

Abstract
Understanding molecular mechanisms mediating epileptogenesis is critical for developing more effective therapies for epilepsy. We recently found that the mammalian target of rapamycin (mTOR) signaling pathway is involved in epileptogenesis, and mTOR inhibitors prevent epilepsy in a mouse model of tuberous sclerosis complex. Here, we investigated the potential role of mTOR in a rat model of temporal lobe epilepsy initiated by status epilepticus. Acute kainate-induced seizures resulted in biphasic activation of the mTOR pathway, as evident by an increase in phospho-S6 (P-S6) expression. An initial rise in P-S6 expression started ∼1 h after seizure onset, peaked at 3–6 h, and returned to baseline by 24 h in both hippocampus and neocortex, reflecting widespread stimulation of mTOR signaling by acute seizure activity. After resolution of status epilepticus, a second increase in P-S6 was observed in hippocampus only, which started at 3 d, peaked 5–10 d, and persisted for several weeks after kainate injection, correlating with the development of chronic epileptogenesis within hippocampus. The mTOR inhibitor rapamycin, administered before kainate, blocked both the acute and chronic phases of seizure-induced mTOR activation and decreased kainate-induced neuronal cell death, neurogenesis, mossy fiber sprouting, and the development of spontaneous epilepsy. Late rapamycin treatment, after termination of status epilepticus, blocked the chronic phase of mTOR activation and reduced mossy fiber sprouting and epilepsy but not neurogenesis or neuronal death. These findings indicate that mTOR signaling mediates mechanisms of epileptogenesis in the kainate rat model and that mTOR inhibitors have potential antiepileptogenic effects in this model.
Introduction
Epilepsy affects ∼1% of people and is associated with significant morbidity and mortality. Approximately one-third of epileptic patients are intractable to currently available treatments (Schuele and Lüders, 2008). In responsive cases, medications can suppress seizures symptomatically, but there is minimal evidence that existing “antiepileptic” drugs correct the underlying brain abnormalities causing epilepsy (epileptogenesis) or alter the natural history of epilepsy. Thus, it is now recognized that novel therapeutic approaches are needed with true antiepileptogenic actions that can prevent or reverse the cellular and molecular mechanisms of epileptogenesis (Dichter, 2006; Löscher and Schmidt, 2006; Stefan et al., 2006). To develop such treatments, a better understanding of the biological processes mediating epileptogenesis is required.
In many epilepsy models, an initial brain insult triggers a cascade of cellular events, which, after a latent period of epileptogenesis, leads to chronic hyperexcitability and seizures. For example, in pharmacological seizures models in rodents, an episode of status epilepticus is induced by chemoconvulsant agents, such as kainate or pilocarpine, and, after recovery from the initial provoked seizures, the animals later develop spontaneous epilepsy (Leite et al., 2002). Numerous cellular and molecular abnormalities have been described during the intervening latent period that may contribute to epileptogenesis, such as immediate early gene activation and changes in gene expression (Dragunow and Robertson, 1987; Elliott et al., 2003; Gorter et al., 2006), alterations in neurotransmitter receptors and ion channels (Brooks-Kayal et al., 1998; Bernard et al., 2004), axonal sprouting and synaptic reorganization (Tauck and Nadler, 1985; Sutula et al., 1988), necrotic and apoptotic neuronal death (Pollard et al., 1994; Sankar et al., 1998; Tuunanen et al., 1999), and neurogenesis (Parent et al., 1997). However, attempts to prevent epileptogenesis by modulating these various cellular and molecular abnormalities have had only limited success (Acharya et al., 2008).
Although numerous downstream mechanisms may mediate epileptogenesis, less is known about initial signaling pathways that trigger the subsequent changes in the brain causing epilepsy. The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that controls protein synthesis related to cell growth and proliferation (Sarbassov et al., 2005; Sandsmark et al., 2007). In brain, mTOR signaling may also regulate neuronal development and synaptic plasticity (Tang et al., 2002; Kumar et al., 2005). Altered mTOR signaling is observed in many human tumors, as well as a variety of cortical malformations and neurodegenerative diseases (Inoki et al., 2005; Tsang et al., 2007). Several of these disorders, including tuberous sclerosis complex (TSC), focal cortical dysplasia, hemimegalencephaly, and ganglioglioma, are highly associated with epilepsy (Wong, 2008), suggesting that mTOR signaling is involved in epileptogenesis.
We recently reported that mTOR signaling is increased in a mouse model of TSC and mTOR inhibition prevents the development of epilepsy in this model (Zeng et al., 2008). Because there could be overlapping mechanisms of epileptogenesis in different types of epilepsy, in the present study, we investigated the role of the mTOR pathway in the kainate model of epilepsy, a model of temporal lobe epilepsy triggered by an initial episode of status epilepticus.
Materials and Methods
Animals and drug treatment.
Male Sprague Dawley rats (Charles River Laboratories) of 6 weeks of age were used in this study. The rats were housed in a controlled environment with ad libitum access to food and water. All animal experiments were performed in accordance with guidelines approved by the Animal Studies Committee at Washington University School of Medicine. Rapamycin (LC Labs) was initially dissolved in 100% ethanol, stored at −20°C, and diluted in a vehicle solution containing 5% Tween 80, 5% PEG 400 (low-molecular-weight grade of polyethylene glycol) (Sigma), and 4% ethanol immediately before injection, as described previously (Zeng et al., 2008). A couple of different rapamycin treatment paradigms were used. For pretreatment studies of rapamycin before onset of status epilepticus, based on pilot studies examining effects of rapamycin on mTOR activation/phospho-S6 (P-S6) expression, rats were treated with rapamycin (6 mg · kg−1 · d−1, i.p.) or vehicle for 3 consecutive days before receiving kainate. On the fourth day, both groups were injected with kainate (12 mg/kg, i.p.; Cambridge Pharma-Chem) to induce acute status epilepticus. Seizure activity was monitored behaviorally and in some cases by EEG. Behavioral seizures were graded according to a modified Racine scale (Racine, 1972; Zeng et al., 2007): stage 1, behavioral arrest with mouth/facial movements; stage 2, head nodding; stage 3, forelimb clonus; stage 4, rearing; stage 5, rearing and falling. The latency to first behavioral seizure activity, total seizure duration, and maximal stage severity were measured. Rats that had stage 4 or 5 seizures for at least 3 h were used for subsequent experiments. Some rats underwent video-EEG studies to correlate behavioral and EEG seizure activity during acute status epilepticus, as well as to monitor for development of chronic epilepsy (see below for video-EEG recording methods). During acute status epilepticus, EEG seizure activity was quantified by the average spike frequency during stage 5 seizures and the total duration of ictal spike activity (in cases in which the frequency of continuous, repetitive spikes gradually decreased toward the end of the status episode, the termination of ictal activity was defined as a spike frequency of <1 spike/s). Other rats were killed for Western blot or histological analysis at predetermined time points, as described below. In addition to vehicle-pretreated and rapamycin-pretreated rats receiving kainate, an additional control group consisted of saline-treated rats that did not receive kainate.
