so-called phylogenetic profiles, formed by family sizes across multiple genomes, are sufficient to infer principal evolutionary trends. The main novelty in our approach is an efficient algorithm to compute the likelihood of a phylogenetic profile in a model of birth-and-death processes acting on a phylogeny.
We examine known gene families in 28 archaeal genomes using a probabilistic model that involves lineage- and family-specific components of gene acquisition, duplication, and loss. The model enables us to consider all possible histories when inferring statistics about archaeal evolution. According to our reconstruction, most lineages are characterized by a net loss of gene families. Major increases in gene repertoire have occurred only a few times. Our reconstruction underlines the importance of persistent streamlining processes in shaping genome composition in Archaea. It also suggests that early archaeal genomes were as complex as typical modern ones, and even show signs, in the case of the methanogenic ancestor, of an extremely large gene repertoire.
Keywords: gene content evolution, maximum likelihood, Last Archaeal Common Ancestor
Introduction
The evolution of homologous gene families, that is, genes of common ancestry, is enmeshed within species histories in a complex manner (Koonin 2005). Concomitantly with the diversification of organismal lineages, gene families expand by duplications, individual genes get eliminated, and new genes arrive by lateral transfer. It is now clear that de novo gene formation and vertical processes (Henikoff et al. 1997, Snel 2002), such as duplication and loss, act in concert with horizontal gene transfer (Boucher et al. 2003, Gogarten and Townsend 2005).
Gene families are identified in current practice by pairwise sequence comparisons, coupled with the clustering of postulated homolog pairs (Tatusov et al. 1997, Alexeyenko et al. 2006) The phylogenetic profile of a gene family comprises the family size across a set of organisms, that is, the number of homologs within the same family in each genome. Such profiles are extremely informative even without taking the gene sequences into account: profile data sets have been used to construct organismal phylogenies (Fitz-Gibbon and House 1999, Snel et al. 1999, Tekaia et al. 1999) and to infer ancestral gene content (Mirkin et al. 2003, Iwasaki and Takagi 2007); similar and complementary profiles hint at functional associations (Tatusov et al. 1997, Pellegrini et al. 1999). Considering various evolutionary processes in a mathematical model of gene family evolution is challenging. One main element that distinguishes the present study from past work is the elaboration of a likelihood framework for phylogenetic profiles that simultaneously accounts for gene duplication, loss, and acquisition. In particular, we describe an algorithm for the exact computation of the likelihood in a phylogenetic gain –loss–duplication model.
The present study uses a gain–loss–duplication model to address gene content evolution in Archaea. Relying on a complete set of known homolog families in 28 sequenced genomes, we inferred lineage- and family-specific statistics. In a precursory step, we constructed a plausible phylogeny using 88 universally conserved proteins, which we believe is a noteworthy result on its own, as the phylogeny resolves some problematic euryarchaeal branching orders (involving Thermoplasmatales, Methanopyrus, and Methanobacteriales) confidently. Gene loss emerges in our analysis as the dominant force that has shaped archaeal genomes throughout their history. Apparently, genome streamlining has been an ongoing process in all lineages with a fairly constant intensity, apart from dramatic genome compactions in endosymbiotic Archaea. Our reconstruction suggests that early Archaea had a comparable genomic complexity to today's organisms. In particular, the euryarchaeal ancestor of two classes of methanogens had a very large genome, resulting from one of the rare upsurges in gene content, similarly to some modern lineages of Methanosarcina and Halobacteria.
Methods
Phylogenetic Profiles in Archaea
Phylogenetic profiles, sequences, and functional annotations were downloaded from the arCOG database of orthologous gene clusters in Archaea (Makarova et al. 2007) at ftp://ftp.ncbi.nih.gov/pub/wolf/ COGs/arCOG. The profiles were amended with data on lineage-specific singletons and inparalog families that have no archaeal homologs outside of one genome (Wolf Y, personal communication), which was produced in the process of compiling the arCOG database.
The following organisms are included in the study: Archæoglobus fulgidus (Arcfu), Haloarcula marismortui ATCC 43049 (Halma), Halobacterium sp. strain NRC-1 (Halsp), Methanosarcina acetivorans (Metac), Methanococcoides burtonii DSM 6242 (Metbu), Methanoculleus marisnigri JR1 (Metcu), Methanospirillum hungatei JF-1 (Methu), Methanocaldococcus jannaschii (Metja), Methanopyrus kandleri (Metka), Methanosarcina mazei (Metma), Methanococcus maripaludis S2 (Metmp), Methanosphaera stadtmanæ (Metst), Methanothermobacter thermoautotrophicus (Metth), Nanoarchæum equitans (Naneq), Picrophilus torridus DSM 9790 (Picto), Pyrococcus abyssi (Pyrab), Pyrococcus furiosus (Pyrfu), Thermoplasma acidophilum (Theac), Thermococcus kodakaraensis KOD1 (Theko), Thermoplasma volcanium (Thevo), Æropyrum pernix (Aerpe), Caldivirga maquilingensis IC-167 (Calma), Cenarchæum symbiosum (Censy), Hyperthermus butylicus (Hypbu), Pyrobaculum ærophilum (Pyrae), Sulfolobus solfataricus (Sulso); Sulfolobus acidocaldarius DSM 639 (Sulac), Thermofilum pendens Hrk 5 (Thepe) with the last eight classified as crenarchaeota. The abbreviations are those used by Makarova et al. (2007) and the arCOG database.
