

Summary
An important goal of gene therapy is to be able to deliver genes so that they express in a pattern that recapitulates the expression of an endogenous cellular gene. Although tissue-specific promoters confer selectivity, in a vector-based system, their activity may be too weak to mediate detectable levels in gene expression studies. We have used a two-step transcriptional activator (TSTA) system to amplify gene expression from lentiviral vectors using the human insulin promoter (HIP). In this system, the HIP drives expression of a potent synthetic transcription activator (the yeast GAL4 DNA binding domain fused to the activation domain of the HSV-1 VP16 activator), which in turn activates a GAL4-responsive promoter driving the enhanced green fluorescent protein reporter gene. Vectors carrying the HIP did not express in non-β cell lines but expressed in murine insulinoma cell lines, indicating that the HIP was capable of conferring cell specificity of expression. The insulin amplifiable-vector was able to amplify gene expression five to nine times over a standard insulin promoter vector. In primary human islets, gene expression from the insulin-promoted vectors was coincident with insulin staining. These vectors will be useful in gene expression studies that require a detectable signal and tissue specificity.
Keywords: lentiviral vector, insulin promoter, transcriptional activation, lineage-specificity, gene amplification
Introduction
An important goal of gene therapy is to be able to deliver genes so that they express in a pattern that recapitulates the expression of an endogenous cellular gene for proper function or for optimal effect of the delivered gene. Regulating transgene expression will also be an important component to reducing risks of adverse advents that may be associated with gene therapy, such as insertional trans-activation of cellular genes. 1,2
Diabetes mellitus, a major world health problem leading to early death and chronic ill health, is caused by the inability of the pancreatic β cells in the islets of Langerhans to secrete adequate insulin to compensate for metabolic demand. Currently, loss of insulin production, whether by immune destruction or impaired compensatory growth of the β cells, is irreversible. Islet cell transplantation is a potential therapy for diabetes mellitus, but routine transplantation has not become standard treatment because of the limited supply of tissue and the problems associated with tissue rejection. Therefore, it is important to develop novel therapies for the treatment of diabetes mellitus. Effective tools to study gene modification of β cells would enhance the development of novel therapies.
Lentivirus-based vectors are attractive gene delivery vehicles for β cells because they can transduce non-dividing or slowly dividing cells and integrate into the host-cell genome for long-term gene expression. 3-5 Recent advances in lentiviral vector design have made it possible to self inactivate (SIN) the native viral LTR enhancer/promoter and to place an internal promoter, such as a tissue-specific promoter, in the expression cassette. 6,7 Tissue-specific promoters are important for targeting gene expression to a particular cell type but can be poor activators of transcription leading to inadequate levels of gene products making gene expression studies in primary cells difficult. To overcome the limitations of low levels of expression from vectors using the insulin gene promoter, we have developed lineage-specific lentiviral vectors that carry the human insulin promoter (HIP) in the two-step transcription amplification (TSTA) system to augment gene expression in pancreatic β cells.
In this TSTA system, a weak promoter controls transcription of a synthetic transactivator composed of the yeast Gal4 DNA binding domain fused to the Herpes Simplex Virus-1 (HSV-1) VP16 immediate early transactivator. 8 9 This synthetic transactivator, in turn, binds to Gal4 DNA responsive elements upstream of a minimal promoter. Wu and colleagues optimized the TSTA system using two tandem copies of HSV-1 VP16 in the transactivator element (Gal4VP2) and five Gal4 DNA binding sites in an adenoviral E4 minimal promoter element (G5E4T). 10 When these elements were cloned into adenoviral11 and lentiviral vectors, 12 the TSTA system successfully increased the level of prostate cancer cell-specific expression from a prostate-specific antigen promoter, while maintaining the lineage-specificity of expression. Transcriptional activation has also worked successfully using neuron and astrocyte-specific promoters. 13,14 In the present study, we have compared gene expression from lentiviral vectors containing the PGK or the HIP using optimized components of the TSTA system. We have found that we can achieve between one half to one log amplification of gene expression using the TSTA system. Importantly, we were able to amplify gene expression using the HIP while preserving beta cell lineage specificity. Additionally, in primary islets, gene expression from lentiviral vectors carrying the HIP was coincident with insulin expression. The vector carrying theTSTA elements was able to amplify gene expression two logs over the standard HIP vector.
Results
Viral titers and Southern blot analysis
We generated a series of SIN lentiviral vectors containing either the constitutive PGK promoter, which has a weak enhancer, or the HIP in the absence or presence of sequences from the TSTA system (Figure 1a). To compare gene expression from the TSTA elements in cis or trans configurations, the PGK promoter driving the transactivator sequences and the minimal promoter driving the eGFP reporter gene were cloned into either the same (cis) or separate (trans) lentiviral vectors. To compare gene expression from a vector containing the TSTA sequences to a vector containing the woodchuck posttranscriptional regulatory element (WPRE), which has been reported to enhance gene expression, 15 HIP lentiviral vectors containing the WPRE sequences were also generated.
Figure 1. Diagram of lentiviral vectors and vector titers.

(A) Lentiviral vectors were produced from the pCCL or SMPU vector backbones that have deletions of the LTR enhancers and promoters (so-called “SIN” configuration symbolized as ΔU3) and contain the HIV-1 central polypurine tract (cPPT) and the Rev response element (RRE). Approximate sizes of vector genomes in kilobase-pairs (kb) are shown to the right of the vector diagrams. (B) Unconcentrated vector titers were determined by qPCR analysis of vector genomes in transduced cells. Error bars represent the standard deviation from the mean of 3 experiments. PGK=phosphoglycerate kinase promoter; Gal4VP2=Gal4 DNA binding domains plus two copies HSV-1 VP16 element; G5E4T=five Gal4 DNA binding sites plus Adenovirus E4 minimal promoter; HIP=human insulin promoter; WPRE=woodchuck post-transcriptional regulatory element; TU/mL=transducing units per milliliter.
Unconcentrated vector titers, as measured by vector copy number, could be obtained in the range of 103 to 106 TU/mL (Figure 1b). The PGK and HIP vectors that contained the TSTA elements in cis configuration had a 2-log- and 1-log-lower titer, respectively, compared to their standard counterpart vectors that did not contain the TSTA elements. This result indicated that the extra TSTA sequences lowered titers, especially in the context of the PGK promoter. Interestingly, when we directly compared the effective titer, as measured by eGFP expression, of the vectors that express eGFP to their DNA titer (Table 1), the titers were similar. This result indicated that, at least of these vectors tested, they do not undergo silencing of eGFP expression.
Table 1.
