

Abstract
There is increasing evidence that a number of cytokines and their receptors are involved in the processes that lead to the development and maintenance of neuropathic pain states. Here we demonstrate that levels of CX3CR1 (the receptor for the chemokine fractalkine) mRNA in lumbar dorsal root ganglia (DRG) increase 5.8-fold 7 days after sciatic nerve axotomy, and 1.7- and 2.9-fold, 3 and 7 days respectively, after the spared nerve injury (SNI) model of neuropathic pain. In contrast, no significant change in the levels of fractalkine mRNA is apparent in the DRG after axotomy or SNI. The increase in CX3CR1 mRNA is paralleled by a 3.9- and 2.1-fold increase in the number of CX3CR1-positive macrophages in the DRG 7 days after axotomy and SNI, respectively. Expression of CX3CR1 in macrophages is also markedly increased in the sciatic nerve proximal to site of injury, by 25.7-fold after axotomy and 16.2-fold after SNI, 7 days after injury. Intra-neural injection into the sciatic nerve of 400 ng or 100 ng of fractalkine in adult 129OlaHsd mice significantly delayed the development of allodynia for 3 days following SNI. Further, CX3CR1 knockout (KO) mice display an increase in allodynia for three weeks after SNI compared to strain-matched Balb/c controls. Taken together, these results suggest an anti-allodynic role for fractalkine and its receptor in the mouse.
Keywords: CX3CL1, CX3CR1, dorsal root ganglia, fractalkine, nerve injury, neuropathic pain
Damage or injury to the peripheral nervous system (PNS) induces alterations in many of the properties of sensory neurons and their central connections, which may lead to allodynia (the perception of pain from a normally innocuous stimulus), hyperalgesia (an exaggerated response to any given pain stimulus) and the development of chronic (neuropathic) pain. Neuropathic pain is hugely detrimental to an individual’s quality of life, causes considerable disability and is an increasing economic burden to health-care systems. Despite intensive research over a number of decades, the mechanisms that cause chronic pain states are still poorly understood (reviewed in Baron 2006).
One approach to elucidate the mechanisms that underlie the response of the PNS to injury is to identify and study factors and/or their receptors whose levels and distribution in the DRG are altered by nerve injury. Using degenerate primer PCR-screening we have previously demonstrated that CX3CR1, the receptor for fractalkine (CX3CL1), is expressed in the mouse DRG and that the levels of this chemokine receptor are markedly up-regulated after sciatic nerve section (axotomy) (Holmes et al. 2004). Fractalkine is the only ligand known to bind and activate the CX3CR1 receptor, and does not bind any other known mammalian chemokine receptors (Stievano et al. 2004). Thus, the normal promiscuity that typifies the chemokine superfamily is absent, suggesting that the roles played by fractalkine and CX3CR1 are non-redundant and may be of critical importance to the nervous system.
Fractalkine and CX3CR1 are widely expressed in the nervous system (Harrison et al. 1994, 1998; Bazan et al. 1997), with fractalkine being constitutively expressed by neurons and CX3CR1 predominantly, but not exclusively, by microglia (Bazan et al. 1997; Nishiyori et al. 1998; Tarozzo et al. 2003), and it has been suggested that fractalkine might act as a specific signal between neurons and glia (Harrison et al. 1998). Recent studies in rat have demonstrated that fractalkine is expressed by most, if not all, neurons of the DRG and dorsal horn of the spinal cord, and levels do not change after peripheral nerve injury or inflammation. In contrast, CX3CR1 seems to be exclusively expressed by microglia in the spinal cord and macrophages in the DRG, and markedly up-regulates in the cord, but apparently not in the rat DRG after nerve injury, and variably after inflammation (Verge et al. 2004; Lindia et al. 2005).
The functional significance of the fractalkine and CX3CR1 interaction in the spinal cord has recently been studied (Milligan et al. 2004, 2005). Acute intrathecal (IT) administration of fractalkine to intact adult rats produced pain facilitation (mechanical allodynia and thermal hyperalgesia), whereas IT injection of an immunoneutralising antiserum to CX3CR1 attenuated the development and maintenance of neuropathic pain (Milligan et al. 2004). Similarly, injection of fractalkine into the periaqueductal grey (PAG) region of the midbrain is also pro-nociceptive (Chen et al. 2007). In contrast, here we show that peripheral intra-neural administration of fractalkine inhibits allodynia in the spared nerve injury (SNI) model of neuropathic pain. Further, CX3CR1 KO mice have increased allodynia in the SNI model of neuropathic pain. These results suggest an anti-allodynic role for fractalkine, and its receptor CX3CR1, in the mouse.
Materials and methods
Animals
Adult 12-14 week old male mice of the 129OlaHsd strain were used in all experiments unless otherwise specified. Animals heterozygous for the loss-of-function mutation in the CX3CR1 gene (Combadière et al. 2003) were backcrossed for six generations with wildtype Balb/c mice. Heterozygotes were crossed and KO and wildtype lines were then derived and bred at the University of Bristol. Animals were housed in standard conditions and fed standard chow and water ad libitum. Repeat serological testing confirmed that the animals used in this study were negative for murine cytomegalovirus. All procedures conformed to the principles of UK animal legislation and animals were humanely sacrificed at the end of each experiment. Tissue from CX3CR1+/GFP mutant mice on the C57BL/6 strain (Jung et al. 2000) were obtained from the colony at OSU, Ohio, USA.
Surgery
For all surgical procedures mice were anaesthetised with Hypnorm (Fentanyl citrate 0.315 mg/mL + Fluanisone 10 mg/mL, VetaPharma Ltd):Hypnovel (Midazolam 5 mg/mL, Roche):water at a ratio of 1:1: 2 at 4 μL/g. For axotomy the sciatic nerve was exposed at lower-thigh level and a ~3 mm segment of nerve resected. For intra-neural injections the sciatic nerve was exposed as above as previously described (Zelenka et al. 2005), and mouse fractalkine (chemokine domain, amino acid residues 25-105, R and D Systems) or vehicle [phosphate-buffered saline (PBS)/0.01% bovine serum albumin] in a volume of 4 μL was slowly injected perineuronally over a 5 min period into the nerve using a pulled microcapillary pipette attached to a Hamilton syringe. In some experiments intrasciatic injections were performed immediately prior to SNI (see below). A modification of the SNI model (Decosterd and Woolf 2000), as previously described (Holmes et al. 2003; Shields et al. 2003) was used as a model of neuropathic pain. An incision was made in the lower thigh, just above the level of the knee, exposing the three terminal branches of the sciatic nerve: the common peroneal, tibial and sural nerves. The common peroneal and sural nerves were tightly ligated with 7/0 silk and sectioned distal to the ligation, leaving the tibial branch intact. Following these procedures the overlying muscle and skin was sutured and the animals allowed to recover. In sham-operated animals the sciatic nerve was exposed at lower thigh level, but not lesioned.
