

Abstract
The notion that the immune system might control the growth of tumors was suggested over 100 years ago by the eminent microbiologist Paul Ehrlich. This concept was refined and expanded by Burnet and Thomas fifty years later with their articulation of the “immune surveillance” hypothesis. In its simplest form, the immune surveillance hypothesis suggests that neoplasms arise spontaneously and express novel antigens that are recognized by the immune system, which either eliminates the tumors or restrains their growth. Within the eye, immune responses are controlled and sometimes profoundly inhibited - a condition known as immune privilege. Immune privilege in the eye is the result of a complex array of anatomical, physiological, and immunoregulatory mechanisms that prevent the induction and expression of many immune responses. Tumors arising in the eye would seem to have an advantage in evading immune surveillance due to ocular immune privilege. Uveal melanoma, the most common and malignant intraocular tumor in adults not only benefits from the immune privilege of the eye, but has adopted many of the mechanisms that contribute to ocular immune privilege as a strategy for protecting uveal melanoma cells once they leave the sanctuary of the eye and are disseminated systemically in the form of metastases. Although the immune system possesses a battery of effector mechanisms designed to rid the body of neoplasms, tumors are capable of rapidly evolving and countering even the most sophisticated immunological effector mechanisms. To date, tumors seem to be winning this arms race, but an increased understanding of these mechanisms should provide insights for designing immunotherapy that was envisioned over half a century ago, but has failed to materialize to date.
Keywords: Anterior Chamber, Immune Escape, Immune Privilege, Immune Surveillance, Metastasis, Natural Killer Cells, Uveal Melanoma
1. Introduction
The notion that the body might be able to defend itself against neoplasms was demonstrated in the century before last by Coley who noted the beneficial effects of bacterial toxins in the treatment of sarcomas (Coley, 1891; Coley, 1893). The responses were probably due to the action of tumor necrosis factor-α (TNF-α), which was undoubtedly present in the bacterial extracts that were used to treat sarcoma patients. In 1909, Paul Ehrlich predicted that the immune system might protect the host from carcinomas (Ehrlich, 1909). However, the absence of intimate knowledge about the immune system and the limited tools for experimental studies prevented Ehrlich from testing this hypothesis. Almost 50 years would pass before Burnet and Thomas would revisit Ehrlich’s prediction and articulate their “immune surveillance” hypothesis (Burnet, 1957; Thomas, 1959). Subsequent studies confirmed the presence of tumor antigens on chemically and virally induced neoplasms in rodents and lent credence to the immune surveillance concept (Klein, 1966; Old and Boyse, 1966). Implicit in the immune surveillance hypothesis is the prediction that neoplasms arise spontaneously and are eliminated by the immune system before they reach clinically detectable sizes. Accordingly, one would expect that mice with defective immune systems should experience a significantly elevated incidence of spontaneous tumors or an accelerated appearance of chemically-induced neoplasms. However, when this hypothesis was tested in athymic nude mice, which lack fully developed T and B cell repertoires, the mice did not differ from immune competent mice in either the incidence of spontaneous tumors or the development of chemically-induced tumors (Stutman, 1974, 1975). As a result, the immune surveillance hypothesis fell into disrepute and languished in the margins of tumor biology for almost two decades. In retrospect we know that although nude mice have defective adaptive immunity, their innate immune responses, especially natural killer (NK) cells, are exceptionally well-developed and can contribute to the immune surveillance of tumors. A number of experimental results led to the re-emergence of the immune surveillance hypothesis in the 1990’s. The finding that administration of neutralizing antibodies to interferon-γ (IFN-γ) resulted in accelerated tumor growth in rodents and the observation that mice deficient in the IFN-γ receptor displayed an increased frequency of chemically- induced cancers resurrected interest in the concept of immune surveillance (Kaplan et al., 1998; Shankaran et al., 2001). Other studies showed that the absence of the gene encoding perforin, a key enzymatic protein involved in cytolysis by cytotoxic T lymphocytes (CTL) and NK cells, resulted in an increased incidence of spontaneous B cell lymphomas in mice (Smyth et al., 2000). Moreover, mice deficient in NK cells and NKT cells were also found to be more susceptible to spontaneous tumors and the accelerated growth of transplanted tumors (Dunn et al., 2006; Smyth et al., 2006). One of the nagging arguments against the immune surveillance hypothesis stems from the observation that animal studies have largely relied on either chemically-induced tumors or transplanted tumors. A convincing rebuttal to this criticism arose from studies in mice lacking the recombination-activating gene-2 (RAG2), which is necessary for the generation of immunoglobulin and T cell receptor rearrangements. These mice lack T cells, B cells, and NKT cells. RAG knockout (KO) mice crossed with mice bearing a mutant form of the p53 tumor suppressor gene have a significantly increased incidence of spontaneous tumors, which provides compelling evidence that elements of the adaptive immune system monitor and restrict the development of spontaneous tumors, a condition that mimics what Burnet and Thomas envisioned over 50 years ago (Liao et al., 1998; Nacht and Jacks, 1998).