Direct comparison of DNA titer to expression titer of gene-amplified vectors.
| Vector | DNA Titer (TU/mL) | Expression Titer (TU/mL) |
|---|---|---|
| PGK | 1.25 × 106 | 3.7 × 106 |
| PGK-A | 2.95 × 104 | 6.2 × 104 |
| HIP | 1.0 × 106 | 1.5 × 106 |
| HIP-LA | 2.85 × 105 | 4.35 × 105 |
| HIP-W | 8 × 105 | 3 × 106 |
TU=transducing units
To determine whether these vectors could be packaged and delivered intact, we transduced HT29 cells and analyzed genomic DNA by Southern blot analysis. Except for the E4min-eGFP vector, we detected integrated proviruses of the appropriate lengths (Figure 2). In addition to the expected 1.8 Kb band for the E4min-eGFP vector, a second band that was smaller than 1.6 Kb was present, indicating that sequences from the E4min-eGFP vector were being deleted. Interestingly, these same sequences in the context of the cis vectors (e.g. PGK-A, HIP-LA) were not being deleted, as bands smaller than full-length were not detected (Figure 2, lanes 3 and 7).
Figure 2. Integrated lentiviral vector proviruses by Southern blot analysis.
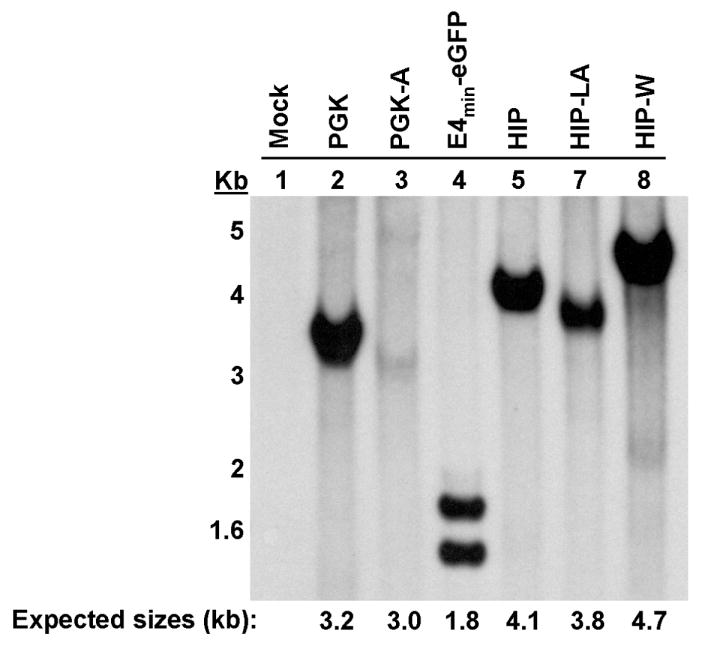
Genomic DNA was isolated 20 days after transduction and expansion of HT-29 cells. Expected provirus sizes following restriction enzyme digestion are shown at the bottom in kilobase-pairs (kb).
Previously, Zhang et al. 10 determined that two VP16 activation domains in the transactivator element and five Gal4 DNA binding sites in the minimal promoter element were optimal in the TSTA system for augmenting gene activity. To determine whether these tandem repeats were rearranging and/or deleting internally, we designed PCR primers to amplify the 345 bp fragment in Gal4VP2, containing the two copies of VP16, and the 170 bp fragment in G5E4T, containing the five copies of the Gal4 DNA binding sites, and amplified these sequences from genomic DNA extracted from transduced cells. Only bands of the appropriate sizes were detected (Supplementary Figure 1), suggesting that these tandem repeats were being carried intact. Overall, lentiviral vectors could be generated that carried the TSTA system elements.
Comparison of gene expression between cis and trans amplified vectors
To investigate the effects of the TSTA elements in cis or trans configuration, we constructed lentiviral vectors that contained the PGK promoter/Gal4VP2 cassette and the G5E4T/eGFP cassette in one single (PGK-A) or two separate (PGK-G and E4min-eGFP) vectors, respectively (Figure 1a). We transduced HT-29 cells and analyzed gene expression by flow cytometry. At an MOI of 1, the E4min-eGFP vector (with the Gal4-binding sites/minimal promoter and eGFP reporter) by itself expressed eGFP at very low mean fluorescence intensity (MFI) in less than 1% of transduced cells (Figure 3, top row, left panel), indicating some basal expression from the minimal promoter in the complete absence of the trans-activating protein. When we transduced with both the PGK-G (expressing the trans-activator protein) and the E4min-eGFP vectors at MOI 1 each, we again observed the eGFPlo population, but approximately 0.1% of the cells expressed eGFP at high MFI (eGFPhi) (Figure 3, top row, middle panel). We surmised that this population was comprised of cells that were transduced with both vectors and the intensity of gene expression was the result of the TSTA elements interacting in trans. In other experiments, we sorted the eGFPhi cells produced from a PGK-G and E4min-eGFP co-transduction and confirmed by Southern blot analysis of genomic DNA that these cells contained both vectors (data not shown). To increase the number of eGFPhi cells, we transduced with both the PGK-G and the E4min-eGFP vectors at MOI 10 each, which resulted in an increase in the number of eGFPlo cells as well as an increase in the number of eGFPhi cells (Figure 3, top row, right panel). However, even at high MOI, only 3% of the cells were eGFPhi, indicating that co-transduction HT-29 cells with two vectors was inefficient.
Figure 3. Comparison of trans- and cis- configurations of amplified lentiviral vectors.

(A) HT-29 cells were transduced with the E4min-eGFP and PGK-G vectors (in trans) at the indicated multiplicity of infections (MOIs) and analyzed for eGFP expression by flow cytometry 5-6 days later. (B) HT-29 cells were transduced with PGK-A at the indicated MOIs and analyzed for eGFP expression 5-6 days later. MFI=mean fluorescent intensity.
To compare gene expression from the cis vector (containing both the transactivator gene and the response element in a single construct) to the trans vectors, we transduced HT-29 cells with PGK-A at MOI 1 and 10. Increasing the MOI led to an appreciable increase in the total number of eGFP positive cells (Figure 3, bottom row, compare middle and right panels). Additionally, with the cis vector, a single MOI 10 was sufficient to obtain the same number of eGFPhi cells as a total MOI 20 (MOI 10 PGK-G vector + MOI 10 E4-min vector) needed with the trans vectors, indicating that transactivation of gene expression is more efficient in the cis vector. Interestingly, at the high MOI, a small population of eGFPlo cells, which was similar to the eGFPlo population from the E4min-eGFP vector, could be seen. This population may represent the basal gene expression from the minimal promoter, although it is unclear why transactivation is not occurring in these cells. Of note, gene expression (i.e. MFI) was approximately four-fold greater from the trans vectors than from the cis vector (Figure 3, top and bottom rows, compare right panels), although we do not know why this is the case. In a separate experiment, we sorted the eGFPhi cells and confirmed that each sorted population contained one vector copy. When we compared the MFI, there was again an approximately four-fold greater MFI from the trans vectors (data not shown). Therefore, the discrepancy in MFI is probably not due to greater efficiency of transduction by the trans vectors. It is possible that the placement of the TSTA elements in the cis vector is not optimal and higher gene expression may be achieved by rearranging the TSTA elements (refer to Discussion). However, because of the inefficiency of using the trans vectors and because the E4min-eGFP vector itself is not carried intact (Figure 2), experiments were continued using only vectors in the cis configuration.
