

Abstract
Hyperphosphorylated tau makes up the filamentous intracellular inclusions of several neurodegenerative diseases, including Alzheimer's disease 1. In the disease process neuronal tau inclusions first appear in transentorhinal cortex, from where they appear to spread to hippocampal formation and neocortex 2. Cognitive impairment becomes manifest when inclusions reach the hippocampus, with abundant neocortical tau inclusions and extracellular β-amyloid deposits being the defining pathological hallmarks of Alzheimer's disease. Abundant tau inclusions, in the absence of β-amyloid deposits, define Pick's disease, progressive supranuclear palsy, corticobasal degeneration and other diseases 1. Tau mutations cause familial forms of frontotemporal dementia, establishing that tau protein dysfunction is sufficient to cause neurodegeneration and dementia 3-5. Thus, transgenic mice expressing mutant (e.g. P301S) human tau in nerve cells exhibit the essential features of tauopathies, including neurodegeneration and abundant filaments made of hyperphosphorylated tau protein 6,7. In contrast, mouse lines expressing single isoforms of wild-type human tau do not produce tau filaments or display neurodegeneration 7,8. Here we have used tau-expressing lines to investigate whether experimental tauopathy can be transmitted. We show that the injection of brain extract from mutant P301S tau-expressing mice into the brain of transgenic wild-type tau-expressing animals induces the assembly of wild-type human tau into filaments and the spreading of pathology from the site of injection to neighbouring brain regions.
Transgenic mouse lines ALZ17 and P301S tau were used 6,8. Mice from line ALZ17, which express the longest human brain tau isoform (441 amino acids) do not exhibit filamentous tau aggregates (Supplementary Information, Fig. S1a). By contrast, mice from line P301S tau, which express the 383 amino acid human tau isoform with the P301S mutation that causes inherited frontotemporal dementia, develop abundant filamentous tau inclusions (Supplementary Information, Fig. S1a). Both the 383 and 441 amino acid tau isoforms contain 4 microtubule-binding repeats, but they differ by the presence of 2 alternatively spliced N-terminal inserts of 29 amino acids each 9.
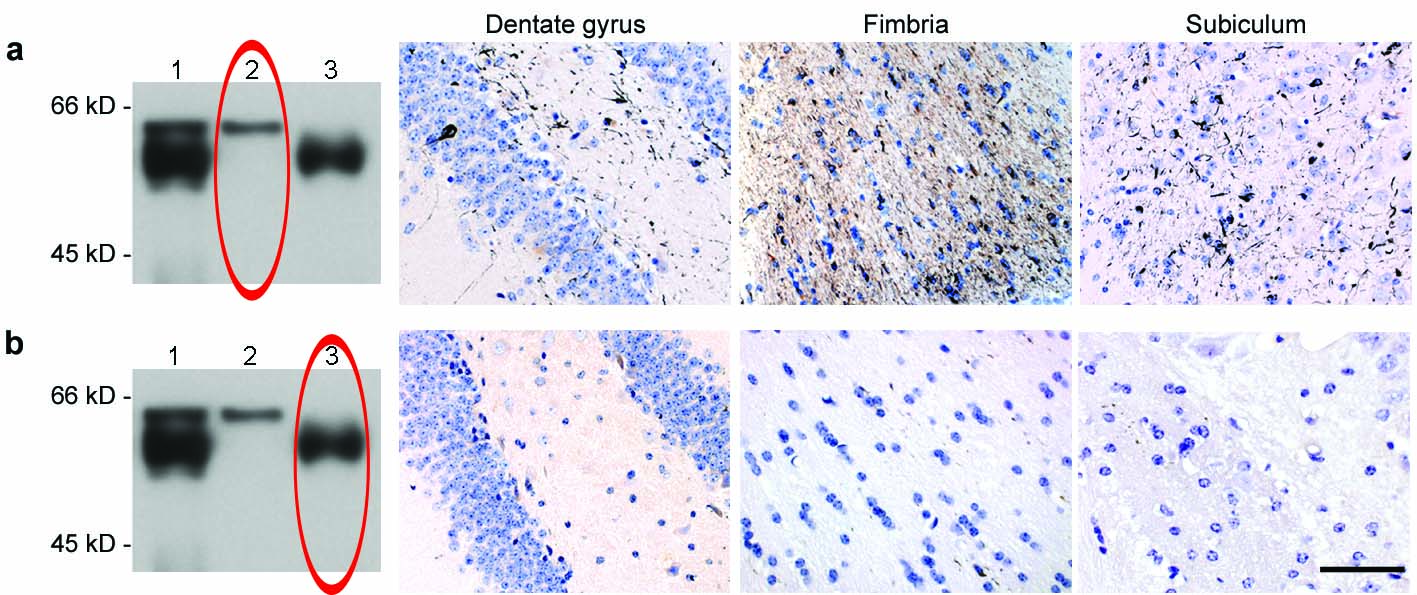
To investigate whether aggregation of tau can be transmitted, we injected diluted extracts of brain homogenates from 6 month-old human P301S tau mice into the hippocampus and the overlying cerebral cortex of 3 month-old ALZ17 mice. Prior to injection, the homogenates were analyzed by immunoblotting and immunoelectron microscopy. Human tau protein bands of 55-64 kDa were detected by Western blotting (Supplementary Information, Fig. S1b). The slowest migrating tau species were immunoreactive with antibody AT100 (Supplementary