Quantitative RT-PCR
CX3CR1 expression was originally detected in mouse DRG poly(A)+ RNA during screening with the degenerate primers 2F [5′-GC ACC G(CT)I AT(GCT) AG(CT) GTI GAC CG(AC) TAT-3′, where I is deoxyinosine] and 1R [5′-G CAG GAA G(GC)C (AG)TA GA(GT) (AG)A(GCT) GGG GTT-3′], designed to amplify G-protein coupled receptor sequences encoding transmembrane domains 3-7 (TM3-TM7) (N.K., unpublished). Quantitative RT-PCR was performed as described previously (Kerr et al. 2004). Briefly, ipsilateral L4 and L5 DRGs were collected from three groups of six animals after sciatic axotomy, SNI or sham surgery. Total RNA was purified using a Qiagen RNeasy Mini Kit, and treated with 10 units of RQ1 RNase-free DNase (Promega) which was inactivated during re-extraction of RNA following RNA cleanup. For each RNA sample, duplicate reactions with reverse transcriptase (RT+) and one without enzyme (RT-), were performed with 1 μg RNA per 50 μL reverse transcription (RT) reaction using Applied Biosystems TaqMan Reverse Transcription Reagents Kit. Triplicate PCRs were performed on each of these samples. The sequence detection primers were designed using Primer Express software (Applied Biosystems). Primers to detect CX3CR1 were forward primer 5′-TCCGCAACTCGGAAGTCAAC-3′ and reverse primer 5′-GAAGTAGCAAAAGCTCATGATAAGCA-3′ corresponding, respectively, to nt 791-810 and 858-833 of the published CX3CR1 cDNA sequence (AF074912) (Combadière et al. 1998a). Primers to detect fractalkine were forward primer 5′-CTCATCCGCTATCAGCTAAACCA-3′ and reverse primer 5′-CCTTCGGGTCAGCACAGAAG-3′ corresponding, respectively, to nt 227-249 and 320-301 of the published fractalkine cDNA sequence (NM_009142). SYBR green was used as reporter dye. Endogenous control gene GAPDH cDNA primers were forward primer 5′-GCAGTGGCAAAGTGGAGATTG-3′ and reverse primer 5′-CTGGAACATGTAGACCATGTAGTTGA-3′ and TaqMan probe (VIC label, plus TAMRA quencher) 5′-CCATCAACGACCCCTTCATTGAC-3′, that correspond, respectively, to nt 111-131, 184-159 and 135-157 (M32599). Relative expression levels were determined by the comparative Ct method. Results are presented as mean of three separate groups of DRG ± SEM of log transformed data. Levels of expression in ipsilateral L4 and L5 DRG after axotomy and SNI were compared to that of ipsilateral L4 and L5 DRG after sham surgery.
Behavioural testing
In all tests the examiner was blind to the treatment group or genotype of the mice and the animals were tested in constant conditions. Thermal thresholds were measured according to the method of Hargreaves et al. (Hargreaves et al. 1988). Animals were habituated to the testing environment for at least 30 min. The hind paws were exposed to a beam of radiant heat through a transparent Perspex holding box (Ugo Basile) and the latency of withdrawal recorded automatically. Each hind paw was measured three times with an interval of at least 5 min between measurements.
Mechanical thresholds were measured with a series of calibrated von Frey filaments from 0.005 g to a maximum of 3.63 g. Animals were placed in Perspex enclosures placed on an elevated grid (Ugo Basile) and habituated for 30 min prior to testing. Mechanical sensitivity was assessed on each mid-plantar hind paw, employing the up-down testing paradigm to determine the threshold force required to elicit a withdrawal response to 50% of stimulations (Dixon 1980; Chaplan et al. 1994). Mice were tested on three separate days prior to surgery (with the third set of measurements used as baseline values), and then at regular intervals after surgery.
Immunohistochemistry
Mice were intracardially perfused with 4% paraformaldehyde/PBS and the spinal columns and sciatic nerves removed and post-fixed for 4 h at 22°C. Spinal cords, L4 and L5 DRG were dissected and all tissues equilibrated in 20% sucrose overnight at 4°C, embedded in Tissue-Tek® OCT mounting medium, frozen on dry ice and then cryostat sectioned (8 μM sections) onto slides. Sections were blocked and permeabilised in 10% normal donkey serum/PBS 0.2% Triton-X-100 (PBST) for 1 h at 22°C. Sections were then incubated in rabbit anti-CX3CR1 (Nanki et al. 2004) (1 : 1000) overnight at 22°C. Sections were then washed 3 × 10 min in PBS and incubated in donkey anti-rabbit FITC-conjugated secondary antibody (Jackson Labs) for 3 h at 22°C. After washing, sections were mounted in Vectashield (Vector Labs). For double labelling, sections were washed as above then incubated in either goat anti-Iba-1 (1 : 500) (Abcam), mouse anti-glial fibrillary acidic protein (1 : 400) (Sigma) or mouse anti-neuronal nuclei (NeuN) (1 : 500; Chemicon), overnight at 22°C. Sections were washed as above then incubated in the appropriate Cy3-conjugated donkey secondary antibody. Images were taken using a Leica fluorescent microscope with DC500 camera and IM50 software. Double labelled, high magnification images were taken using a Leica TCS-NT confocal laser-scanning microscope. For quantification of the numbers of CX3CR1-positive cells, 8-12 sections from 129OlaHsd L5 DRG and sciatic nerve were imaged and captured (using the Leica image capture system described above) at x200 magnification, imported into Adobe Photoshop (v7 for Windows) and then further magnified 4-fold. The number of CX3CR1-positive profiles/500 μm2 for each section was then counted (two areas per section). At least 770, 450 and 280 positive profiles were counted for each axotomised, SNI and sham operated animal, respectively (n = 4 for each group) and for sciatic nerves, cells were counted in 8-12 sections from each nerve, in an area of 500 μm2 mid nerve, proximal to the lesion site. At least 1000, 700 and 100 positive profiles were counted for each axotomised, SNI and sham operated animal, respectively (n = 4 for each group).