Comparison of gene expression between standard and amplified vectors
Next, we studied further the level of gene expression enhancement from the amplified vectors compared to the standard vectors. We transduced cells at low MOI to yield single-low vector copy numbers per cell and compared gene expression using MFI when the pool of eGFP-expressing cells was between 1 to 10%. Gene expression from the amplified PGK vector (PGK-A) compared to the standard PGK vector was increased approximately two-and-a-half to four-fold in HT-29 and Hep G2 cells, respectively, ten-fold in HEK293 and NIT-1 cells, and eighteen-fold in Beta-TC-6 cells (Figure 4). Thus, for the vector with the non-specific PGK promoter, the TSTA elements increased expression levels in all cell types assayed.
Figure 4. Quantification of the increase in gene expression from amplified vectors compared to standard vectors.
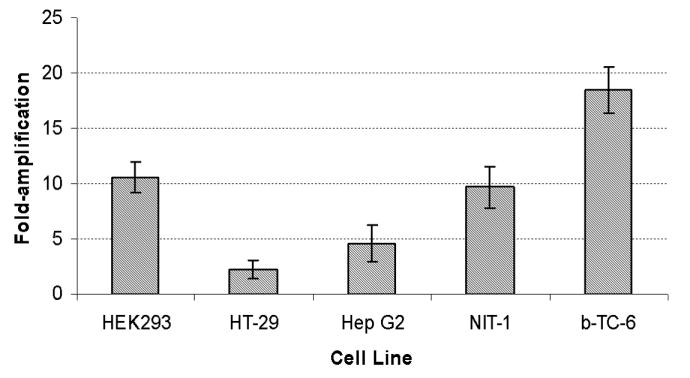
The indicated cell lines were transduced with the standard PGK vector (PGK) or the PGK vector carrying the TSTA elements (PGK-A). MFI was recorded when 1-10% of the transduced pool was eGFP positive at 5-7 days after transduction. Fold increase was calculated by dividing MFI of PGK-A by MFI of PGK. Values shown represent the mean of two experiments and error bars indicate the standard deviation from the mean.
The HIP vectors express in a lineage-specific manner
To determine whether the HIP vectors express in a lineage-specific pattern, we compared gene expression in insulin-producing beta cells (Beta-TC-6, NIT-1, and Min6), non-insulin-producing cells (HEK293, Hep G2, and HT-29), and non-insulin-producing pancreatic cells (ARIP and Panc-1). Insulin mRNA production from the endogenous insulin genes by the beta cell lines, but not by the non-insulin-producing cell lines, was first confirmed by RT-PCR (Supplementary Figure 2). Cells were transduced with the various vectors and MFI of eGFP expression was analyzed by flow cytometry when the cell population was between 1 to 10% eGFP positive. To control for the differences in gene expression intensity between each cell line, we also transduced each cell line with the non-specific PGK promoter vector and normalized gene expression from each HIP vector to this internal control. We consistently detected gene expression from the HIP vectors in the β cell insulinoma cell lines but not in other cell lines (Figure 5). The presence of the WPRE slightly enhanced intensity of gene expression (less than two-fold), but the lineage-amplified HIP vector (HIP-LA) was able to increase intensity of gene expression five to nine-fold over the standard HIP vector. We found a low level of gene expression from the HIP-LA vector in Hep G2 cells, possibly because Hep G2 cells contain similar transcription factors as β cells that regulate the HIP promoter, and this low level of expression was amplified by the TSTA elements.
Figure 5. Gene expression from the human insulin promoter (HIP) is restricted to pancreatic β cells.
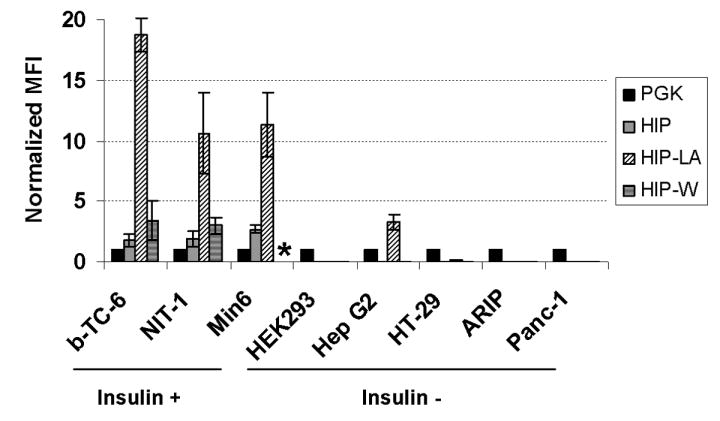
Cell lines tested were transduced with the standard HIP vector (HIP), the HIP vector carrying the TSTA elements (HIP-LA), or the HIP vector carrying the WPRE (HIP-W). MFI was recorded when 1-10% of the transduced pool was eGFP positive at 5-7 days after transduction. MFI from each vector was normalized to the MFI from the PGK vector, which was also tested in each cell line, by dividing the MFI of the vector of interest by the MFI of the PGK vector. Experiments were repeated 2-4 times, and error bars represent the standard deviation from the mean. * HIP-W was not tested in Min6 cells.
To demonstrate that the lack of gene expression was not due to the lack of transduction, we extracted genomic DNA from transduced HEK293, HT-29, and NIT-1 cells and determined vector copy number by quantitative PCR (qPCR). We found that, although HIP vectors did not express in HEK293 and HT-29, the cells carried similar vector copy numbers as cells that were transduced with the constitutively-expressing PGK vector (Table 2). The HIP vectors were functional because they were capable of expressing and transferring similar vector copy numbers to NIT-1 cells. Therefore, gene expression from the HIP vectors was limited to β-type cells even though different cell types could be transduced, and the TSTA elements could enhance gene expression from the HIP without compromising the cell-type specificity of this promoter.
Table 2.
Non-expressing cells (HEK293 and HT-29) have similar vector copy numbers compared to eGFP-expressing cells (NIT-1).
| Vector | Cell Type | %eGFP (MFI) | Copy/cell |
|---|---|---|---|
| PGK | HEK293 | 2.2 (122) | 0.011 |
| HT-29 | 2.4 (254) | 0.014 | |
| NIT-1 | 2.4 (583) | 0.021 | |
| HIP | HEK293 | <1 | 0.049 |
| HT-29 | <1 | 0.017 | |
| NIT-1 | 4.4 (557) | 0.055 | |
| HIP-LA | HEK293 | <1 | 0.047 |
| HT-29 | <1 | 0.018 | |
| NIT-1 | 2.7 (3498) | 0.026 | |
| HIP-W | HEK293 | <1 | 0.005 |
| HT-29 | <1 | 0.004 | |
| NIT-1 | 1.4 (1075) | 0.010 |
MFI=mean fluorescent intensity.