For posttreatment studies of rapamycin after status epilepticus, rapamycin injection (6 mg · kg−1 · d−1, i.p.) or vehicle was started 24 h after the onset of kainate status epilepticus for 6 consecutive days and every other day for the following weeks until the predefined end point of the study. Other experimental groups involved phenobarbital pretreatment (30 mg/kg, i.p.; Sigma) before kainate to suppress status epilepticus and phenobarbital treatment (30 mg · kg−1 · d−1, i.p.) starting the day after kainate status epilepticus for 6 consecutive days to suppress early spontaneous seizures.
Western blot analysis.
Rapamycin and vehicle-treated rats were killed for Western blot analysis of markers of mTOR activation at various time points (1 h, 3 h, 6 h, 16 h, 24 h, 3 d, 5 d, 1 week, 3 weeks, and 5 weeks) after onset of status epilepticus. Western blot analysis of phospho-S6 and S6 was performed using standard methods, as described previously (Zeng et al., 2008). In brief, protein extracts from neocortex and hippocampi were separated by SDS-PAGE and transferred to nitrocellulose membrane. After blocking with 5% skim milk, the membranes were incubated with the rabbit anti-phospho-S6 antibody (1:1000; Cell Signaling Technology), followed by peroxidase-conjugated secondary antibody. After the signals were visualized with ECL reagent (Pierce), the membranes were reprobed and incubated with the rabbit anti-S6 antibody (1:1000; Cell Signaling Technology). Signals were quantitatively analyzed with NIH ImageJ software.
Neuronal death assays.
Rapamycin- and vehicle-treated rats were killed for histological analysis of neuronal death by Fluoro-Jade B and terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) staining, 7 d after kainate status epilepticus. Rats were anesthetized with isoflurane and transcardially perfused with PBS, followed by 4% paraformaldehyde. The brains were removed immediately and postfixed with 4% paraformaldehyde overnight at 4°C. After dehydrating in 30% sucrose for at least 24 h, the brains were sectioned coronally at a thickness of 50 μm with a vibratome. Three sections selected from a one-in-six series were collected from each animal at the same level of hippocampus, starting at 2.8 mm posterior to bregma, and were stained separately for Fluoro-Jade B and TUNEL staining.
Staining for Fluoro-Jade B (FJB) (Histo-Chem) was performed as described previously (Schmued and Hopkins, 2000; Zeng et al., 2007). In brief, the sections were mounted on gelatin-coated slides and dried at room temperature. After rehydration in 100% ethanol (EtOH) (5 min), 70% EtOH (2 min), and distilled water (dH2O) (2 min), the sections were oxidized in 0.06% potassium permanganate for 10 min, washed with water, and then immersed in 0.0004% FJB solution for 20 min in the dark. Thereafter, slides were washed in dH2O, air dried, cleared, and coverslipped. TUNEL staining was performed using the kit obtained from Millipore Bioscience Research Reagents according to the instructions of the manufacturer and as done previously (Zeng et al., 2007). A Carl Zeiss LSM PASCAL confocal microscope with a 10×/0.3 numerical aperture (NA) objective was used to acquire images (920 × 920 μm fields) within CA1, CA3, and dentate hilus at a similar location in different animals. The number of FJB- and TUNEL-positive cells per image field in the hippocampal CA1, CA3, and hilus were counted in each of the three sections per animal by an examiner blind to experimental conditions.
5-Bromodeoxyuridine injection and labeling.
Rapamycin- and vehicle-treated rats were killed for analysis of hippocampal neurogenesis by bromodeoxyuridine (BrdU) staining, 7 d after kainate status epilepticus. BrdU (Sigma) was dissolved in 0.9% NaCl. From day 3 after kainate, each group of rats received daily injection of BrdU for 4 d (50 mg/kg at a concentration of 15 mg/ml, i.p.). Twenty-four hours after the last dose, the rats were killed. Tissue sections of 50 μm were cut with a vibratome. Three sections selected from a one-in-six series were collected from each animal at the same level of hippocampus, starting at 2.8 mm posterior to bregma, and were stained separately for BrdU. Sections were denatured in 2N HCl for 20 min at 37°C and then neutralized in 0.1 m borate buffer, pH 8.5, for 10 min. After thoroughly washing with PBS, the sections were blocked with 5% normal goat serum and permeated by 0.2% Triton X-100. Sections were then incubated with anti-mouse BrdU (1:500; Sigma) for 48 h at 4°C, followed by an anti-mouse IgG conjugated with Alexa Fluor-488 (1:500) for 2 h at room temperature. A Carl Zeiss LSM PASCAL confocal microscope with a 10×/0.3 NA objective was used to acquire images (920 × 920 μm fields) within dentate hilus at a similar location in different animals. BrdU-positive cells located in the granule cell layer of the dentate gyrus (DG) were counted in three sections per animal and averaged.
Timm's staining.
Rapamycin- and vehicle-treated rats were killed for histological analysis of mossy fiber sprouting (MFS) by Timm's staining, 4 weeks after kainate status epilepticus. Rats were anesthetized and transcardially perfused with sulfide solution as described previously (Buckmaster, 2004). In brief, coronal serial sections of 50 μm were cut with a vibratome, and one-in-six series of sections were mounted on slides, dried, and developed for 60–80 min until the molecular layer in the DG was clearly stained in 120 ml of 50% gum arabic, 20 ml of 2 m citrate buffer, 60 ml of 0.5 m hydroquinone, and 1 ml of 19% silver nitrate. The sections were then observed and photographed with a Carl Zeiss Axioskop microscope and AxioCamHR digital camera. Mossy fiber sprouting was assessed according to previously published criteria (Cavazos et al., 1991) as follows: 0, no granules between the tip and crests of the dentate gyrus; 1, sparse granules in the supragranular region in a patchy distribution between the tip and crests of the dentate gyrus; 2, more numerous granules in the supragranular region in a continuous distribution between the tip and crests of the dentate gyrus; 3, prominent granules in the supragranular region in a continuous pattern between the tip and crests, with occasional patches of confluent granules between the tip and crests; 4, prominent granules in the supragranular region that form a confluent dense laminar band between the tip and crests; and 5, a confluent dense laminar band of granules in the supragranular region that extends into the inner molecular layer. The Timm's scores of the individual sections were assigned by an investigator blinded to the experimental conditions and were averaged for each animal.
Video-EEG recording.