A crucial tenet of the immune surveillance concept is that patients with underlying immune deficiencies should experience an elevated incidence or an accelerated progression of tumors. Circumstantial evidence suggests that this is the case. Individuals with acquired or hereditary immune deficiencies experience a higher than normal incidence of virally-associated and carcinogen-associated cancers and organ transplant recipients who are subjected to long-term immunosuppressive drugs have a 3- to 8-fold increase in the incidence of neoplasms (Reiman et al., 2007; Swann and Smyth, 2007). In the case of kidney transplant patients, there are reports of 2- to 5-fold increases in cancers of the colon, lung, bladder, prostate, and a 30-fold increase in skin cancers and kidney cancers (Birkeland et al., 1995). Likewise, the risk of melanoma doubles in organ transplant patients (Penn, 1996). The innate immune system also appears to play a role in immune surveillance, as patients with Chediak Higashi syndrome, a condition that results in severe impairment of NK cell-mediated cytotoxicity, have a 200-fold increase in their risk for developing cancer (Kobayashi, 1985). Thus, both experimental and clinical data support the notion that the immune system can monitor the development and control the outgrowth of cancers in both humans and animals.
2. Immunoediting: Cancer’s Answer to Immune Surveillance
Although the evidence supporting immune surveillance of cancer is compelling, the battle between the immune system and cancer is not one-sided. The mere fact that cancer remains a major cause of morbidity and mortality is a testament to the imperfections of immune surveillance. In fact, there are some who suggest that under some conditions, the immune system might unwittingly contribute to tumor development. This concept was raised almost 40 years ago by Prehn who proposed that the immune system might stimulate, rather than inhibit, tumor growth (Prehn, 1972). Prehn suggested that a “weak” immune response stimulated tumor growth, while more intense immune responses controlled tumors. Consistent with this is the time-honored observation that chronic inflammation is associated with an increased risk of cancer (Balkwill et al., 2005). Moreover, the use of anti-inflammatory drugs to treat chronic inflammatory diseases is associated with a reduced risk for cancer (Dannenberg and Subbaramaiah, 2003). It is clear that cancers arise in the face of an intact immune system. Growing evidence suggests that a “weak” immune response not only favors tumor outgrowth, but also positively selects those tumor cell populations that are either inherently resistant to immune effector elements or that are invisible to both the adaptive and innate arms of the immune response. This “immune editing” concept expands the original immune surveillance hypothesis and proposes that the immune response to tumors sculpts the tumor’s antigenic phenotype by deleting those tumor cells that are immunogenic, while allowing non-immunogenic tumor cells to escape immune elimination (Dunn et al., 2002). This ongoing selection is somewhat analogous to the process that leads to antibiotic-resistant bacteria. Dunn et al. have proposed that this immunoediting is comprised of three stages: a) elimination, b) equilibrium, and c) escape (Dunn et al., 2002). The elimination stage corresponds to the original immune surveillance concept, which implied an “all or none” response in which a nascent tumor was either eliminated entirely or grew in an unrestrained manner. There is extensive evidence from experimental animal studies demonstrating that elements of the innate and adaptive immune systems can contribute to tumor elimination (Dunn et al., 2004a, b; Swann and Smyth, 2007).
The second stage of immunoediting is the equilibrium phase in which immune surveillance prevents the outgrowth of tumors without eliminating all of the tumor cell populations. This is tantamount to what has been labeled “tumor dormancy”. There are numerous clinical examples of tumor dormancy in which cancer patients experience relapses after long periods (sometimes decades) of quiescence (Callaway and Briggs, 1989; Demicheli et al., 1996; Herrlinger et al., 2005; Matsui et al., 2006; Sagalowsky and Molberg, 1999). Moreover, disseminated tumor cells can be detected in the blood of some patients who have been in clinical remission for over 20 years (Meng et al., 2004). However, unlike true tumor dormancy, in which tumor cells enter a G0–G1 arrest or the tumor vasculature is insufficient for tumor expansion, the equilibrium phase of immunoediting is believed to be a dynamic interaction between the immune system and tumor cells that results in the emergence of tumor cell populations that express either reduced immunogenicity or resistance to immune effector mechanisms. Animal studies have provided the most compelling evidence suggesting that immune elements “edit” the immunogenic phenotype of tumors. Tumors arising in mice with immunodeficient backgrounds elicit immune responses and undergo rejection when transplanted to immunocompetent recipients. Chemically-induced tumors in IFN-γ or T cell-deficient mice and lymphomas from perforin −/− mice undergo immune rejection when transplanted to wild-type immunocomptetent recipients, but grow progressively if transplanted to immunodeficient hosts (Teng et al., 2008). These findings suggest that tumors arising in immunoincompetent mice can express potent antigens that render the tumors highly immunogenic and potentially vulnerable to immune elimination. However, in the absence of immune selection pressure, the tumors survive. By contrast, when these highly immunogenic tumors are transplanted to an immunocompetent host, they are rejected.