Reconstruction of Archaeal Phylogeny
The phylogeny was constructed using concatenated multiple alignments of selected orthologous protein sequences. The sequences were chosen from the arCOG database based on phylogenetic profiles: we selected all arCOG groups where every studied genome contained exactly one homolog. There are 88 such groups (see Supplemental Material for sequences), and 46 of those correspond to ribosomal proteins (r-proteins). Alignments were done using the program Muscle (Edgar 2004). Phylogenies were built by likelihood maximization using PhyML (Guindon and Gascuel 2003), with the Jones–Taylor–Thornton substitution model and eight discrete gamma categories and invariant sites. The expected number of substitutions per amino acid site was computed on each edge for the r-proteins in the JTT+I+Γ 8 model by PhyML. Bootstrap support values for the branches were computed by PhyML, using 500 replicates.
Inference of Gene Content Evolution
We maximized the likelihood (see below for the likelihood computation) of the data set using a gain–loss–duplication model with a Poisson distribution at the root and four discrete gamma categories capturing rate variation across families, for edge length tf and duplication λf each. For a given set of model parameters (three parameters— —per edge, one for the root's Poisson parameter Γ and two gamma shape parameters for rate variation), the likelihood of each family was computed using (1) with the described methods of manipulating rate variation and correcting for absent profiles. The data set's likelihood (i.e., the product of family likelihoods) was then maximized numerically as a function of the model parameters, using custom-made software implementing the Broyden– Fletcher–Goldfarb–Shanno conjugate gradient method and Brent's one-dimensional optimization method (Press et al. 1997). Family sizes and lineage-specific events (gains,losses,expansions, and contractions) were computed using posterior probabilities in the optimized gain–loss–duplication model.
—per edge, one for the root's Poisson parameter Γ and two gamma shape parameters for rate variation), the likelihood of each family was computed using (1) with the described methods of manipulating rate variation and correcting for absent profiles. The data set's likelihood (i.e., the product of family likelihoods) was then maximized numerically as a function of the model parameters, using custom-made software implementing the Broyden– Fletcher–Goldfarb–Shanno conjugate gradient method and Brent's one-dimensional optimization method (Press et al. 1997). Family sizes and lineage-specific events (gains,losses,expansions, and contractions) were computed using posterior probabilities in the optimized gain–loss–duplication model.
Phylogenetic Birth-and-Death Model
A phylogenetic birth-and-death model formalizes the evolution of an organism-specific census variable along a rooted phylogeny T. We consider only binary phylogenies here; the full set of methods applicable to multifurcating phylogenies is described in the Supplementary Material. The model specifies edge lengths, as well as birth-and-death processes (Ross 1996, Kendall 1949) acting on the edges. Populations of identical individuals evolve along the tree from the root toward the leaves by Galton–Watson processes. At nonleaf nodes of the tree, populations are instantaneously copied to evolve independently along the adjoining descendant edges. Let the random variable ξ(x)∈{0,1,2,…} denote the population count at every node x∈𝒱(T). Every edge xy is characterized by a loss rate μxy, a duplication rate λxy, and a gain rate κxy. If (X(t):t ≥ 0) is a linear birth-and-death process (Kendall 1949, Takács 1962) with these rate parameters, then ℙ{ξ(y) = m|ξ(x) = n} = ℙ{X(txy) = m|X(0) = n}, where txy > 0 is the edge length, which defines the time interval during which the birth-and-death process runs. The joint distribution of (ξ(x):x∈𝒱(T)) is determined by the phylogeny, the edge lengths and rates, along with the distribution at the root ρ, denoted as γ(n) = ℙ{ξ(ρ) = n}.
It is assumed that one can observe the population counts at the terminal nodes (i.e., leaves) but not at the inner nodes of the phylogeny. As individuals are considered identical, we are also ignorant of the ancestral relationships between individuals within and across populations. The population counts at the leaves form a phylogenetic profile, which is formally a function φ:ℒ(T)↦{0,1,2,…}, where ℒ(T)⊂𝒱(T) denotes the set of leaf nodes. Our central problem is to compute the likelihood of a profile, that is, the probability of the observed counts for fixed model parameters. Define the notation φ(ℒ′) = (φ(x):x∈ℒ′) for the partial profile within a subset ℒ′⊆ℒ(T). Similarly, let ξ(ℒ′) = (ξ(x):x∈ℒ′) denote the vector-valued random variable composed of individual population counts. The likelihood of φ is the probability L = ℙ{ξ(ℒ(T)) = φ}. Let Tx denote the subtree of T rooted at node x. Define the survival count range Mx for every node x as Mx = ∑y∈ℒ(Tx)φ(y). Clearly, the ranges can be calculated easily in a postorder traversal.
For our discussion, we borrow standard terminology applied to homologous genes (Sonnhammer and Koonin 2002). For every edge xy, the population of node y can be split by ancestry at node x: inparalog groups are formed by the progenies of each individual at x and a xenolog group is formed by the individuals whose ancestor immigrated into the population. When ξ(x) = n on the edge xy, then ξ(y) = η + ∑i = 1nζi, where η is the xenolog group size, and ζi are the independent and identically distributed inparalog group sizes. The distribution of xenolog and inparalog group sizes is the well-characterized transient distribution of the appropriate linear birth-and-death processes (Kendall 1949, Karlin and McGregor 1958, Takács 1962; see Supplemental Material). Namely, each ζi has a shifted geometric distribution, and for κ > 0, η has a negative binomial or Poisson distribution. The distributions’ parameters are known functions of the edge length txy and rates κxy,λxy,μxy.
