

1. Introduction
In 1956 Eleuterio invoked what is now known as an olefin metathesis reaction in order to explain how certain heterogeneous Mo catalysts could produce polyethylene from propylene; he proposed that propylene was converted into 2-butene and ethylene and that ethylene was then selectively polymerized.i An intense interest in the catalytic olefin metathesis reaction developed in the 1970's as a consequence of the ability to synthesize various polymers from cyclic olefins using tungsten, molybdenum, and rhenium catalysts (prepared using a variety of recipes) and as people became aware of potential applications of olefin metathesis in organic chemistry.
The accepted mechanism of the olefin metathesis reaction, the basic version of which is shown in equation 1, was proposed in 1971 by Hérrison and Chauvin (equation 2),ii but no
| (1) |
 |
(2) |
M=CHR complexes were known at that time that would react with olefins catalytically in a metathesis-like fashion. Therefore a search began for M=C complexes that would carry out the metathesis reaction rapidly and catalytically, could be isolated and studied, could be varied in great detail, and could tolerate various functional groups of interest in catalytic organic transformations.
Developments in organometallic chemistry that led to practical catalysts began with the Cp2Ti(Cl)(CH2AlMe2) chemistry discovered by Tebbe,iii followed by chemistry derived from Cp2Ti(Cl)(CH2AlMe2) by Grubbs,iv and tantalum chemistry, starting with the discovery of the remarkably thermally stable (Me3CCH2)3Ta=CHCMe3 (1),v which is believed to be formed through an intramolecular α-hydrogen abstraction in unstable Ta(CH2CMe3)5. Compound 1 is an example of how relatively sterically bulky covalently bound ligands can stabilize pseudotetrahedral and electron-deficient species (10 electrons in this case) toward ligand redistribution or alkylidene coupling. The 18 electron methylene complex, Cp2Ta(CH2)Me (2), is stable enough at room temperature to isolate and characterize, although it decomposes slowly in solution in a bimolecular fashion to yield an ethylene complex.vi The principle of employing bulky covalently bound ligands in order to stabilize electron deficient pseudotetrahedral species against bimolecular decompositions, and the synthesis of neopentylidene (or neophylidene, CHCMe2Ph) complexes in particular as the initial alkylidenes, continued to be a central feature during the development of high oxidation state alkylidene chemistry and alkene metathesis catalysts.vii
Various tantalum alkylidenes were found to react with ethylene, but gave only products of rearrangement of an intermediate tantalacyclobutane complex, not metathesis products.viii A key observation was that although Ta(CH-t-Bu)Cl3(PMe3)2 reacted with olefins to yield products of rearrangement of unobservable intermediate tantalacyclobutane complexes, Ta(CH-t-Bu)Cl(PMe3)(O-t-Bu)2 led to productive metathesis of cis-2-pentene.ix This was the first time that facile productive metathesis of a simple olefin starting with a high oxidation state alkylidene complex had been observed, and the first indication that alkoxides might be desirable ligands in metathesis catalysts.x Bimolecular coupling of alkylidenes and rearrangement of a metallacyclobutane to an olefin are still believed to be the major modes of decomposition of high oxidation state alkylidene complexes.
Over a period of years what was hinted at in Ta chemistry was realized in high oxidation state Mo, W, and Re chemistry. That process and the many alkylidenes that have been prepared have been described in previous reviews.xi (The reader should consult these reviews for details that are not covered here.) The most practical and versatile of these catalysts so far have been W and Mo imido alkylidene bisalkoxide catalysts, M(NR)(CHR’)(OR”)2. In this review I summarize some of the important characteristics of high oxidation state catalysts of Mo and W for the metathesis of olefins, but will concentrate on relatively recent findings in the last few years that have not been covered in other review articles.xi I will focus on organometallic chemistry rather than applications of metathesis reactions.
Catalysts based on ruthenium were developed by Grubbs in the mid 1990's.xii Since Ru catalysts are less sensitive to air and to functionalities than Mo or W catalysts, they became the catalysts of choice for many organic chemists in the laboratory. While Ru and Mo (or W) catalysts have much in common in terms of the details of metathesis chemistry, there are some significant differences in reactivities, efficiencies, and decomposition pathways. Loss of a donor ligand from a 16e Ru complex usually is necessary in order to generate the relatively reactive four-coordinate 14e core, whereas no ligand must be lost from the four-coordinate 14e core of Mo and W catalysts.
It is increasingly unlikely that a small number of metathesis catalysts will carry out a wide variety of metathesis reactions. Therefore the ability to prepare hundreds of catalyst variations and to take advantage of detailed mechanistic knowledge of catalyst activity will be the key to the success of optimally tuned catalysts in the long run. Molybdenum and tungsten alkylidene chemistry has progressed to the point where thousands of highly active and tunable catalysts are now available.
2. Principles of tungsten and molybdenum imido alkylidene chemistry
Formation of the 18e compound shown in equation 3,xiii and its activity as a catalyst for the metathesis of terminal and internal olefins in the presence of a trace of AlCl3, led to a search
 |
(3) |
for four-coordinate 14e imido alkylidene complexes. The hope was that a bulky imido group would be much less likely to allow bimolecular decomposition reactions than an oxo ligand for simple steric reasons.
The N-2,6-i-Pr2C6H3 (NAr) ligand was chosen in order to maximize steric bulk close to the metal without increasing the chance of intramolecular CH activation (e.g., in an ortho t-butyl group). The triethylamine-catalyzed conversion of W(C-t-Bu)(NHAr)Cl2(dme) into 4xiv (equation 4) illustrated just how mobile α protons can be in certain transformations in which the total
 |
(4) |
number of metal-ligand π bonds does not change between the starting material and the product. Addition of bulky alkoxide ligands to 4 (OR = OCMe3, OCMe(CF3)2, O-2,6-i-Pr2C6H3, etc.) produced 14 electron (assuming formation of a W=N triple bond) W(NAr)(CH-t-Bu)(OR)2 species (5), adducts thereof (e.g., with THF or dimethyoxyethane), or “ate” complexes (anionic trialkoxide species) if the alkoxide is relatively small.
Compounds of type 5 proved to be relatively active catalysts for the metathesis of ordinary internal olefins (e.g., cis-2-pentene). New alkylidene complexes (e.g., W(N-2,6-i-Pr2C6H3)(CHPh)[OCMe(CF3)2]2) could be isolatedxv and two types of tungstacyclobutane intermediates could be observed under a variety of circumstances, either trigonal bipyramidal (e.g., 6a) or square pyramidal (e.g., 6b) geometries. The olefin is believed to approach
 |
one of the two CNO faces of pseudotetrahedral 5 and bind weakly to the metal before or as the metallacyclobutane ring forms. All five-coordinate metallacycles are believed to be relatively fluxional. In any system in which ethylene is generated, unsubstituted metallacyclobutanes can form. Any metallacycle that has a β proton may decompose through rearrangement of the metallacycle to an olefin.
Attention shifted to molybdenumxvi in the belief that molybdenum would be more tolerant of functionalities than tungsten. Molybdacyclobutane species are rarely observed, either because they lose an olefin more readily than does a tungstacyclobutane species, or because they rearrange to olefins more readily than tungstacyclobutanes. The interplay between metallacyclobutane rearrangement, loss of olefin from a metallacyclobutane (productive metathesis), and bimolecular decomposition of alkylidenes are strong determinants of the overall efficiency of a metathesis catalyst. Elucidation of modes of decomposition of alkylidenes other than metallacyclobutane rearrangement and bimolecular decomposition has been rare. When the imido ligand, alkoxides, and alkylidene are relatively small, intermolecular ligand scrambling (disproportionations) could open up a variety of other decomposition pathways.
A relatively general three step synthesis of Mo(NAr)(CHCMe2R)(OTf)2(dme) (7; dme = 1,2-dimethoxyethane)xvii set the stage for rapid progress. A variety of imido groups (R’ = 2,6-i-Pr2C6H3, 2,6-Me2C6H3, 2,6-Cl2C6H3, 2-t-BuC6H4, 2-(CF3)C6H4, 1-adamantyl, and others) can be installed in the first two facile synthetic steps and therefore a variety of catalyst precursors of type 7 can be prepared.xviii Compound 7 and analogous species that contain different imido groups react readily with a variety of nucleophiles, even LiOCMe(CF3)2. Many catalysts that contain C2-symmetric chiral biphenolates or binaphtholates have been prepared and have been highly successful for asymmetric metathesis reactions of interest in organic chemistry.xid In general biphenolates or binaphtholates must contain substituents in the 3 and 3' positions in order to stabilize imido alkylidene complexes against bimolecular decompositions and in order to maximize the efficiency of an asymmetric metathesis reaction.
The use of steric bulk to prevent bimolecular decompositions of intermediates in metathesis reactions has limits. For example, it has been shown that ligands can be so sterically demanding that a 14e species simply does not react readily even with ethylene. A rare example of this type is Mo(NAdamantyl)(CHCMe2Ph)(Silox)2 (Silox = OSi(t-Bu)3), which is unchanged after being heated in toluene-d8 under five atm of ethylene at 120 °C for several hours, in contrast to facile reactions of many other siloxide, aryloxide, or alkoxide derivatives.xix However, Mo(NAd)(CHCMe2Ph)(pyrrolide)(Silox) (vide infra) does react with ethylene and terminal olefins. The potential of a reaction of this type will be covered in section 8.
Some Mo(NAr)(CH-t-Bu)X2 species in which X is not an alkoxide appear to be relatively unreactive towards olefins and acetylenes. Examples are Mo(NAr)(CH-t-Bu)(neopentyl)2,xx Mo(NAr)(CH-t-Bu)(NPh2)2,xxi Mo(NAr)(CHCMe2R')(pyrrolide)2 speciesxxii (vide infra), Mo(NAr)(CHCMe2Ph)(η1-C5H5)(η5-C5H5),xxiii and Mo(NR)(CH-t-Bu)(R'CO2)2 species (in which the carboxylates are bidentate).xxiv In discussing reactivity the substrate must also be considered. For example, Mo(NAd)(CH-t-Bu)(Ph3CCO2)2 is an 18e species that does not react readily with ethylene or norbornene. Nevertheless, it will slowly catalyze the cyclopolymerization of diethyldipropargylmalonate; evidently only a terminal acetylene is reactive enough to capture an intermediate with less than 18e, e.g., one in which one of the bidentate carboxylates is monodentate.
An important feature of all Mo and W imido alkylidene chemistry is the formation of syn alkylidene or anti alkylidene isomers, the rate of interconversion of which and relative reactivities of which are of fundamental importance in metathesis reactions.xxv (The syn alkylidene substituent points toward the imido group, the anti substituent points away.) Since syn and anti isomers can be formed as a consequence of metathesis, precise details of the mechanism of a given reaction are often elusive. In many cases the presence of syn and anti isomers and interconversion of them either thorough rotation about the Mo=C bond or through the metathesis process itself is beneficial overall as it adds a new layer of flexibility and therefore potentially further control of any catalyst system.
Monoadducts of imido alkylidene complexes (e.g., with trimethylphosphine, pyridine, or quinuclidine), either in solution or in the solid state, usually have a structure in which the donor has added to the metal from a direction perpendicular to the C=M=N plane to yield a trigonal bipyramidal species in which the donor is located in the axial position (equation 5). It is proposed that an olefin binds to the metal weakly in the same position and quickly combines with the alkylidene to form a metallacyclobutane species. Interconversion of syn and anti
 |
(5) |
isomers does not appear to be facile in 16e adducts, i.e., the base must first be lost to yield the parent 14e species. All evidence so far suggests that 16e five-coordinate adducts that do not lose a donor ligand are inactive for olefin metathesis. More reactive anti isomers bind 2e donors more strongly than syn isomers. Other types of adducts have been observed in solution, but not all types have been isolated. It seems least likely that an olefin can attack trans to the imido ligand, since the alkylidene would have to rotate by 90° in one direction or the other in order to form the metallacyclobutane ring. Ligand dissociation and reassociation is a potentially complicating feature of reactions that involve four-coordinate alkylidene complexes. However, 2e donors also can be employed in a beneficial manner, e.g., to stabilize otherwise unstable 14e species.xxvi,xxvii,xxviii Finally, binding of some functionality in a substrate to an intermediate alkylidene, especially in an intramolecular fashion, can slow or even halt metathesis turnover.
DFT(B3PW91) calculations have been carried out in order to rationalize the structural, electronic, and spectroscopic properties of Mo and W imido M(NR)(CHR')(X)(Y) olefin metathesis catalysts.xxix The calculated structures, energetics (syn and anti isomers), and spectroscopic properties (JCH coupling constants) are in good agreement with the available experimental data. These quasi-tetrahedral complexes have a linear imido group and a CHα agostic interaction, which stabilizes the syn isomer. The triply bonded imido ligand is a weaker electron donor than the alkylidyne in a Re(CR)(CHR)(X)(Y) analog of a Mo or W imido alkylidene system.xxx Therefore, the CHα agostic interaction in the syn species is stronger in Mo and W imido complexes compared to Re alkylidyne complexes and the weaker imido π bonds allow the alkylidene to rotate more readily.