Information, Fig. S1b) and other phosphorylation-dependent anti-tau antibodies (not shown). By immunoelectron microscopy, tau filaments were present in the tissue extracts (Supplementary Information, Fig. S1c). Injection of brain extract from human P301S tau mice induced filamentous tau pathology in ALZ17 mice, as indicated by the appearance of Gallyas-Braak silver staining 10,11 (Fig. 1a) and the presence of tau filaments by immunoelectron microscopy (Fig. 1b). Gallyas-Braak staining was present intracellularly 6, 12 and 15 months after the injection of brain extract (n=5 per group). In addition to silver-positive nerve cell bodies and processes, the injection of brain extract from P301S tau mice resulted in the appearance of immunoreactivity with antibody AT100 (Fig. 1a), indicative of tau filaments 6. In contrast, no silver-positive lesions were observed at corresponding levels of the hippocampus (Supplementary Information, Fig. S2a) of 18 month-old non-injected ALZ17 mice or in ALZ17 mice 15 months after the injection of brain extract from non-transgenic control mice. ALZ17 animals injected with P301S extract immunodepleted of tau did not reveal any Gallyas- or AT100-positive structures 6 months post-injection (Fig. 1c), demonstrating that the presence of tau in the P301S extract was necessary to induce filamentous tauopathy.
Figure 1. Induction of filamentous tau pathology in ALZ17 mice injected with brain extract from mice transgenic for human P301S tau.
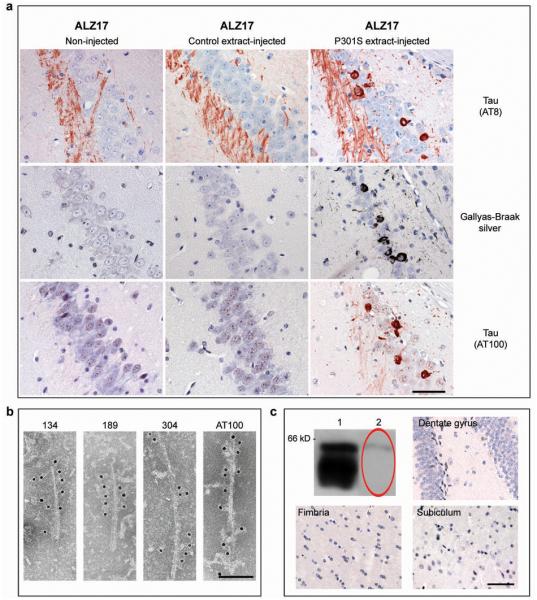
(a) Staining of the hippocampal CA3 region from 18 month-old ALZ17 mice with anti-tau antibody AT8, Gallyas-Braak silver or anti-tau antibody AT100. Non-injected (left), 15 months after injection with brain extract from non-transgenic control mice (middle) and 15 months after injection with brain extract from 6 month-old mice transgenic for human P301S tau protein (right). The sections were counterstained with haematoxylin. Scale bar, 50 μm (same magnification in all panels). (b) Immunoelectron microscopy of filaments extracted from the brain of an ALZ17 mouse 15 months after the injection of brain extract from mice transgenic for human P301S tau. Labelling with anti-tau sera 134, 189 and 304, and antibody AT100. Scale bar, 100 nm. (c) No filamentous tau pathology in ALZ17 mice injected with tau-immunodepleted human P301S tau brain extract. Top left: Western blot with anti-tau antibody HT7 of P301S brain extract before (lane 1) and after (lane 2) immunodepletion. Clockwise: Gallyas-Braak staining of dentate gyrus, subiculum, and fimbria of ALZ17 mice 6 months after injection with tau-immunodepleted P301S brain extract. The sections were counterstained with haematoxylin. Scale bar, 50 μm (same magnification in all panels). The full scan of the Western blot data is available in the Supplementary Information, Fig. S7.