Surviving Lineages
A key factor in inferring the likelihood formulas is the probability that a given individual at a tree node x has no descendants at the leaves within the subtree rooted at x. The corresponding extinction probability is denoted by Dx, which can be computed in a postorder traversal (Csűrös and Miklós 2006). An individual at node x is referred to as surviving if it has at least one progeny at the leaves descending from x. Let Ξ(x) denote the number of surviving individuals at each node x. The number of surviving xenologs and inparalogs follow the same class of distributions as the total number of xenologs and inparalogs (see Supplemental Material). Consequently, if ξ(x) = n on edge xy, then Ξ(y) = η + ∑i = 1nζi, where η is the surviving xenolog count with a Poisson or negative binomial distribution, and ζi are surviving paralog counts, with shifted geometric distributions. The distributions’ parameters can be computed explicitly using the process parameters and the extinction probabilities. In the formulas to follow, we use the probabilities wy*[m∥n] = ℙ{η + ∑i = 1nζi = m;∀ζi > 0}, which can be computed by dynamic programming for all n,m ≤ My in O(My2) time (see Supplemental Material).
Computing the Likelihood
We compute the likelihood using conditional survival likelihoods defined as the probability of observing the partial profile within Tx given the number of surviving individuals Ξ(x): Lx[n] = ℙ{ξ(ℒ(Tx)) = φ(ℒ(Tx))|Ξ(x) = n}. For m > Mx, Lx[m] = 0. For values m = 0,1,…,Mx, the conditional survival likelihoods can be computed recursively as shown below.
If node x is a leaf, then
 |
If x is an inner node with children x1,x2, then Lx[n] can be expressed using Lxi[·] and auxiliary values Bi;·,· for i = 1,2 in the following manner. Auxiliary values Bi;t,s are defined for i = 1,2 and s = 0,…,Mxi as follows.
 |
where Gxi(k) = ℙ{ζ = k} for a surviving inparalog group at xi. In the above equations, 0 < t ≤ Mx1. For all n = 0,…,Mx
 |
The complete likelihood is computed as
For some parametric distributions γ, there is a closed formula for ℙ{Ξ(ρ) = m}. In particular, if γ is the stationary distribution for a gain –loss–duplication or a gain–loss models, then Ξ(ρ) has a negative binomial or Poisson distribution, respectively. The likelihood for a Poisson distribution at the root is
| (1) |
where Γ is the mean family size at the root.
The likelihood formula (1) is corrected to account for the fact that the data set does not contain all-absent profiles with φ(x) = 0 for all leaves x, in a manner analogous to Felsenstein (1992).
Family-specific rate variation is considered by computing the likelihood values for each discrete rate category c characterized by factors (tc,κc,μc,λc). The factors in our analysis are either constant 1, or correspond to the expected values within the four quartiles of a gamma distribution with mean 1.
Results and Discussion
Computational Analysis of Phylogenetic Profiles
Birth-and-death processes are commonly used to model a population of identical individuals (Kendall 1949, Karlin and McGregor 1958) and waiting queues (Takács 1962). Their use in modeling gene family evolution is justified by the fact that losses and duplications seem to occur independently between the members of multigene families (Nei and Rooney 2005). The most general process we consider is a gain–loss–duplication process that is characterized by the rates of gain κ, loss μ, and duplication λ: a population of size n grows by a rate of (λn + κ) and decreases by a rate of μn. In our context, the population comprises homologs of a given family in the genome. Gene acquisition occurs with a rate of κ, combining various means such as innovation and lateral transfer. We model gene family evolution in a phylogenetic setting by associating gain–loss–duplication processes with the branches of a phylogenetic tree. The corresponding phylogenetic birth-and-death model defines a probabilistic framework for the evolution of gene family size. The observed family sizes at the terminal nodes form a phylogenetic profile. In principle, a phylogenetic birth-and-death model suits likelihood-based inference since it is a probabilistic graphical model (Jordan 2004) with a tree structure. The mathematical difficulties stem from the fact that the state space of the processes (i.e., family size) is infinitely large. Consequently, routine computational techniques used to analyze molecular sequence evolution Felsenstein 1981) are not applicable. Previously proposed likelihood methods (Hahn et al. 2005, Spencer et al. 2006, Iwasaki and Takagi 2007) have sidestepped the infinity problem by using approximative calculations with bounds on maximal family size.
We have introduced (Csűrös and Miklós 2006) a procedure for computing the likelihood in a restricted gain–loss–duplication model (assuming 0 < κ and 0 < λ < μ), without imposing artificial size bounds. The weakness of that procedure is potential numerical instability, due to the use of alternating sums in the formulas. We found practical cases (such as the archaeal gene content study we report below), where the numerical instability led to serious errors. The novel procedure presented here is numerically stable, as well as computationally efficient. It applies to arbitrary gain–loss–duplication models, including degenerate cases such as the one of Hahn et al. (2005) with λ = μ and κ = 0. The algorithm takes O(M2n) time to complete for a phylogenetic profile over n species and M total number of genes (see Supplemental Material).
Gene Content Evolution in Archaea
Archaea constitute one of the three main domains of cellular life, and are notable for a spectacular diversity of adaptive strategies to extreme environments (Garrett and Klenk 2006). We examined gene content evolution in Archaea. For the purposes of the study, we have selected 28 completely sequenced genomes covering all major physiological and metabolic groups recognized in cultured Archaea: thermophiles, halophiles, acidophiles, nitrifiers, and methanogens (Valentine 2007). Homolog gene families were extracted from the arCOG (archaeal clusters of orthologous groups) database (Makarova et al. 2007), and combined with groupings of genes that have no archaeal homologs outside of single genomes. The complete data set consists of 14,216 families, of which 7,461 are among the arCOGs.