Statistics
Data are presented as means ± SEM. Quantitative PCR and CX3CR1-positive cell count data were analysed by Student’s t-test. Behavioural data were analysed by one-way repeated measures of anova (Kruskal-Wallis test), followed by Dunn’s multiple comparison post-hoc test (using version 4 of GraphPad Prism software, San Diego, USA). A p value of < 0.05 was taken to be significant.
Results
Quantification of CX3CR1 and fractalkine mRNA in the DRG after nerve injury
The CX3CR1 cDNA was cloned from an adult mouse (129OlaHsd) DRG cDNA library, and using quantitative RT-PCR (Taqman) analysis we found that the mRNA levels in the DRG increased 5.8 ± 0.5-fold at 7 days after sciatic nerve axotomy, compared to sham-operated DRG. Similarly, DRG expression of CX3CR1 rose 1.7 ± 0.1- and 2.9 ± 0.2-fold, at 3 and 7 days after the SNI model of neuropathic pain compared to sham-operated DRG (Fig. 1a). In contrast, levels of fractalkine mRNA in the DRG did not significantly change after axotomy or SNI, compared to sham-operated DRG.
Fig. 1.
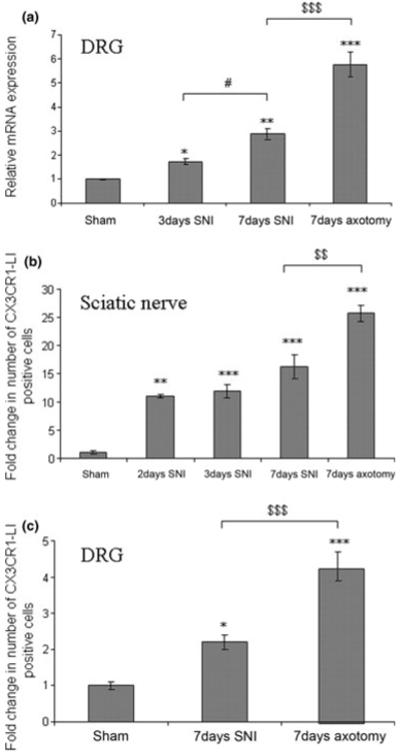
Up-regulation of CX3CR1 expression in the 129OlaHsd DRG and sciatic nerve after peripheral nerve injury. (a) Quantitative RT-PCR of CX3CR1 mRNA in L4/L5 DRG after SNI or axotomy, normalised to sham operated (at day 7) L4/L5 DRG (n = 3). Sham vs. 3 day SNI, p < 0.05 (*); sham vs. 7 day SNI, p < 0.01 (**); sham vs. axotomy, p < 0.001 (***); 7 day SNI vs. 7 day axotomy, p < 0.001 ($$$); 3 day SNI vs. 7 day SNI, p < 0.05 (#). (b) Fold change in the number of CX3CR1-LI-cells in 500 μm2 sciatic nerve proximal to the injury site after SNI or axotomy, normalised to equivalent sham operated (at day 7) sciatic nerve (n = 4), Sham vs. 2 day SNI, p < 0.01 (**); sham vs. 3 day and 7 day SNI, p < 0.001 (***); sham 7 day axotomy, p < 0.001 (***). 7 day SNI vs. 7 day axotomy, p < 0.01 ($$). (c) Fold change in the number of CX3CR1-LI-cells in the ipsilateral L5 DRG after SNI or axotomy, normalised to sham operated (at day 7) L5 DRG (n = 4). Sham vs. 7 day SNI, p < 0.05 (*); sham vs. 7 day axotomy, p < 0.001 (***); 7 day SNI vs. 7 day axotomy, p < 0.001 ($$$).
Distribution of CX3CR1-positive cells in the sciatic nerve, DRG and spinal cord after nerve injury
The upregulation of CX3CR1 mRNA in the DRG after axotomy and SNI was reflected in a marked increase in CX3CR1 protein, as determined by immunohistochemistry. A significant increase in CX3CR1 expression was also observed in the sciatic nerve, especially proximal to the site of injury, and in the dorsal horn of the spinal cord, albeit to a lesser extent than the sciatic nerve.
Sciatic nerve
Longitudinal sections of sciatic nerve from 129OlaHsd mice demonstrated a marked increase in the numbers of CX3CR1-like immunoreactive (LI) expressing cells after axotomy and SNI (Fig. 2b and c, respectively) compared to the uninjured nerve (Fig. 2a), particularly proximal to the injury site. Quantification of CX3CR1-LI positive cells showed a 25.7 ± 1.3-fold increase in the number of CX3CR1-LI positive cells in the sciatic nerve proximal to the injury site 7 days after axotomy (Fig. 1b, CX3CR1 positive profiles/500 μm2: sham 6 ± 2 vs. axotomy 154 ± 8). Similarly a 11.1 ± 0.3, 11.9 ± 1.2 and 16.2 ± 2.0-fold increase was observed in the sciatic nerve proximal to the injury site 2, 3 and 7 days after SNI, respectively, compared to shamoperated sciatic nerve (Fig. 1, CX3CR1 positive profiles/500 μm2: sham 6 ± 2 vs. SNI 67 ± 2, 71 ± 7, and 97 ± 12 at 2, 3 and 7 days, respectively). The majority of the CX3CR1-LI-postive cells co-localised with the macrophage marker Iba-1 (Ito et al. 1998) (Fig. 3 top panel). No significant differences were noted in the number of CX3CR1-LI-positive cells in the sciatic nerve of sham operated and naïve animals.
Fig. 2.
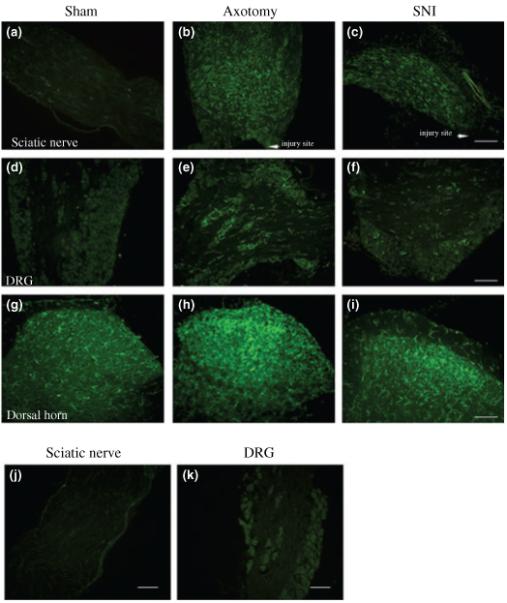
CX3CR1 in 129OlaHsd mice is present at very low levels in the intact adult sciatic nerve (a), with higher expression observed in the intact DRG (d) and dorsal horn of the spinal cord (g). 7 days after axotomy or SNI, CX3CR1-LI staining is markedly increased in the sciatic nerve (b and c) and DRG (e and f), and to a lesser extent in the ipsilateral dorsal horn of the spinal cord (h and i), as compared to sham operated controls. Bars = 100 μm. No CX3CR1 staining was noted in the sciatic nerve (j) or DRG (k) of CX3CR1 KO animals, 7 days after axotomy.