Rapamycin- and vehicle-treated rats were monitored for spontaneous seizures by weekly video-EEG recording sessions from weeks 1 to 6 after kainate administration (outlined in Fig. 6A). For surgical implantation of epidural and hippocampal depth electrodes, rats were anesthetized with 1–2% isoflurane in a stereotaxic frame 3 d before kainate administration. Bilateral anterior and posterior epidural cortical screw electrodes (−1.60 mm bregma, 1.80 mm lateral and −4.0 mm bregma, 3.0 mm lateral) and reference (+2.0 mm bregma, 1.0 mm lateral) and ground (−10 mm bregma, 1.0 mm lateral) electrodes were inserted in the skull, soldered on electronic pins, and secured with dental cement (Parkell). In some experiments, insulated wires were positioned bilaterally into hippocampus (−4.5 mm bregma, 3.5 mm lateral, 3.5 mm deep) in lieu of the posterior epidural electrodes. Rats were acclimated in cylindrical 10-inch-diameter acrylic cages for at least 1 d before monitoring with a digital video-EEG acquisition system (XLTEK). Multiple-channel EEG was acquired using standard alternating current amplifiers with 1–70 Hz bandpass filters. Time-locked digital video was recorded with a day–night video camera (12 h light/dark cycle). Rats were monitored continuously for the first week and then an epoch of 48 h for each week thereafter. Because of technical issues, not all rats were monitored during every weekly time period.
Figure 6.

Rapamycin pretreatment reduces KA-induced epilepsy in rats. A, Experimental design demonstrating timing of drug treatment and video-EEG monitoring. B, Representative EEG background and interictal epileptiform spikes in vehicle- and rapamycin-pretreated rats. Note there are significantly more epileptiform spikes in vehicle-pretreated rats. **p < 0.01, ***p < 0.001 by t test. C, Number of spontaneous seizures per day in vehicle- and rapamycin-pretreated rats during the first week after status epilepticus. D, Representative electrographic seizure is shown (top). Rapamycin postponed spontaneous seizure onset and reduced seizure frequency. Seizures started to develop within 1–2 weeks after kainate administration and became more frequent in vehicle-pretreated rats, whereas only rare seizures occurred in rapamycin-pretreated rats. *p < 0.05 by two-way ANOVA (n = 6–8 rats per time point and group). KA, Kainate; Rap, rapamycin; Veh, vehicle.
Video-EEG data were analyzed by two independent trained observers. All EEG data from each monitoring session was reviewed for electrographic seizures, and video was analyzed as needed to confirm behavioral correlates of electrographic seizures and to rule out sources of artifact. Electrographic seizures were clearly identifiable as discrete periods of repetitive, evolving spike discharges that lasted at least 10 s and usually originated in hippocampus but quickly had secondary generalization to neocortical electrodes. In addition, interictal spikes were identified and defined as fast (<200 ms) epileptiform waveforms that were at least twice the amplitude of the background activity. Seizure frequency (number of seizures per 48 h period) and interictal spike frequency (number of spikes per minute) were calculated from each 48 h epoch.
Statistics.
All statistical analysis was performed using SigmaStat (Systat Software). Quantitative differences between rapamycin- and vehicle-treated rats were analyzed by Student's t test when comparing two groups and by one-way ANOVA with Tukey's multiple comparisons post hoc tests when comparing more than two groups. Quantitative data are expressed as mean ± SEM. A value of p < 0.05 was considered significant.
Results
mTOR pathway is activated by kainate-induced seizures in a biphasic manner
To determine whether acute kainate-induced seizures trigger mTOR pathway signaling, the ratio of phospho-S6 to total S6 protein expression was used as a marker of activity of the mTOR pathway and assayed by Western blotting at different time intervals after seizure onset. Kainate status epilepticus resulted in a biphasic activation of the mTOR pathway, including both acute and chronic periods of increased P-S6 expression, correlating with acute seizure activity and chronic epileptogenesis, respectively (Fig. 1). In the acute phase (Fig. 1A,B), a significant increase in the ratio of P-S6/S6 was initiated within 1 h of the onset of kainate-induced seizures. This increase in mTOR activity peaked ∼3 h and then returned to baseline after 6 h, which correlated closely with the resolution of acute kainate seizures (seizure duration, 6.0 ± 1.3 h). The kainate seizure-induced mTOR activity was observed both in hippocampus and neocortex, reflecting the widespread involvement of the brain with the seizures. After kainate status epilepticus, mTOR activity remained at preseizure baseline for at least 2 d, but then subsequent assays of P-S6 expression revealed a second period of mTOR activation, starting ∼3 d after kainate injection (Fig. 1C,D). This increase in mTOR activity reached a maximum by ∼5 d and gradually decreased over the following few weeks, returning to baseline by 5 weeks after kainate status epilepticus. Interestingly, the secondary increase was only observed in hippocampus (Fig. 1C, top), with no corresponding change in neocortex (Fig. 1C, bottom), suggesting that chronic mTOR activation was spatially and temporally correlated with hippocampal epileptogenesis in the kainate model. To distinguish whether the initial mTOR pathway activation was a result of the kainate-induced seizure activity or a direct pharmacological effect of kainate, other rats were pretreated with phenobarbital (30 mg/kg, i.p.; Sigma) before kainate injection to suppress seizures. No seizure activity was observed in phenobarbital-pretreated rats, and there was no increase in the expression of P-S6 (data not shown), indicating that acute seizures themselves caused the initial mTOR activation.
Figure 1.

Kainate-induced seizures results in biphasic activation of the mTOR pathway. A, In the acute phase of kainate status epilepticus, Western blotting shows P-S6 and total S6 expression in hippocampus (top; Hip) and neocortex (bottom) at different time intervals within the first 24 h after the onset of seizures (which lasted 6.0 ± 1.3 h). B, Quantitative summary demonstrates that phosphorylation of S6 was increased within 1 h of the onset of kainate-induced seizures, peaked at 3 h, and returned to baseline after 6 h in both hippocampus and neocortex. The ratio of P-S6/S6 was normalized to rats without kainate (0 h). C, In the chronic phase after kainate status epilepticus, Western blotting shows P-S6 and total S6 expression in hippocampus (top) and neocortex (bottom) at longer time intervals over several weeks. D, Quantitative summary demonstrates that phosphorylation of S6 increased again at 3 d in hippocampus, but not neocortex, peaked at 5 d, and decreased to baseline by 5 weeks. The ratio of P-S6/S6 was normalized to rats without kainate (0d). *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA (n = 6 rats for each time point and group).
Rapamycin pretreatment blocks kainate seizure-induced mTOR activation
We attempted to block the kainate seizure-induced mTOR activation with the mTOR inhibitor rapamycin. After a series of pilot experiments testing various doses and durations of rapamycin, we found that pretreatment of rapamycin at a dose of 6 mg · kg−1 · d−1 intraperitoneally for 3 consecutive days before kainate injection was effective in preventing the initial increase in P-S6 activated over several hours by kainate-induced seizures both in hippocampus (Fig. 2A, top) and neocortex (Fig. 2A, bottom). Similarly, rapamycin pretreatment almost completely blocked the delayed increase in P-S6 over several weeks in hippocampus (Fig. 2B). In controlling for the potential direct effects of rapamycin on kainate-induced seizures, we found that there was no difference in behavioral seizure latency, duration, and severity between rats with vehicle and rapamycin pretreatment before kainate (Fig. 2C). In addition, EEG ictal spike activity lasted slightly longer than behavioral seizures but also showed no significant difference between vehicle- and rapamycin-treated rats in average spike frequency during stage 5 seizures (2.3 ± 0.1 vs 2.4 ± 0.1 spikes/s) and total ictal spike duration (7.7 ± 0.7 vs 7.3 ± 0.5 h). This indicates that rapamycin directly inhibited kainate seizure-induced mTOR activation without affecting the properties of the kainate seizures themselves.