Phylogenetic Relationships
Archaeal phylogenetic relationships have been resolved to an increasing degree of confidence (Forterre et al. 2006) with the aid of accumulating sequence data. Figure 1 shows our consensual phylogeny based on maximum likelihood trees for concatenated alignments of 46 r-proteins and 88 unique conserved proteins (uc-proteins), which are precisely those that have exactly one homolog in each sampled genome. Congruent phylogenies were proposed before (Forterre et al. 2006, Gribaldo and Brochier-Armanet 2006), based on complete phylogenomics evidence. In our study, r-proteins and uc-proteins show solid support for most recognized phylogenetic relationships but provide contradictory signals for the placement of some euryarchaeal groups. Notably, both sequence data sets support the basal position of N. equitans, which was originally thought to be a specimen of a separate group from Euryarchaeota and Crenarchaeota (Waters et al., 2003), but is more likely an early-branching euryarchaeal organism (Makarova and Koonin 2005, Forterre et al. 2006). The data also support the early-branching position of nonthermophilic crenarchaea represented by C. symbiosum. In fact, nonthermophilic crenarchaea may constitute a separate phylum from Euryarchaota and Crenarchaeota, tentatively named Thaumarchaeota (Brochier-Armanet et al. 2008).
FIG. 1.—

Consensus evolutionary tree of Archaea in the study. The consensus is based on maximum likelihood trees for concatenated alignments of ribosomal and unique conserved proteins. Branch lengths are set by maximum likelihood for the r-proteins. Recognized archaeal orders are highlighted. The boxed triples on the left show the percentage of bootstrap samples supporting the particular edges in three data sets (from 500 replicates for each set): r-proteins, uc-proteins, and uc-proteins without C. symbiosum(Censy) and N. equitans(Naneq). All other edges have >97% bootstrap support in all data sets. Numbers next to the terminal taxa denote genome size in million base pairs.
The observed uncertainties about euryarchaeal groups concern the placement of Thermoplasmata, and so-called Class I methanogens (Bapteste et al. 2005) comprising Methanopyrales, Methanobacteriales, and Methanococcales. Thermoplasmata were originally thought to be a an early-branching lineage of Euryarchaeota (Forterre et al. 2006), but analyses of r-proteins (Matte-Tailliez et al. 2002) have provided strong evidence for their late-branching position after Class I methanogens as in fig. 1. R-proteins in our study support the late-branching of Thermoplasmatales (89% bootstrap value), but a maximum likelihood tree built from uc-proteins places Thermoplasmatales between Nanoarchaea and Thermococcales (66% BV). It has been argued that this placement is due to long-branch attraction Matte-Tailliez et al. 2002, Brochier et al. 2004, a frequent systematic bias of sequence evolution models (Rodríguez-Ezpeleta et al. 2007). Indeed, after we removed N. equitans and C. symbiosum from the uc-protein data set, the late-branching position of Thermoplasmatales regained solid support (100% BV).
The correct phylogenetic position of M. kandleri (Metka) is one of the remaining puzzles in archaeal evolution. The existence of close phylogenetic relationships between Class I methanogens is fairly certain, but different protein sets and taxonomic sampling give conflicting or weak indications (Slesarev et al. 2002, Brochier et al. 2004, Brochier et al. 2005, GaoGupta.methanogenesis) about the exact branching order among Methanopyrales, Methanobacteriales, and Methanococcales. R-proteins in our study give a weak support for the monophyly of Methanococcales and Methanobacteriales at the exclusion of Methanopyrales (49% BV) and faintly favor the paraphyly of Class I methanogens (37% BV for the immediate split of Methanopyrales between Thermococcales and Methanobacteriales/Methanococcales; see Supplemental Material). Uc-proteins, however, solidly point to the monophyly of Class I methanogens ( > 97% BV). Interestingly, the maximum likelihood trees built from uc-proteins do not resolve well the relationships between Halobacteriales, Methanosarcinales, and Methanomicrobiales (see Supplemental Material), but there is little reason to doubt that r-proteins provide a genuine phylogenetic signal about the monophyly of Class II methanogens (Bapteste et al. 2005, Brochier-Armanet et al. 2008), uniting Methanosarcinales and Methanomicrobiales.
We conclude that based on protein sequences, Thermoplasmatales constitute a late-branching euryarchaeal lineage, and their early-branching status is a long-branch attraction artifact. Furthermore, the sequences provide evidence of the monophyly of both Class I and Class II methanogens.
Evolutionary Rates: Correlations Between Sequence and Gene Content Evolution
We experimented with models of increasing complexity that combine lineage- and gene-specific factors in the gain–loss–duplication processes. Specifically, we assumed that the process for family f on branch e is characterized by the rates  , and runs for a duration of
, and runs for a duration of  . Here,
. Here, 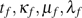 are branch-specific process parameters, and tf,κf,μf,λf are family-specific rate variation coefficients. Starting with simple models with invariant family-specific coefficients, we introduced rate variation in a model hierarchy with increasing complexity. In more complex models, some coefficients were drawn randomly from a discretized gamma distribution (Yang 1994). Different family-specific coefficients do not have the same impact on the model fit. We found the largest improvement when introducing variation in edge length (tf), followed by duplication–rate variation (λf). Further variation in loss and gain rates led to insignificant improvements in the model fit and were not assumed in the analysis.
are branch-specific process parameters, and tf,κf,μf,λf are family-specific rate variation coefficients. Starting with simple models with invariant family-specific coefficients, we introduced rate variation in a model hierarchy with increasing complexity. In more complex models, some coefficients were drawn randomly from a discretized gamma distribution (Yang 1994). Different family-specific coefficients do not have the same impact on the model fit. We found the largest improvement when introducing variation in edge length (tf), followed by duplication–rate variation (λf). Further variation in loss and gain rates led to insignificant improvements in the model fit and were not assumed in the analysis.