Amplified gene expression is associated with increased levels of mRNA
We next studied the eGFP mRNA species produced from the different vectors. Whereas one eGFP mRNA is expected from each of the standard vectors with or without the WPRE, two eGFP mRNA species are possible from the amplified vectors: one smaller mRNA that is initiated from the TATA box in the adenoviral E4 minimal promoter and a second, larger mRNA that is initiated from the upstream PGK or HIP internal promoter (Figure 6a). We extracted total RNA from PGK or PGK-A transduced Beta-TC-6 cells that were cell sorted for their eGFP expression. We detected the appropriate 1.16 Kb eGFP mRNA from PGK-transduced cells (Figure 6b, lane 2). The predominant eGFP mRNA detected from PGK-A-transduced cells was the shorter, 1.18 Kb, amplified message produced from the E4 minimal promoter. We detected a larger, but weaker, eGFP mRNA of approximately 2.27 Kb, which corresponded to the size expected for the mRNA initiating from the PGK promoter (Figure 6b, lane 3).
Figure 6. Northern blot analysis of eGFP mRNAs produced from standard and amplified lentiviral vectors.

(a) Diagram of expected eGFP mRNA species produced from the internal, full-length or minimal promoter. Expected sizes in kilobase-pairs (kb) are given to the right of each mRNA, (b) eGFP mRNA species detected in transduced Beta-TC-6 cells. Cells were transduced with the indicated vector, eGFP positive cells were isolated by fluorescence-assisted cell sorting (FACS) and expanded, and total RNA were extracted. Black arrow indicates putative 2.27 kb eGFP mRNA initiating from the full-length promoter, (c) Flow cytometric and vector copy analysis of Beta-TC-6 cells transduced with the indicated vectors at low MOI and analyzed at 5 days, c=vector copies, MFI=mean fluorescence intensity, (d) Northern blot (top panel) and densitometry analysis (bottom panel) of eGFP mRNA produced from cells shown in “(c).” Bars of graph represent the intensity of the eGFP mRNA bands, which have been corrected for background and normalized for loading, based on the intensity of β-actin mRNA bands.
In order to directly correlate the levels of eGFP mRNA to MFI produced from the different vectors, we transduced Beta-TC-6 cells with the HIP, HIP-LA, or HIP-W vector at the same MOI. Each of the transduced pools had a similar number of eGFP positive cells and vector copy number, which verified that the cells were transduced similarly (Figure 6c). Similar to previous results (Figure 5), the HIP-LA vector amplified gene expression approximately five-fold over the HIP vector, while the HIP-W vector showed increased gene expression approximately 2.5-fold over the HIP vector. Total RNA was extracted from these low copy number-transduced pools and analyzed by Northern blot for eGFP mRNA. Notably, the intensity of the eGFP bands correlated with MFI. The HIP vector, which had the lowest MFI, had the lowest intensity band; the HIP-W vector, which had an intermediate MFI, had an intermediate intensity band; and the HIP-LA vector, which had the highest MFI, had the highest intensity band (Figure 6d, upper panel). This result was quantified by measuring the intensities of the bands by densitometry (Figure 6d, lower panel). Similar to the comparison of the MFIs, there was approximately two-fold more HIP-W eGFP mRNA and seven-fold more HIP-LA eGFP mRNA compared to HIP eGFP mRNA. We also measured the relative quantities of the eGFP mRNAs from each vector by quantitative reverse transcription PCR (qRT-PCR) with comparable results (data not shown). Therefore, the intensity of eGFP gene expression is directly proportional to the amount of eGFP mRNA produced, and while the presence of the WPRE increased eGFP mRNA production, the presence of the TSTA elements greatly increased eGFP mRNA production through amplification.
Optimal gene expression in primary human islets is achieved from the amplified HIP vector
Next, we studied gene expression from the different vectors in primary human islets. Human islets were received from the Islet Cell Resource Centers (ICR) Basic Science Islet Distribution Program or the National Disease Research Interchange (NDRI). Islet clusters were dissociated using dispase and then cultured onto matrix-coated wells, 16 which allowed the cells to be transduced as a monolayer the next morning. The islets were transduced with PGK, HIP, HIP-LA, or HIP-W vectors at a final vector concentration of 3 × 106 TU/mL and analyzed for eGFP expression by flow cytometry five days later. Islets were typically 85% viable by trypan blue exclusion after this transduction procedure (data not shown). As expected, cells transduced with the PGK vector had the highest percentage of eGFP positive cells, as this promoter is active in insulin positive and insulin negative cells (Figure 7). Similar to the insulinoma cell lines, the intensity of eGFP expression was lowest in HIP transduced cells, intermediate in HIP-W transduced cells, and highest in HIP-LA transduced cells. The HIP-LA vector was able to amplify gene expression more than sixteen-fold over the standard HIP vector and six-fold over the HIP-W vector in primary human islets. In HIP-LA transduced islets, we observed two populations of eGFP positive cells: an eGFPlo-expressing population and an eGFPhi-expressing population (Figure 7, fourth panel). There was an approximate twenty-fold difference in MFI between the two populations, and no difference in viability was observed. The eGFPlo population may represent leaky gene expression from the amplified vector in β cells that have de-differentiated, from other non-insulin-producing cells of the islets, such as the α, δ, or PP cells. An alternative explanation is that some vectors have integrated into unfavorable sites, while other vectors have integrated into more favorable sites. Studies are on-going in our lab to characterize gene expression from HIP vectors in primary cells.
Figure 7. eGFP expression in transduced human islets.

Human islets were transduced with the indicated vectors at a final concentration of 3 × 106 TU/mL and analyzed by flow cytometry 5 days later. Panels from a representative experiment are shown. MFI=mean fluorescent intensity.
To determine whether eGFP-expression correlated with insulin-expression, an aliquot of each sample of the transduced cells was subjected to immunohistochemistry analysis. As expected, the PGK vector drove eGFP expression in insulin-positive and insulin-negative cells, whereas gene expression from the lineage-specific HIP vectors was restricted to insulin-positive cells (Figure 8). In islets that were transduced with the HIP-LA vector, we detected two-intensities of eGFP expression, which correlated with the flow cytometry data. Very bright eGFP-expression correlated with insulin expression (Figure 8, fourth column, long arrows), while very dim eGFP expression did not (Figure 8, fourth column, short arrow). Additionally, bright eGFP expression from the HIP-LA vector was so intense that it required a ten-fold shorter exposure time, compared to HIP and HIP-W vectors, to visualize the green cells (data not shown).