Figure 2.

Rapamycin pretreatment abolishes mTOR activation from kainate-induced seizures both acutely and chronically but does not alter the acute properties of kainate status epilepticus. A, In the acute phase of kainate status epilepticus, Western blotting shows P-S6 and S6 expression at different time interval after kainate-induced status epilepticus in rapamycin-pretreated (KA+, Rap+) and vehicle-pretreated (KA+, Rap−) rats in hippocampus and neocortex. Vehicle-treated rats that did not receive kainate (KA−, Rap−; 0 h) serve as an additional control. B, In the chronic phase after kainate status epilepticus, Western blotting shows P-S6 and total S6 expression at longer time intervals over several weeks in rapamycin- and vehicle-pretreated rats in hippocampus. Note that pretreatment with rapamycin almost completely inhibits the activation of P-S6 both acutely and chronically after kainate status epilepticus (n = 6 rats for each time point and group). C, Rapamycin pretreatment had no effect on the latency, severity (stage), or duration of kainate-induced status epilepticus based on behavioral analysis (n = 10 rats per group), as well as ictal EEG spike discharges (see Results). Veh, Vehicle.
Rapamycin pretreatment decreases kainate seizure-induced cell death, neurogenesis, and mossy fiber sprouting
We next determined whether rapamycin could prevent the activation of cellular/molecular mechanisms that have been implicated in the process of chronic epileptogenesis in the kainate model, such as neuronal death, neurogenesis, and mossy fiber sprouting. Consistent with previous reports of neuronal death in the kainate model, all vehicle-pretreated rats (n = 10 of 10) demonstrated evidence of neuronal death by Fluoro-Jade B staining after kainate status epilepticus in neurons from the dentate hilus, CA3, and CA1 pyramidal regions (Fig. 3A–C,G). In contrast, in rats pretreated with rapamycin, 7 of 10 did not exhibit any Fluoro-Jade B-positive neurons after status epilepticus, whereas the remaining three had only sparse positive degenerating pyramidal neurons (Fig. 3D–G). Fluoro-Jade B labeling was also never observed in saline-injected control rats that did not receive kainate (n = 0 of 3). Similar protective effects of rapamycin against neuronal death were observed with TUNEL staining (data not shown).
Figure 3.
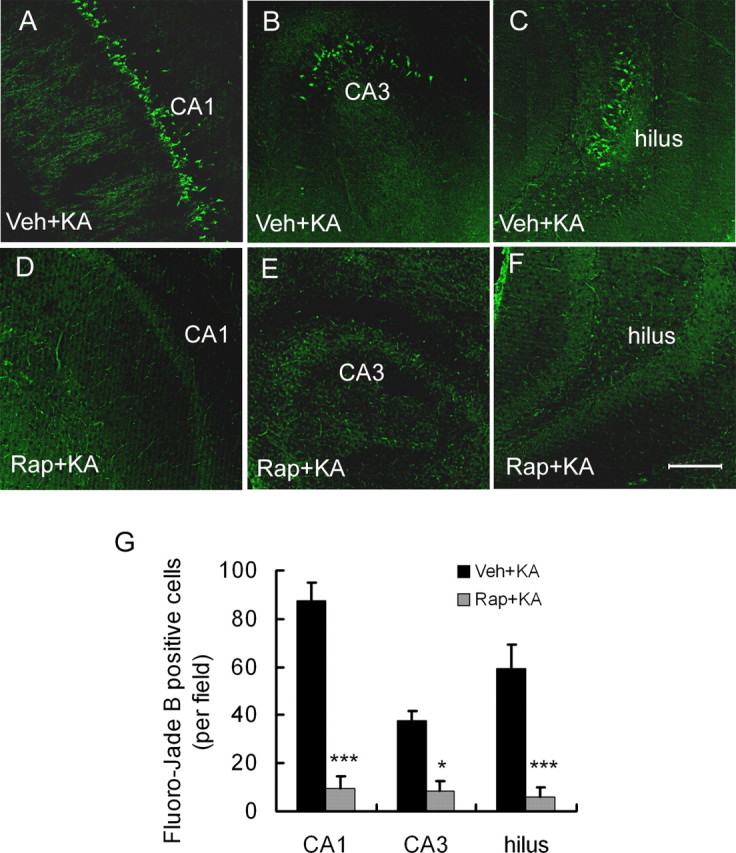
Rapamycin pretreatment decreases neuronal cell death after kainate-induced seizures. Representative sections of Fluoro-Jade B staining in hippocampus of vehicle- (Veh+KA; A–C) and rapamycin-pretreated (Rap+KA; D–F) rats are shown 7 d after kainate status epilepticus. Abundant Fluoro-Jade B-positive neurons can be found in vehicle-pretreated rats in CA1, CA3, and hilus but not in rapamycin-pretreated rats. G, Quantitative analysis demonstrates a significant decrease in Fluoro-Jade B-positive neurons in rapamycin-pretreated rats. Scale bar, 200 μm. *p < 0.05, ***p < 0.001 by t test (n = 10 rats per group).
The effect of rapamycin on kainate seizure-induced neurogenesis of dentate granule cells was assessed by BrdU labeling. There were a small number of BrdU-positive nuclei in the granular cell layer in saline-injected control animals that did not receive kainate. In vehicle-pretreated rats, kainate status epilepticus caused a significant increase in the number of BrdU-positive cells in this region, whereas in the rapamycin-treated group, the number of BrdU-positive cells was significantly lower (Fig. 4).
Figure 4.
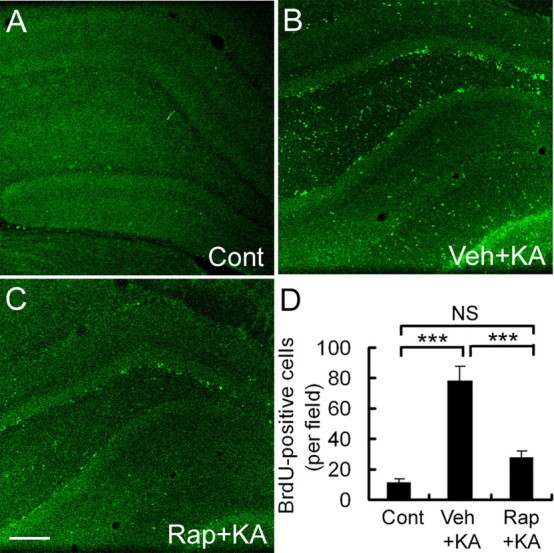
Rapamycin pretreatment decreases neurogenesis after kainate-induced seizures. Representative sections show BrdU staining in control (Cont) rats that did not receive kainate (A; KA) and vehicle-pretreated (B; Veh) and rapamycin-pretreated (C; Rap) rats that received kainate. D, Quantitative analysis demonstrates a significant decrease in BrdU-positive cells in the dentate gyrus of rapamycin-pretreated rats compared with vehicle-pretreated rats after kainate status epilepticus. Scale bar, 200 μm. ***p < 0.001 by one-way ANOVA (n = 3 in control rats; n = 6 in kainate-treated rats).