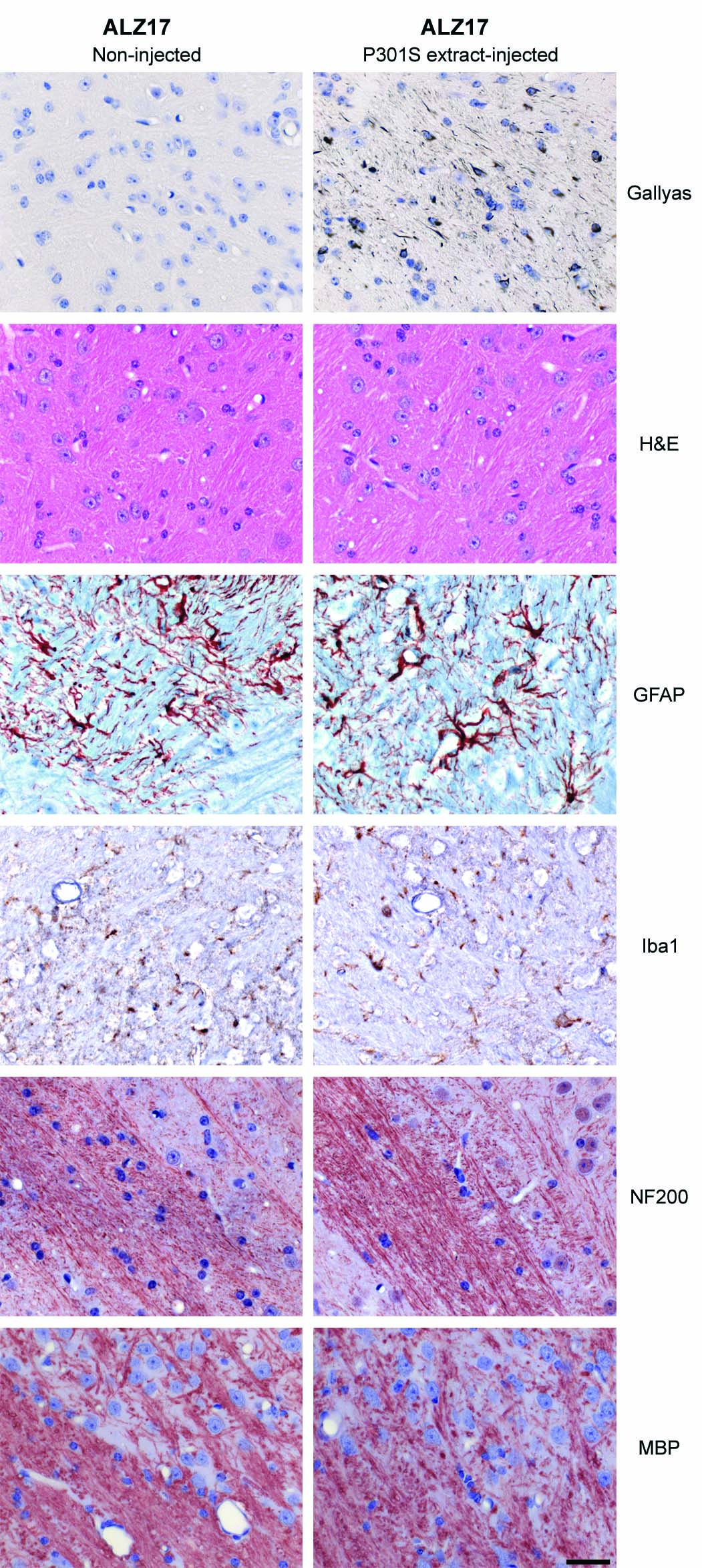
AT8 immunoreactivity (reflecting tau hyperphosphorylation), but not AT100 staining, was present in the hippocampus of 18 month-old ALZ17 mice, as previously reported 8. Following the injection of brain extract from human P301S tau mice, AT8 immunoreactivity became more widespread, indicating the promotion of tau hyperphosphorylation (Fig. 1a). Filamentous tau pathology in P301S tau-injected ALZ17 mice was induced in different cell types. Silver-positive structures morphologically indistinguishable from those found in human tauopathies were observed in the brains of injected ALZ17 mice (Supplementary Information, Fig. S2b). They included neurofibrillary tangles (arrows in panels 1 and 2), neuropil threads (arrowheads in panels 1 and 2) and oligodendroglial coiled bodies (arrows in panels 3 and 4). The silver-positive structures were also immunoreactive for phosphorylated tau (Supplementary Information, Fig. S2b panels 2 and 3). Filaments extracted from injected brains were decorated by phosphorylation-dependent anti-tau antibodies and by antibodies specific for tau isoforms with N-terminal inserts (Fig. 1b). It follows that the filaments had formed from the wild-type 441 amino acid human tau isoform expressed in line ALZ17 and were not derived from the injected material. This is also supported by the finding that inclusions in ALZ17 mice injected with P301S brain extract were immunoreactive with antibodies specific for tau with N-terminal inserts (Supplementary Information, Fig. S3). No silver staining was observed in ALZ17 mice 1 day after the injection of P301S tau brain extract (not shown). No signs of neuronal loss, astrogliosis, inflammation, axonal damage or myelin breakdown were observed in ALZ17 mice 15 months after the injection with P301S tau brain extract when compared to non-injected ALZ17 animals (Supplementary Information, Fig. S4).
The induction of filamentous tau in ALZ17 mice was time- and brain region- dependent. Quantitative assessment in the hippocampus revealed a significant increase in the number of silver-positive lesions between 6, 12 and 15 months after injection (Fig. 2). Neuropil threads were most abundant, followed by coiled bodies and neurofibrillary tangles (Fig. 2b). The same was true for the cerebral cortex, although fewer silver-positive structures developed there over time (Fig. 2a).