There are several aspects of metathesis reactions worth pointing out in which ethylene is either generated or employed as one of the substrates. One is that intermediate methylene species are the most reactive alkylidene complexes, do not have syn and anti isomers, and are the most likely to decompose bimolecularly. Recent studies suggest that bimolecular decomposition of methylenes can be avoided when catalysts are prepared on silica surfaces (see section 7), although metallacyclobutanes on silica surfaces can still rearrange to olefins. It is important to note that recent evidence suggests that metallacyclobutane rearrangement is accelerated by ethylene in some circumstances.xxxi,xxxii (See also below.)
The arylimido or alkoxide ligand is an attractive means of attaching some functionality in a relatively remote manner. For example, polymerizeable groups within biphenolate ligands have created the opportunity to incorporate catalysts into organic supports.xxxiii,xxxiv The arylimido group has also been employed as a means of attaching a catalyst to a polymer support.xxxv Applications employing these methods have included surface modificationxxxvi and the synthesis of stationary phases for chromatography.xxxvii Imido alkylidenes have been synthesized in which the alkylidene is tethered to the imido group through the ortho position,xxxviiia with the sterically more
 |
(6) |
protected imido ligand shown in equation 6 allowing more stable catalysts to be formed.xxxviiib The species shown in equation 6 is stable only as a quinuclidine adduct, whereas Mo(NAr)(CHCMe2R)(ORF6)2 and Mo(NAr')(CHCMe2R)(ORF6)2 (Ar’ = 2,6-Me2C6H3) are stable in the absence of a 2e donor. The potential benefits of intramolecular tethering remain to be explored.
It can be desirable to change a neopentylidene or neophylidene species to one that is still relatively stable, but more reactive. Alkylidenes can be switched through a reaction such as that shown in equation 7 (E = CO2Et). This approach has also been employed for synthesizing bimetallic species, which are useful initiators for the preparation of triblock copolymers.xxxix,xl
 |
(7) |
 |
(8) |
An example is shown in equation 8. The ability to switch alkylidenes is a sensitive function of the alkoxide, with hexafluoro-t-butoxides having been the most useful so far. In order to encourage formation of the new alkylidene the monomer that is employed also must not be homometathesized readily and reaction conditions must be employed that minimize any significant back reaction between the desired alkylidene species and the initial metathesis product.
In the last several years many ROMP catalysts have been developed for the synthesis of advanced materials,xli among them catalysts for regioselectively polymerizing 1-alkynes and cyclopolymerizing 1,6-heptadiynes to give polyenes that contain largely five- or six-membered rings.xlii Biscarboxylate complexes, both of Moxxiv and Ru,xliii have proven especially useful for cyclopolymerizing 1,6-heptadiynes. Thick, optical quality films of substituted polyacetylenes have been prepared with W(NAr)(CH-t-Bu)(ORF6)2 catalysts in the presence of THF or hexafluoro-t-butanol. These have large, ultrafast χ(3) nonlinearities and applications to image correlation.xliv
3. Precursors to bisalkoxides and monoalkoxides
The availability of a wide variety of imido alkylidene variations and the necessity of searching for the desirable variation (e.g., for asymmetric reactionsxid) suggests that a large number of potential catalysts would have to be prepared, purified, and stored. Therefore, we explored the possibility of employing Mo(NR)(CHR')X2 species (prepared from Mo(NR)(CHR')(triflate)2(dme)) that would yield Mo(NR)(CHR’)(OR”)2 variations in situ through addition of two equivalents of ROH or one equivalent of diol. The byproduct (HX) formed upon addition of ROH to Mo(NR)(CHR')X2 should not block activity of the resulting bisalkoxide complex.
Addition of ROH to Mo(NAr)(CH-t-Bu)(neopentyl)2xlv was found to lead often to monoalkoxide complexes, Mo(NAr)(CH-t-Bu)(CH2-t-Bu)(OR), instead of bisalkoxides, consistent with the reaction shown in equation 9. It was recognized that Mo(NAr)(CH-t-Bu)(CH2-t-Bu)(OR) is chiral at the metal by virtue of having four different groups bound to the
 |
(9) |
metal and therefore of potential interest in terms of asymmetric metathesis reactions (vide infra). It was proposed that the second equivalent of ROH did not react readily with the second neopentyl group because ROH adds trans to the neopentyl group (equation 10) and therefore leads only to degenerate alkoxide exchange through 1,3 proton migration. Although dineopentyl (or dineophyl) species are the most likely to be stable enough to be employed as catalyst precursors, dibenzyl species that have been prepared recently are also potentially useful.xxiii In any monoalkyl monoalkoxide species there is some concern that the alkyl ligand could be involved in α proton migration or CH activation or olefin insertion reactions at some stage during a metathesis reaction.
 |
(10) |
Bisamido species such as Mo(NAr)(CH-t-Bu)(NPh2)2 were explored as precursors to bisalkoxides since the diphenylamido groups should be readily protonated and since diphenylamine should not interfere to a significant degree with subsequent metathesis reactions.xxi Unfortunately, bisamido species in general were found to be relatively difficult to isolate in pure form. They also were found to be relatively resistant to protonation in certain circumstances. For example, Mo(NAr)(CH-t-Bu)(NPh2)2 did not react readily with many of the more sterically demanding biphenols and binaphthols. Slow protonolysis of Mo-NR2 bonds can be attributed to steric problems that prevent formation of an ROH adduct in which H is relatively easily transferred to another ligand, but also to the fact that all π electron pairs on nitrogens can be donated to the metal. Therefore, Mo(NAr)(CH-t-Bu)(NR2)2 species could be said to have 18 electron counts. Complexes that contain ligands such as [N(t-Bu)(3,5-(CF3)2C6H3)]− were at least as difficult to prepare and isolate as Mo(NAr)(CH-t-Bu)(NPh2)2 species and were even less reactive as precursors to bisalkoxide species.
We then turned to the pyrrolide ligand, C4H4N−, and substituted versions thereof. A variety of pyrrolide complexes have been known in the literature for decades, but almost none that contain W or Mo.xlvi Initial interest in pyrrolides in transition metal chemistry in general probably stemmed from the fact that C4H4N− is isoelectronic with C5H5− and therefore can bind to a metal in an η5 fashion. However, a pyrrolide ligand also can bind in an η1 fashion, and probably much more readily than a cyclopentadienyl ligand can bind in an η1 fashion. Therefore η1 pyrrolide complexes that contain <18 electrons and interconversion of η1 and η5 pyrrolide ligands might be anticipated. One might also suspect that a η1 pyrrolide, in contrast to ordinary amido ligands, to be a relatively poor π electron donor as a consequence of the lone pair on the nitrogen being part of a 6π aromatic system. Since pyrroles bind to early metals in relatively low oxidation states in an η2 fashion at one of the C-C bonds,xlvii pyrroles are unlikely to block subsequent metathesis reactions by coordinating to the metal strongly in high oxidation state imido alkylidene complexes.
A variety of bispyrrolide complexes can be prepared readily from 7 in relatively high yields, in contrast to bisamido complexes. The structure of one bispyrrolidexxii (where the pyrrolide is the parent pyrrolide, Pyr) has been shown to be a dimer, {Mo(NAr)(syn-CHCMe2Ph)(η5-Pyr)(η1-Pyr)}{Mo(NAr)(syn-CHCMe2Ph)(η1-Pyr)2}. This species is highly fluxional in solution as a consequence of breaking up readily to give monomeric, 18e Mo(NAr)(syn-CHCMe2Ph)(η5-Pyr)(η1-Pyr) and 14e Mo(NAr)(syn-CHCMe2Ph)(η1-Pyr)2, which interconvert rapidly with each other. On the other hand, bis-2,5-dimethylpyrrolides, Mo(NR)(CHCMe2Ph)(η1-Me2Pyr)(η5-Me2Pyr) (R = Ar, Ad, Ar', 2-CF3C6H4, and 2,6-Cl2C6H3), are highly fluxional monomers, as confirmed through a solid state X-ray structure in which R = 1-Adamantyl.xlviii Mo(NAr)(CHCMe2Ph)(η1-2,5-Ph2NC4H2)(η5-2,5-Ph2NC4H2), Mo(NAr)(CHCMe2Ph)(η1-2,3,4,5-NC4Me4)(η5-2,3,4,5-NC4Me4), and Mo(NAr)(CHCMe2Ph)(η1-2,5-i-Pr2NC4H2)(η5-2,5-i-Pr2NC4H2) have also been prepared and characterized crystallographically.xlviii Some pyrrolides are prone for steric reasons to bind in an η1 fashion, as found in Mo(NAr)(CHCMe2Ph)(η1-2-MesitylNC4H3)2.xlix Mo(NAr)(CHCMe2Ph)(indolide)2 has been shown to be a 14 electron species, Mo(NAr)(CHCMe2Ph)(η1-indolide)2.xlviii In all bispyrrolide complexes observed so far the alkylidene has been found to be in the syn confirmation in the solid state and in solution. Bispyrrolide complexes that contain alkylidenes that are linked (e.g., as shown in equation 8) have also been prepared recently.l
Several tungsten imido alkylidene bispyrrolide complexes have been prepared, namely W(NR)(CHCMe2Ph)(η1-Pyr)2(dme) (R = Ar or ArCl, where ArCl = 2,6-Cl2C6H3) and W(NR)(CHCMe2Ph)(η1-Me2Pyr)(η5-Me2Pyr) (R = Ar or ArCl) and characterized crystallographically.li Although bispyrrolides in general do not react readily with olefins, methylene species, W(NR)(CH2)(η1-Me2Pyr)(η5-Me2Pyr) (R = Ar or ArCl), could be prepared in good yield in a reaction between the neopentylidene species and ethylene at 60 °C (e.g., as shown in equation 11). This result is unusual since methylene species usually cannot be isolated after a reaction of a neopentylidene or neophylidene complex with ethylene, especially at 60 °C. There is a possibility that formation of an 18e η1,η5-bispyrrolide complex slows bimolecular decomposition of the methylene species. The bispyrrolide methylene species creates the possibility of accessing mono- or bisalkoxide methylene species through addition of alcohols (vide infra), and evaluating their stabilities and reactivities in the absence of ethylene. In contrast, Mo(NAr)(CHCMe2Ph)(η1-Me2Pyr)(η5-Me2Pyr) reacts with ethylene only extremely slowly and only the ethylene complex, Mo(NAr)(CH2CH2)(η1-Me2Pyr)(η5-Me2Pyr), has been observed.lii
 |
(11) |
Bispyrrolides react with a variety of monoalcohols and diols, often relatively cleanly. Biphenolate or binaphtholate species are of special interest since they have been employed for a wide variety of asymmetric metathesis reactions. It has been shown that a representative number of asymmetric Mo catalysts can be prepared in situ, and that they deliver results that are virtually identical to those found for isolated catalysts.liii These and related results provide good evidence that the pyrrole formed upon protonolysis of the pyrrolide does not interfere with subsequent metathesis reactions. Interestingly, it has also been shown that catalysts can be prepared from bispyrrolide precursors that cannot be prepared via the bistriflate route. One example is Mo(N-2,6-Br2-4-MeC6H2)(CHCMe3)[Biphen] (where [Biphen]2− = 3,3'-di-t-butyl-5,5',6,6'-tetramethyl-1,1'-biphenyl-2,2'-diolate).xxii Previous attempts to prepare this species through addition of K2[Biphen] to Mo(N-2,6-Br2-4-MeC6H2)(CHCMe3)(OTf)2(DME) failed to produce the desired species in pure form and in a practical yield.liv Unsubstituted pyrrolide complexes are the most reactive toward alcohols; complexes that contain substituted pyrrolides react more slowly. Precise details of the protonolyses have not yet been elucidated. An η5-pyrrolide could be protonated directly at the nitrogen if the alcohol has a sufficiently low pKa, although formation of an initial ROH adduct is probably required followed by proton migration. Proton migration is likely to result in formation of a pyrrolenine complex,liii,lv since a pyrrolenine complex has been isolated as a product of the reaction between an imido alkylidene bispyrrolide and a relatively strong acid (equation 12; ArF = 3,5-(CF3)2C6H3).li If pyrrolenine dissociates or is displaced from the metal, it would rearrange rapidly to the pyrrole.lvi
 |
(12) |
Mo bispyrrolides have been shown to be useful for synthesizing rare cationic Mo alkylidene complexes.xlix For example, addition of one equivalent of [HNMe2Ph]BArF4 to Mo(NAr)(CHCMe2Ph)(Pyr)2 in THF produced the cationic pyrrolide species shown in equation 13). Other species that were prepared include [Mo(NAr)(CHCMe2Ph)(Me2Pyr)(THF)2]BArF4 and [Mo(NAr)(CHCMe2Ph)(Me2Pyr)(2,4-lutidine)]BArF4. The THF complexes served as precursors to cationic species such as {Mo(NAr)(CHCMe2Ph)[OC(CF3)2Me](THF)3}BArF4, [Mo(NAr)(CHCMe2Ph)(O-2,6-i-Pr2C6H3)(THF)]BArF4, and [Mo(NAr)(CHCMe2Ph)(OAdamantyl)(THF)2]BArF4. Some of the cationic species react readily with olefins, but the resulting alkylidenes do not seem to be stable and long-lived in typical metathesis reactions that were explored.