Figure 8. Immunohistochemistry analysis for eGFP and insulin in transduced human islets.

Aliquots of transduced islets from Figure 7 were cytospun and simultaneously stained for eGFP (green) and insulin (red). Cell nuclei were stained with DAPI (blue). Islets transduced with HIP-LA (fourth column) had eGFP bright cells that were also insulin positive (compare second and third rows, long arrows) as well as eGFP dim cells that were insulin negative (short arrow).
We have demonstrated that lentiviral vectors carrying the HIP express in a lineage-specific manner, and we have produced a transcriptionally-amplified HIP vector that can specifically amplify gene expression in primary human β cells. This development will facilitate gene expression studies in primary cells previously hindered by undetectable or weak signals from the insulin promoter.
Discussion
Developing vectors with better regulated expression is important to advancing the field of gene therapy. One method to regulate gene expression is to use tissue-specific gene regulatory elements, such as enhancers and promoters. The intact, 1.4 kb HIP contains all of the functional elements necessary for tissue-specific expression and glucose responsiveness. 17 Although full promoter activity can be defined and preserved, tissue-specific promoters tend to be poor activators of transcription, especially outside the context of their native, chromosomal environment, making gene expression studies difficult. We have developed lentiviral vectors that amplify gene expression from the HIP using a TSTA method. TSTA elements have been used to boost gene expression from weak cancer specific promoters18,19 20 and have been useful for non-invasive imaging studies in developmental biology21 and cancer therapy. 11,12,19,22 Here, we have demonstrated that gene amplification from the HIP is superior in gene expression in primary human islets.
We developed cis and trans amplifying vectors to determine the optimal functional context for the TSTA elements. Interestingly, the sequences that comprised the minimal E4 promoter and eGFP gene were not carried intact in the context of the trans vector (E4min-eGFP) but were carried intact in the context of the cis vectors (PGK-A and HIP-LA). For this reason, studies were continued using only the cis vectors. It is possible that cryptic splice donor (SD) and/or splice acceptor (SA) sites were created during construction of the E4min-eGFP vector and not of the cis vectors.
In HT-29 cells, gene expression from the trans vectors (PGK-G and E4min-eGFP) was approximately three-fold greater than from the cis vector (PGK-A; Figure 3). It is possible that, in the context of a lentiviral vector, the placement of the TSTA elements in the cis vector was not optimal for amplifying gene expression compared to the trans vectors. Although we do not know the reason for this difference in expression levels, it is possible that expression from our cis vector is affected by “transcriptional interference,” which occurs when one transcription unit represses an adjacent unit when the two lay head-to-tail in tandem in a single construct. 23-25 Transcriptional interference is believed to be caused by inaccessibility or competitive binding of the transcriptional machinery to the two proximal promoters. Whether this is the case or not, we believe that the affect of transcriptional interference is low in our system because the second transcriptional unit is comprised of a minimal promoter, which would minimize binding of non-specific or unnecessary transcription factors.
We observed a basal level of expression from the vector carrying only the adenoviral E4 minimal promoter. This “leaky” expression could be due to the activity of nearby enhancer elements in the chromosome where the vector integrated or from residual activity of the SIN LTR of the lentiviral vector itself26 on the E4 minimal promoter. It may be possible to decrease the basal promoter activity by using an alternative minimal promoter, such as that from the HSV-1 thymidine kinase (TK) gene , the long terminal repeat (LTR) of the mouse mammary tumor virus (MMTV), 18 or the fish E1b basal promoter, as others have done in conjunction with the Gal4/VP16 transactivator. 21 The addition of insulator elements to block out exogenous enhancer activity would also be beneficial for decreasing basal level expression.
The WPRE has been reported to increase gene expression from onco- and lentiviral vectors by augmenting RNA processing. 27,28 In agreement with a study conducted by Zufferey et al., 15 we also observed an increase in gene expression levels when the WPRE was present, which corresponded to an increase in RNA levels. When compared directly, the presence of the TSTA elements increased gene expression and RNA levels several-fold more than the WPRE. Whereas the WPRE improves RNA processing to increase gene expression, we believe the TSTA elements produce large amounts of mRNA for translation, through transcriptional amplification, resulting in increased gene expression.
In primary human islets, we observed the same step-wise increase in gene expression from the standard, to the WPRE-containing, to the amplified vector, as we observed in the insulinoma cell lines. We are perplexed that two different populations of eGFP-expressing cells are present in the islets that were transduced with the HIP-LA vector. That the very bright eGFP cells correlated with insulin staining, while the very dim eGFP cells did not, indicated that gene expression was amplified in a cell-specific manner. It is possible that the E4 minimal promoter was weakly active in the non-β cells of the islet cultures. However, this non-specific expression was dimmer than expression from the standard HIP vector. We estimate that gene expression from the HIP vector in primary β cells is ten- to twenty-times lower than gene expression from the HIP-LA vector based on the flow cytometry and immunofluorescence studies. Therefore, we do not think that the very low, non-specific expression from the HIP-LA vector precludes its usefulness in gene expression studies.
The islet experiments were repeated with a different batch of primary human islets, and similar results were obtained. Although it is expected that gene expression would persist long-term, we did not keep the cultures for more than 7 days after transduction, as the cells would have de-differentiated in long-term culture. At 7 days, the viability of the cultures remained high (>80%) with no decrease in eGFP expression (data not shown). Future experiments will be to determine whether labeled β cells can be isolated by sorting and cultured viably to obtain a pure population of gene-modified cells.
In summary, we have developed a lentiviral vector that enhances gene expression from the human insulin promoter while preserving lineage-specificity in β-cells. In primary human islets, compared to the non-amplified lineage-specific vectors, gene expression was superior. Gene amplified lineage-specific vectors will be useful tools for studying gene modification of primary cells.
Materials and Methods
Cell lines
The human embryonic kidney cell line HEK293, human colorectal adenocarcinma cell line HT-29, human hepatocellular carcinoma cell line Hep G2, rat pancreatic tumor cell line ARIP, human pancreatic epithelial carcinoma cell line Panc-1, and murine pancreatic insulinoma cell lines Beta-TC-6 and NIT-1 were obtained from the American Type Culture Collection (ATCC, Manassas, VA). The murine insulinoma cell line Min6 was provided by Gay Crooks at Children’s Hospital Los Angeles. The HEK293 and HT-29 cell lines were maintained in Dulbecco’s modified Eagle medium (DMEM; Mediatech, Inc., Herndon, VA) containing 10% fetal bovine serum (FBS; Omega Scientific, Inc., Tarzana, CA), 2mM L-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin (Gemini Bio-products, West Sacramento, CA). The Hep G2 cell line was maintained in Eagle’s minimum essential medium (ATCC) containing 10% FBS, L-glutamine, and penicillin-streptomycin. The ARIP cell line was maintained in F-12K medium (ATCC) containing 10% FBS, L-glutamine, and penicillin-streptomycin. The Beta-TC-6, NIT-1, Min6, and Panc-1 cell lines were maintained in DMEM modified to contain 1.5 g/L sodium bicarbonate (ATCC), 15% FBS, L-glutamine, and penicillin-streptomycin. All cell lines were maintained in a 37° C incubator with 5% CO2.