Figure 2. Temporal increase in the number of Gallyas-Braak-positive structures at the injection sites (− 2.5 mm from bregma) in ALZ17 mice.

(a) A statistically significant increase in silver-positive structures was observed in hippocampus between 6 and 12 months and between 12 and 15 months. In cerebral cortex, a significant increase was present between 12 and 15 months. The results are expressed as means ± S.E.M. (n=5). *p<0.05. (b) In hippocampus, significant increases in neuropil threads and coiled bodies were observed between 6 and 12 months, and between 12 and 15 months. For neurofibrillary tangles, a significant increase was observed between 12 and 15 months. The results are expressed as means ± S.E.M. (n=5). *p<0.05; **p<0.001; ***p<0.0001. (c) Gallyas staining of the hippocampal region (CA1, stratum radiatum and subiculum) of ALZ17 mice injected with human P301S tau brain extract 6 (top), 12 (middle) and 15 (bottom) months after injection. The sections were counterstained with haematoxylin. Scale bar, 50 μm (same magnification in all panels).
Positive silver staining did not remain confined to the injected areas, but spread also to neighbouring brain regions, as visualized in three coronal brain sections encompassing the injection sites and the levels 1.7 mm anterior and 1.3 mm posterior to the injection site (Fig. 3, Table 1). At the injection level, abundant silver staining was present in the hippocampus, fimbria, optic tract and thalamus with fewer abnormal structures in more distant regions, such as medial lemniscus, zona incerta and cerebral peduncle. The hypothalamus was the most distant region where filamentous tau pathology developed (4 mm ventral from the injection sites; an adult mouse brain measures approximately 5.5 mm in height and 13 mm in length). Almost 2 mm anterior to the injection level, filamentous tau pathology was found in the fimbria, thalamus, internal capsule, caudate-putamen, somatosensory cortex, hypothalamus, and the amygdala 15 months after the injection of brain extract from human P301S tau mice (Table 1, Supplementary Information, Fig. S5a). More than 1 mm posterior to the injection level, the cerebral peduncle, hippocampus, superior colliculus, substantia nigra, entorhinal cortex, deep mesencephalic nucleus, and the pontine nuclei exhibited filamentous lesions 15 months after injection (Table 1, Supplementary Information, Fig. S5b). Moderate filamentous tau pathology was observed in some brain regions of the contralateral, non-injected hemisphere (Supplementary Information, Fig. S5c).
Figure 3. Spreading of filamentous tau pathology in ALZ17 mice injected with brain extract from mice transgenic for human P301S tau.
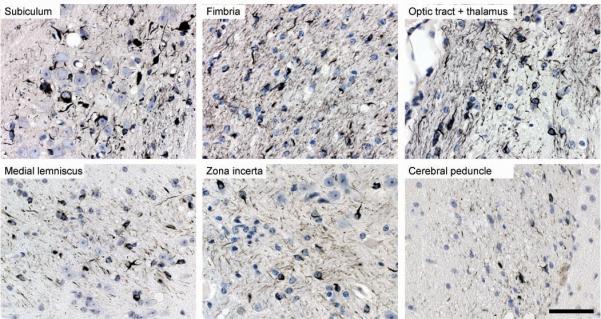
Gallyas-Braak silver staining of brain regions at a distance from the injection sites 15 months post-injection. The sections were counterstained with haematoxylin. Scale bar, 50 μm (same magnification in all panels).
Table1.
Semi-quantitative grading of filamentous tau pathology in ALZ17 mice injected with brain extract from mice transgenic for human P301S tau (n=5 for each time point).
| Anterior (1.7 mm from injection level) | |||
|---|---|---|---|
| Time |
6 months | 12 months | 15 months |
| Region | |||
|
| |||
| fimbria | ++ | +++ | +++ |
| thalamus | + | ++ | ++ |
| internal capsule | − | + | ++ |
| caudate putamen | − | + | + |
| somatosensory cortex | − | + | + |
| hypothalamus | − | − | + |
| amygdala | − | − | + |
|
Injection level | |||
|
Time |
6 months | 12 months | 15 months |
| Region | |||
|
| |||
| hippocampus *1 | ++ | +++ | +++ |
| optic tract | ++ | ++ | +++ |
| fimbria | + | ++ | +++ |
| medial lemniscus | + | ++ | +++ |
| zona incerta | + | ++ | +++ |
| thalamus | + | ++ | ++ |
| cerebral peduncle | + | ++ | ++ |
| visual cortex 2 | + | + | ++ |
| hypothalamus | + | + | + |
| amygdala | − | + | + |
|
Posterior (1.3 mm from injection level) | |||
|
Time |
6 months | 12 months | 15 months |
| Region | |||
|
| |||
| cerebral peduncle | ++ | +++ | +++ |
| hippocampus * | + | ++ | +++ |
| superior colliculus | + | ++ | ++ |
| substantia nigra | + | + | + |
| entorhinal cortex | − | + | ++ |
| deep mesencephalic nucleus | − | + | ++ |
| pontine nuclei | − | − | + |
−: no Gallyas-Braak-positive structures; +: 0-20; ++: 20-40; +++: >40
CA1, CA2, CA3, dentate gyrus and subiculum
injection sites
injection sites
To determine which kind of tau species was responsible for the induction of aggregated tau in ALZ17 mice, P301S extracts containing either soluble or insoluble tau were injected into ALZ17 mice. Injection of insoluble tau induced a large number of Gallyas-Braak-positive structures in ALZ17 animals (Supplementary Information, Fig. S6), similar to what was observed after injection of crude P301S extracts. In addition, there was a similar increase and spreading of fibrillar tau pathology over time (data not shown). Injection of soluble tau induced Gallyas-Braak-positive structures to a much lesser extent (<5%), when compared with insoluble tau (Supplementary Information, Fig. S6). Thus, it is predominantly insoluble tau species that induce tau aggregation.