In the absence of extraneous scaling, we set  to examine the total rates of gene content change on each edge e. We found a conspicuous correlation across branches between the rate of sequence evolution (expected numbers of substitutions per site for r-proteins) and the component rates of gene content evolution: on this point, see Figure 2 for loss, and the Supplemental Material for duplication and gain. More precisely, the correlation holds for the lineage-specific components of loss, duplication, and gain rates in a decreasing order of strength (P values of 1.1×10 − 11, 8.2×10 − 6, and 1.6×10 − 4, respectively, by Student's t-test for Spearman rank order correlation coefficient).
to examine the total rates of gene content change on each edge e. We found a conspicuous correlation across branches between the rate of sequence evolution (expected numbers of substitutions per site for r-proteins) and the component rates of gene content evolution: on this point, see Figure 2 for loss, and the Supplemental Material for duplication and gain. More precisely, the correlation holds for the lineage-specific components of loss, duplication, and gain rates in a decreasing order of strength (P values of 1.1×10 − 11, 8.2×10 − 6, and 1.6×10 − 4, respectively, by Student's t-test for Spearman rank order correlation coefficient).
FIG. 2.—

Branch-specific loss rates  compared with expected numbers of substitutions (or edge length) for each branch e. Pairs of sibling terminal taxa are connected by lines.
compared with expected numbers of substitutions (or edge length) for each branch e. Pairs of sibling terminal taxa are connected by lines.
The apparent correlations between gene content and sequence evolution rates imply that a steady balance has been maintained between drift and natural selection in almost all lineages. Loss and duplication rates, in particular, have similar vagaries as amino acid substitution rates and provide thus comparable molecular clocks. We measured each terminal node's depth by summing the rates along branches from the root to the node in question. Excluding N. equitans and C. symbiosum, the coefficient of variation of the depth is 26% for protein sequences, 23% for gene loss rates, and 20% for duplication rates. Depths by gene gain rates span about a 4-fold range: for substitution, loss, and duplication, the span is close to 2-fold.
Genes have thus been eliminated in all archaeal lineages with a fairly universal constancy, apart from occasional accelerations. In other words, genome degradation processes seem to persist at a fairly common intensity in every lineage (Mira et al. 2001). Conceivably, genome decay is counterbalanced by natural selection that eliminates deleterious mutations. The root cause of dramatically increased gene loss in obligate symbionts such as N. equitans (Makarova and Koonin 2005) may be reduced selection (Hershberg et al. 2007, Koonin and Wolf 2008). Principles of population genetics imply that changes in population size alone can explain rate changes (Lynch 2006): selection power is weaker in a smaller population, which should manifest in accelerated evolution of sequences (Ohta 1972) and gene content.
We examined the differences between evolutionary rates in sibling terminal taxa for signs of natural selection. Figure 2 shows that gene loss and amino acid substitution rates differ in a concerted fashion for three pairs, that is, for M. stadtmanæ–M. thermoautotrophicus, Halobacterium sp.–H. marismortuimi, and S. acidocaldarius–S. solfataricus. In seven other pairs, loss rates are essentially the same, even if substitution rates may differ. The agreements between substitution and gene loss rate changes attest to common selection forces and mutation processes acting on different forms of genome decay and are predicted by population-genetic arguments (Lynch 2006).
In the lineage leading to M. stadtmanæ, a human commensal (Fricke et al. 2006), all rates are simultaneously larger when compared with its sibling lineage M. thermoautotrophicus, which may be attributed to a smaller population size for the former, which has a smaller habitat. Gene gain and duplication rates behave in general less predictably: numerical differences between loss, gain, and duplication rates on sibling lineages occur in almost all possible sign combinations. The observed fluctuations corroborate the intuition that selection pressures acting on gain and duplication are strong and variable (Wolf et al. 2002). It is plausible that during episodes of massive adaptation, the selective advantages of gene acquisition may outweigh possible negative consequences of an increased genome, and thus drive elevated gene gains, especially if coupled with small population sizes. In our case, unusually large gain rates are inferred on some of the deepest branches (such as the one leading to node E1 in fig. 1 or to the halobacterial ancestor), as well as on the terminal branches leading to M. acetivorans (Metac), H. marismortuimi (Halma) and P. ærophilum (Pyrae).
History of Archaeal Gene Census: Streamlining and Surges
We inferred a probable history of archaeal gene content using posterior probabilities for ancestral family sizes and family size changes, computed from the phylogenetic profiles in the fitted model. Figure 3 summarizes the results by lineages. (See Supplemental Material for bootstrap confidence intervals: the uncertainty in ancestral family counts is estimated to be within ±19% for all nodes. We note that alternate phylogenies for the Class I methanogens give similar results that fall within those confidence intervals.)