Fig. 3.
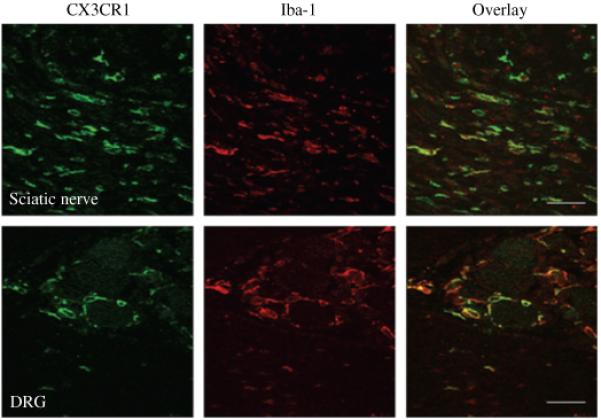
Co-localisation of CX3CR1-LI (green) with Iba-1 (red), a macrophage marker, is noted in the sciatic nerve (top panel) and DRG (bottom panel), one week after SNI in 129OlaHsd mice. Bars = 25 μm.
DRG
Immunohistochemistry for CX3CR1 in 129OlaHsd mice demonstrated that both the staining intensity and number of CX3CR1-LI-positive cells in the DRG increased after axotomy (Fig. 2e), and SNI (Fig. 2f). This was confirmed by analysis of green fluorescent protein (GFP)-labelled cells in CX3CR1+/GFP mutant mice, in which a GFP reporter gene was inserted into the CX3CR1 locus (Jung et al. 2000) (data not shown). Quantification of CX3CR1-LI cells revealed a 3.9 ± 0.5- and 2.2 ± 0.2-fold increase in the number of CX3CR1-LI positive cells in the DRG 7 days after axotomy and SNI respectively, compared to sham-operated DRG (Fig. 1c, CX3CR1-LI-positive profiles/500 μm2: sham 12 ± 1 vs. axotomy 49 ± 5 vs. SNI 25 ± 2). CX3CR1 was solely expressed in non-neuronal cells adjacent to neuronal soma in the DRG, and co-localised with Iba-1 (Fig. 3, bottom panel) but not with the neuronal marker NeuN (data not shown). No significant differences were noted in the number of CX3CR1-LI-positive cells in the DRG of sham operated and naïve animals.
Spinal cord
An increase in CX3CR1-LI cells was seen in 129OlaHsd animals after axotomy and SNI, ipsilateral to the side of the lesion, particularly in the medial two thirds of laminae I-III where the majority of injured primary afferents terminate (Fig. 2h and i, respectively). This finding was identical to that observed in the GFP-positive cells in the lumbar spinal cord of CX3CR1+/GFP mice (data not shown).
No specific CX3CR1 staining was observed in the sciatic nerve or DRG of CX3CR1 KO mice (Combadière et al. 2003), 7 days after axotomy (Fig. 2j and k). The upregulation in CX3CR1 expression in the sciatic nerve, DRG and spinal cord of Balb/c mice 7 days after SNI, was very similar to that observed in the 129OlaHsd mice (data not shown). No specific staining was observed in sections in which the primary antibody was omitted (data not shown).
Effect of intra-neural injection of fractalkine into the sciatic nerve on allodynia
In order to study the nociceptive effects of peripheral administration of fractalkine, we first performed intra-neural injection of the chemokine or vehicle into the sciatic nerve at the mid-thigh level of uninjured adult 129OlaHsd mice. The previous studies had used central administration of fractalkine varying between 30-100 ng (Milligan et al. 2005; Chen et al. 2007), and we therefore used a similar dose range (30-400 ng) by intra-neural injection. Baseline responses were measured prior to and at 2 days after 400 ng fractalkine injection and no significant effects on mechanical or thermal withdrawal thresholds were noted (1.69 ± 0.21 g vs. 1.48 ± 0.57 g; and 16.8 ± 4.5 vs. 15.9 ± 1 s, pre-injection and day 2, respectively). Similarly, no effects were noted on mechanical or thermal withdrawal thresholds in intact mice after an injection of 100 ng or 30 ng fractalkine (data not shown).
We then repeated the fractalkine or vehicle intra-sciatic injections immediately prior to performing the SNI model of neuropathic pain. In all experiments vehicle-injected mice developed significant mechanical allodynia by 2 days post-SNI/injection (Fig. 4, p < 0.05, one-way repeated measures anova followed by Dunn’s multiple comparison test). However, when 400 ng or 100 ng of fractalkine were administered into the sciatic nerve at the same time as SNI surgery, there was a significant delay in the development of allodynia by 3 days, when compared to the vehicle-injected group (Fig. 4a and b). When 30 ng of fractalkine was injected into the sciatic nerve at the same time as SNI surgery, no significant difference in allodynia was observed (data not shown). As expected, the sham SNI group did not develop allodynia (data not shown). Of note, mice do not develop significant or consistent thermal hyperalgesia following SNI (Holmes et al. 2003).
Fig. 4.