Kainate status epilepticus can lead to the development of supragranular MFS, which may also contribute to the development of chronic epilepsy. To test whether rapamycin can block kainate-induced MFS, we conducted Timm's staining to detect evidence of this aberrant synaptic circuit. In vehicle-treated rats, the staining in the supragranular region was very intense after kainate status epilepticus. Rapamycin pretreatment significantly attenuated this MFS induced by kainate (Fig. 5). Quantitative analysis showed that the severity score of MFS was significantly decreased in the rapamycin-treated group (Fig. 5D).
Figure 5.

Rapamycin pretreatment reduces mossy fiber sprouting after kainate-induced seizures. Timm's staining shows mossy fiber staining from control (Cont) rats that did not receive kainate (A; KA) and vehicle-pretreated (B; Veh) and rapamycin-pretreated (C; Rap) rats 28 d after kainate-induced status epilepticus. A1–C1 are higher magnification of boxed regions in A–C, respectively. m, Molecular layer; g, granule cell layer. D, Quantitative analysis demonstrates a significant decrease in Timm's score in rapamycin-pretreated rats compared with vehicle-pretreated rats after kainate-induced status epilepticus. Scale bars: A–C, 200 μm; A1–C1, 50 μm. ***p < 0.001 by one-way ANOVA (n = 3 in control rats; n = 6 in kainate-treated rats).
Rapamycin pretreatment decreases kainate-induced spontaneous seizures
Vehicle- and rapamycin-pretreated rats were monitored for spontaneous seizures from weeks 1 to 7 after kainate status epilepticus using the protocol as in Figure 6A. On EEG analysis, interictal epileptiform spikes were observed frequently in vehicle-pretreated rats after kainate status epilepticus but were significantly decreased in rapamycin-pretreated rats (Fig. 6B). In addition, whereas none of the rapamycin-pretreated rats had seizures in the first 7 d after kainate status epilepticus, four of seven vehicle-pretreated rats developed spontaneous motor (stage 4 or above) seizures, which started within a few days and increased in frequency by the end of the first week after kainate status epilepticus (Fig. 6C). Over the following several weeks, all the vehicle-treated rats had at least one spontaneous seizure, with an average of 4.8 ± 1.6 seizures per 48 h period. In contrast, no seizures were recorded in three of eight rapamycin-pretreated rats from weeks 1 to 7, and the other rapamycin-pretreated rats had a very low seizure frequency of 0.2 ± 0.1 seizures per 48 h period (Fig. 6D).
Posttreatment with rapamycin after status epilepticus blocks late mTOR activation and decreases mossy fiber sprouting and spontaneous seizures
We also examined the ability of late rapamycin treatment initiated 24 h after onset of kainate status epilepticus to block the delayed increase in P-S6 in hippocampus. Late rapamycin treatment was able to inhibit the second phase of mTOR activation. By comparison, similar late phenobarbital treatment did not block the delayed increased in P-S6 expression in the week after kainate status epilepticus (Fig. 7A,B), despite suppressing the onset of spontaneous seizures during this period. Late rapamycin also significantly decreased mossy fiber sprouting as assayed by Timm's staining (Fig. 7C–E). However, in contrast to the effects of rapamycin pretreatment, posttreatment with rapamycin after status epilepticus had no significant effect on hippocampal neuronal death or neurogenesis. No difference was observed in neuronal cell death in CA1, CA3, and hilus between vehicle-treated and late rapamycin-treated rats (Fig. 7F). There appeared to be a slight decrease in BrdU-positive cells in dentate gyrus in rapamycin-treated rats compared with vehicle-treated rats after status epilepticus (61.3 ± 10.1 vs 68.6 ± 11.9 per field), but this was not statistically significant. In chronic video-EEG monitoring studies, rapamycin posttreatment reduced the frequency of chronic spontaneous seizures. At 6 weeks after status epilepticus, late rapamycin-treated rats averaged 0.7 ± 0.3 seizures per 48 h epoch compared with 6.0 ± 1.4 seizures per 48 h in vehicle-treated controls (Fig. 7G).
Figure 7.

Rapamycin posttreatment after status epilepticus blocks the late phase on mTOR activation and reduces mossy fiber sprouting and epilepsy but not cell death. A, B, Rats were treated with vehicle (KA+, Rap−) or rapamycin (KA+, Rap+) starting 24 h after onset of kainate status epilepticus for 6 consecutive days. As assayed at 1 week after status epilepticus, rapamycin posttreatment blocked the late phase of mTOR activation. In contrast, as an additional control, phenobarbital, which can suppress acute seizure activity, had no effect of kainate-induced mTOR activation (KA+, PB+). *p < 0.05 compared with KA+Veh and KA+PB by one-way ANOVA (n = 4 rats per group). C–E, Rapamycin posttreatment (KA+Rap) reduced mossy fiber sprouting and Timm's score in dentate gyrus after kainate status epilepticus compared with controls (KA+Veh). *p < 0.05 by t test (n = 6 rats per group). F, Rapamycin posttreatment had no significant effect on neuronal death in hippocampus after kainate status epilepticus. G, Rapamycin posttreatment reduced frequency of spontaneous seizures up to 6 weeks after kainate status epilepticus. *p < 0.05 by one-way ANOVA (n = 7 rats per time point for rapamycin group; n = 7–10 rats per time point for vehicle group).
Discussion
The molecular mechanisms that lead to the development of epilepsy after an initial precipitating injury of the brain are incompletely understood. In the present study, we implicate involvement of the mTOR signaling pathway in mediating epileptogenesis in a rat model of temporal lobe epilepsy after an episode of status epilepticus. We first demonstrated that the mTOR pathway is strongly activated with a biphasic time course after kainate-induced status epilepticus, and this mTOR activation could be blocked by rapamycin treatment. Second, although not altering the acute characteristics of status epilepticus, rapamycin pretreatment counteracted known effects of kainate status epilepticus on neuronal death, neurogenesis, and mossy fiber sprouting, which may represent cellular mechanisms causing epileptogenesis in this model. Finally, both pretreatment and posttreatment with rapamycin significantly decreased the development of chronic epilepsy, indicating that mTOR signaling is involved in epileptogenesis and that mTOR inhibitors could be useful as “antiepileptogenic” therapy.