 |
(13) |
Monoalkoxide monopyrrolide species are formed and can be isolated in some circumstances upon addition of a monoalcohol to bispyrrolide complexes.lvii If a bisalkoxide species also forms, the MonoAlkoxide Pyrrolide (MAP) species often can be separated from the bisalkoxide readily through selective crystallization. Some examples are shown in equation 14. It was shown in an X-ray study of Mo(NAr)(CHCMe2Ph)(OAr)(Me2Pyr) that the
 |
(14) |
dimethylpyrrolide is bound in an η1 fashion. An η5-Me2Pyr ligand appears to be untenable in this circumstance for steric reasons. The electron deficiency of the metal also may be attenuated through π bonding from the alkoxide ligand. An η5-Me2Pyr ligand might be tenable if smaller more electron withdrawing alkoxides or smaller alkylidenes or smaller imido ligands are present, but formation of Mo(NR)(CHR’)(η1-Me2Pyr)(OR”) would allow facile reaction with an olefin. So far the only MAP species that undergoes any intermolecular ligand scrambling (e.g., to yield bispyrrolide and bisalkoxide species) is the one where OR = OC6F5.
4. Studies concerning monoalkoxide pyrrolide (MAP) species
There are several attractive features of species analogous to those shown in equation 14 that are potentially interesting as far as metathesis reactions are concerned. The first is that their reactivity towards olefins is not intermediate between bispyrrolides (minimal reactivity) and bisalkoxides (often highly active), but often much greater than even bisalkoxides. The second is that the number of variations of MAP species is large, i.e., thousands of catalysts should be accessible, often in situ, through variation of all four ligands plus the metal (Mo or W). Third, the metal is a stereogenic center as a consequence of four different ligands being covalently attached to it. (Covalently bound ligands eliminate the possibility that the metal can invert through dissociation of a donor ligand to give a planar three-coordinate species.) Therefore of the two possible approaches of a substrate perpendicular to the N=Mo=C plane one would be trans to the pyrrolide while the other would be trans to the alkoxide. The approach that is preferred therefore may be determined for a combination of electronic and steric reasons. Fourth, if the alkoxide is enantiomerically pure then diastereomers should be formed that could have different reactivities.lvii Fifth, preliminary studies have shown that remarkably active and long-lived MAP catalysts can be prepared on dehydroxylated silica surfaces (vide infra); in this case OR is a surface siloxide. Sixth, no structure at any point is restricted because of two or more ligands being connected to one another. Finally, theoretical studies suggest that Mo(NR)(CHR')(X)(Y) catalysts in which X and Y are quite different types of ligands, in particular a “donor” (e.g., an alkyl) and an “acceptor” (e.g., an alkoxide), in fact are predicted to be more efficient than bisalkoxide species.lviii (In MAP species the alkoxide might be considered to be a poorer donor than a pyrrolide, but whether the “donor” and “acceptor” labels actually could be applied to a pyrrolide and an alkoxide, respectively, remains to be seen.)
A study of the reactivity of MAP complexes involved (inter alia) desymmetrization of 8 to 9 in the context of a synthesis of the adrenergic blocker quebrachamine (equation 15).lix,lx It was possible to convert 8 to (racemic) 9 with Mo(NAr)(CHCMe2Ph)(ORF6)2, but the
 |
(15) |
conversion was not practical (30 mol% Mo, 59% yield, C6H6, 22 °C, 2 h), and of course not asymmetric. Interestingly, Mo(NAr)(CHCMe2Ph)(Me2Pyr)(ORF6) catalyzed this transformation with an efficiency ∼30× greater than Mo(NAr)(CHCMe2Ph)(ORF6)2 (1 mol% Mo, 79% yield, C6H6, 22 °C, 1 h). However, no asymmetric Mo-based catalyst that has been employed in past asymmetric reactions,xid nor any known Ru-based asymmetric catalyst, carried out this transformation (<5% yield with up to 50 mol% catalyst).
Addition of bulky enantiomerically pure alcohols to Mo(NAr)(CHCMe2Ph)(Me2Pyr)2 led to diastereomers, e.g., as in equation 16 for 10. Both diastereomers could be isolated. X-ray
 |
(16) |
studies showed the major one to have the SMo configurationlix (Figure 1), while the minor diastereomer was shown to have the RMo configurationlxi (Figure 2). In (SMo)-10 the space trans to the pyrrolide is occupied by one of the bromides in a 3' position, while in (RMo)-10 that space is occupied by the pendant half of the phenoxide (OR*) ligand. An X-ray structure of a PMe3 adduct of (RMo)-10 showed it to be closest to a SP with the PMe3 trans to the pyrrolide (Figure 3).lxii The Mo-P1 distance (2.5703(11) Å) is relatively long and the Mo-P bond likely to be relatively weak, as found experimentally in solution, i.e., PMe3 dissociates readily to yield an equilibrium of (RMo)-10(PMe3), (RMo)-10, and free PMe3. PMe3 then catalyzes the inversion of configuration at Mo through formation and rearrangement of a five-coordinate adduct and an equilibrium between (SMo)-10/(RMo)-10 = 2.0 is established. Finally, an unsubstituted tungstenacyclobutane species was shown to be a TBP with axial imido and phenoxide ligands.lxiii The distances and angles in this TBP tungstacyclobutane are indistinguishable from those found in tungstacyclobutane rings in bisalkoxide complexes. These and other data support the proposition that an olefin substrate is likely to approach trans to the pyrrolide and to invert the configuration at the metal as a consequence of metathesis. Inversion is likely to be fastest with ethylene, the most reactive olefin.
Figure 1.
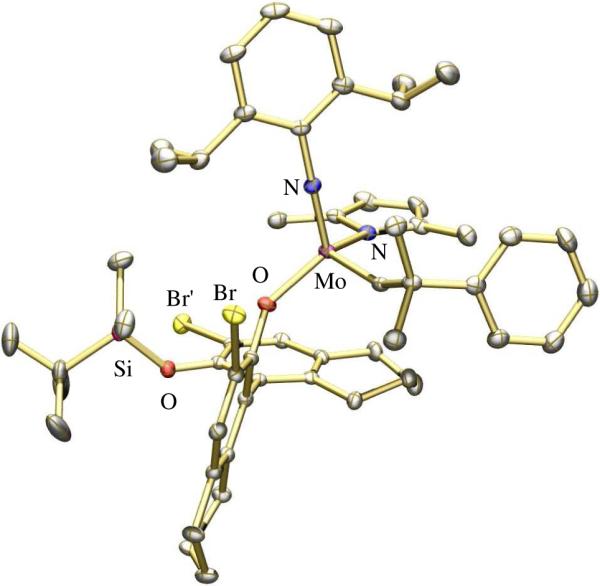
The X-ray structure of (SMo)-10.
Figure 2.
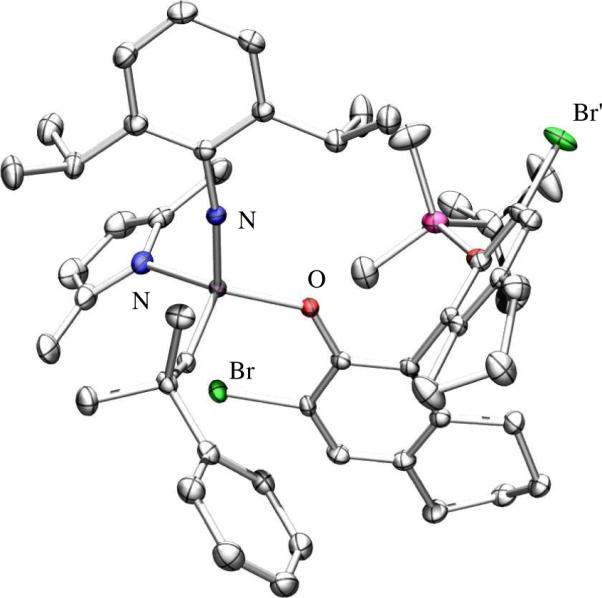
The X-ray structure of (RMo)-10.
Figure 3.
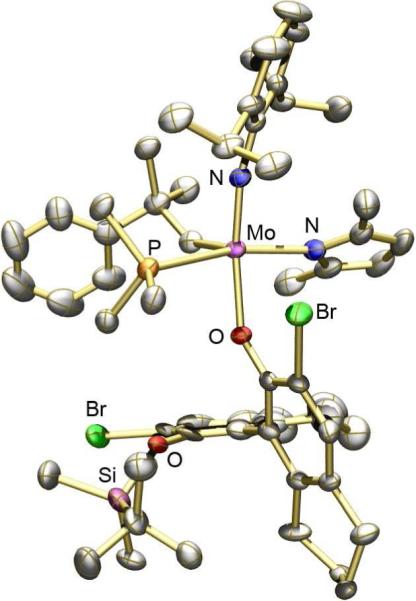
Structure of (RMo)-10(PMe3).
Pure (SMo)-10, as well as the 7:1 diastereomeric mixture, was shown to catalyze the conversion shown in equation 16 rapidly and with high %ee. Catalyst 11 (equation 17) was
 |
(17) |
shown to be slightly more efficient than 10. A variety of other ring-closing reactions build a strong case for the relatively high reactivity of 10, e.g., the conversion of 12 to 13 (equation 18). The optimum previous catalyst for this transformation was Mo(NAr')(CHCMe2Ph)(Biphen) (Ar’ = 2,6-Me2C6H3), which produced 13 in 30 %ee with 15 mol% Mo in 24 h (75% conversion), i.e., catalyst 10 is approximately two orders of magnitude faster than Mo(NAr')(CHCMe2Ph)(Biphen) in the reaction in equation 18 and produces a far superior %ee.
 |
(18) |
The successful use of MAP catalysts (generated in situ as diastereomeric mixtures) for carrying out difficult asymmetric metathesis transformations with high %ee touches on several fundamental issues in asymmetric catalysis in general and could redefine the approach to asymmetric olefin metathesis reactions. The most important finding is that the metal center is stereogenic as a result of the arrangement of four different ligands in a tetrahedral arrangement and that the configuration at the metal is exploited in asymmetric metathesis reactions. The MAP species contrast dramatically with related 18e (usually monocyclopentadienyl) complexes prepared by H. Brunnerlxiv and virtually all other potential catalysts that are chiral at the metal center by virtue of the ligand arrangement alone.lxv MAP species appear to be the only examples of <18e pseudotetrahedral stereogenic-at-metal species that are efficient catalysts for a metal-based reaction.lxiv,lxv Brunner stated in his 1999 reviewlxiv that “active use of chirality at the metal atom in enantioselective catalysts remains a challenge for the future.” It should be noted that catalysts 10 and 11 are relatively highly optimized for the substrate in question. Variations in any ligand (e.g., adamantyl instead of diisopropylphenylimido, etc.) can lead to dramatically different results for a given substrate. Many further detailed studies clearly will be required.