We next injected brain extract from human P301S tau mice into non-transgenic control mice. Interestingly, the wild-type mice showed Gallyas-Braak- and AT100-positive threads and coiled bodies (but no NFTs) 6 months (Fig. 4) and 12 months after injection (data not shown). They remained confined to the injection site and did not increase in number between 6 and 12 months post-injection (data not shown). Gallyas-Braak-positive structures were stained with anti-murine tau antibody MT1, but not with human tau-specific antibody T14, demonstrating that filamentous tau induced in wild-type mice was made of murine tau (Fig. 4). No tau pathology was observed in sham-lesioned mice (Fig. 4). These results suggest that the expression of human tau in ALZ17 mice was essential for the temporal increase and spreading of filamentous tau pathology after injection of human P301S tau brain extract. However, the presence of a small number of mouse tau filaments in the extract could be responsible for the aggregation of murine tau in wild-type animals and explain the modest amount of filamentous tauopathy. It remains to be clarified to what extent tau expression levels influence spreading.
Figure 4. Induction of filamentous tau pathology in non-transgenic C57BL/6 mice injected with brain extract from mice transgenic for human P301S tau.

Gallyas-Braak silver staining and immunostaining with phosphorylation-dependent anti-tau antibody AT8, murine tau-specific antibody MT1 and human tau-specific antibody T14 in the fimbria of a non-transgenic mouse 6 months after the injection with the human P301S tau brain extract (left column). Gallyas-Braak-, AT8- and MT1-positive, T14-negative, tau deposits were observed in neurites (arrows) and in oligodendrocytes (arrowheads). No tau pathology was observed in age-matched sham-lesioned animals (right column). The sections were counterstained with haematoxylin. Scale bar, 50 μm (same magnification in all panels).
The present findings show that the intracerebral injection of brain extract from mice with a filamentous tau pathology induces the formation and spreading of silver-positive aggregates made of hyperphosphorylated tau in mice transgenic for human wild-type tau, demonstrating the experimental transmission of tauopathy. During the process leading to Alzheimer's disease, neuronal tau pathology forms in a stereotypical fashion in transentorhinal cortex, from where it appears to spread to hippocampal formation and neocortex 2, consistent with a uniform biological process. We also show the appearance over time of silver-positive structures at sites that are at a considerable distance from the injection sites in hippocampus and cerebral cortex. Brain regions that develop pathology are connected anatomically to the injection sites or to each other. This, together with the stereotypical appearance of silver-positive structures in defined brain regions over time in all animals studied (Table 1), is clearly indicative of the active induction and spreading of pathology rather than the passive diffusion of tau aggregates from the injection sites to more distant regions. The lack of obvious signs of neurodegeneration in ALZ17 mice 15 months after the injection with brain extract from mice transgenic for human P301S tau contrasts with the nerve cell loss that characterizes the P301S tau mice 6. This suggests that the molecular tau species responsible for transmission and neurotoxicity are not identical. It remains to be seen whether neurodegenerative changes appear at later time points.
A combined neuronal and glial tau pathology is the defining feature of a number of human neurodegenerative diseases characterized by the assembly of wild-type four-repeat tau isoforms into filaments. They include progressive supranuclear palsy, corticobasal degeneration and argyrophilic grain disease 12. By contrast, in Pick's disease, three-repeat tau isoforms are found in the mostly neuronal inclusions and in Alzheimer's disease, both three- and four-repeat tau isoforms make up neurofibrillary tangles 12. In addition, the morphologies of tau filaments vary widely in different tauopathies 13. Together with the findings reported here showing transmission and spreading of filamentous tau pathology, this is reminiscent of mammalian and yeast prions, for which different strains have been described, based on the existence of separate conformers of assembled protein 14. It is tempting to speculate that distinct tau strains may underlie the pathogenesis of different sporadic tauopathies. This hypothesis can now be tested experimentally by injecting ALZ17 mice with brain extracts from patients with sporadic tauopathies.