mTOR is a serine/threonine kinase that activates a cascade of downstream effectors involved in numerous biological processes, including protein synthesis, ribosomal biogenesis, and cell growth and proliferation (Sandsmark et al., 2007). Through these cellular actions, mTOR signaling has been implicated in controlling physiological responses to environmental and metabolic stresses, is dysregulated in a variety of disorders, including diabetes, obesity, cardiovascular disease, cancer, and neurological diseases (Kwiatkowski, 2003; Inoki et al., 2005; Sarbassov et al., 2005; Tsang et al., 2007), and may be well suited for mediating mechanisms of epileptogenesis. In the present study, the initial injurious stimulus of status epilepticus leads to a biphasic activation of mTOR, with the first peak of activation occurring in both hippocampus and neocortex acutely within a few hours during the seizure activity and a second, delayed peak occurring several days later within hippocampus only. Because mTOR can be triggered by glutamate receptor stimulation (Lenz and Avruch, 2005; Gong et al., 2006), it is not surprising that the initial widespread mTOR activation occurs with status epilepticus, which causes massive glutamate release. The trigger for the second, delayed peak in mTOR activation is less clear, but the timing and selective hippocampal localization correlate with initial cellular changes that have been implicated in epileptogenesis within hippocampus during the latent period after status epilepticus and before the onset of limbic epilepsy in the kainate model. Thus, both the temporal and spatial distribution of mTOR activation suggests that mTOR could be central to epileptogenesis in this model. Furthermore, because mTOR inhibition by rapamycin attenuated the development of epilepsy and other downstream cellular abnormalities implicated in epileptogenesis in the kainate model, these data support that mTOR plays a primary causal role in epileptogenesis and may serve as an initial central signaling trigger that activates multiple downstream epileptogenic pathways and processes.
The known biochemical functions of the mTOR pathway are well suited for mediating mechanisms of epileptogenesis. Cellular and molecular changes in neurotransmitter receptors and ion channels, synaptic reorganization, programmed cell death, and neurogenesis have been proposed to promote the development of epilepsy in the kainate model. Most of these putative mechanisms of epileptogenesis are dependent on protein synthesis and other biochemical processes that could be regulated by mTOR signaling. In support of this idea, we found that inhibition of mTOR with rapamycin pretreatment could reduce the amount of apoptosis, neurogenesis, and mossy fiber sprouting after status epilepticus. mTOR signaling has been implicated in apoptosis in other systems and may have both proapoptotic and anti-apoptotic actions (Castedo et al., 2002; Asnaghi et al., 2004a), but, to our knowledge, a relationship of mTOR in programmed cell death has not been reported previously in epilepsy models per se. Although mTOR activation may be neuroprotective in some situations, such as ischemic brain injury (Koh et al., 2008), in many other neuronal and non-neuronal systems, mTOR-mediated protein synthesis promotes apoptosis (Castedo et al., 2001; Asnaghi et al., 2004b; Thyrell et al., 2004; Shacka et al., 2007; Chen et al., 2008), consistent with our finding that rapamycin protects against neuronal death in the kainate model. The inhibition of cell death by rapamycin indicates that mTOR functions upstream from cell death mechanisms in these systems. Similarly, the established role of mTOR in regulating cell proliferation has been related to neurogenesis within the brain (Han et al., 2008) but again not specifically linked to epilepsy. In comparison, a recent preliminary study indicated a connection between mTOR and mossy fiber sprouting after pilocarpine status epilepticus (Buckmaster and Ingram, 2007). Interestingly, although rapamycin can attenuate mossy fiber sprouting, previous attempts to block mossy fiber sprouting by using other protein synthesis inhibitors have failed (Williams et al., 2002; Toyoda and Buckmaster, 2005). Thus, mTOR signaling may have a relatively specific role in some brain abnormalities occurring during epileptogenesis.
Despite the documented effects of status epilepticus in causing cellular and molecular changes in the brain, the causal role of these abnormalities in leading to the development of chronic epilepsy is still controversial, and attempts to block epileptogenesis by regulating these processes have had only limited success. For example, NMDA glutamate receptor antagonists, administered just before status epilepticus, can block neuronal death and chronic epilepsy in the pilocarpine model (Rice and DeLorenzo, 1998). However, NMDA antagonists applied immediately after status epilepticus in the kainate model could only block neuronal death and not chronic epileptogenesis (Brandt et al., 2003). Similarly, many clinically used seizure medications have been tested for antiepileptogenic effects in animal models, as well as in people. A few animal studies suggest that available seizure drugs, such as phenobarbital, valproate, and topiramate, may have antiepileptogenic properties (Bolanos et al., 1998; Prasad et al., 2002; Kudin et al., 2004; Suchomelova et al., 2006), but in most of these cases, it is difficult to differentiate between the direct anticonvulsant (seizure-suppressing) actions of these drugs versus true antiepileptogenic effects. Many other animal studies have found no evidence that current seizure medications can prevent epileptogenesis (Mikati et al., 1994; Rigoulot et al., 2004; Brandt et al., 2006, 2007), despite sometimes having neuroprotective effects against cell death or other cellular effects of the initial precipitating injury. Furthermore, clinical trials in people have also shown that conventional seizure medications, such as phenytoin and valproate, can suppress acute symptomatic seizures but do not prevent the development of chronic epilepsy in head trauma patients (Temkin et al., 1990, 1999). Thus, there is still a dire need for the identification of better, clinically available, antiepileptogenic drugs.
In the present study, rapamycin most likely has true antiepileptogenic actions rather than simply an anticonvulsant effect. First of all, rapamycin pretreatment had no measureable effect on the severity or duration of acute kainate-induced seizures, and a relatively brief duration of rapamycin administration was required. Previous studies have also shown minimal to no direct effects of rapamycin on neuronal excitability (Daoud et al., 2007; Rüegg et al., 2007). From a pathophysiological standpoint, the most likely mechanism of action of rapamycin, involving inhibition of mTOR-mediated protein synthesis, could have long-term consequences on epileptogenesis, without acute, direct anticonvulsant actions. However, it is difficult to rule out other actions of rapamycin that affect either neuronal excitability or epileptogenesis. The ability of rapamycin pretreatment to decrease or block the effects of kainate seizures on neuronal death, neurogenesis, and mossy fiber sprouting is evidence that mTOR functions as an upstream regulator of these cellular processes but does not prove that reversal of these downstream mechanisms causally mediates the antiepileptogenic effects of rapamycin. In contrast to pretreatment, rapamycin applied after status epilepticus blocked the late phase of mTOR activation and reduced mossy fiber sprouting but not neuronal death or neurogenesis. Because rapamycin posttreatment was able to reduce chronic spontaneous seizures, this suggests that mossy fiber sprouting may be more important for epileptogenesis and is more dependent on the late phase of mTOR activation. However, the relationship between mossy fiber sprouting and epilepsy in these studies is only correlative. It will be important for future studies to more specifically identify the necessary downstream targets mediating epileptogenesis that are reversed by rapamycin. Independent of the specific downstream mechanisms involved, the partial effectiveness of late rapamycin treatment is encouraging from a clinical standpoint in suggesting that mTOR inhibitors could have antiepileptogenic actions even when administered after the onset of the initial precipitating injury.