Lentiviral vectors
All HIV-1-based lentiviral vectors used in this study were based on the pCCL self-inactivating (SIN) vector backbone developed by Luigi Naldini 7 or the SMPU SIN vector backbone developed by Paula Cannon (Childrens Hospital Los Angeles, Los Angeles, CA) 29. These vectors contain the central polypurine tract (cPPT) and rev response element (RRE) sequences. In our hands, gene expression was similar between pCCL and SMPU vectors that carried the same gene regulatory elements (data not shown). Two internal promoters were used in these studies: the 570 basepair (bp) murine phosphoglycerate kinase promoter (PGK) and the 1.4 kilo-basepair (kb) human insulin promoter (HIP). The standard vectors contain either PGK or HIP (the 1.4 kb Bcl I to Hind III fragment from pFOXCAT1.4,17 kindly provided by Michael German, University of California at San Francisco) upstream of the enhanced green fluorescent protein (eGFP; BD Biosciences, Mountainview, CA). The amplified vectors contain the two-step transcriptional transactivation (TSTA) sequences, which includes a synthetic transactivator element composed of the yeast Gal4 DNA binding domain fused to two tandem copies of the Herpes Simplex Virus-1 (HSV-1) VP16 immediate early transactivator (Gal4VP2, 810 bp) and a minimal promoter element composed of five Gal4 DNA binding sites fused to the TATA box of the E4 gene of adenovirus (G5E4T, 240 bp). Gal4VP2 and G5E4T elements were removed from pBCVP2G5-L 10 (kindly provided by Lily Wu, University of California at Los Angeles)) and cloned into SIN lentiviral vectors. Cis amplified vectors contain either the PGK or insulin promoter driving Gal4VP2 and G5E4T sequences upstream of eGFP in one vector construct. Trans amplified vectors contain the PGK promoter upstream of Gal4VP2 in one vector construct and G5E4T upstream of eGFP in a separate vector construct.
Vector supernatant production and titers
Lentiviral vector supernatants were produced by triple transfection of HEK293T cells. To produce concentrated vector supernatant, 5 × 107 cells were seeded onto a 500-cm square plate, 1 day prior to transfection. Cells were transfected by DNA/calcium phosphate co-precipitation with 20 μg pMD.G (VSV-G), 100 μg pRΔ8.9 packaging plasmid 30, and 100 μg of vector plasmid. Forty-eight to seventy-two hours post-transfection, vector supernatants were harvested, clarified by passing through a 0.45 μm membrane, and crudely concentrated using Centricon plus 70 ultrafiltration filters (Millipore, Bredford, MA). Crude supernatants were ultracentrifuged at 500 × g for 2 h, and concentrated supernatants were stored at -70° C until used. Vector supernatants could be concentrated to between107 to109 TU/mL, as determined by vector copy analysis by quantitative polymerase chain reaction (qPCR, data not shown).
Comparison of titers was determined from unconcentrated vector supernatants, which were produced in small scale by calcium phosphate triple transfection of 1.8 × 106 HEK293T cells seeded in 35 mm dishes with 3.6 μg pRΔ8.9, 3.6 μg vector plasmid, and 1 μg pMD.G. Following exposure of sub-confluent HT-29 cells to 10-fold serial dilutions of unconcentrated vector supernatant for 6 days, genomic DNA was extracted and the vector copy number was determined by qPCR. The number of transducing units per milliliter (TU/mL) was calculated by multiplying the vector copy number, which was extrapolated from a standard curve of known vector copy number, the number of cells at the time of supernatant addition, and the supernatant dilution.
For the experiment to compare copy number titer to expression titer, small scale supernatants were produced as described above. Sub-confluent HT-29 cells were exposed to 10-fold serial dilutions of unconcentrated PGK or PGK-A vector supernatants for 6 days. Sub-confluent Beta-TC-6 cells were exposed to 10-fold serial dilutions of unconcentrated HIP, HIP-LA, or HIP-W vector supernatants for 6 days. Following exposure, the cells were collected and one-half was aliquoted for genomic DNA extraction and vector copy number analysis, as described above, and the second half was aliquoted for eGFP expression analysis by flow cytometry. The expression titer was calculated by multiplying the number of eGFP-positive cells, the number of cells at the time of supernatant addition, and the supernatant dilution.
Transduction of cell lines
To facilitate evaluation of expression in cells with a single copy of vector, aliquots of cells were transduced with a series of half-log serial dilutions of each supernatant. Cells were transduced by the addition of diluted supernatant in the presence of 8 μg/mL polybrene (Sigma, St. Louis, MO) to aliquots of 105 cells in a total volume of 1 ml. The proportion of cells expressing eGFP was determined by flow cytometry 5 to 7 days later with detailed expression analysis performed on cells transduced by the supernatant dilution that yielded less than 10% positive cells.
Human islet culture and transduction
Human pancreatic islets were received from the Islet Cell Resource Centers (ICR) Basic Science Islet Distribution Program and the National Disease Research Interchange (NDRI). Immediately after receipt, islets were dissociated into smaller cell clusters using Dispase (BD Biosciences, San José, CA) and were plated onto 96-well plates coated with HTB-9 matrix16 at approximately 200 islets per well. Following an overnight recovery period, cells were transduced with the selected vectors using a final vector concentration of 3×106 TU/mL in a total volume of 50 μL. Cells were maintained in RPMI-1640 containing 5.5 mM glucose 5 days before analysis.
Southern Blot Analysis
For each vector, 1 × 105 HT-29 cells in a 35 mm tissue culture dish were transduced and grown to confluency. The transduced cells were expanded until confluent in a 100 mm tissue culture dish, and genomic DNA were extracted using a standard phenol/chloroform procedure. Ten micrograms of genomic DNA was digested overnight with the appropriate restriction enzymes (single or double digestions with Afl II, Bgl II, EcoR V, Kpn I, Nar I; New England Biolabs, Ipswich, MA; Supplementary Figure 1) to release an intact proviral fragment that spanned 5’ of the internal promoter to 3’ of the eGFP gene. Digested DNA was separated on an agarose gel by electrophoresis, blotted onto Hybond-XL nylon membrane (Amersham, Buckinghamshire, UK), hybridized with 32P-labeled DNA probe spanning 741 bp of the eGFP gene, and exposed to Kodak X-Omat film.