Recent studieslxi suggest that only (SMo)-10 will initiate ring-closing of sterically demanding substrates such as that shown in equation 18. (RMo)-10 relatively ineffective until a mixture of (SMo)-10 and (RMo)-10 is established through reactions with of (RMo)-10 with relatively small olefins, especially ethylene. Preliminary studies confirm that (SMo)-10 or (RMo)-10 react instantly with ethylene to yield mixtures of (SMo)-10, (RMo)-10, both diastereomers of their methylene analogs, and an unsubstituted metallacyclobutane species that all interconvert (along with ethylene) at rates of the order of the NMR time scale (∼100 s−1).lxii Therefore the configuration at the metal inverts readily as a consequence of a single olefin metathesis step. The alkylidene would not be changed if the metathesis reaction were degenerate. It is relatively easy to imagine how the configuration at the metal can invert. An example is shown in equation 19. Ethylene attacks the alkylidene/imido/alkoxide (CNO) face of a (R) species trans to the pyrrolide and the RCH=CH2 olefin product leaves the intermediate metallacyclobutane species trans to the pyrrolide to give an (S) species having a new alkylidene species with the opposite configuration at the metal center. (Square pyramidal intermediates with the imido group in an apical position, which have been observed and crystallographically characterized for bisalkoxides,lxvi and which are considered to be intermediates by Eisenstein,lviii are not shown in equation 19, but the conclusion would not change.)
 |
(19) |
Eisenstein's calculations would suggest that addition of the olefin to the metal is made possible through a preferential “opening” of the CNO face (the rear face of the initial alkylidene as drawn in equation 19) as the olefin approaches, that the olefin binds relatively weakly, and that weak olefin binding is the most favorable for formation and breakup of the metallacyclobutane and subsequent loss of the olefin from M. However, it is also possible that decomposition pathways (vide infra), e.g., bimolecular decomposition of methylene species and/or rearrangement of intermediate metallacyclobutane species, are slowed dramatically in stereogenic-at-metal species. The possible role of (reversible) η5-pyrrolide binding in metathesis reactions in general is not known.
5. Catalyst decomposition in solution
Even in the absence of water, air, and reactive solvent impurities, the long term stability of a high oxidation state metathesis catalyst in solution is limited by bimolecular coupling of alkylidenes, especially methylene species, and by rearrangement of metallacyclobutanes to olefins in competition with loss of an olefin from a metallacyclobutane (equation 20). Both lead to “reduced” metal species, among them olefin complexes (or metallacyclopentane species in equilibrium with olefin complexes) or M=M dimers.lxvii,lxviii Some of these reduced metal species may catalyze other reactions that involve olefins. For example, unsubstituted d0 metallacyclopentane complexes behave as (slow) catalysts for the dimerization of ethylene to 1-butene. Ethylene is one of the most efficient “reducing” agents since it generates unstable methylene species, but also because recent evidence suggests that it promotes rearrangement of a metallacyclobutane to an olefin.lxix
 |
(20) |
Decomposition pathways have been explored in the greatest detail in tungsten systems. For example, an investigation of reactions of tungsten complexes that contain a chiral biphenoxide ligandxxxi with ethylene revealed that tungstacyclobutane complexes, an ethylene complex, and a tungstacyclopentane complex are formed. Perhaps the most interesting finding was observation (in 13C NMR studies) of a heterochiral bimetallic methylene complex in which the methylenes are bridging asymmetrically (equation 21). Interestingly, the homochiral
 |
(21) |
version of this dimer could not be observed in the system in which the enantiomerically pure Biphen2− ligand was employed. This result raises the possibility that alkylidenes could decompose at dramatically different rates in racemic versus enantiomerically pure systems.lxx In related studies of molybdenum systems that contain chiral ligands, molybdacyclobutanes, olefin complexes, molybdacyclopentane complexes, and base-free methylene complexes Mo(NR)(CH2)(diolate) were all observed in NMR studies.lxxi A dimer analogous to that shown in equation 21 was not observed in the Mo system.
Perhaps the most complete study that involves tungsten complexes is one in which the complexes contain the 2,6-dichlorophenylimido ligand.lxxii W(NArCl)(CHCMe3)(Biphen)(Tetrahydropyran) reacts with excess ethylene to give the tungstacyclobutane complex, W(NArCl)(Biphen)(C3H6), which yields W(NArCl)(Biphen)(C2H4)(THF) upon addition of THF. The tungstacyclopentane complex, W(NArCl)(Biphen)(C4H8), forms upon treatment of W(NArCl)(Biphen)(C2H4)(THF) with ethylene. W(NArCl)(Biphen)(C3H6) decomposes to yield (inter alia) the dimeric, heterochiral methylidene complex, [W(NArCl)(Biphen)(μ-CH2)]2, which decomposes further to yield homochiral [W(NArCl)(Biphen)]2(μ-CH2CH2). Finally, [W(NArCl)(Biphen)]2(μ-CH2CH2) loses ethylene reversibly to yield a dimer of W(NArCl)(Biphen), which could not be isolated and identified. A number of important species were crystallographically characterized. The structure of [W(NArCl)(Biphen)(μ-CH2)]2 reveals that the methylene ligands are in fact asymmetrically bridging, as proposed in a molybdenum complex (vide supra) and as shown in equation 21 for a tungsten complex. It is interesting to note that [W(NArCl)(Biphen)(μ-CH2)]2 is heterochiral, while [W(NArCl)(Biphen)]2(μ-CH2CH2) is homochiral, which suggests that [W(NArCl)(Biphen)(μ-CH2)]2 dissociates into monomeric W(NArCl)(Biphen)(μ-CH2) species that then recombine. The interatomic W(1)-W(1') distance (3.1211(6) Å) in [W(NArCl)(Biphen)(μ-CH2)]2 suggests that no W-W bond is present, while the C(1)-C(1)' distance (2.913(12) Å) suggests that no C-C bond is present. Although the methylene ligands are slightly distorted from a planar structure, as one would expect as a consequence of their behavior as “donors” to another tungsten, they retain many of the structural and spectroscopic characteristics we would expect of the methylene ligand in the unobservable monomeric species, W(NArCl)(CH2)(Biphen). This study serves as a model for formation of bimetallic species from other alkylidenes.
The first characterized bimetallic decomposition product with the empirical formula M(NR)X2 (e.g., X = alkoxide) was derived from complexes of the type Mo(NR)(CHR’)(OR”)2. It was found to contain bridging imido ligands in a symmetric, planar Mo2N2 core, and a pseudotetrahedral arrangement about each Mo.lxxiii It was first proposed that [M(NR)(OR”)2]2 complexes of the same general type also contain bridging imido groups.lxxiv However, it was shown later that certain tungsten complexes decompose to yield W=W compounds in which imido ligands are not bridging.lxvii,lxviii One type of W=W species that was observed in both heterochiral and homochiral forms is [W(NR)(CH2-t-Bu)(OR')]2 (R = 2,6-dimethylphenyl (Ar') or 2,6-diisopropylphenyl (Ar); R’ = pentafluorophenyl or hexafluoroisopropyl). The other type is [W(NR)(OR')2]2 (R’ = trifluoro-t-butyl or hexafluoro-t-butyl). In the synthesis shown in equation 22
 |
(22) |
the reaction of the neopentylidene complex with 2-pentene yields intermediate ethylidene and propylidene species, which then decompose bimolecularly because of the relatively small size of these alkylidenes. Only internal olefins are present in this system, which bind too weakly to form stable metallacylopentane or olefin complexes, and therefore allow the W=W species to form, usually in contrast to reactions in which ethylene or terminal olefins are employed. When the imido ligand is 2,6-dichlorophenylimido,lxxii species that contain bridging imido ligands are formed upon reaction with internal olefins, for example, as shown in equation 23. As
 |
(23) |
noted earlier, the reaction between W(NArCl)(CHCMe3)(Me2Pyr)2 and ethylene leads to a monomeric methylene complex. When the pyrrolide is unsubstituted, then a dimeric species is formed (equation 24). The rate of formation of μ-NArCl species may be accelerated as a consequence of interaction of one of the ortho chlorides with a second metal.
 |
(24) |
When complexes with the formula [M(NR)X2]2 contain bridging imido ligands and when they contain terminal imido ligands has not been settled. The situation is most dramatically revealed in unpublished studies that involve Mo(NAd)(ORF6)2 species.lxxv A crystal recovered from a >10-year old sample of the reaction between Mo(NAd)(CHCMe2Ph)[OCMe(CF3)2]2 and two equivalents of 2,3-bis(trifluoromethyl)norbornadiene in toluene-d8 in a sealed NMR tubelxxvi was shown in an X-ray study to be the dimeric species, {Mo[OCMe(CF3)2]2}2(μ-NAd)2, which contains symmetrically bridging imido ligands, a planar Mo2N2 ring, and pseudotetrahedral arrangement of ligands around each metal. On the other hand, addition of cis-2-pentene to Mo(NAd)(CHCMe2Ph)[OCMe(CF3)2]2 gave [OCMe(CF3)2]2(NAd)Mo=Mo(NAd)[OCMe(CF3)2]2 in which the adamantylimido ligands are terminal. The isolation and structural characterization of both unbridged and bridged {Mo(NAd)[OCMe(CF3)2]2}2 species establish that the two isomers are formed in different circumstances and do not interconvert readily. All attempts to interconvert the two isomers (photochemically and thermally) so far have failed.
A recent study of (Me2CF3CO)2(Ar'N)W=W(NAr')(OCMe2CF3)2 (Ar’ = 2,6-Me2C6H3) has shown some of the complexity of its reactions.lxxvii It reacts immediately at room temperature with one equivalent of CCl4 to give sparingly soluble [W(OCMe2CF3)2Cl]2(μ-NAr')2. [W(NAr')(OCMe2CF3)2]2 forms asymmetric 1:1 adducts with simple donor ligands such as PMe3, PPhMe2, and pyridine, and reacts with ethylene to yield [W(NAr')(OCMe2CF3)2]2(μ-C2H4), which could be argued contains a 1,2-dimetallacyclobutane ring. Heating a toluene solution of [W(NAr')(OCMe2CF3)2]2(μ-C2H4) produces (OCMe2CF3)2(Et)W(μ-NAr')(μ-N-2-CH2-6-MeC6H3)W(OCMe2CF3)2, in which the proton required to form the ethyl ligand arises through CH activation in one of the methyl groups of the NAr’ ligand. Treatment of [W(NAr')(OCMe2CF3)2]2 with 2-butyne yields [W(NAr')(OCMe2CF3)2]2(μ-MeCCMe), while addition of four equivalents of CH3CN to [W(NAr')(OCMe2CF3)2]2 yields a mixture of two products with the empirical formula W(NAr')(OCMe2CF3)2(CH3CN), one of which was found to contain the [μ-NC(Me)=C(Me)N]4− ligand in an unsymmetric dimer. The general pattern seemed to be a tendency for imido and/or alkoxide groups to transfer from one metal to the other at some stage. Therefore it seems unlikely in general that the M=M bond could be cleaved in high yield in a series of reactions that are the reverse of those that lead to M=M bonded species from monomeric Mo=CHR species, a type of reaction that would be a way to reform alkylidenes (vide infra). Reactions of M=M species that are related to a large number of reactions of M-M triple bonds that have been described primarily by Chisholm.lxxviii
Among the reasons why MAP species are relatively efficient catalysts is that bimolecular decomposition of methylenes might be inherently slower than when two alkoxides are present. Slowing decomposition of the methylene prolongs catalyst life and increases efficiency, by definition.
6. Regeneration of alkylidenes
High oxidation state alkylidenes are generated through α hydrogen abstraction reactions, usually in neopentyl or neophyl complexes. If alkylidenes could be regenerated from decomposition products that form cleanly (e.g., monomeric olefin complexes), especially under catalytic conditions, then turnovers might be increased dramatically. Such processes may already be leading to increased turnover without our knowledge.
One way to regenerate a catalyst is through transfer of an alkylidene from a phosphorus or sulfur ylide, which was first demonstrated for tantalum(III), and then for W(IV), with an alkylidene phosphorane.lxxix,lxxx However, in neither case was the resulting alkylidene species a known metathesis catalyst. Of course the alkylidene transfer agent must not react destructively with the resulting alkylidene that has formed. For example, reactions between the prototypical metathesis catalyst, Mo(NAr)(CHCMe3)[OCMe(CF3)2]2 (Ar = 2,6-i-Pr2C6H3) and Ph3P=CH2 was found to give an anionic imido alkylidyne complex, {Ph3PMe}{Mo(NAr)(CCMe3)[OCMe(CF3)2]2},lxxxi one of many possible unwanted consequences.
A second attractive way to generate alkylidenes is through rearrangement of olefins to alkylidenes.lxxxii The first example of rearrangement of an olefin complex to give its alkylidene analog was conversion of [N3N]Ta(C2H4) into [N3N]Ta(CHMe) with phenylphosphine as a catalyst ([N3N]3− = [(TMSNCH2CH2)3N]3−).lxxxiii The reaction mechanism was proposed to consist of conversion of the olefin into an alkyl phosphido complex through addition of an H from the phosphine and then removal of an α hydrogen from the alkyl by the phosphide to reform the phosphine and form the alkylidene. [N3N]Ta(CHMe) is not a catalyst for the facile metathesis of olefins.
An exhaustive study of rearrangement of olefins bound to (Silox)3M framework (M = Nb or Ta, Silox− = (t-Bu)3SiO−) has been published by Wolczanski.lxxxiv Compounds of the type (Silox)3Nb(olefin) rearrange upon being heated in solution to their (Silox)3Nb(alkylidene) isomers. In some cases equilibria are observed between (Silox)3Nb(olefin) and (Silox)3Nb(alkylidene) isomers. The mechanism is believed to consist of formation of an alkyl intermediate through CH abstraction in a t-butyl group, as shown in equation 25.
 |
(25) |
(Silox)3M(alkylidene) compounds do not react with olefins for steric reasons. These studies establish one pathway of forming alkylidenes from olefins if there is a source of H that can form an intermediate alkyl reversibly.