The potential clinical applications of rapamycin for preventing epileptogenesis are significant and immediate. Similar to the present study, rapamycin was shown recently to have strong antiepileptogenic effects in a mouse model of TSC (Zeng et al., 2008). As a leading genetic cause of epilepsy and a disease that can often be identified before the onset of seizures, TSC represents an ideal candidate for antiepileptogenic treatment with rapamycin. Although TSC involves genetic mutation of proteins that directly regulate mTOR, it is probable that other types of epilepsy attributable to other etiologies may share similar downstream mechanisms of epileptogenesis. Thus, the potential clinical utility of mTOR inhibitors for preventing epileptogenesis should be considered in many types of epilepsy, such as TSC, symptomatic temporal lobe epilepsy, and posttraumatic epilepsy. Given the current lack of proven antiepileptogenic agents, mTOR inhibitors could ultimately have significant benefits for many epilepsy patients.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants K02 NS045583 and R01 NS056872 (M.W.), the Tuberous Sclerosis Alliance (M.W.), and NIH Neuroscience Blueprint Center Core Grant P30 NS057105 (to Washington University).
References
- Acharya MM, Hattiangady B, Shetty AK. Progress in neuroprotective strategies for preventing epilepsy. Prog Neurobiol. 2008;84:363–404. doi: 10.1016/j.pneurobio.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnaghi L, Bruno P, Priulla M, Nicolin A. mTOR: a protein kinase switching between life and death. Pharmacol Res. 2004a;50:545–549. doi: 10.1016/j.phrs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Asnaghi L, Calastretti A, Bevilacqua A, D'Agnano I, Gatti G, Canti G, Delia D, Capaccioli S, Nicolin A. Bcl-2 phosphorylation and apoptosis activated by damaged microtubules require mTOR and are regulated by Akt. Oncogene. 2004b;23:5781–5791. doi: 10.1038/sj.onc.1207698. [DOI] [PubMed] [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- Bolanos AR, Sarkisian M, Yang Y, Hori A, Helmers SL, Mikati M, Tandon P, Stafstrom CE, Holmes GL. Comparison of valproate and phenobarbital treatment after status epilepticus in rats. Neurology. 1998;51:41–48. doi: 10.1212/wnl.51.1.41. [DOI] [PubMed] [Google Scholar]
- Brandt C, Potschka H, Löscher W, Ebert U. N-methyl-d-aspartate receptor blockade after status epilepticus protects against limbic brain damage but not against epilepsy in the kainate model of temporal lobe epilepsy. Neuroscience. 2003;118:727–740. doi: 10.1016/s0306-4522(03)00027-7. [DOI] [PubMed] [Google Scholar]
- Brandt C, Gastens AM, Sun M, Hausknecht M, Löscher W. Treatment with valproate after status epilepticus: effects on neuronal damage, epileptogenesis, and behavioral alterations in rats. Neuropharmacology. 2006;51:789–804. doi: 10.1016/j.neuropharm.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Brandt C, Glien M, Gastens AM, Fedrowitz M, Bethmann K, Volk HA, Potschka H, Löscher W. Prophylactic treatment with levetiracetam after status epilepticus: lack of effect on epileptogenesis, neuronal damage, and behavioral alterations in rats. Neuropharmacology. 2007;53:207–221. doi: 10.1016/j.neuropharm.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS. Prolonged infusion of tetrodotoxin does not block mossy fiber sprouting in pilocarpine-treated rats. Epilepsia. 2004;45:452–458. doi: 10.1111/j.0013-9580.2004.67103.x. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Ingram E. Prolonged infusion of rapamycin suppresses mossy fiber sprouting in a model of temporal lobe epilepsy. Epilepsia. 2007;48(Suppl 6) [Google Scholar]
- Castedo M, Ferri KF, Blanco J, Roumier T, Larochette N, Barretina J, Amendola A, Nardacci R, Métivier D, Este JA, Piacentini M, Kroemer G. Human immunodeficiency virus 1 envelope glycoprotein complex-induced apoptosis involves mammalian target of rapamycin/FKBP12-rapamycin-associated protein-mediated p53 phosphorylation. J Exp Med. 2001;194:1097–1110. doi: 10.1084/jem.194.8.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castedo M, Ferri KF, Kroemer G. Mammalian target of rapamycin (mTOR): pro- and anti-apoptotic. Cell Death Differ. 2002;9:99–100. doi: 10.1038/sj.cdd.4400978. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Golarai G, Sutula TP. Mossy fiber synaptic reorganization induced by kindling: time course of development, progression, and permanence. J Neurosci. 1991;11:2795–2803. doi: 10.1523/JNEUROSCI.11-09-02795.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu L, Luo Y, Huang S. MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J Neurochem. 2008;105:251–261. doi: 10.1111/j.1471-4159.2007.05133.x. [DOI] [PubMed] [Google Scholar]
- Daoud D, Scheld HH, Speckmann EJ, Gorji A. Rapamycin: brain excitability studied in vitro. Epilepsia. 2007;48:834–836. doi: 10.1111/j.1528-1167.2006.00976.x. [DOI] [PubMed] [Google Scholar]
- Dichter MA. Models of epileptogenesis in adult animals available for antiepileptogenesis drug screening. Epilepsy Res. 2006;68:31–35. doi: 10.1016/j.eplepsyres.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Robertson HA. Kindling stimulation induces c-fos protein(s) in granule cells of the rat dentate gyrus. Nature. 1987;329:441–442. doi: 10.1038/329441a0. [DOI] [PubMed] [Google Scholar]
- Elliott RC, Miles MF, Lowenstein DH. Overlapping microarray profiles of dentate gyrus gene expression during development- and epilepsy-associated neurogenesis and axon outgrowth. J Neurosci. 2003;23:2218–2227. doi: 10.1523/JNEUROSCI.23-06-02218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong R, Park CS, Abbassi NR, Tang SJ. Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J Biol Chem. 2006;281:18802–18815. doi: 10.1074/jbc.M512524200. [DOI] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA, Aronica E, Breit T, Rauwerda H, Lopes da Silva FH, Wadman WJ. Potential new antiepileptogenic targets indicated by microarray analysis in a rat model for temporal lobe epilepsy. J Neurosci. 2006;26:11083–11110. doi: 10.1523/JNEUROSCI.2766-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Wang B, Xiao Z, Gao Y, Zhao Y, Zhang J, Chen B, Wang X, Dai J. Mammalian target of rapamycin (mTOR) is involved in the neuronal differentiation of neural progenitors induced by insulin. Mol Cell Neurosci. 2008;39:118–124. doi: 10.1016/j.mcn.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- Koh PO, Cho JH, Won CK, Lee HJ, Sung JH, Kim MO. Estradiol attenuates the focal cerebral ischemic injury through mTOR/p70S6 kinase signaling pathway. Neurosci Lett. 2008;436:62–66. doi: 10.1016/j.neulet.2008.02.061. [DOI] [PubMed] [Google Scholar]