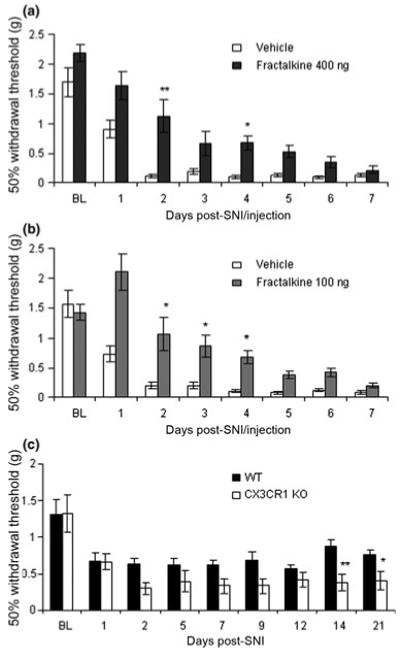
Intra-sciatic fractalkine delays the development of allodynia in 129OlaHsd mice. Responses to mechanical stimuli were assessed prior to (baseline, BL), and after SNI surgery in which (a) vehicle (n = 8) or fractalkine at 400 ng (n = 8) and (b) vehicle (n = 10) or 100 ng fractalkine (n = 9) was injected into the sciatic nerve at the time of SNI surgery. In both groups vehicle but not fractalkine-treated mice developed significant allodynia by 2 days post-SNI (p < 0.01 repeated measures anova with Dunn’s multiple comparison post-hoc test). The injection of fractalkine had a highly significant effect in both groups (p < 0.0001, Kruskal-Wallis, one-way anova), and the development of allodynia was significantly delayed by 3 days in both the 400 ng and 100 ng fractalkine-treated groups (using Kruskal-Wallis one-way anova with Dunn’s multiple comparison post-hoc test). Key: * and ** refer to p < 0.05 and p < 0.01. (c) CX3CR1 KO mice have increased allodynia after SNI. Withdrawal thresholds to mechanical stimulation were measured before (baseline values, BL) and after SNI. Both Balb/c (n = 12) and CX3CR1 KO mice (n = 13) developed significant allodynia by 2 days post-SNI (p < 0.05 Balb/c and p < 0.001 CX3CR1 KO, using one-way repeated measures anova with Dunn’s multiple comparison post-hoc test). Genotype had a highly significant effect on withdrawal thresholds (p < 0.0001, one-way anova Kruskal-Wallis test). The KO mice had reduced mechanical withdrawal thresholds compared to Balb/c mice for the duration of the experiment, but it was only at the later time points, once the allodynia had become chronic, that the differences were significant using Dunn’s multiple comparison post-hoc test. Key: * and ** refer to p < 0.05 and p < 0.01.
These results imply that peripheral CX3CR1 activation by fractalkine can play an anti-nociceptive role after nerve injury when the expression of the receptor in the sciatic nerve and DRG is up-regulated.
Development of allodynia in CX3CR1 KO mice
Since the intra-neural administration of exogenous fractalkine attenuates neuropathic pain-related behaviour, we next examined the behavioural nociceptive phenotype of CX3CR1 KO compared to sex- and age-matched Balb/c animals. Baseline thermal and mechanical withdrawal thresholds were measured prior to SNI surgery and neither were significantly different between genotypes. Thermal withdrawal thresholds were 14.0 ± 0.8 s and 12.9 ± 0.8 s for Balb/c and CX3CR1 KO animals, respectively. Mechanical withdrawal thresholds were 1.31 ± 0.19 g and 1.32 ± 0.25 g for Balb/c and KO animals, respectively (Fig. 4c, BL). Following SNI both groups developed significant allodynia by day 2 (p < 0.05 wild-type mice and p < 0.001 CX3CR1 KO mice, one-way repeated measures anova followed by Dunn’s multiple comparison test). The CX3CR1 KO mice developed greater allodynia compared to Balb/c animals at all time points after day 2, for the 3 weeks of testing (Fig. 4c). Genotype had a significant effect on withdrawal thresholds (p < 0.0001, one-way anova). It was only at the later time points once the allodynia had become chronic, that the differences were significant using Dunn’s multiple comparison test. Of note, the Balb/c mice developed allodynia at day 1 post-SNI surgery compared to day 2 for the 129OlaHsd animals (Fig. 4). This strain variability in responses to nociceptive stimuli is in keeping with previous work (Mogil et al. 1999).
Discussion
There is an increasing body of published work demonstrating that a number of chemokines are involved in the development of neuropathic pain states. They are often proinflammatory and increase pain-related behaviour in a number of different neuropathic and/or inflammatory pain models, reviewed in (Abbadie 2005; White et al. 2005; Scholz and Woolf 2007). The chemokine fractalkine, acting via its cognate G-protein coupled receptor CX3CR1, has recently been proposed to be involved in signalling between neurons and glia. Unlike other chemokines, fractalkine is a relatively large multimodular protein consisting of a N-terminal chemokine domain with a mucin-like stalk connected to a transmembrane domain (which tethers the molecule to the plasma membrane), and an intracellular cytoplasmic domain (Bazan et al. 1997; Pan et al. 1997). The soluble, secreted, chemotactic form of fractalkine is produced by cleavage from the transmembrane domain (Garton et al. 2001; Tsou et al. 2001). Both forms of fractalkine activate CX3CR1 (Imai et al. 1997; Combadière et al. 1998b; Harrison et al. 1998; Meucci et al. 2000).
The present study has focused on the potential effect of intra-neural administration of fractalkine in the modulation of pain-related behaviour, following injury to the PNS. Here we show that there is an increase in CX3CR1 expression in the DRG, spinal cord and, most markedly, in the sciatic nerve after nerve injury. The CX3CR1-positive cells in the spinal cord most likely represent resident microglia as previously described in the rat (Verge et al. 2004; Lindia et al. 2005; Cardona et al. 2006), though it is possible that invading peripheral macrophages may also contribute to the observed staining, as suggested by Hu et al. (Hu et al. 2007). The majority of CX3CR1-positive cells in the DRG and sciatic nerve appear to be macrophages, though the possibility that a minority might represent Schwann and/or satellite cells cannot at present be excluded. We observed no evidence for neuronal expression of CX3CR1 in either the DRG or spinal cord, similar to that reported in the rat (Verge et al. 2004). However, it has previously been shown that fractalkine can invoke an immediate (within milliseconds) increase in the levels of intracellular calcium and electrical excitability in a subpopulation of cultured (and thus axotomised) neonatal DRG neurons (Oh et al. 2001), implying that some sensory neurons may express the receptor, at least in the neonatal period. In contrast to CX3CR1, levels of fractalkine mRNA did not change after axotomy or SNI in the DRG, which is also in keeping with previous findings in the rat (Verge et al. 2004).