A third possible way to regenerate alkylidenes is from reactions between a reduced metal species and 3,3-diphenylcyclopropene, which Binger first employed to prepare a vinylalkylidene species from a Ti(II) species.lxxxv More relevant are reactions between W(O)L3Cl2 or W(NR)L3Cl2 species (e.g., L = PMe2Ph, or P(OR)3) and 3,3-diphenylcyclopropene to give first cyclopropene complexes, which were isolated and crystallographically characterized, and then vinylalkylidene complexes through rearrangement of the cyclopropene.lxxxvi Because the L ligands are strongly bound to the metal, the reactivity of the resulting imido alkylidene species towards olefins was poor. The precise mechanism of cyclopropene rearrangement is not known. The possibility of regenerating alkylidenes through reactions between (e.g.) M(IV) olefin complexes and 3,3-diphenylcyclopropane has not been tested.
A fourth potential method of forming alkylidenes is through formation and contraction of a metallacyclopentane to a metallacyclobutane ring, which has been observed for Talxxxvii or Re,lxxxviii followed by a metathesis of the MC3 ring. An unusual homologation of a vinyl tin species to an allyl tin species in the presence of ethylene and Mo(NAr)(C2H4)[Biphen]lxxi,lxxxix was proposed to involve a metallacyclobutane formed from an allyl tin species and ethylene (equation 26). If this mechanism is correct, and if other “special” olefins could be found that will become involved in such reactions, then alkylidenes potentially could be regenerated from d2 olefin complexes, which are common decomposition products in metathesis reactions with Mo and W catalysts. There are some significant hurdles that would have to be overcome. For example, the “special” olefin could itself not be involved in irreversible catalytic olefin metathesis reactions.
 |
(26) |
There is some evidence that Mo and W metathesis catalysts can be formed from reduced Mo or W species.xc For example, complexes that contain an unsupported M=M double bond (M = Mo or W) such as [Mo(NAr)(CH2-t-Bu)(OC6F5)]2 (Ar = 2,6-i-Pr2C6H3) or {W(NAr')[OCMe2(CF3)]2}2 (Ar’ = 2,6-Me2C6H3) will slowly catalyze olefin metathesis reactions. However, no alkylidene species has been observed and it is estimated that only a relatively small amount (<2−3%) of the M=M species is “activated” by the olefin. Monomeric Mo(N-2,6-Cl2C6H3)[rac-Biphen](CH2=CH2)(ether) has also been found to metathesize certain olefins. The metathesis reactions are slow and the alkylidene species responsible for metathesis have not yet been identified. They are proposed to be imido alkylidene bisalkoxide species of the type being discussed here.
The method of formation of alkylidenes in “classical” Mo or W systems when alkylating agents are employed is assumed to be analogous to reactions that have given rise to well-defined imido alkylidene species,i primarily α hydrogen abstraction reactions. However, small amounts of catalysts could be formed from certain olefins such as norbornenes in monometallic or bimetallic mechanisms related to those discussed in this section. Alternatively, an olefin can react with an oxo (a “reverse Wittig” reaction), which could be the manner in which alkylidenes are formed when metal oxides (Mo, W, Re) are treated with olefins.i
7. Catalysts supported on silica
For decades scientists have been trying to synthesize and characterize transition metal catalysts that are covalently attached to “hard” supports such as silica. Such “immobilized homogeneous catalysts” are attractive for several reasons. One is that from an engineering perspective the product (if the supported catalyst does not become entrained in a polymer) often can readily be separated from the catalyst. Another reason that is potentially beneficial in metathesis chemistry is that bimolecular coupling of alkylidenes should be minimized if the metal centers are too far apart. Considerable headway has been made in the last few years in preparing supported catalysts on silica that can be identified through solid state NMR techniques.xci Recently efforts have turned toward the preparation of olefin metathesis catalysts based on Mo and W that are attached to silica. Methods of preparing catalysts that are attached to organic polymer supports were mentioned at the end of section 2.xxxiii-xxxv Bimolecular decompositions are not eliminated in swellable polymers with a relatively low degree of crosslinking because polymer mobility is still significant and the catalyst concentration within the polymer is often relatively high compared to the concentration in a typical homogeneous reaction.xxxiii
It has been proposed that Mo(N)(CH2-t-Bu)3 sublimed onto SiO2(500) yields first Mo(NH)(CH2-t-Bu)3(OSi500) and then neopentane (which was observed and quantified) and Mo(NH)(CH-t-Bu)(CH2-t-Bu)(OSi500) upon α hydrogen abstraction.xcii The formulation is based on the observation of a νNH IR stretch at 3370 cm−1, reaction of the supported species with acetone to give 2,4,4-trimethyl-pent-2-ene, and polymerization of norbornene at an initial frequency of 3000 h−1. In contrast, the reaction between two equivalents of Mo(N)(CH2-t-Bu)3 and (HO)Ph2SiOSiPh2(OH) yielded Mo(NH)(CH2-t-Bu)3(OPh2SiOSiPh2O)Mo(NH)(CH2-t-Bu)3. The alkylidene carbon atom was not detected in a solid state 13C NMR spectrum.
The reaction between SiO2(700) and Mo(N)(CH2-t-Bu)3 in benzene was explored several years later.xciii Again protonation of the nitrido ligand to form intermediate Mo(NH)(CH2-t-Bu)3(OSi700) followed by loss of neopentane to form Mo(NH)(CH-t-Bu)(CH2-t-Bu)(OSi500) was observed, which was found to be a highly active catalyst. Reactions carried out in dichloromethane or THF led to poorly active or inactive catalysts. Use of a molecular silanol (c-C5H9)7Si7O12SiOH, a silsesquioxane) led to Mo(NH)(CH2-t-Bu)3(POSS), which did not behave as a metathesis catalyst in solution.
Impregnation of SiO2(700) with Mo(NAr)(CHCMe3)(CH2CMe3)2 in pentane produced the monoattached species shown in equation 27, according to IR, elemental analyses, and various solid state NMR studies.xciv,xcv The corresponding molecular siloxy complexes were prepared in
| (27) |
reactions between Mo(NAr)(CHCMe3)(CH2CMe3)2 and (c-C5H9)7Si7O12SiOH or (t-BuO)3SiOH in C6D6 or pentane and shown to have NMR characteristics similar to those of Mo(NAr)(CHCMe3)(OSi700)(CH2CMe3). Exposure of Mo(NAr)(CHCMe3)(OSi700)(CH2CMe3) to 1-octene at 25 °C in a batch reactor produced metathesis products with a turnover frequency (TOF) about three times greater than found in solution for the homogeneous analogs. Similar results were obtained for the analogous W system.xcvi
Delayed acquisition methods (echo and constant-time acquisition) were found to improve resolution in the proton solid state NMR spectra of surface organometallic catalysts such as Mo(NAr)(CHCMe3)(OSi700)(CH2CMe3).xcvii Therefore all proton resonances could be observed, which is not possible with simple magic angle spinning techniques. Combining constant-time acquisition with heteronuclear carbon-proton correlation spectroscopy also improves resolution of the 2D HETCOR spectra.
Reaction of W(NAr)(CHCMe3)(CH2-t-Bu)2 with SiO2(200) led to a mixture of W(NAr)(CHCMe3)(OSi200)(CH2CMe3), W(NAr)(CHCMe3)(OSi200)2, and W(NAr)(CH2CMe3)2(OSi200)2, since the surface density of siloxy OH groups in SiO2(200) is much higher than in SiO2(700).xcviii All surface species were characterized by various spectroscopic techniques and compared with homogeneous models that contain silsesquioxanes. W(NAr)(CHCMe3)(OSi200)(CH2CMe3) and W(NAr)(CHCMe3)(OSi200)2 are both active toward the metathesis of cis-2-pentene and propene, while W(NAr)(CH2CMe3)2(OSi200)2 is inactive. W(NAr)(CH2CMe3)2(OSi200)2 also does not readily eliminate neopentane to yield W(NAr)(CHCMe3)(OSi200)2 under the reaction conditions.
Addition of Mo(NAr)(CHCMe2R)(X)2 (X = pyrrolide or diphenylamido; R = Me or Ph) to SiO2(700) leads to formation of HX and Mo(NAr)(CHCMe2R)(X)(OSi700).xcix The resulting surface-supported catalyst is active for the metathesis of propene and ethyl oleate and the ring-closing metathesis of diallylmalonate. The remaining nitrogen-based ligand (X) in the supported catalyst is believed to be responsible for the greater stability and selectivity of the surface catalysts compared to those in which X is neopentyl.
When Mo(NAr)(CHCMe2R)(Me2Pyr)2 (Me2Pyr = 2,5-dimethylpyrrolide) is added to SiO2(700), both Mo(NAr)(CHCMe2R)(Me2Pyr)(OSi700) (∼80%) and Mo(NAr)(CH2CMe2R)(Me2Pyr)2(OSi700) are formed, according to mass balance analysis, IR, and NMR studies.c Metathesis of propene in a flow reactor (∼400 mL m−1; 4800 mol of propene per mol of Mo per min) gave selectively ethene and 2-butenes at a rapid initial rate (5.2 s−1). In a period of 1500 min over 105 turnovers were achieved. While the initial rate is similar to that obtained for the parent pyrrolide system, the 2,5-dimethylpyrrolide catalyst appears to deactivate more slowly. The difference between these two systems is even more pronounced in the self metathesis of ethyl oleate. Similar results were obtained when the imido ligand is adamantylimido or 2-trifluoromethylphenylimido; the initial turnover frequencies are faster than with Mo(NAr)(CHCMe2R)(Me2Pyr)2(OSi700) as a catalyst for some substrate/catalyst combinations.
In a recent report, room temperature molecular dynamics in a series of silica supported organometallic complexes could be determined experimentally using a combination of solid state nuclear magnetic resonance (NMR) spectroscopy and density functional theory calculations (DFT).ci Carbon chemical shift anisotropies (CSA) and proton/carbon dipolar coupling constants were determined experimentally. Dynamics were validated using a method that integrates static values for the CSA obtained computationally from DFT calculations, thereby obviating the need for low temperature measurements. Dynamics could be quantified and were discussed in terms of the molecular structure of the surface organometallic complexes.
The reaction between Mo(NAr)(CHCMe2R)(O-t-Bu)2 and SiO2(700) has been shown to yield a highly active silica-supported heterogeneous alkene metathesis catalyst of the type Mo(NAr)(CHCMe2R)(O-t-Bu)(OSi700).cii These studies demonstrate the dramatic enhancement of catalytic activity upon replacement of a t-butoxide by a surface siloxide, which is relatively electron withdrawing. They may also lend support to the theory of a higher activity of catalysts that are asymmetric at the metal, although it is difficult at this stage to deconvolute the various reasons for increased activity.
Key intermediates in catalytic metathesis reactions catalyzed by surface-attached species have only recently been observed in a study involving W(NAr)(CH-t-Bu)(2,5-Me2Pyr)2 that has been grafted onto SiO2(700).ciii Addition of W(NAr)(CH-t-Bu)(2,5-Me2Pyr)2 to SiO2(700) yields W(NAr)(CH-t-Bu)(2,5-Me2Pyr)(OSi700) as the major species (∼90%) along with W(NAr)(CH2-t-Bu)(2,5-Me2Pyr)2(OSi700), according to mass balance analysis, IR, and NMR studies. This catalyst displays good activity and stability for the metathesis of propene. In reactions that involve ethylene, solid state NMR spectroscopy allowed observation of methylene complex as well as unsubstituted metallacyclobutane complexes, which appear as a mixture of square based pyramidal and trigonal bipyramidal species. Observation of these key reaction intermediates in alkene metathesis will play a key role in the design of future catalysts. This work is the first in which the intermediates that have been observed in homogeneous processes have also been observed on a silica surface and lends strong support to the proposal that catalysts can be designed on a silica surface that function in a manner that is entirely analogous to what is observed in solution.
8. New reactions
One of the most interesting new reactions that has been observed with imido alkylidene catalysts is “alkane metathesis” (AM). Alkane metathesis was first observed in 1973 by Burnett and Hughes.civ who showed that when butane was passed over a mixture of a heterogeneous platinum on alumina catalyst (a dehydrogenation catalyst) and tungsten oxide on silica (an olefin metathesis catalyst) at ∼400 °C, lower and higher MW alkanes were formed, predominantly propane and pentane (25% and 16%, respectively). Supported Ta and W hydride catalysts that function as alkane metathesis catalysts at much lower temperatures have been reported by the Basset group.cv,cvi For example, propane can be converted in low yields to a mixture of C1 to C6 alkanes using silica-supported or alumina-supported Ta hydride catalysts. These catalysts are believed to operate through reactions that involve formation of alkylidene intermediates and olefins from alkanes. Both branched and linear products are formed, as well as methane.