Northern Blot Analysis
For bulk eGFP mRNA analysis in Beta-TC-6 cells, 1 × 105 cells were transduced with the appropriate vector and expanded to 2 × 106 cells. Pools of eGFP positive cells were isolated by fluorescence-activated cell sorting (FACS) using a FACSVantage cytometer (Becton Dickinson, San Diego, CA) using CellQuest software, and sorted cells were grown to confluency in a 100 mm tissue culture dish. For quantitative comparison of eGFP mRNA from HIP vectors, Beta-TC-6 cells were transduced with a series of half-log serial dilutions of each supernatant to obtain transduced pools with similar, low vector copy number. Transduced pools having similar numbers of eGFP positive cells were expanded to confluency in 100 mm tissue culture dishes. Total cellular RNA were isolated using an RNeasy Midi Kit (Qiagen Inc., Valencia, CA) according to manufacturer’s protocol. Five micrograms of total cellular RNA were separated on an agarose gel by electrophoresis, blotted onto Hybond-XL nylon membrane (Amersham, Buckinghamshire, UK), hybridized with 32P-labeled DNA probe spanning 741 bp of the eGFP gene, and exposed to Kodak X-Omat film. To validate equal RNA gel loading for each sample, blotted nylon membranes were stripped of radioactively labeled eGFP probes using 0.1 M NaOH, and re-probed with 32P-labeled human β-actin cDNA probe (Clontech, Mountain View, CA) for HT-29 RNA or 32P-labeled mouse β-actin DECAprobe (Ambion, Austin, TX) for Beta-TC-6 RNA.
Flow cytometric analysis and cell sorting
For flow cytometry analysis of eGFP expression in cell lines, cells were harvested using trypsin, resuspended in cell culture medium, washed and resuspended in phosphate buffered saline (PBS). For flow cytometry analysis of eGFP expression in primary human islets, islets were washed and resuspended in PBS. The samples were analyzed on a FACScan using CellQuest software (Becton Dickinson, San Diego, Calif.). Transduced cells for eGFP mRNA analysis were sorted using a FACSVantage cytometer (Becton Dickinson, San Diego, CA) based on eGFP expression, collected in the appropriate cell culture medium containing 30% FBS, and cultured as indicated above.
Vector copy number analysis in transduced cell lines
The vector copy number was determined by real-time PCR analysis. Genomic DNA were extracted using the DNeasy Tissue Kit (Qiagen Inc., Valencia, CA) according to the manufacturer’s protocols. Lentiviral vector sequences were amplified using a 200 nM concentration each of a sense primer (5’-ACCTGAAAGCGAAAGGGAAAC-3’) and an antisense primer (5’-CGCACCCATCTCTCTCCTTCT-3’), which spans 146 bp in the vector genomic packaging signal (Ψ). Each PCR amplification reaction was performed with 150-300 ng of genomic DNA in Taqman Universal PCR Master Mix (Applied Biosystems Inc., Foster City, CA). Amplified vector sequences were detected with 50 nM probe (5’–6FAMAGCTCTCTCGACGCAGGACTCGGCTAMRA–3’), which is homologous to vector sequences internal to the primers. Samples and standards were run in duplicate. Each PCR run included a standard curve of genomic DNA with known lentiviral vector copy numbers of 2, 0.2, 0.02, and 0.002 vector copies per cell. PCR amplification was performed in an Applied Biosystems GeneAmp 7700 sequence detection system instrument with denaturation at 95°C for 10 min and followed by 40 cycles of amplification, which included denaturation at 95°C for 15 s and annealing and extension at 60°C for 60 s. Analysis was performed using the Sequence Detection software (version 1.9.1).
Densitometry analysis
Radiographic bands produced on an X-ray film were converted to a photographic image at 600 dots per inch (dpi) resolution using a flatbed scanner. The densities of the bands of interest were calculated using Kodak 1D 3.6 software (Rochester, NY) and corrected for background by subtracting out a “mock” reading and normalized for loading by dividing by densities of β-actin bands in those lanes.
Immunohistochemistry
Cytospin slides were prepared from an aliquot of sample that was used for flow cytometry analysis. Cells were fixed with 4% Paraformaldehyde solution for 15 min., followed by three, 5 min. washes with phosphate buffered saline containing 0.05% Tween-20 (PBS-T; Sigma-Aldrich, St Louis, MO). The sample was “blocked” with PBS-T containing 10% (v/v) normal donkey serum (Jackson ImmunoResearch, West Grove, PA) and 0.1% Triton X-100 (Sigma-Aldrich, St Louis, MO). The slides were incubated overnight at 4C° in the presence of the following primary antibodies: 1:200 dilution of polyclonal guinea pig anti-insulin antibody (Dako, Carpinteria, CA) and 1:1000 dilution of polyclonal rabbit anti-green fluorescent protein antibody (Invitrogen, Carlsbad, CA). Following the overnight incubation, slides were washed three times with PBS-T and incubated for 45 min. with secondary antibody [1:250 dilution of Cy3-conjugated donkey anti-guinea pig (Jackson ImmunoResearch, West Grove, PA) and 1:500 dilution of Alexa-488-conjugated donkey anti-rabbit antibody (Invitrogen, Carlsbad, CA)]. The slides were washed several times with PBS-T and mounted using Vectashield™ mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Fluorescent images were obtained with a Leica DM RXA upright fluorescent microscope utilizing EasyFISH™ software (Applied Spectral Imaging, Vista, CA).
Acknowledgments
We thank Denise A. Carbonaro-Sarracino for technical assistance with Taqman assays, Ewa Zielinska and Lora Barsky for technical assistance with cell sorting, Karen A. Pepper for assistance with molecular assays, and Xingchao Wang for technical assistance with immunohistochemistry assays. We also thank the Cellular Image Core of Childrens Hospital Los Angeles. We acknowledge use of tissues procured by ICR Basic Science Islet Distribution Program and the NDRI with support from the National Institutes of Health (NIH) grant 5 U42 RR006042-17. Additional support was provided by awards from the NIH: 5F32 AI056894 (K.L.S.), 1R21 DK62649 (D.B.K.), 1RO1 DK68719 (G.M.C.), and from the Juvenile Diabetes Research Foundation: 17-2006-1137 (D.B.K.).