The functional significance of the fractalkine/CX3CR1 interaction in the spinal cord has recently been studied by Watkins and co-workers. They demonstrated that acute injection (IT) of 30 ng fractalkine induced a rapid and short lived alteration in pain-related behaviour characterised by a reduction in thermal and mechanical thresholds in intact adult rats, whereas IT injection of 0.3 ng fractalkine only decreased mechano-sensory withdrawal thresholds (Milligan et al. 2004). The pro-nociceptive effect of fractalkine was blocked by the simultaneous IT administration of an anti-CX3CR1 antibody (Milligan et al. 2004). This antibody also reduced the allodynia and thermal hyperalgesia that occurred after chronic constriction injury or sciatic inflammatory injury (Milligan et al. 2004). Two groups have shown that the pro-nociceptive effects of IT fractalkine administration were blocked by the co-administration of minocycline (an inhibitor of microglial activation), an IL-1 receptor antagonist, immunoneutralisation of IL-6, or a nitric oxide (NO) synthase inhibitor (Milligan et al. 2005; Owolabi and Saab 2006). Most recently, injection of fractalkine into the periaqueductal grey (PAG) region of the midbrain has been shown to be pro-nociceptive (Chen et al. 2007) similar to that observed when injected IT. Of note, the endogenous membrane-bound form of fractalkine has been shown to be released by cathepsin S, a cysteine protease expressed by activated microglia, after peripheral nerve injury. Furthermore, pharmacological inhibition of cathepsin S can reverse neuropathic pain-like behaviour (Clark et al. 2007).
Taken together, these results suggest that central administration of fractalkine, or release of membrane-bound fractalkine by cathepsin S, appears to induce a pro-nociceptive effect in the intact rat and after peripheral nerve injury by activating microglia in the spinal cord.
In contrast to the above, our findings suggest that exogenous fractalkine exerts an anti-allodynic effect when administered intra-neurally after SNI, and that this effect is mediated via activation of CX3CR1-bearing macrophages. The absence of effect of exogenous fractalkine in the intact adult animal presumably reflects the low levels of CX3CR1 expression in the uninjured sciatic nerve and DRG. The source of the CX3CR1-expressing cells is most likely a combination of an influx of non-resident macrophages into the sciatic nerve in response to injury, which is further increased by the injection of fractalkine, since it is a leukocyte chemotactic factor (Imai et al. 1997; Nanki et al. 2004). In addition, it is possible that the exogenously administered fractalkine either passively diffuses, or is retrogradely axonally transported, to the cell bodies of the sensory neurons, where the chemokine may be released and in turn activate the increased number of CX3CR1-bearing macrophages in the DRG.
The work of Watkins and others demonstrates that a single IT or PAG injection of fractalkine modulates nociception for up to 90 min (Milligan et al. 2004, 2005; Chen et al. 2007; Sun et al. 2007). Similarly, the effects of exogenous fractalkine in a range of other paradigms that include synaptic plasticity (Bertollini et al. 2006), neuroprotection (Limatola et al. 2005), microglial cell migration (Lauro et al. 2006) and leukocyte adhesion (Kerfoot et al. 2003) are also short lived, with effects usually lasting less than 2 h. Compatible with these findings, a number of chemokines have short half-lives (Saccani et al. 2000; Hillyer and Male 2005), though the half-life of fractalkine has not been reported. These data therefore imply that the prolonged antiallodynic effects observed after an intra-neural injection of fractalkine are likely to be indirect and mediated by the release from peripheral CX3CR1-positive macrophages of down-stream effectors, which in combination inhibit allodynia for a prolonged period. A diverse range of cytokines and other inflammatory mediators are known to be secreted by activated glia, many of which have been shown to modulate nociception/allodynia (reviewed in Scholz and Woolf 2007; White et al. 2007). These include the pronociceptive cytokines IL-1b, IL-12, IL-18, IFNγ and TNFα (Verri et al. 2005, 2007; Wolf et al. 2006; Xu et al. 2006; Liu et al. 2007) and the anti-nociceptive cytokines IL-2, IL-4, and IL-10 (Yao et al. 2002; Vale et al. 2003). To date, there is no direct evidence that fractalkine can mediate the release of any of the above cytokines from glia/macrophages in the peripheral nervous system. In contrast in the central nervous system, the effects of fractalkine appear to be dependent upon the release of IL-1, IL-6 or NO from the spinal cord (Milligan et al. 2005). Further, fractalkine inhibits the release of the pro-nociceptive cytokine TNFα from cultured glia obtained from newborn rat brain (Zujovic et al. 2000; Mizuno et al. 2003). Other examples of chemokines modulating the release or activity of known pain-mediators include the regulation of calcitonin generelated peptide by CCL2 and CXCL1 (Qin et al. 2005) and transient receptor potential vanilloid I by CCL3 (Zhang et al. 2005). In addition to the above mechanisms by which fractalkine might modulate allodynia it is also possible that the chemokine might recruit opioid-containing immune cells to the site of injury and/or DRG, which would induce local opioid release and would in turn reduce allodynia (Szabo et al. 2002; Brack et al. 2004; Rittner and Stein 2005).
In the context of previous studies, our results suggest that fractalkine may play opposing and site-dependent nociceptive roles dependent on the route of administration, though the summation of the two seems to be inhibitory, at least in the mouse, on the basis of the increased allodynia in the CX3CR1 KO animals. Consistent with this, Chen found an unequivocal pro-nociceptive response with a single PAG injection of 100 ng of fractalkine (Chen et al. 2007), whilst we found the opposite effect when the same dose was administered by intra-neural injection. It should however be noted that the experiments by the Watkins and Chen groups were performed in rat whereas ours were undertaken in the mouse. Although the distribution of the receptor appears to be similar in the spinal cord in both species, the absence of an upregulation of CX3CR1 in the rat DRG after nerve injury (Verge et al. 2004), may in part explain the observed differences in nociception. Further, it is possible that there may be differences in the immunomodulatory systems between these two species which might alter the behavioural effects of the fractalkine/CX3CR1 interaction. Compatible with this, distinct inflammatory reactions after spinal cord injury have been noted in mice and rats (Sroga et al. 2003).
In summary, we have demonstrated that the expression of the fractalkine receptor CX3CR1 is markedly up-regulated after nerve injury in macrophages, in the adult mouse sciatic nerve and DRG. Intra-neural administration of fractalkine delays the development of allodynia in the SNI model of neuropathic pain, and consistent with this, CX3CR1 KO mice have an increase in allodynia in the same pain model. Further investigations are now warranted to confirm that the effects of exogenous fractalkine on allodynia are mediated by CX3CR1, and to identify the down-stream effectors of CX3CR1 activation that modulate allodynia.
Acknowledgement
This work was supported by the Medical Research Council, the Medical Research Committee of the Charitable Trusts for the United Bristol Hospitals, and The National Institutes of Health (NINDS).