AM systems that have been published by Goldman and Brookhartcvii involve two homogeneous catalysts (“tandem catalysts”) operating independently in the same solution (Scheme 1). One is an Ir-based pincer-type dehydrogenation/hydrogenation catalystcviii while the second is a homogeneous Mo-based catalyst such as Mo(NAr)(CHCMe2Ph)(ORF6)2 or a heterogeneous Re-based catalyst (Re2O7 on alumina). Tandem catalytic reactions are relatively rare, in part because the two catalysts must not destructively interfere with one another. For example, Ru metathesis catalysts of the type developed by Grubbs that were tried yielded no alkane metathesis products, presumably because Ru catalysts deactivated the Ir catalysts. The reactions are run in neat alkane (e.g., octane or decane) at a minimum temperature of 125 °C and at that temperature require days to approach the equilibrium distribution of even and odd linear alkanes with chains longer and shorter than the original. No branched product or methane is observed. At this stage the limiting reaction is standard decomposition of the Mo-based metathesis catalyst, although there is evidence (e.g., formation of diisopropylaniline) that suggests that more destructive non-standard decomposition pathways may be operating. The low concentration of ethylene, which is believed to be involved in rearrangement of metallacyclobutane intermediates (vide supra), as well as the low rate of bimolecular coupling of alkylidenes at low catalyst concentrations, are believed to be factors in sustaining metathesis activity for extended periods at 125 °C.
Scheme 1.

Alkane metathesis by a (pincer)Ir transfer-dehydrogenation catalyst and a Mo-based olefin metathesis catalyst illustrated with conversion of two mol C(n) n-alkane to C(2n-2) n-alkane plus ethane.
In an initial survey of approximately forty molybdenum- and tungsten-based homogeneous catalysts for AM, tungsten was found to outperform molybdenum in most cases.cix For example, W(NAr)(CHCMe2Ph)[OC(CF3)3]2 was found to be ∼50% more efficient than Mo(NAr)(CHCMe2Ph)(ORF6)2. The greatest activity was obtained using W(NAr)(CHCMe2Ph)(OSiPh3)2. Triphenylsiloxide and variations are not common auxiliary ligands in high oxidation state metathesis chemistry. The success of triphenyl siloxide is somewhat surprising in view of the possibility of aryl CH activation and formation of an alkyl from an alkylidene, an example of which is known.cx Silicon is likely to reduce the amount of donation of π electron density to the metal and make the metal much more electron deficient than in a typical alkoxide, possibly even nonafluoro-t-butoxide.
Enyne metathesis reactionscxi are widely practiced with Grubbs-type Ru-based catalysts, but they have not been successful with Mo bisalkoxide species. The reason is not known, but a greater tendency to polymerize the terminal acetylene in competition with ring-closing is a possibility. Recently it has been demonstrated for the simple substrates shown in Figure 4 (E = CO2Et) with Mo(NAr)(CHCMe2Ph)(Me2Pyr)(OR) (MAP) catalysts, where R = t-Bu, i-Pr, Ar, i-PrF6, or t-BuF6.cxii The products resulted largely or exclusively from initial addition of the triple bond to produce the metallacyclobutene intermediate in which the olefinic substituent is located in the β position (“β addition”; equation 28). Ruthenium catalysts yield almost exclusively the α addition product for these and other similar substrates.cxi These are the first enyne metatheses to be reported for Mo catalysts.
 |
(28) |
Polymerization of 3,3-disubstituted cyclopropenes by several molybdenum alkylidene initiators of the type Mo(NAr)(CHR)(OR')2 to give all trans polymers has recently been reported (equation 29).cxiii Surprisingly, this appears to be the first report of polymerization of a cyclopropene.i The presence of two different groups on C(3) of the cyclopropene (e.g., Me and Ph) creates the possibility that tactic polymers could be prepared with the appropriate catalyst. That appears to be possible with Mo(NR)(CHR')(diolate) species that contain electron withdrawing diolate ligands analogous to those that have been employed for asymmetric metathesis reactions.cxiv The tacticity (iso or syndio) of the (tactic) polymers is not known.
 |
(29) |
A stoichiometric method of synthesizing oligoenes of diisopropyldipropargylmalonate, [CH2][bx][CH2] (x = 2,3,4,5), in which the “b” repeat unit is identical to the five-membered ring formed in a tail-to-tail cyclopolymerization of diisopropyldipropargylmalonate, and methylene groups are capping the ends has been reported.cxv For example, [Mo][b][Mo] can be prepared as shown in equation 30. A “Mo-Wittig” reaction between [Mo][b][Mo] and two equivalents of the aldehyde, [CH2][b][O], then gives the “trimer”, [CH2][b3][CH2]. Treatment of [CH2][b3][CH2] with two equivalents of [Mo][CHCMe2Ph] gives [Mo][b3][Mo], which upon treatment with two equivalents of [CH2][b][O] gives the “pentamer”, [CH2][b5][CH2], which contains 11 double bonds in conjugation in a molecule in which the repeat units are connected by trans C=C bonds. The oligoenes are not always obtained in high yield in pure form because (inter alia and surprisingly) C=C metathesis reactions compete with the Mo-Wittig reaction of the aldehyde (e.g., to give [CH2][b2][CH2] also in the synthesis of [CH2][b3][CH2]). These complications could be minimized by employing adducts, (L)[Mo][bx][Mo](L) where L = PMe3 or quinuclidine, instead of base-free [Mo][bx][Mo] species in the Mo-Wittig reaction.
 |
(30) |
 |
(31) |
Asymmetric metatathesis has been dominated by reactions in which an asymmetric center at a carbon atom is established.xid It has now been shown that Mo-based biphenolates and binaphtholate catalysts can be employed for the enantioselective synthesis of P-stereogenic phosphinates and phosphine oxides (e.g., equation 32).cxvi Enantioselectivity is currently still modest, but is expected to increase with the advent of new MAP catalysts. Synthesis of molecules through catalytic asymmetric olefin metathesis in which the stereogenic center is not carbon is still in its infancy.
 |
(32) |
A notable shortcoming of advances in catalytic olefin metathesis is a paucity of methods that selectively furnish Z alkenes. For example, nearly all ring-opening/cross-metathesis (ROCM) reactions afford E olefin products predominantly or exclusively.xid With the advent of MAP complexes in which (apparently) an anti orientation of the alkylidene is not favorable, and in which metallacyclobutane intermediates have a well-defined TBP structure, it would seem possible to restrict metallacyclobutane intermediates to those in which a substituent points “up,” as shown in equation 33, if the imido group is “small” and the OR’ group is “large.” This is the crux of the argument as to why Z- and enantioselective ring-opening/cross-metathesis reactions catalyzed by stereogenic-at-Mo adamantylimido complexes are successful;cxvii an example is shown in equation 34. This is the first report of enantioselective and Z-selective ring-opening
 |
(33) |
 |
(34) |
cross metathesis. It should be noted that the conditions chosen for preparing the MAP catalysts in situ produces only a 60−70% yield in some cases, but the MAP catalysts simply are much more reactive than the bispyrrolide or bisalkoxide impurities and therefore yield results derived from reactions that involve them.
9. Concluding Remarks
In this review I have concentrated on advances in Mo and W imido alkylidene chemistry that have been reported relatively recently. The ability to synthesize a large variety of complexes from bispyrrolide precursors, especially MAP complexes, often in situ, has pushed imido alkylidene chemistry in new directions in terms of fundamentals as well as applications. The fact that MAP catalysts can be orders of magnitude more reactive than bisalkoxide, biphenolate, or biphenoxide catalysts hints at the possibility that steric effects are likely to become increasingly subtle, but significant, and that electronic effects at the metal may prove to be increasingly powerful in terms of asymmetric metathesis reactions; the interplay between the two may lead to increasingly fine control of metathesis reactions. Efficient asymmetric ring-closing reactions, metathesis of alkanes, new ROMP polymerizations, enyne metatheses, Z- and enantioselective ring-opening cross metathesis, and reactions involving silica-supported catalysts seem to point to a continuing expansion of useful applications of metathesis with well-defined alkylidene catalysts.
Figure 5.
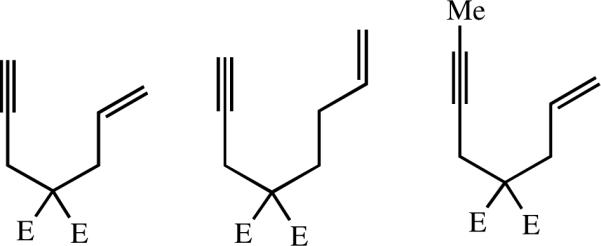
Selected enyne substrates explored with MAP catalysts.
Acknowledgements
I thank the National Science Foundation (currently CHE-0554734 to R. R. S.) for supporting research on multiple metal-carbon bonds, and the National Institutes of Health (currently GM-59426 to R. R. S. and A. H. Hoveyda) for supporting work on applications of asymmetric metathesis to organic chemistry. I thank the many coworkers who have been involved in this research for their hard work and enthusiasm.
References
- i.Ivin KJ, Mol JC. Olefin Metathesis and Metathesis Polymerization. Academic Press; San Diego: 1997. [Google Scholar]
- ii.Hérrison JL, Chauvin Y. Makromol. Chem. 1971;141:161. [Google Scholar]
- iii.Tebbe FN, Parshall GW, Reddy GS. J. Am. Chem. Soc. 1978;100:3611. [Google Scholar]
- iv.a Howard TR, Lee JB, Grubbs RH. J. Am. Chem. Soc. 1980;102:6876. [Google Scholar]; b Lee JB, Gajda GJ, Schaefer WP, Howard TR, Ikariya T, Straus DA, Grubbs RH. J. Am. Chem. Soc. 1981;103:7358. [Google Scholar]; c Straus DA, Grubbs RH. Organometallics. 1982;1:1658. [Google Scholar]; d Ikariya T, Ho SCH, Grubbs RH. Organometallics. 1985;4:199. [Google Scholar]; e Gilliom LR, Grubbs RH. Organometallics. 1986;5:721. [Google Scholar]; f Anslyn EV, Grubbs RH. J. Am. Chem. Soc. 1987;109:4880. [Google Scholar]
- v.a Schrock RR. J. Am. Chem. Soc. 1974;96:6796. [Google Scholar]; b Schrock RR, Fellmann JD. J. Am. Chem. Soc. 1978;100:3359. [Google Scholar]
- vi.Schrock RR, Sharp PR. J. Am. Chem. Soc. 1978;100:2389. [Google Scholar]
- vii.Since the alkylidene ligand changes in a metathesis reaction, the original alkylidene containing species is more properly called an “initiator.” Nevertheless, we will continue to call it a catalyst.