From the increase in silver-positive structures over time, it appears likely that nerve cell processes and oligodendrocytes are major sites of filament induction. These findings are comparable to previous work using mouse models of prion diseases, β-amyloid deposition and some peripheral amyloidoses 15-22. However, unlike the extracellular location of filamentous deposits in those other diseases, tau inclusions are intracellular. Mechanisms must therefore exist by which tau aggregates can either gain access to the inside of cells or activate filament-inducing cascades from the outside. Inside cells, tau aggregates could function as seeds for the ordered assembly of transgenic wild-type human tau into silver- and AT100-positive filamentous deposits. Intracellular filamentous tau pathology could also be induced by extracellular tau aggregates. Previous work has indeed shown potentiation of tau pathology by extracellular β-amyloid aggregates in mice transgenic for mutant human tau 18,19. However, no such effect was observed in mice transgenic for wild-type human tau 23.
It has been suggested that misfolded superoxide dismutase, which causes a subset of familial forms of amyotrophic lateral sclerosis, is secreted from motor neurons 24. By analogy, spreading of tau pathology may also result from the release of aggregates from affected nerve cells and glial cells. It will be important to investigate whether such mechanisms exist for misfolded tau. The formation of tau aggregates in a single brain cell and their subsequent spreading may therefore be at the origin of sporadic tauopathies. Consequently, the presence of tau aggregates in the extracellular space would be an obligatory step in the events leading to disease. Immunisation strategies may prevent this process 25.
The present findings demonstrate the transmission of tauopathy between transgenic mouse lines and describe an experimental system in which to investigate the spreading of pathology and the existence of tau strains. It will be important to identify the molecular tau species capable of inducing transmission, aggregation and spreading. Similar mechanisms may also underlie Parkinson's disease and dementia with Lewy bodies, where α-synuclein pathology appears to spread from brainstem areas to midbrain and neocortex 26. Furthermore, in Parkinson's disease patients who had undergone transplantation of fetal midbrain neurons, filamentous α-synuclein pathology appeared to spread from affected host tissue to the grafted neurons 27. Unlike prion diseases 28, human tauopathies are not believed to be infectious. Experimental model systems of the type described here now make it possible to dissect the similarities and differences between tauopathies and prion diseases.
METHODS
Mice
Homozygous human wild-type tau ALZ17 transgenic mice 8, homozygous human P301S tau transgenic mice 6 and non-transgenic control mice, all females on a C57BL/6 background, were used. Experiments were performed in compliance with protocols approved by the local Basel Committee for Animal Care and Animal Use.
Preparation of brain extracts
Six month-old mice transgenic for human P301S tau and age-matched non-transgenic control mice were deeply anaesthetized with pentobarbital (100 mg/kg) and killed by decapitation. Brainstems were dissected, snap-frozen in liquid nitrogen and stored at −80° C. Brainstems from 3 mice were combined and homogenized at 10% (w/v) in sterile phosphate-buffered saline (PBS), briefly sonicated (Branson 450, output 2, 5 times 0.9 sec) and centrifuged at 3,000×g at 4° C for 5 min. The supernatant was aliquoted, snap-frozen and kept at −80° C until use.
Separation of soluble and insoluble tau
Brainstem extracts from mice transgenic for human P301S tau were centrifuged at 100,000×g at 4°C for 20 min. The supernatants containing soluble tau were aliquoted and stored at −80°C until use. The pellets containing insoluble tau were resuspended in the original volume of PBS, sonicated (Branson 450, output 1.5, 3 times 0.9 sec), aliquoted and stored at −80°C until use.
Immunodepletion of tau
A mixture of anti-tau antibodies HT7 (Pierce, Rockford, IL), AT8 (Pierce), and TAU-5 (Biosource, Camarilla, CA) (20 μg of each antibody) was incubated for 2 h with 10 μl of Protein G Sepharose 4 Fast flow (PGS, Pharmacia). Sepharose beads were washed twice with 1 ml PBS and the liquid removed with a glass capillary. P301S brain extract (100 μl, prepared as described above) was added to semi-dry beads and allowed to react overnight at 4°C on an overhead mixer. The beads were settled by centrifugation (1 min, 200×g) and 70 μl of the tau-depleted supernatant were removed. Five μl were used for Western blot analysis and the remainder stored at −80°C until use. As a control, P301S extract was treated as above, but in the absence of anti-tau antibodies.
Western blotting
Brain extracts were run on 8% Tris-glycine SDS-PAGE, transferred onto a PVDF membrane (Immobilon P, Millipore, Zug, Switzerland) and probed with anti-tau antibodies HT7 (1:1,000), AT8 (1:1,000) and AT100 (1:200, Pierce). HT7 recognizes human tau, but not mouse tau 29, AT8 labels tau phosphorylated at S202 and T205 30 and AT100 detects tau phosphorylated at T212, S214 and T217 31. The secondary antibody was goat anti-mouse IgG (Chemicon, Temecula, CA). For signal detection, ECL plus (Amersham, Piscataway, NJ) was used in conjunction with Amersham Hyperfilm ECL.