Footnotes
Supplementary information is available at Gene Therapy ’s website, http://www.nature.com/gt/index.html
References
- 1.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. Science. Vol. 302. New York, NY: 2003. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1; pp. 415–419. [DOI] [PubMed] [Google Scholar]
- 2.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. The New England journal of medicine. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 3.Leibowitz G, Beattie GM, Kafri T, Cirulli V, Lopez AD, Hayek A, et al. Gene transfer to human pancreatic endocrine cells using viral vectors. Diabetes. 1999;48:745–753. doi: 10.2337/diabetes.48.4.745. [DOI] [PubMed] [Google Scholar]
- 4.Gallichan WS, Kafri T, Krahl T, Verma IM, Sarvetnick N. Lentivirus-mediated transduction of islet grafts with interleukin 4 results in sustained gene expression and protection from insulitis. Human gene therapy. 1998;9:2717–2726. doi: 10.1089/hum.1998.9.18-2717. [DOI] [PubMed] [Google Scholar]
- 5.Ju Q, Edelstein D, Brendel MD, Brandhorst D, Brandhorst H, Bretzel RG, et al. Transduction of non-dividing adult human pancreatic beta cells by an integrating lentiviral vector. Diabetologia. 1998;41:736–739. doi: 10.1007/s001250050977. [DOI] [PubMed] [Google Scholar]
- 6.Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. Journal of virology. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. Journal of virology. 1998;72:9873–9880. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 9.Emami KH, Carey M. A synergistic increase in potency of a multimerized VP16 transcriptional activation domain. The EMBO journal. 1992;11:5005–5012. doi: 10.1002/j.1460-2075.1992.tb05607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L, Adams JY, Billick E, Ilagan R, Iyer M, Le K, et al. Molecular engineering of a two-step transcription amplification (TSTA) system for transgene delivery in prostate cancer. Mol Ther. 2002;5:223–232. doi: 10.1006/mthe.2002.0551. [DOI] [PubMed] [Google Scholar]
- 11.Sato M, Johnson M, Zhang L, Zhang B, Le K, Gambhir SS, et al. Optimization of adenoviral vectors to direct highly amplified prostate-specific expression for imaging and gene therapy. Mol Ther. 2003;8:726–737. doi: 10.1016/j.ymthe.2003.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iyer M, Salazar FB, Lewis X, Zhang L, Carey M, Wu L, et al. Noninvasive imaging of enhanced prostate-specific gene expression using a two-step transcriptional amplification-based lentivirus vector. Mol Ther. 2004;10:545–552. doi: 10.1016/j.ymthe.2004.06.118. [DOI] [PubMed] [Google Scholar]
- 13.Liu BH, Yang Y, Paton JF, Li F, Boulaire J, Kasparov S, et al. GAL4-NF-kappaB fusion protein augments transgene expression from neuronal promoters in the rat brain. Mol Ther. 2006;14:872–882. doi: 10.1016/j.ymthe.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 14.Liu B, Paton JF, Kasparov S. Viral vectors based on bidirectional cell-specific mammalian promoters and transcriptional amplification strategy for use in vitro and in vivo. BMC biotechnology. 2008;8:49. doi: 10.1186/1472-6750-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zufferey R, Donello JE, Trono D, Hope TJ. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. Journal of virology. 1999;73:2886–2892. doi: 10.1128/jvi.73.4.2886-2892.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beattie GM, Lappi DA, Baird A, Hayek A. Functional impact of attachment and purification in the short term culture of human pancreatic islets. J Clin Endocrinol Metab. 1991;73:93–98. doi: 10.1210/jcem-73-1-93. [DOI] [PubMed] [Google Scholar]
- 17.Odagiri H, Wang J, German MS. Function of the human insulin promoter in primary cultured islet cells. The Journal of biological chemistry. 1996;271:1909–1915. doi: 10.1074/jbc.271.4.1909. [DOI] [PubMed] [Google Scholar]
- 18.Segawa T, Takebayashi H, Kakehi Y, Yoshida O, Narumiya S, Kakizuka A. Prostate-specific amplification of expanded polyglutamine expression: a novel approach for cancer gene therapy. Cancer research. 1998;58:2282–2287. [PubMed] [Google Scholar]
- 19.Iyer M, Wu L, Carey M, Wang Y, Smallwood A, Gambhir SS. Two-step transcriptional amplification as a method for imaging reporter gene expression using weak promoters. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14595–14600. doi: 10.1073/pnas.251551098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiao J, Doubrovin M, Sauter BV, Huang Y, Guo ZS, Balatoni J, et al. Tumor-specific transcriptional targeting of suicide gene therapy. Gene therapy. 2002;9:168–175. doi: 10.1038/sj.gt.3301618. [DOI] [PubMed] [Google Scholar]
- 21.Koster RW, Fraser SE. Tracing transgene expression in living zebrafish embryos. Developmental biology. 2001;233:329–346. doi: 10.1006/dbio.2001.0242. [DOI] [PubMed] [Google Scholar]
- 22.Iyer M, Salazar FB, Lewis X, Zhang L, Wu L, Carey M, et al. Non-invasive imaging of a transgenic mouse model using a prostate-specific two-step transcriptional amplification strategy. Transgenic research. 2005;14:47–55. doi: 10.1007/s11248-004-2836-1. [DOI] [PubMed] [Google Scholar]
- 23.Kadesch T, Berg P. Effects of the position of the simian virus 40 enhancer on expression of multiple transcription units in a single plasmid. Molecular and cellular biology. 1986;6:2593–2601. doi: 10.1128/mcb.6.7.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villemure JF, Savard N, Belmaaza A. Promoter suppression in cultured mammalian cells can be blocked by the chicken beta-globin chromatin insulator 5’HS4 and matrix/scaffold attachment regions. Journal of molecular biology. 2001;312:963–974. doi: 10.1006/jmbi.2001.5015. [DOI] [PubMed] [Google Scholar]
- 25.Hasegawa K, Nakatsuji N. Insulators prevent transcriptional interference between two promoters in a double gene construct for transgenesis. FEBS letters. 2002;520:47–52. doi: 10.1016/s0014-5793(02)02761-8. [DOI] [PubMed] [Google Scholar]
- 26.Logan AC, Haas DL, Kafri T, Kohn DB. Integrated self-inactivating lentiviral vectors produce full-length genomic transcripts competent for encapsidation and integration. J Virol. 2004;78:8421–8436. doi: 10.1128/JVI.78.16.8421-8436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kraunus J, Schaumann DH, Meyer J, Modlich U, Fehse B, Brandenburg G, et al. Self-inactivating retroviral vectors with improved RNA processing. Gene therapy. 2004;11:1568–1578. doi: 10.1038/sj.gt.3302309. [DOI] [PubMed] [Google Scholar]
- 28.Higashimoto T, Urbinati F, Perumbeti A, Jiang G, Zarzuela A, Chang LJ, et al. The woodchuck hepatitis virus post-transcriptional regulatory element reduces readthrough transcription from retroviral vectors. Gene therapy. 2007;14:1298–1304. doi: 10.1038/sj.gt.3302979. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Rosol M, Ge S, Peterson D, McNamara G, Pollack H, et al. Dynamic tracking of human hematopoietic stem cell engraftment using in vivo bioluminescence imaging. Blood. 2003;102:3478–3482. doi: 10.1182/blood-2003-05-1432. [DOI] [PubMed] [Google Scholar]
- 30.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]