- viii.McLain SJ, Wood CD, Schrock RR. J. Am. Chem. Soc. 1979;101:4558. [Google Scholar]
- ix.a Schrock RR, Rocklage SM, Wengrovius JH, Rupprecht G, Fellmann J. J. Molec. Catal. 1980;8:73. [Google Scholar]; b Rocklage SM, Fellmann JD, Rupprecht GA, Messerle LW, Schrock RR. J. Am. Chem. Soc. 1981;103:1440. [Google Scholar]
- x.Schrock RR. Polyhedron. 1995;14:3177. [Google Scholar]
- xi.a Schrock RR. Chem. Rev. 2002;102:145. doi: 10.1021/cr0103726. [DOI] [PubMed] [Google Scholar]; b Schrock RR. In: Carbene Chemistry. Bertrand G, editor. FontisMedia/Marcel Dekker; Lausanne and New York: 2002. p. 205. [Google Scholar]; c Schrock RR. In: Handbook of Metathesis. Grubbs RH, editor. Wiley-VCH; Weinheim: 2003. p. 8. [Google Scholar]; d Schrock RR, Hoveyda AH. Angew. Chem. Int. Ed. 2003;42:4592. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]; e Schrock RR. J. Molec. Catal A. 2004;213:21. [Google Scholar]; f Schrock RR. Chem. Comm. 2004:2773. doi: 10.1039/b504541j. [DOI] [PubMed] [Google Scholar]; g Schrock RR. Angew. Chem. Int. Ed. 2006;45:3748. doi: 10.1002/anie.200600085. [DOI] [PubMed] [Google Scholar]; h Schrock RR. Le Prix Nobel 2005. Almqvist & Wiksell International; Stockholm, Sweden: 2006. p. 206. [Google Scholar]; i Schrock RR, Czekelius CC. Adv. Syn. Catal. 2007;349:55. [Google Scholar]
- xii.Grubbs RH, editor. Handbook of Metathesis. Catalyst Development. Vol. 1; Applications in Organic Synthesis. Vol. 2.; Applications in Polymer Synthesis. Vol. 3. Wiley-VCH; Weinheim: 2003. [Google Scholar]
- xiii.Wengrovius JH, Schrock RR. Organometallics. 1982;1:148. [Google Scholar]
- xiv.a Schaverien CJ, Dewan JC, Schrock RR. J. Am. Chem. Soc. 1986;108:2771. [Google Scholar]; b Schrock RR, DePue R, Feldman J, Schaverien CJ, Dewan JC, Liu AH. J. Am. Chem. Soc. 1988;110:1423. [Google Scholar]
- xv.a Feldman J, Schrock RR. Prog. Inorg. Chem. 1991;39:1. [Google Scholar]; b Schrock RR. In: Topics in Organometallic Chemistry. Alkene Metathesis in Organic Synthesis. Fürstner A, editor. Vol. 1. Springer; Berlin: 1998. p. 1. [Google Scholar]; c Schrock RR, DePue R, Feldman J, Schaverien CJ, Dewan JC, Liu AH. J. Am. Chem. Soc. 1988;110:1423. [Google Scholar]
- xvi.a Murdzek JS, Schrock RR. Organometallics. 1987;6:1373. [Google Scholar]; b Schrock RR, Krouse SA, Knoll K, Feldman J, Murdzek JS, Yang DC. J. Molec. Catal. 1988;46:243. [Google Scholar]
- xvii.Schrock RR, Murdzek JS, Bazan GC, Robbins J, DiMare M, O'Regan M. J. Am. Chem. Soc. 1990;112:3875. [Google Scholar]
- xviii.Oskam JH, Fox HH, Yap KB, McConville DH, O'Dell R, Lichtenstein BJ, Schrock RR. J. Organometal. Chem. 1993;459:185. [Google Scholar]
- xix.Wampler KM, Hock AS, Schrock RR. Organometallics. 2007;26:6674. [Google Scholar]
- xx.Sinha A, Lopez LPH, Schrock RR, Hock AS, Müller P. Organometallics. 2006;25:1412. doi: 10.1021/om060430j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxi.Sinha A, Schrock RR, Müller P, Hoveyda AH. Organometallics. 2006;25:4621. doi: 10.1021/om060430j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxii.Hock A, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2006;128:16373. doi: 10.1021/ja0665904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxiii.Wampler K. unpublished results.
- xxiv.a Schattenmann FJ, Schrock RR, Davis WM. J. Am. Chem. Soc. 1996;118:3295. [Google Scholar]; b Schrock RR, Tonzetich ZJ, Lichtscheidl AG, Müller P. Organometallics. 2008;27:3986. [Google Scholar]
- xxv.Oskam JH, Schrock RR. J. Am. Chem. Soc. 1993;115:11831. [Google Scholar]
- xxvi.Schrock RR, Luo S, Lee JCJ, Zanetti NC, Davis WM. J. Am. Chem. Soc. 1996;118:3883. [Google Scholar]
- xxvii.Wu Z, Wheeler DR, Grubbs RH. J. Am. Chem. Soc. 1992;114:146. [Google Scholar]
- xxviii.Teng X, Cefalo DR, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2002;124:10779. doi: 10.1021/ja020671s. [DOI] [PubMed] [Google Scholar]
- xxix.a Poater A, Solans-Monfort X, Clot E, Copéret C, Eisenstein O. Dalton Trans. 2006:3077. doi: 10.1039/b604481f. [DOI] [PubMed] [Google Scholar]; b Solans-Monfort X, Clot E, Copéret C, Eisenstein O. Organometallics. 2005;24:1586. [Google Scholar]
- xxx.a Toreki R, Schrock RR. J. Am. Chem. Soc. 1992;114:3367. [Google Scholar]; b Toreki R, Vaughan GA, Schrock RR, Davis WM. J. Am. Chem. Soc. 1993;115:127. [Google Scholar]
- xxxi.Tsang WCP, Hultzsch KC, Alexander JB, Bonitatebus PJ, Jr., Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2003;125:2652. doi: 10.1021/ja0210603. [DOI] [PubMed] [Google Scholar]
- xxxii.Leduc A-M, Salameh A, Soulivong D, Chabanas M, Basset J-M, Copéret C, Solans-Monfort X, Clot E, Eisenstein O, Boehm VPW, Roeper M. J. Am. Chem. Soc. 2008;130:6288. doi: 10.1021/ja800189a. [DOI] [PubMed] [Google Scholar]
- xxxiii.a Hultzsch KC, Jernelius JA, Hoveyda AH, Schrock RR. Angew. Chem. Int. Ed. 2002:589. [Google Scholar]; b Dolman SJ, Hultzsch KC, Pezet F, Teng X, Hoveyda AH, Schrock RR. J. Am. Chem. Soc. 2004;129:10945. doi: 10.1021/ja047663r. [DOI] [PubMed] [Google Scholar]
- xxxiv.a Kroell RM, Schuler N, Lubbad S, Buchmeiser MR. Chem. Comm. 2003:2742. doi: 10.1039/b306427a. [DOI] [PubMed] [Google Scholar]; b Buchmeiser MR. Polymer. 2007;48:2187. [Google Scholar]; c Buchmeiser MR. Adv. Polym. Sci. 2006;197:137. [Google Scholar]; d Buchmeiser MR. New J. Chem. 2004;28:549. [Google Scholar]; e Buchmeiser MR. J. Chrom. A. 2004:1060, 43. doi: 10.1016/j.chroma.2004.04.027. [DOI] [PubMed] [Google Scholar]
- xxxv.Wang D, Kroell R, Mayr M, Wurst K, Buchmeiser MR. Adv. Synth. Catal. 2006;348:1567. [Google Scholar]
- xxxvi.Buchmeiser MR. Monatsh. Chem. 2006;137:825. [Google Scholar]
- xxxvii.Buchmeiser MR. J. Sep. Sci. 2008;31:1907. doi: 10.1002/jssc.200800104. [DOI] [PubMed] [Google Scholar]
- xxxviii.a Ciszewski JT, Xie B, Cao C, Odom AL. Dalton Trans. 2003:4226. [Google Scholar]; b Lokare KS, Staples RJ, Odom AL. Organometallics. 2008;27:5130. [Google Scholar]
- xxxix.Schrock RR, Gabert AJ, Singh R, Hock AS. Organometallics. 2005;24:5058. [Google Scholar]
- xl.Mayershofer MG, Nuyken O, Buchmeiser MR. Macromolecules. 2006;39:2452. [Google Scholar]
- xli.Buchmeiser MR. Chem. Rev. 2000;100:1565. doi: 10.1021/cr990248a. [DOI] [PubMed] [Google Scholar]
- xlii.a Vygodskii YS, Shaplov AS, Lozinskaya EI, Vlasov PS, Malyshkina IA, Gavrilova ND, Kumar PS, Buchmeiser MR. Macromolecules. 2008;41:1919. [Google Scholar]; b Buchmeiser MR. Adv. Polym. Sci. 2005;176:89. [Google Scholar]; c Buchmeiser MR. Monatsh. Chem. 2003;134:327. [Google Scholar]
- xliii.Buchmeiser MR, Wang D, Naumov S, Wurst K. J. Organomet. Chem. 2006;691:5391. [Google Scholar]
- xliv.Chi S-H, Hales JM, Fuentes-Hernandez C, Tseng S-Y, Cho J-Y, Odom S, Zhang Q, Barlow S, Schrock RR, Marder SR, Kippelen B, Perry JW. Adv. Mat. 2008;17:3199. [Google Scholar]
- xlv.Sinha A, Schrock RR. Organometallics. 2004;23:1643. [Google Scholar]
- xlvi.a Kershner DL, Basolo F. Coord. Chem. Rev. 1987;79:279. [Google Scholar]; b DuBois MR. Coord. Chem. Rev. 1998;174:191. [Google Scholar]; c Senge MO. Angew. Chem. Int. Ed. 1996;35:1923. [Google Scholar]
- xlvii.Myers WH, Welch KD, Graham PM, Keller A, Sabat M, Trindle CO, Harman WD. Organometallics. 2005;24:5267. [Google Scholar]
- xlviii.Marinescu SC, Singh R, Hock AS, Wampler KM, Schrock RR, Müller P. Organometallics. 2008;27:6570. [Google Scholar]
- xlix.Jiang AJ, Schrock RR, Müller P. Organometallics. 2008;27:4428. [Google Scholar]
- l.Gabert AJ, Schrock RR, Müller P. Chem. Asian J. 2008;3:1535. doi: 10.1002/asia.200800076. [DOI] [PubMed] [Google Scholar]
- li.Kreickmann T, Arndt S, Schrock RR, Müller P. Organometallics. 2007;26:5702. and references therein.
- lii.Marinescu S. unpublished results.
- liii.Pilyugina T. Ph.D. Thesis. Massachusetts Institute of Technology; 2007. [Google Scholar]
- liv.Jamieson JY. Ph.D. thesis. Massachusetts Institute of Technology; 2002. [Google Scholar]
- lv.a Myers WH, Koontz JI, Harman WD. J. Am. Chem. Soc. 1992:5684. [Google Scholar]; b Hodges DM, Gonzalez J, Koontz JI, Myers WH, Harman WD. J. Org. Chem. 1995:2125. [Google Scholar]; c Johnson TJ, Arif AM, Gladysz JA. Organometallics. 1993:4728. [Google Scholar]; d Myers WH, Welch KD, Graham PM, Keller A, Sabat M, Trindle CO, Harman WD. Organometallics. 2005:5267. [Google Scholar]; e Aumann R, Fröhlich R, Zippel F. Organometallics. 1997:2571. [Google Scholar]; f Rakowski DuBois M, Vasquez LD, Peslherbe L, Noll BC. Organometallics. 1999:2230. [Google Scholar]; g Guidotti S, Camurati I, Focante F, Angellini L, Moscardi G, Resconi L, Leardini R, Nanni D, Mercandelli P, Sironi A, Beringhelli T, Maggioni D. J. Org. Chem. 2003:5445. doi: 10.1021/jo020647x. [DOI] [PubMed] [Google Scholar]
- lvi.a Elguero J, Marzin C, Katritzaky AR, Linda P. The Tautomerism of Heterocycles. In: Katritzky AR, Boulton AJ, editors. Advances in Heterocyclic Chemistry. Suppl 1. Academic Press; New York: 1976. [Google Scholar]; b Dubnikova F, Lifshitz A. J. Phys. Chem. A. 1998;102:10880. [Google Scholar]; c Somers KRF, Kryachko ES, Ceulemans A. J. Phys. Chem. A. 2003;107:5427. [Google Scholar]; d Martoprawiro M, Bacskay GB, Mackie JC. J. Phys. Chem. A. 1999;103:3923. [Google Scholar]; e Karpfen A, Schuster P, Berner H. J. Org. Chem. 1979;44:374. [Google Scholar]
- lvii.Singh R, Schrock RR, Müller P, Hoveyda AH. J. Am. Chem. Soc. 2007;129:12654. doi: 10.1021/ja075569f. [DOI] [PubMed] [Google Scholar]
- lviii.Poater A, Solans-Monfort X, Clot E, Copéret C, Eisenstein O. J. Am. Chem. Soc. 2007;129:8207. doi: 10.1021/ja070625y. [DOI] [PubMed] [Google Scholar]
- lix.Malcolmson SJ, Meek SJ, Sattely ES, Schrock RR, Hoveyda AH. Nature. 2008;456:933. doi: 10.1038/nature07594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lx.Sattely ES, Meek SJ, Malcolmson SJ, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:943. doi: 10.1021/ja8084934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxi.Meek SJ, Malcolmson SJ, Li B, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. doi: 10.1021/ja907805f. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxii.Marinescu S, Schrock RR, Li B. J. Am. Chem. Soc. 2009;131:58. doi: 10.1021/ja808308e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxiii.Jiang A. unpublished results.