Abbreviations used
- DRG
dorsal root ganglia
- GFP
green fluorescent protein
- IT
intrathecal
- KO
knockout
- PAG
periaqueductal grey
- PBS
phosphate-buffered saline
- SNI
spared nerve injury
References
- Abbadie C. Chemokines, chemokine receptors and pain. Trends Immunol. 2005;26:529–534. doi: 10.1016/j.it.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Baron R. Mechanisms of disease: neuropathic pain-a clinical perspective. Nat. Clin. Pract. Neurol. 2006;2:95–106. doi: 10.1038/ncpneuro0113. [DOI] [PubMed] [Google Scholar]
- Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- Bertollini C, Ragozzino D, Gross C, Limatola C, Eusebi F. Fractalkine/CX3CL1 depresses central synaptic transmission in mouse hippocampal slices. Neuropharmacology. 2006;51:816–821. doi: 10.1016/j.neuropharm.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Brack A, Rittner HL, Machelska H, Leder K, Mousa SA, Schafer M, Stein C. Control of inflammatory pain by chemokine-mediated recruitment of opioid-containing polymorphonuclear cells. Pain. 2004;112(3):229–238. doi: 10.1016/j.pain.2004.08.029. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen X, Geller EB, Rogers TJ, Adler MW. The chemokine CX3CL1/fractalkine interferes with the antinociceptive effect induced by opioid agonists in the periaqueductal grey of rats. Brain Res. 2007;1153:52–57. doi: 10.1016/j.brainres.2007.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Grist J, et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. PNAS. 2007 doi: 10.1073/pnas.0610811104. 0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combadière C, Gao J, Tiffany HL, Murphy PM. Gene cloning, RNA distribution, and functional expression of mCX3CR1, a mouse chemotactic receptor for the CX3C chemokine fractalkine. Biochem. Biophys. Res. Commun. 1998a;253:728–732. doi: 10.1006/bbrc.1998.9849. [DOI] [PubMed] [Google Scholar]
- Combadière C, Salzwedel K, Smith ED, Tiffany HL, Berger EA, Murphy PM. Identification of CX3CR1. A chemotactic receptor for the human CX3C chemokine fractalkine and a fusion coreceptor for HIV-1. J. Biol. Chem. 1998b;273:23799–23804. doi: 10.1074/jbc.273.37.23799. [DOI] [PubMed] [Google Scholar]
- Combadière C, Potteaux S, Gao JL, Esposito B, Casanova S, Lee EJ, Debre P, Tedgui A, Murphy PM, Mallat Z. Decreased atherosclerotic lesion formation in CX3CR1/apolipo-protein E double knockout mice. Circulation. 2003;107:1009–1016. doi: 10.1161/01.cir.0000057548.68243.42. [DOI] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, Raines EW. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) J. Biol. Chem. 2001;276:37993–38001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Barber CM, Lynch KR. cDNA cloning of a G-protein-coupled receptor expressed in rat spinal cord and brain related to chemokine receptors. Neurosci. Lett. 1994;169:85–89. doi: 10.1016/0304-3940(94)90362-x. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl Acad. Sci. USA. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillyer P, Male D. Expression of chemokines on the surface of different human endothelia. Immunol. Cell Biol. 2005;83:375–382. doi: 10.1111/j.1440-1711.2005.01345.x. [DOI] [PubMed] [Google Scholar]
- Holmes FE, Bacon A, Pope RJ, Vanderplank PA, Kerr NC, Sukumaran M, Pachnis V, Wynick D. Transgenic overexpression of galanin in the dorsal root ganglia modulates pain-related behavior. Proc. Natl Acad. Sci. USA. 2003;100:6180–6185. doi: 10.1073/pnas.0937087100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes FE, Arnott N, Isles N, Thomas CK, Combadière C, Wynick D. The chemokine fractalkine modulates nociceptive behaviour and axonal regeneration after peripheral nerve injury. Soc. Neurosci. 2004 abstract 64.13. [Google Scholar]
- Hu P, Bembrick AL, Keay KA, McLachlan EM. Immune cell involvement in dorsal root ganglia and spinal cord after chronic constriction or transection of the rat sciatic nerve. Brain Behav. Immun. 2007;21:599–616. doi: 10.1016/j.bbi.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Imai T, Hieshima K, Haskell C, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res. Mol. Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerfoot SM, Lord SE, Bell RB, Gill V, Robbins SM, Kubes P. Human fractalkine mediates leukocyte adhesion but not capture under physiological shear conditions; a mechanism for selective monocyte recruitment. Eur. J. Immunol. 2003;33:729–739. doi: 10.1002/eji.200323502. [DOI] [PubMed] [Google Scholar]
- Kerr NC, Holmes FE, Wynick D. Novel isoforms of the sodium channels Nav1.8 and Nav1.5 are produced by a conserved mechanism in mouse and rat. J. Biol. Chem. 2004;279:24826–24833. doi: 10.1074/jbc.M401281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauro C, Catalano M, Trettel F, Mainiero F, Ciotti MT, Eusebi F, Limatola C. The chemokine CX3CL1 reduces migration and increases adhesion of neurons with mechanisms dependent on the beta1 integrin subunit. J. Immunol. 2006;177:7599–7606. doi: 10.4049/jimmunol.177.11.7599. [DOI] [PubMed] [Google Scholar]
- Limatola C, Lauro C, Catalano M, Ciotti MT, Bertollini C, Di AS, Ragozzino D, Eusebi F. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J. Neuroimmunol. 2005;166:19–28. doi: 10.1016/j.jneuroim.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Lindia JA, McGowan E, Jochnowitz N, Abbadie C. Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J. Pain. 2005;6:434–438. doi: 10.1016/j.jpain.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Liu YL, Zhou LJ, Hu NW, Xu JT, Wu CY, Zhang T, Li YY, Liu XG. Tumor necrosis factor-alpha induces longterm potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK. Neuropharmacology. 2007;52:708–715. doi: 10.1016/j.neuropharm.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl Acad. Sci. USA. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Zapata V, Chacur M, et al. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur. J. Neurosci. 2004;20:2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x. [DOI] [PubMed] [Google Scholar]
- Milligan E, Zapata V, Schoeniger D, Chacur M, Green P, Poole S, Martin D, Maier SF, Watkins LR. An initial investigation of spinal mechanisms underlying pain enhancement induced by fractalkine, a neuronally released chemokine. Eur. J. Neurosci. 2005;22:2775–2782. doi: 10.1111/j.1460-9568.2005.04470.x. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003;979:65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Wilson SG, Bon K, et al. Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. Pain. 1999;80:67–82. doi: 10.1016/s0304-3959(98)00197-3. [DOI] [PubMed] [Google Scholar]