FIG. 3.—

A digest of gene content evolution in Archaea. The bar graphs plot posterior means for number of families. The chart on the left shows the number of families with at least one homolog; the fatter part of the bar is proportional to the number of multigene families. The chart in the middle plots the families acquired and lost on the branch leading to the indicated node. The net change is highlighted by the solid part of the bars. The chart on the right shows how many families underwent a contraction from multigene to single-gene composition, or expanded from a single homolog to multiple paralogs. For instance, the common ancestor of Methanococcales is inferred to have had 1723 gene families, out of which 156 were gained after the split with Methanobacteriales. During the same time, 586 families present at the common ancestor M1 were lost, and the solid bar indicates the net loss of 430 ( = 586-156) families. Among multimember families retained from M1, 68 contracted to a single homolog, and 13 single-member families expanded. Note that scaling is the same on the left-hand side and in the middle,but different on the right-hand side.
Our reconstruction suggests a recurrent theme in archaeal evolution: a major physiological or metabolic invention leads to a successful founding population in a new environment, which then further diversifies by genomic streamlining. We can see notably that fig. 3 shows only a few branches where gains prevail over losses (i.e., at least twice as many gains as losses): such is the case for some deep crenarchaeal and euryarchaeal branches, and the terminal lineages for M. acetivorans and H. marismortuimi. About half of the remaining terminal lineages and two-thirds of remaining deep lineages are dominated by loss. Moreover, there is only one ancestral node (the crenarchaeal ancestor) in the entire tree for which gain is dominant in both descendant lineages.
Why would gene loss be so prevalent? We speculate that the versatility of a large genome in such extant lineages as M. acetivorans (Galagan et al. 2002) and H. marismortuimi (Baliga et al. 2004) can be upheld for only relatively short time periods. Genetic drift already leads to the diversification of descendant lineages, which are frequently isolated, given the disconnectedness of the extreme environments they dwell in (Whitaker et al. 2003, Escobar-Páramo et al. 2005). Specialization and the loss of dispensable functions should be favorable in the descendants that are typically under significant energy stress (Valentine 2007). Genomic streamlining should also be favored by population-size effects due to the isolation (Lynch 2006), even in the case of slightly deleterious loss of function.
After the crenarchaeal split, the main euryarchaeal lineage has been characterized by the accumulation of new families, culminating in a large surge on the branch leading to node E1, where many new families appeared. The time interval (judging by sequence divergence in fig. 1) and the extent of gene gain is similar to what is seen with H. marismortuimi (Halma) and M. acetivorans (Metac). The inference of large gains in the E1 lineage is due to the large number of gene families shared between multiple descendant lineages, and especially between the two classes of methanogens (Slesarev et al. 2002, Bapteste et al. 2005, Gao and Gupta 2007, Makarova et al. 2007). In fact, this lineage may very well have been where hydrogenotrophic methanogenesis was invented, which then underwent modifications, extensions, and degradations in subsequent lineages. It was noted in previous genome-scale comparisons (Bapteste et al. 2005, Gao and Gupta 2007) that it is likely that euryarchaeal lineages acquired methanogenesis predominantly by vertical inheritance because the associated pathways are fairly complex, and neither the sequences nor the phylogenetic profiles show evidence of substantial amounts of lateral gene transfer LGT. Figure 3 suggests that methanogenesis appeared after the split of Thermococcales in the company of more than 760 genes. Based on extant examples of archaea with such swelled genomes (Galagan et al. 2002, Baliga et al. 2004), it is plausible that the corresponding archaeal organisms were extremely versatile.
Our inference of ancestral gene content is quite different from previous reconstructions based on parsimony principles (Makarova et al. 2007, Csűrös 2008): at deep nodes, we postulate larger genomes. Parsimonious reconstructions (Mirkin et al. 2003, Kunin et al. 2005, Csűrös 2008) aim to minimize the number of implied loss and gain events. As a consequence, parsimony inherently underestimates the age of gene families.
A major concern in ancestral gene content reconstruction is that “patchy” profiles arise from a combination of lineage-specific loss events and LGT. Frequent LGT imply smaller ancestral genome sizes (Dagan and Martin 2007). Our reconstruction reveals the prevalence of differential loss, but LGT events are far from uncommon. Lineage-specific gains (“Gain column” in fig. 3) account to more than 14% of families (“Families in the genome”) at half of all the lineages. A probabilistic framework, such as a phylogenetic birth-and-death model, makes it feasible to take all possible gene family histories into consideration in a mathematically sound way. A case in point is the last archaeal common ancestor (LACA), where only about 1300 families are inferred to have been present with a posterior probability of at least 90%, which is close to a parsimony-based inference of about 1000 families (Makarova et al. 2007). Given the uncertainties of most family histories, the exact genome composition of LACA is hard to estimate, but the fractional probabilities point to a genome with slightly more than 2000 families, which is similar to such extant organisms as S. solfataricus. Such a large genome size implies that LACA's genomic complexity was even greater than previously imagined (Makarova et al. 2007), on a par with modern, moderately sized archaeal genomes.
Supplementary Material
Supplementary Material are available online at Molecular Biology and Evolution (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work has been supported by a grant from the Natural Sciences and Engineering Research Council of Canada to M.Cs., and the EU FP6 Marie Curie grant MTKD-CT-2006-042794. Part of the study was done while M.Cs. was a sabbatical visitor at the Rényi Institute of Mathematics, supported by a Marie-Curie Transfer-of-Knowledge fellowship. We are grateful to Yuri Wolf for providing data on lineage-specific gene families. We thank Igor Rogozin, Csaba Pál and Balázs Papp for informative discussions.