- lxiv.Brunner H. Angew. Chem. Int. Ed. 1999;38:1194. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1194::AID-ANIE1194>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- lxv.a Fontecave M, Hamelin O, Ménage S. Top. Organomet. Chem. 2005;15:271. [Google Scholar]; b Therrien B, Ward TR. Angew. Chem. Int. Ed. 1999;38:405. doi: 10.1002/(SICI)1521-3773(19990201)38:3<405::AID-ANIE405>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- lxvi.Feldman J, Schrock RR. Prog. Inorg. Chem. 1991;39:1. [Google Scholar]
- lxvii.Lopez LPH, Schrock RR. J. Am. Chem. Soc. 2004;126:9526. doi: 10.1021/ja0400988. [DOI] [PubMed] [Google Scholar]
- lxviii.Lopez LPH, Schrock RR, Müller P. Organometallics. 2006;25:1978. doi: 10.1021/om060430j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxix.Tsang WCP, Schrock RR, Hoveyda AH. Organometallics. 2001;20:5658. [Google Scholar]
- lxx.Merrifield JH, Lin G-Y, Kiel WA, Gladysz JA. J. Am. Chem. Soc. 1983;105:5811. [Google Scholar]
- lxxi.Tsang WCP, Jamieson JY, Aeilts SA, Hultzsch KC, Schrock RR, Hoveyda AH. Organometallics. 2004;23:1997. [Google Scholar]
- lxxii.Arndt S, Schrock RR, Müller P. Organometallics. 2007;26:1279. [Google Scholar]
- lxxiii.Robbins J, Bazan GC, Murdzek JS, O'Regan MB, Schrock RR. Organometallics. 1991;10:2902. [Google Scholar]
- lxxiv.Schrock RR, DePue RT, Feldman J, Yap KB, Yang DC, Davis WM, Park LY, DiMare M, Schofield M, Anhaus J, Walborsky E, Evitt E, Krüger C, Betz P. Organometallics. 1990;9:2262. [Google Scholar]
- lxxv.Lopez LPH. Ph.D. Thesis. Massachusetts Institute of Technology; 2005. [Google Scholar]
- lxxvi.Oskam J. Ph.D. Thesis. Massachusetts Institute of Technology; 1993. [Google Scholar]
- lxxvii.Lopez LPH, Schrock RR, Müller P. Organometallics. 2008;27:3857. [Google Scholar]
- lxxviii.a Chisholm MH. Acc. Chem. Res. 1990;23:419. [Google Scholar]; b Chisholm MH, Huffman JC, Hoffman DM. Chem. Soc. Rev. 1985;14:69. [Google Scholar]; c Chisholm MH. J. Chem. Soc. Dalton Trans. 1996:1781. [Google Scholar]
- lxxix.Sharp PR, Schrock RR. J. Organometal. Chem. 1979;171:43. [Google Scholar]
- lxxx.a Johnson LK, Virgil SC, Grubbs RH, Ziller JW. J. Am. Chem. Soc. 1990;112:5384. [Google Scholar]; b Johnson LK, Frey M, Ulibarri TA, Virgil SC, Grubbs RH, Ziller JW. J. Am. Chem. Soc. 1993;115:8167. [Google Scholar]
- lxxxi.Tonzetich ZJ, Schrock RR, Müller P. Organometallics. 2006;25:4301. doi: 10.1021/om060430j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxxxii.Miller GA, Cooper NJ. J. Am. Chem. Soc. 1985;107:709. [Google Scholar]
- lxxxiii.Freundlich JS, Schrock RR, Davis WM. J. Am. Chem. Soc. 1996;118:3643. [Google Scholar]
- lxxxiv.Hirsekorn KF, Veige AS, Marshak MP, Koldobskaya Y, Wolczanski PT, Cundari TR, Lobkovsky EB. J. Am. Chem. Soc. 2005;127:4809. doi: 10.1021/ja046180k. [DOI] [PubMed] [Google Scholar]
- lxxxv.Binger P, Mueller P, Benn R, Mynott R. Angew. Chem., Int. Ed. Engl. 1989;28:610. [Google Scholar]
- lxxxvi.a De la Mata FJ, Grubbs RH. Organometallics. 1996;15:577. [Google Scholar]; b Johnson LK, Grubbs RH, Ziller JW. J. Am. Chem. Soc. 1993;115:8130. [Google Scholar]
- lxxxvii.McLain SJ, Sancho J, Schrock RR. J. Am. Chem. Soc. 1979;101:5451. [Google Scholar]
- lxxxviii.Yang GK, Bergman RG. Organometallics. 1985;4:129. [Google Scholar]
- lxxxix.Schrock RR, Duval-Lungulescu M, Peter Tsang WCP, Hoveyda AH. J. Am. Chem. Soc. 2004;126:1948. doi: 10.1021/ja039173p. [DOI] [PubMed] [Google Scholar]
- xc.Schrock RR, Lopez LPH, Hafer J, Singh R, Sinha A, Müller P. Organometallics. 2005;24:5211. [Google Scholar]
- xci.Copéret C, Chabanas M, Petroff Saint-Arroman R, Basset J-M. Angew. Chem. Int. Ed. 2003;42:156. doi: 10.1002/anie.200390072. [DOI] [PubMed] [Google Scholar]
- xcii.Herrmann WA, Stumpf AW, Priermeier T, Bogdanovic S, Dufaud V, Basset J-M. Angew. Chem. Int. Ed. 1997;35:2803. [Google Scholar]
- xciii.Blanc F, Chabanas M, Copéret C, Fenet B, Herdweck E. J. Organometal. Chem. 2005;690:5014. [Google Scholar]
- xciv.Blanc F, Copéret C, Thivolle-Cazat J, Basset J-M, Lesage A, Emsley L, Sinha A, Schrock RR. Angew. Chem. Int. Ed. 2006;45:1216. doi: 10.1002/anie.200503205. [DOI] [PubMed] [Google Scholar]
- xcv.Blanc F, Salameh A, Thivolle-Cazat J, Basset J-M, Copéret C, Sinha A, Schrock RR. C. R. Chim. 2008;11:137. [Google Scholar]
- xcvi.Rhers B, Salameh A, Baudouin A, Quadrelli EA, Taoufik M, Copéret C, Lefebvre F, Basset J-M, Solans-Monfort X, Eisenstein O, Lukens WW, Lopez LPH, Sinha A, Schrock RR. Organometallics. 2006;25:3554. [Google Scholar]
- xcvii.Blanc F, Copéret C, Thivolle-Cazat J, Basset J-M, Lesage A, Emsley L, Sinha A, Schrock RR. Inorg. Chem. 2006;45:9587. doi: 10.1021/ic061222m. [DOI] [PubMed] [Google Scholar]
- xcviii.Rhers B, Quadrelli EA, Baudouin A, Taoufik M, Copéret C, Lefebvre F, Basset J-M, Fenet B, Sinha A, Schrock RR. J. Organometal. Chem. 2006;691:5448. [Google Scholar]
- xcix.Blanc F, Thivolle-Cazat T, Basset J-M, Copéret C, Hock AS, Tonzetich ZJ, Sinha A, Schrock RR. J. Am. Chem. Soc. 2007;129:1044. doi: 10.1021/ja068249p. [DOI] [PubMed] [Google Scholar]
- c.Blanc F, Berthoud R, Salameh A, Basset J-M, Copéret C, Singh R, Schrock RR. J. Am. Chem. Soc. 2007;129:8434. doi: 10.1021/ja073095e. [DOI] [PubMed] [Google Scholar]
- ci.Blanc F, Basset J-M, Copéret C, Sinha A, Tonzetich ZJ, Schrock RR, Solans-Monfort X, Clot E, Eisenstein O, Lesage A, Emsley L. J. Am. Chem. Soc. 2008;130:5886. doi: 10.1021/ja077749v. [DOI] [PubMed] [Google Scholar]
- cii.Blanc F, Rendón N, Berthoud R, Basset J-M, Copéret C, Tonzetich ZJ, Schrock RR. J. Chem. Soc., Dalton Trans. 2008:3156. doi: 10.1039/b805686m. [DOI] [PubMed] [Google Scholar]
- ciii.Blanc F, Berthoud’ R, Copéret’ C, Lesage A, Emsley L, Singh R, Kreickmann T, Schrock RR. Proc. Nat. Acad. Sci. 2008;105:12123. doi: 10.1073/pnas.0802147105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- civ.Burnett RL, Hughes TR. J. Catal. 1973;31:55. [Google Scholar]
- cv.a Basset JM, Copéret C, Lefort L, Maunders BM, Maury O, Le Roux E, Saggio G, Soignier S, Soulivong D, Sunley GJ, Taoufik M, Thivolle-Cazat J. J. Am. Chem. Soc. 2005;127:8604. doi: 10.1021/ja051679f. [DOI] [PubMed] [Google Scholar]; b Le Roux E, Taoufik M, Copéret C, de Mallmann A, Thivolle-Cazat J, Basset J-M, Maunders BM, Sunley GJ. Angew. Chem., Int. Ed. 2005;44:6755. doi: 10.1002/anie.200501382. [DOI] [PubMed] [Google Scholar]; c Vidal V, Theolier A, Thivolle-Cazat J, Basset J-M. Science. 1997;276:99. doi: 10.1126/science.276.5309.99. [DOI] [PubMed] [Google Scholar]; d Thieuleux C, Copéret C, Dufaud V, Marangelli C, Kuntz E, Basset J-M. J. Mol. Catal. A. 2004;213:47. [Google Scholar]
- cvi.a Schinzel S, Chermette H, Copéret C, Basset J-M. J. Am. Chem. Soc. 2008;130:7984. doi: 10.1021/ja800474h. [DOI] [PubMed] [Google Scholar]; b Copéret C, Maury O, Thivolle-Cazat J, Basset J-M. Angew. Chem., Int. Ed. 2001;40:2331. doi: 10.1002/1521-3773(20010618)40:12<2331::AID-ANIE2331>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; c Basset J-M, Copéret C, Soulivong D, Taoufik M, Thivolle-Cazat J. Angew. Chem., Int. Ed. 2006;45:6082. doi: 10.1002/anie.200602155. [DOI] [PubMed] [Google Scholar]; d Blanc F, Copéret C, Thivolle-Cazat J, Basset J-M. Angew. Chem., Int. Ed. 2006;45:6201. doi: 10.1002/anie.200602171. [DOI] [PubMed] [Google Scholar]
- cvii.a Goldman AS, Roy AH, Huang Z, Ahuja R, Schinski W, Brookhart M. Science. 2006;312:257. doi: 10.1126/science.1123787. [DOI] [PubMed] [Google Scholar]; b Ahuja R, Kundu S, Goldman AS, Brookhart M, Vincente BC, Scott SL. Chem. Commun. 2008:253. doi: 10.1039/b712197k. [DOI] [PubMed] [Google Scholar]
- cviii.a Göttker-Schnetmann I, White P, Brookhart M. J. Am. Chem. Soc. 2004;126:1804. doi: 10.1021/ja0385235. [DOI] [PubMed] [Google Scholar]; b Göttker-Schnetmann I, Brookhart M. J. Am. Chem. Soc. 2004;126:9330. doi: 10.1021/ja048393f. [DOI] [PubMed] [Google Scholar]; c Liu F, Pak EB, Singh B, Jensen CM, Goldman AS. J. Am. Chem. Soc. 1999;121:4086. [Google Scholar]; d Xu W, Rosini GP, Krogh-Jespersen K, Goldman AS, Gupta M, Jensen CM, Kaska WC. Chem. Commun. 1997:2273. [Google Scholar]; e Zhu K, Achord PD, Zhang X, Krogh-Jespersen K, Goldman AS. J. Am. Chem. Soc. 2004;126:13044. doi: 10.1021/ja047356l. [DOI] [PubMed] [Google Scholar]
- cix.Bailey BC, Schrock RR, Kundu S, Goldman AS, Huang Z, Brookhart M. Organometallics. 2009;28:355. [Google Scholar]
- cx.Schrock RR, DePue RT, Feldman J, Yap KB, Yang DC, Davis WM, Park L, DiMare M, Schofield M, Anhaus J, Walborsky E, Evitt E, Krüger C, Betz P. Organometallics. 1990;9:2262. [Google Scholar]
- cxi.Diver ST, Giessert AJ. Chem. Rev. 2004;104:1317. doi: 10.1021/cr020009e. [DOI] [PubMed] [Google Scholar]
- cxii.Singh R, Schrock RR, Müller P, Hoveyda AH. J. Am. Chem. Soc. 2007;129:12654. doi: 10.1021/ja075569f. [DOI] [PubMed] [Google Scholar]
- cxiii.Singh R, Czekelius C, Schrock RR. Macromolecules. 2006;39:1316. [Google Scholar]
- cxiv.Singh R, Schrock RR. Macromolecules. 2008;41:2990. [Google Scholar]
- cxv.Scriban C, Schrock RR, Müller P. Organometallics. 2008;27:6202. [Google Scholar]
- cxvi.Harvey JS, Malcolmson SJ, Dunne KS, Meek SJ, Thompson AL, Schrock RR, Hoveyda AH, Gouverneur V. Angew. Chem. Int. Ed. 2009;48:762. doi: 10.1002/anie.200805066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- cxvii.Ibrahem I, Yu M, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131 doi: 10.1021/ja900097n. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]