- Nanki T, Urasaki Y, Imai T, Nishimura M, Muramoto K, Kubota T, Miyasaka N. Inhibition of fractalkine ameliorates murine collagen-induced arthritis. J. Immunol. 2004;173(11):7010–7016. doi: 10.4049/jimmunol.173.11.7010. [DOI] [PubMed] [Google Scholar]
- Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, Kume T, Akaike A, Satoh M. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett. 1998;429:167–172. doi: 10.1016/s0014-5793(98)00583-3. [DOI] [PubMed] [Google Scholar]
- Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J. Neurosci. 2001;21(14):5027–5035. doi: 10.1523/JNEUROSCI.21-14-05027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owolabi SA, Saab CY. Fractalkine and minocycline alter neuronal activity in the spinal cord dorsal horn. FEBS Lett. 2006;580:4306–4310. doi: 10.1016/j.febslet.2006.06.087. [DOI] [PubMed] [Google Scholar]
- Pan Y, Lloyd C, Zhou H, et al. Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation. Nature. 1997;387:611–617. doi: 10.1038/42491. [DOI] [PubMed] [Google Scholar]
- Qin X, Wan Y, Wang X. CCL2 and CXCL1 trigger calcitonin gene-related peptide release by exciting primary nociceptive neurons. J. Neurosci. Res. 2005;82:51–62. doi: 10.1002/jnr.20612. [DOI] [PubMed] [Google Scholar]
- Rittner HL, Stein C. Involvement of cytokines, chemokines and adhesion molecules in opioid analgesia. Eur. J. Pain. 2005;9:109–112. doi: 10.1016/j.ejpain.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Saccani A, Saccani S, Orlando S, Sironi M, Bernasconi S, Ghezzi P, Mantovani A, Sica A. Redox regulation of chemokine receptor expression. Proc. Natl Acad. Sci. USA. 2000;97:2761–2766. doi: 10.1073/pnas.97.6.2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat. Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Shields SD, Eckert WA, III, Basbaum AI. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J. Pain. 2003;4:465–470. doi: 10.1067/s1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- Sroga JM, Jones TB, Kigerl KA, McGaughy VM, Popovich PG. Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J. Comp. Neurol. 2003;462(2):223–240. doi: 10.1002/cne.10736. [DOI] [PubMed] [Google Scholar]
- Stievano L, Piovan E, Amadori A. C and CX3C chemokines: cell sources and physiopathological implications. Crit. Rev. Immunol. 2004;24:205–228. doi: 10.1615/critrevimmunol.v24.i3.40. [DOI] [PubMed] [Google Scholar]
- Sun S, Cao H, Han M, Li TT, Pan HL, Zhao ZQ, Zhang YQ. New evidence for the involvement of spinal fractalkine receptor in pain facilitation and spinal glial activation in rat model of monoarthritis. Pain. 2007;129:64–75. doi: 10.1016/j.pain.2006.09.035. [DOI] [PubMed] [Google Scholar]
- Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ, Rogers TJ. Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc. Natl Acad. Sci. USA. 2002;99(16):10276–10281. doi: 10.1073/pnas.102327699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarozzo G, Bortolazzi S, Crochemore C, Chen SC, Lira AS, Abrams JS, Beltramo M. Fractalkine protein localization and gene expression in mouse brain. J. Neurosci. Res. 2003;73:81–88. doi: 10.1002/jnr.10645. [DOI] [PubMed] [Google Scholar]
- Tsou CL, Haskell CA, Charo IF. Tumor necrosis factor-alpha-converting enzyme mediates the inducible cleavage of fractalkine. J. Biol. Chem. 2001;276:44622–44626. doi: 10.1074/jbc.M107327200. [DOI] [PubMed] [Google Scholar]
- Vale ML, Marques JB, Moreira CA, Rocha FA, Ferreira SH, Poole S, Cunha FQ, Ribeiro RA. Antinociceptive effects of interleukin-4, -10, and -13 on the writhing response in mice and zymosan-induced knee joint incapacitation in rats. J. Pharmacol. Exp. Ther. 2003;304:102–108. doi: 10.1124/jpet.102.038703. [DOI] [PubMed] [Google Scholar]
- Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 2004;20:1150–1160. doi: 10.1111/j.1460-9568.2004.03593.x. [DOI] [PubMed] [Google Scholar]
- Verri WA, Jr, Molina RO, Schivo IR, Cunha TM, Parada CA, Poole S, Ferreira SH, Cunha FQ. Nociceptive effect of subcutaneously injected interleukin-12 is mediated by endothelin (ET) acting on ETB receptors in rats. J. Pharmacol. Exp. Ther. 2005;315:609–615. doi: 10.1124/jpet.105.089409. [DOI] [PubMed] [Google Scholar]
- Verri WA, Jr, Cunha TM, Parada CA, Poole S, Liew FY, Ferreira SH, Cunha FQ. Antigen-induced inflammatory mechanical hypernociception in mice is mediated by IL-18. Brain Behav. Immun. 2007;21:535–543. doi: 10.1016/j.bbi.2006.11.005. [DOI] [PubMed] [Google Scholar]
- White FA, Bhangoo SK, Miller RJ. Chemokines: integrators of pain and inflammation. Nat. Rev. Drug Discov. 2005;4:834–844. doi: 10.1038/nrd1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Jung H, Miller RJ. Chemokines and the pathophysiology of neuropathic pain. PNAS. 2007;104:20151–20158. doi: 10.1073/pnas.0709250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G, Gabay E, Tal M, Yirmiya R, Shavit Y. Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain. 2006;120:315–324. doi: 10.1016/j.pain.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Xu JT, Xin WJ, Zang Y, Wu CY, Liu XG. The role of tumor necrosis factor-alpha in the neuropathic pain induced by Lumbar 5 ventral root transection in rat. Pain. 2006;123:306–321. doi: 10.1016/j.pain.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Yao MZ, Gu JF, Wang JH, Sun LY, Lang MF, Liu J, Zhao ZQ, Liu XY. Interleukin-2 gene therapy of chronic neuropathic pain. Neuroscience. 2002;112:409–416. doi: 10.1016/s0306-4522(02)00078-7. [DOI] [PubMed] [Google Scholar]
- Zelenka M, Schafers M, Sommer C. Intraneural injection of interleukin-1beta and tumor necrosis factor-alpha into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain. 2005;116:257–263. doi: 10.1016/j.pain.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Zhang N, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ. A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc. Natl Acad. Sci. USA. 2005;102:4536–4541. doi: 10.1073/pnas.0406030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zujovic V, Benavides J, Vige X, Carter C, Taupin V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia. 2000;29:305–315. [PubMed] [Google Scholar]