Stereotaxic surgery
Three month-old ALZ17 mice and non-transgenic C57BL/6 control mice were anaesthetized with a mixture of ketamine (10 mg/kg) and xylazine (20 mg/kg). When deeply anaesthetized, the mice were placed on a heating pad to maintain body temperature during surgery. Using a Hamilton syringe, the hippocampus (A/P, −2.5 mm from bregma; L, +/− 2.0 mm; D/V, −1.8 mm) and the overlying cerebral cortex (A/P, −2.5 mm from bregma; L, +/− 2.0 mm; D/V, −0.8 mm) each received a unilateral (right hemisphere) stereotaxic injection of 2.5 μl brain extract, at a speed of 1.25 μl/min. Following injection, the needle was kept in place for an additional 3 min before gentle withdrawal. The surgical area was cleaned with sterile saline and the incision sutured. Mice were monitored until recovery from anaesthesia and checked weekly following surgery. For sham-lesioned wild-type mice (n=3), the same procedure was applied without injection of brain extract.
Histology and immunohistochemistry
ALZ17 mice were deeply anaesthetized with pentobarbital (100 mg/kg) and killed by transcardial perfusion with 20 ml cold PBS, followed by 20 ml 4% paraformaldehyde in PBS. The brains were dissected and post-fixed overnight. Following paraffin embedding, 5 μm coronal sections were prepared. Sections were silver-impregnated following the method of Gallyas-Braak to visualize filamentous tau pathology 10,11. Haematoxylin and eosin staining (H&E) was performed for morphological analysis. For immunohistochemistry, the following anti-tau antibodies were used: T14 (1:1,000, Zymed, San Francisco, CA), AT8 (1:1,000), AT100 (1:1,000), BR189 (1:500), BR304 (1:500) and MT1 (1:500). T14 is specific for human tau 32. MT1 was raised against amino acids 114-127 of the longest brain isoform of mouse tau. It is specific for mouse tau and does not recognize human tau 6. BR189 and BR304 were raised against amino acids 76-87 and 45-73 of tau, respectively (in the numbering of the longest human brain tau isoform). They are specific for tau isoforms with one (BR304) or two (BR304 and BR189) N-terminal inserts 9. Antibody Olig2 (1:500, Chemicon, Temecula, CA) was used to stain oligodendrocytes. Antibodies NF200 (1:100, DakoCytomation, Glostrup, Denmark), Iba1 (1:100, Abcam, Cambridge, UK), GFAP (1:100, Abcam), and MBP (1:500, Abcam) were used to visualize neurofilaments, astrocytes, microglia, and myelin basic protein, respectively. Secondary antibodies were from Vector Laboratories, Burlingame, CA (Vectastain ABC kit).
Immunoelectron microscopy
Immunoelectron microscopy of injected P301S brain extract using anti-tau antibody BR134 was performed as described 33. BR134 was raised against a synthetic peptide corresponding to amino acids 428-441 of tau 9. In some experiments, sarkosyl-insoluble tau was prepared from injected ALZ17 brains, as described 34. Anti-tau antibodies BR134, BR189, BR304 and AT100 were used to identify tau filaments.
Quantitative analysis of tau pathology
ALZ17 mice injected with P301S brain extract were analyzed at 6, 12 and 15 months post-injection (five mice per time point). Gallyas-Braak-stained coronal sections were selected at identical brain coordinates 35 (anterior to the injection level, − 0.8 mm from bregma; injection level, − 2.5 mm from bregma; posterior to the injection level, − 3.8 mm from bregma). Silver-positive structures (neurofibrillary tangles, neuropil threads and coiled bodies) were counted at these 3 levels with a ×20 objective (Zeiss, Feldbach, Switzerland). At the injection level (− 2.5 mm from bregma), total counts of silver-positive structures were assessed in the injection sites hippocampus (CA1, CA2, CA3, dentate gyrus and subiculum) and visual cortex. In the visual cortex, silver-positive lesions developed just around and at a very short distance from the needle tract. For semi-quantitative assessment, Gallyas-Braak staining was graded as follows: (−), no silver-positive structures; (+), < 20 silver-positive structures; (++), 20-40 silver-positive structures; (+++), > 40 silver-positive structures. For each group there was only slight variation between individual animals.
Statistical analysis
Statistical analysis of quantitative tau pathology data was performed using the Poisson regression algorithm with the help of the SAS 8.02 software. Link function was the logarithm and scale was estimated by deviance (to account for overdispersion). Significance tests for differences between mice at 12 months and at 6 months following inoculation with brain extract and between mice at 15 months and at 12 months following inoculation were obtained. No multiplicity correction was applied. The results of the significance tests are reported in Figure 2 (panels a and b), as follows: *p<0.05; **p<0.001; ***p<0.0001.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the Swiss National Science Foundation (3100AO-120261) (M.T.); the Alzheimer Association (ZEN-06-27341), the German National Genome Network (NGFN2) and the German Competence Network in Degenerative Dementias (01GI0705) (M.J.); the U.K. Medical Research Council (R.A.C, G.F., M.G.) and the U.K. Alzheimer's Research Trust (M.G.). We thank K.H. Wiederhold (Novartis Institutes for Biomedical Research, Basel) and N. Schaeren-Wiemers (University Hospital Basel) for antibodies and helpful discussions.