References
- Alexeyenko A, Tamas I, Liu G, Sonnhammer ELL. Automatic clustering of orthologs and inparalogs shared by multiple genomes. Bioinformatics. 2006;22:e9–e15. doi: 10.1093/bioinformatics/btl213. [DOI] [PubMed] [Google Scholar]
- Baliga NS, Bonneau R, Facciotti MT, et al. 15 co-authors. Genome sequence of Haloarcula morismurtuimi: a halophilic archaeon from the Dead Sea. Genome Res. 2004;14:2221–2234. doi: 10.1101/gr.2700304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste É, Brochier C, Boucher Y. Higher-level classification of the Archaea: evolution of methanogenesis and methanogens. Archaea. 2005;1:353–363. doi: 10.1155/2005/859728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher Y, Douady CJ, Papke RT, Walsh DA, Boudreau MER, Nesbo CL, Case RJ, Doolittle WF. Lateral gene transfer and the origin of prokaryotic groups. Ann Rev Genet. 2003;37:283–328. doi: 10.1146/annurev.genet.37.050503.084247. [DOI] [PubMed] [Google Scholar]
- Brochier C, Forterre P, Gribaldo S. Archaeal phylogeny based on proteins of the transcription and translation machineries: tackling the Methanopyrus paradox. Genome Biol. 2004;5:R17. doi: 10.1186/gb-2004-5-3-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier C, Forterre P, Gribaldo S. An emerging phylogenetic core of Archaea: phylogenies of transcription and translation machineries converge following addition of new genome sequences. BMC Evol Biol. 2005;5:36. doi: 10.1186/1471-2148-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier-Armanet C, Boussau B, Gribaldo S, Forterre P. Mesophilic crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat Rev Microbiol. 2008;6:245–252. doi: 10.1038/nrmicro1852. [DOI] [PubMed] [Google Scholar]
- Csűrös M. Ancestral reconstruction by asymmetric Wagner parsimony over continuous characters and squared parsimony over distributions . Springer Lect Notes Bioinform. Proceedings of the Sixth RECOMB Comparative Genomics Satellite Workshop. 2008:72–86. 5267. [Google Scholar]
- Csűrös M, Miklós I. Proceedings of the Tenth Annual International Conference on Research in Computational Molecular Biology (RECOMB) Vol. 3909. Springer Lect Notes Bioinform; 2006. A probabilistic model for gene content evolution with duplication, loss, and horizontal transfer; pp. 206–220. [Google Scholar]
- Dagan T, Martin W. Ancestral genome sizes specify the minimum rate of lateral gene transfer during prokaryote evolution. Proc Natl Acad Sci USA. 2007;104(3):870–875. doi: 10.1073/pnas.0606318104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Páramo P, Gosh S, DiRuggiero J. Evidence for genetic drift in the diversification of a geographically isolated population of the hyperthermophylic archaeon Pyrococcus. Mol Biol Evol. 2005;22(11):2297–2303. doi: 10.1093/molbev/msi227. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies from restriction sites, a maximum likelihood approach. Evolution. 1992;46:159–173. doi: 10.1111/j.1558-5646.1992.tb01991.x. [DOI] [PubMed] [Google Scholar]
- Fitz-Gibbon ST, House CH. Whole genome-based phylogenetic analysis of free-living microorganisms. Nucleic Acids Res. 1999;27(21):4218–4222. doi: 10.1093/nar/27.21.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forterre P, Gribaldo S, Brochier-Armanet C. Natural history of the archaeal domain. In: Garrett RA, Klenk H-P, editors. Chapter 2. Malden (MA): Blackwell Publishing; 2006. pp. 17–28. Archaea: evolution, physiology, and molecular biology. [Google Scholar]
- Fricke WF, Seedorf H, Henne A, KrŁuer M, Liesegang H, Hedderich R, Gottschalk G, Thauer RK. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J Bacteriol. 2006;188(2):642–658. doi: 10.1128/JB.188.2.642-658.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, Nusbaum C, Roy A, et al. The genome of M. acetivorans reveals extensive metabological and physiological diversity. Genome Res. 2002;12:532–542. doi: 10.1101/gr.223902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Gupta RS. Phylogenomic analysis of proteins that are distinctive of Archaea and its main subgroups and the origin of methanogenesis. BMC Genomics. 2007;8:86. doi: 10.1186/1471-2164-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett RA, Klenk H-P, editors. Archaea: evolution, physiology, and molecular biology. Malden (MA): Blackwell Publishing; 2006. [Google Scholar]
- Gogarten JP, Townsend JP. Horizontal gene transfer, genome innovation and evolution. Nat Rev Microbiol. 2005;3:679–687. doi: 10.1038/nrmicro1204. [DOI] [PubMed] [Google Scholar]
- Gribaldo S, Brochier-Armanet C. The origins and evolution of Archaea: a state of the art. Philos Trans R Soc Lond B. 2006;361:1007–1022. doi: 10.1098/rstb.2006.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hahn MW, De Bie T, Stajich JE, Nguyen C, Cristianini N. Estimating the tempo and mode of gene family evolution from comparative genomic data. Genome Res. 2005;15:1153–1160. doi: 10.1101/gr.3567505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Greene EA, Pietrokovski S, Bork P, Atwood TK, Hood L. Gene families: the taxonomy of protein paralogs and chimeras. Science. 1997;278:609–614. doi: 10.1126/science.278.5338.609. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Tang H, Petrov DA. Reduced selection leads to accelerated gene loss in Shigella. Genome Biol. 2007;8:R164. doi: 10.1186/gb-2007-8-8-r164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki W, Takagi T. Reconstruction of highly heterogeneous gene-content evolution across the three domains of life. Bioinformatics. 2007;23(13):i230–i239. doi: 10.1093/bioinformatics/btm165. [DOI] [PubMed] [Google Scholar]