- Kudin AP, Debska-Vielhaber G, Vielhaber S, Elger CE, Kunz WS. The mechanism of neuroprotection by topiramate in an animal model of epilepsy. Epilepsia. 2004;45:1478–1487. doi: 10.1111/j.0013-9580.2004.13504.x. [DOI] [PubMed] [Google Scholar]
- Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DJ. Tuberous sclerosis: from tubers to mTOR. Ann Hum Genet. 2003;67:87–96. doi: 10.1046/j.1469-1809.2003.00012.x. [DOI] [PubMed] [Google Scholar]
- Leite JP, Garcia-Cairasco N, Cavalheiro EA. New insights from the use of pilocarpine and kainate models. Epilepsy Res. 2002;50:93–103. doi: 10.1016/s0920-1211(02)00072-4. [DOI] [PubMed] [Google Scholar]
- Lenz G, Avruch J. Glutamatergic regulation of the p70S6 kinase in primary mouse neurons. J Biol Chem. 2005;280:38121–38124. doi: 10.1074/jbc.C500363200. [DOI] [PubMed] [Google Scholar]
- Löscher W, Schmidt D. New horizons in the development of antiepileptic drugs: innovative strategies. Epilepsy Res. 2006;69:183–272. doi: 10.1016/j.eplepsyres.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikati MA, Holmes GL, Chronopoulos A, Hyde P, Thurber S, Gatt A, Liu Z, Werner S, Stafstrom CE. Phenobarbital modifies seizure-related brain injury in the developing brain. Ann Neurol. 1994;36:425–433. doi: 10.1002/ana.410360314. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard H, Charriaut-Marlangue C, Cantagrel S, Represa A, Robain O, Moreau J, Ben-Ari Y. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63:7–18. doi: 10.1016/0306-4522(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Prasad A, Williamson JM, Bertram EH. Phenobarbital and MK-801, but not phenytoin, improve the long-term outcome of status epilepticus. Ann Neurol. 2002;51:175–181. doi: 10.1002/ana.10085. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rice AC, DeLorenzo RJ. NMDA receptor activation during status epilepticus is required for the development of epilepsy. Brain Res. 1998;782:240–247. doi: 10.1016/s0006-8993(97)01285-7. [DOI] [PubMed] [Google Scholar]
- Rigoulot MA, Koning E, Ferrandon A, Nehlig A. Neuroprotective properties of topiramate in the lithium pilocarpine model. J Pharmacol Exp Ther. 2004;308:787–795. doi: 10.1124/jpet.103.057091. [DOI] [PubMed] [Google Scholar]
- Rüegg S, Baybis M, Juul H, Dichter M, Crino PB. Effects of rapamycin on gene expression, morphology, and electrophysiological properties of rat hippocampal neurons. Epilepsy Res. 2007;77:85–92. doi: 10.1016/j.eplepsyres.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandsmark DK, Pelletier C, Weber JD, Gutmann DH. Mammalian target of rapamycin: master regulator of cell growth in the nervous system. Histol Histopathol. 2007;22:895–903. doi: 10.14670/HH-22.895. [DOI] [PubMed] [Google Scholar]
- Sankar R, Shin DH, Liu H, Mazarati A, Pereira de Vasconcelos A, Wasterlain CG. Patterns of status epilepticus-induced neuronal injury during development and long-term consequences. J Neurosci. 1998;18:8382–8393. doi: 10.1523/JNEUROSCI.18-20-08382.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Schuele SU, Lüders HO. Intractable epilepsy: management and therapeutic alternatives. Lancet Neurol. 2008;7:514–524. doi: 10.1016/S1474-4422(08)70108-X. [DOI] [PubMed] [Google Scholar]
- Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth KA, Zhang J. Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci Lett. 2007;414:57–60. doi: 10.1016/j.neulet.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan H, Lopes da Silva FH, Löscher W, Schmidt D, Perucca E, Brodie MJ, Boon PA, Theodore WH, Moshé SL. Epileptogenesis and rational therapeutic strategies. Acta Neurol Scand. 2006;113:139–155. doi: 10.1111/j.1600-0404.2005.00561.x. [DOI] [PubMed] [Google Scholar]
- Suchomelova L, Baldwin RA, Kubova H, Thompson KW, Sankar R, Wasterlain CG. Treatment of experimental status epilepticus in immature rats: dissociation between anticonvulsant and antiepileptogenic effects. Pediatr Res. 2006;59:237–243. doi: 10.1203/01.pdr.0000196333.16608.30. [DOI] [PubMed] [Google Scholar]
- Sutula T, He XX, Cavazos J, Scott G. Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science. 1988;239:1147–1150. doi: 10.1126/science.2449733. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin NR, Dikmen SS, Wilensky AJ, Keihm J, Chabal S, Winn HR. A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med. 1990;323:497–502. doi: 10.1056/NEJM199008233230801. [DOI] [PubMed] [Google Scholar]
- Temkin NR, Dikmen SS, Anderson GD, Wilensky AJ, Holmes MD, Cohen W, Newell DW, Nelson P, Awan A, Winn HR. Valproate therapy for prevention of posttraumatic seizures: a randomized trial. J Neurosurg. 1999;91:593–600. doi: 10.3171/jns.1999.91.4.0593. [DOI] [PubMed] [Google Scholar]
- Thyrell L, Hjortsberg L, Arulampalam V, Panaretakis T, Uhles S, Dagnell M, Zhivotovsky B, Leibiger I, Grandér D, Pokrovskaja K. Interferon alpha-induced apoptosis in tumor cells is mediated through the phosphoinositide 3-kinase/mammalian target of rapamycin signaling pathway. J Biol Chem. 2004;279:24152–24162. doi: 10.1074/jbc.M312219200. [DOI] [PubMed] [Google Scholar]
- Toyoda I, Buckmaster PS. Prolonged infusion of cycloheximide does not block mossy fiber sprouting in a model of temporal lobe epilepsy. Epilepsia. 2005;46:1017–1020. doi: 10.1111/j.1528-1167.2005.04605.x. [DOI] [PubMed] [Google Scholar]
- Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12:112–124. doi: 10.1016/j.drudis.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Tuunanen J, Lukasiuk K, Halonen T, Pitkänen A. Status epilepticus-induced neuronal damage in the rat amygdaloid complex: distribution, time-course and mechanisms. Neuroscience. 1999;94:473–495. doi: 10.1016/s0306-4522(99)00251-1. [DOI] [PubMed] [Google Scholar]
- Williams PA, Wuarin JP, Dou P, Ferraro DJ, Dudek FE. Reassessment of the effects of cycloheximide on mossy fiber sprouting and epileptogenesis in the pilocarpine model of temporal lobe epilepsy. J Neurophysiol. 2002;88:2075–2087. doi: 10.1152/jn.2002.88.4.2075. [DOI] [PubMed] [Google Scholar]
- Wong M. Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development. Epilepsia. 2008;49:8–21. doi: 10.1111/j.1528-1167.2007.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Rensing NR, Sinatra PM, Rothman SM, Wong M. Kainate seizures cause acute dendritic spine loss and actin depolymerization in vivo. J Neurosci. 2007;27:11604–11613. doi: 10.1523/JNEUROSCI.0983-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]