Footnotes
Supplementary Information
REFERENCES
- 1.Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 3.Poorkaj P, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 4.Hutton M, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 5.Spillantini MG, et al. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. USA. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen B, et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Probst A, et al. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000;99:469–481. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- 8.Frank S, Clavaguera F, Tolnay M. Tauopathy models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:39–53. doi: 10.1007/s00401-007-0291-9. [DOI] [PubMed] [Google Scholar]
- 9.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 10.Gallyas F. Silver staining of Alzheimer's neurofibrillary changes by means of physical development. Acta Morphol. Acad. Sci. Hung. 1971;19:1–8. [PubMed] [Google Scholar]
- 11.Braak H, Braak E, Ohm T, Bohl J. Silver impregnation of Alzheimer's neurofibrillary changes counterstained for basophilic material and lipofuscin pigment. Stain Technol. 1988;63:197–200. doi: 10.3109/10520298809107184. [DOI] [PubMed] [Google Scholar]
- 12.Lee VM-Y, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 13.Crowther RA, Goedert M. Abnormal tau-containing filaments in neurodegenerative diseases. J. Struct. Biol. 2000;130:271–279. doi: 10.1006/jsbi.2000.4270. [DOI] [PubMed] [Google Scholar]
- 14.Prusiner SB. Prions. Proc. Natl. Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Legname G, et al. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 16.Kane MD, et al. Evidence for seeding of β-amyloid by intracerebral infusion of Alzheimer brain extracts in β̄amyloid precursor protein-transgenic mice. J. Neurosci. 2000;15:3606–3611. doi: 10.1523/JNEUROSCI.20-10-03606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer-Luehmann M, et al. Exogenous induction of cerebral beta-amyloidosis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 18.Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 19.Bolmont T, et al. Induction of tau pathology by intracerebral infusion of amyloid-β-containing brain extract and by amyloid-β deposition in APP X tau transgenic mice. Am. J. Pathol. 2007;171:2012–2020. doi: 10.2353/ajpath.2007.070403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xing Y, et al. Transmission of mouse senile amyloidosis. Lab. Invest. 2001;81:493–499. doi: 10.1038/labinvest.3780257. [DOI] [PubMed] [Google Scholar]
- 21.Lundmark K, et al. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc. Natl. Acad. Sci. USA. 2002;99:6979–6984. doi: 10.1073/pnas.092205999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker LC, LeVine H, III, Mattson MP, Jucker M. Inducible proteopathies. Trends Neurosci. 2006;29:438–443. doi: 10.1016/j.tins.2006.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boutajangout, et al. Characterisation of cytoskeletal abnormalities in mice transgenic for wild-type human tau and familial Alzheimer's disease mutants of APP and presenilin-1. Neurobiol. Dis. 2004;15:47–60. doi: 10.1016/j.nbd.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Urushitani M, et al. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nature Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- 25.Asuni AA, Boutajangout A, Quartermain D, Sigurdson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 2007;27:9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braak H, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 27.Brundin P, Li J-Y, Holton JL, Lindvall O, Revesz T. Research in motion: the enigma of Parkinson's disease pathology spread. Nature Rev. Neurosci. 2008;9:741–745. doi: 10.1038/nrn2477. [DOI] [PubMed] [Google Scholar]
- 28.Brown P, et al. Human spongiform encephalopathy: The National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann. Neurol. 1994;35:513–529. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- 29.Mercken M, et al. Affinity purification of human tau proteins and the construction of a sensitive sandwich enzyme-linked immunosorbent assay for human tau detection. J. Neurochem. 1992;58:548–553. doi: 10.1111/j.1471-4159.1992.tb09754.x. [DOI] [PubMed] [Google Scholar]
- 30.Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci. Lett. 1995;189:167–170. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- 31.Yoshida H, Goedert M. Sequential phosphorylation of tau protein by cAMP-dependent protein kinase and SAPK4/p38δ or JNK2 in the presence of heparin generates the AT100 epitope. J. Neurochem. 2006;99:154–164. doi: 10.1111/j.1471-4159.2006.04052.x. [DOI] [PubMed] [Google Scholar]
- 32.Kosik KS, et al. Epitopes that span the tau molecule are shared with paired helical filaments. Neuron. 1988;1:817–825. doi: 10.1016/0896-6273(88)90129-8. [DOI] [PubMed] [Google Scholar]
- 33.Crowther RA. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc. Natl. Acad. Sci. USA. 1991;88:2288–2292. doi: 10.1073/pnas.88.6.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 35.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic Press; New York: 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.