- Jordan MI. Graphical models. Stat Sci. 2004;19(1):140–155. [Google Scholar]
- Karlin S, McGregor J. Linear growth, birth, and death processes. J Math Mech. 1958;7(4):643–662. [Google Scholar]
- Kendall DG. Stochastic processes and population growth. J R Stat Soc Series B. 1949;11(2):230–282. [Google Scholar]
- Koonin EV. Orthologs, paralogs, and evolutionary genomics. Ann Rev Genet. 2005;39:309–338. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI. Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008;36(21):6688–6719. doi: 10.1093/nar/gkn668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, Goldovsky L, Darzentas N, Ouzounis CA. The net of life: reconstructing the microbial phylogenetic network. Genome Res. 2005;15(7):954–959. doi: 10.1101/gr.3666505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. Streamlining and simplification of microbial genome achitecture. Ann Rev Microbiol. 2006;60:327–349. doi: 10.1146/annurev.micro.60.080805.142300. [DOI] [PubMed] [Google Scholar]
- Makarova K, Koonin EV. Evolutionary and functional genomics of the Archaea. Curr Opin Microbiol. 2005;8:586–594. doi: 10.1016/j.mib.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Sorokin AV, Novichkov PS, Wolf YI, Koonin EV. Clusters of orthologous genes for 41 archaeal genomes and implications for evolutionary genomics of archaea. Biol Direct. 2007;2:33. doi: 10.1186/1745-6150-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matte-Tailliez O, Brochier C, Forterre P, Philippe H. Archaeal phylogeny based on ribosomal proteins. Mol Biol Evol. 2002;19(5):631–639. doi: 10.1093/oxfordjournals.molbev.a004122. [DOI] [PubMed] [Google Scholar]
- Mira A, Ochman H, Moran NA. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001;17(10):589–596. doi: 10.1016/s0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- Mirkin BG, Fenner TI, Galperin MY, Koonin EV. Algorithms for computing evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol Biol. 2003;3:2. doi: 10.1186/1471-2148-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Rooney AP. Concerted and birth-and-death evolution of multigene families. Annu Rev Genet. 2005;39(1):121–152. doi: 10.1146/annurev.genet.39.073003.112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. Population size and rate of evolution. J Mol Evol. 1972;1:305–314. [PubMed] [Google Scholar]
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA. 1999;96(8):4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press WH, Teukolsky SA, Vetterling WV, Flannery BP. Numerical recipes in C. 2nd ed. New York: Cambridge University Press; 1997. the art of scientific computing. [Google Scholar]
- Rodríguez-Ezpeleta N, Brinkmann H, Roure B, Lartillot N, Lang BF, Philippe H. Detecting and overcoming systematic errors in genome-scale phylogenies. Syst Biol. 2007;56(3):389–399. doi: 10.1080/10635150701397643. [DOI] [PubMed] [Google Scholar]
- Ross SM. Stochastic Processes. 2nd ed. New York: Wiley & Sons; 1996. [Google Scholar]
- Slesarev AI, Mezhevaya KV, Makarova KS, et al. 16 co-authors. The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens. Proc Natl Acad Sci USA. 2002;99(7):4644–4649. doi: 10.1073/pnas.032671499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nat Genet. 1999;21(1):108–110. doi: 10.1038/5052. [DOI] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA. Genomes in flux: the evolution of archaeal and proteobacterial gene content. Genome Res. 2002;12(1):17–25. doi: 10.1101/gr.176501. [DOI] [PubMed] [Google Scholar]
- Sonnhammer ELL, Koonin EV. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 2002;18(12):619–620. doi: 10.1016/s0168-9525(02)02793-2. [DOI] [PubMed] [Google Scholar]
- Spencer M, Susko E, Roger AJ. Modelling prokaryote gene content. Evol Bioinform Online. 2006;2:165–186. [PMC free article] [PubMed] [Google Scholar]
- Takács L. Introduction to the theory of queues. New York: Oxford University Press; 1962. [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Tekaia F, Lazcano A, Dujon B. The genomic tree as revealed from whole proteome comparisons. Genome Res. 1999;9(6):550–557. [PMC free article] [PubMed] [Google Scholar]
- Valentine DL. Adaptations to energy stress dictate the ecology and evolution of the Archaea. Nat Rev Microbiol. 2007;5:316–323. doi: 10.1038/nrmicro1619. [DOI] [PubMed] [Google Scholar]
- Waters E, Hohn MJ, Ahel I, et al. 22 co-authors. The genome of Nanoarchaeum equitans: insights into early archaeal evolution and derived parasitism. Proc Natl Acad Sci USA. 2003;100(22):12984–12988. doi: 10.1073/pnas.1735403100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker RJ, Grogan DW, Taylor JW. Geographic barriers isolate endemic populations of hyperthermophylic archaea. Science. 2003;301(5635):976–978. doi: 10.1126/science.1086909. [DOI] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Grishin NV, Koonin EV. Genome trees and the Tree of Life. Trends Genet. 2002;18(9):472–479. doi: 10.1016/s0168-9525(02)02744-0. [DOI] [PubMed] [Google Scholar]
- Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol. 1994;39:306–314. doi: 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.