The third stage of immunoediting is the escape phase in which tumor cells have been edited and selected for their capacity to elude immune detection and elimination. A number of processes can contribute to immune escape. Many tumors down regulate or eliminate their expression of major histocompatibility complex (MHC) class Ia molecules, which are necessary restricting elements for CTL and whose absence renders the tumor cells invisible to CTL (Algarra et al., 2000; Marincola et al., 2000). A recent study has shown that 50% of the primary uveal melanomas examined expressed MIC-A/B, which is the ligand that activates NKG2D on NK cells (Vetter et al., 2004). However, none of the 11 metastases expressed MIC-A/B, which suggests that a selection process was invoked and facilitated the outgrowth of disseminated melanoma cells, which unlike the primary tumor, were now invisible to NK cells. Other tumors create an immunosuppressive milieu by secreting immunosuppressive and anti-inflammatory cytokines such as IL-10 and transforming growth factor-β (TGF-β) (Khong and Restifo, 2002). Tumors also have the capacity to disable immune effector elements. Uveal melanoma cells express complement regulatory proteins that inactivate the complement cascade and prevent cytolysis by complement-fixing antibodies (Goslings et al., 1996). Uveal melanomas also produce indoleamine dioxygenase (IDO), which depletes tryptophan resulting in the elimination of T lymphocytes by starvation (Chen et al., 2007).
3. Immune Privilege in the Eye: Fertile Ground for Tumor Growth?
The earliest documented evidence suggesting that the eye possessed unusual immunological properties can be traced to the century before last when the Dutch ophthalmologist van Dooremaal noted the prolonged survival of murine skin allografts placed into the anterior chamber (AC) of the dog eye (van Dooremaal, 1873). The first indication that the human eye possessed similar qualities occurred with the report of the first successful corneal transplant, which was performed on a human subject in 1905 (Zirm, 1906). This remarkable feat occurred 60 years before anti-rejection drugs were developed for organ transplantation and decades before the major histocompatibility complex was defined. In the 1940’s pathologists used the anterior chamber (AC) of the rabbit eye for propagating human tumor biopsy specimens as a putative assay for malignancy. We now know that the survival of such xenografts was the result of ocular immune privilege and not necessarily the malignant potential of the transplanted tissues.
The concept of ocular immune privilege and the term itself can be attributed to Sir Peter Medawar who not only recognized the role of the immune system in the rejection of organ allografts, but also understood the significance of the prolonged survival of organ allografts placed into the eye or the brain (Billingham and Boswell, 1953; Billingham et al., 1951; Medawar, 1948). Medawar believed that the apparent absence of patent lymphatic drainage of the eye acted to sequester foreign allografts and their antigens from the peripheral immune apparatus. Subsequent studies performed in mice have shown that antigens introduced into the AC of the mouse eye are not sequestered as originally proposed by Medawar, but in fact, rapidly reach head and neck lymph nodes (Camelo et al., 2006; Camelo et al., 2004; Egan et al., 1996). Thus, “immunological ignorance” due to anatomical sequestration is not the basis for immune privilege in the AC of the eye.
A flurry of research beginning in the late 1970’s has revealed that immune privilege in the eye is the sum total of anatomical, physiological, and immunoregulatory processes that restrict the induction and expression of immune responses. Immune privilege in the eye is extended to both the innate and adaptive immune responses.
3.1. Anatomical and Structural Features that Contribute to Ocular Immune Privilege
The notion that the AC of the eye lacked lymphatic drainage was originally based on anatomical studies, which consistently failed to detect lymph vessels, and more recent studies that have yet to demonstrate the expression of lymph vessel endothelial-specific surface markers (e.g., LYVE-1) on putative lymph vessels in the AC. Nonetheless, antigens introduced into the AC accumulate in ipsilateral lymph nodes in the heads and necks of mice (Camelo et al., 2006; Camelo et al., 2004) and induce the clonal expansion of T cells bearing the T cell receptor (TCR) specific for the antigen injected into the AC (Egan et al., 1996). In the end, it may be a semantic issue as to whether patent lymph vessels drain the AC of the eye, as it has been shown that the uveal/scleral pathway can direct AC-injected molecules to head and neck lymph nodes and can account for 25–35% of the outflow from the AC of the monkey eye (Sherman et al., 1978). Regardless of whether there are patent lymphatics in the interior of the eye, antigens introduced into the AC reach regional lymph nodes but fail to elicit conventional adaptive immune responses (discussed below).
The unique vasculature of the eye creates a blood:ocular barrier that restricts the movement of macromolecules and leukocytes from the vasculature into the eye. The vascular endothelial cells in the retina and iris are non-fenestrated and have tight junctional complexes that contribute to this anatomical barrier (Bill, 1970). This blood:ocular barrier is believed to reduce the likelihood that leukocytes will enter the eye. It also restricts the movement of T lymphocytes that recognize antigens unique to the eye. For example, experimental autoimmune uveitis (EAU) is mediated by CD4+ T lymphocytes that recognize retina-specific epitopes such as interphotoreceptor retinoid-binding peptide (IRBP) or retinal S-antigen. Although rodents can be immunized with IRBP or retinal S-antigen, ocular inflammation is not provoked unless the hosts’ blood:ocular barrier is breached by the administration of pertussis toxin.
Although MHC class Ia antigens are expressed on virtually all nucleated cells in the body, their expression is reduced or absent on many cells within the eye, especially those cells with a limited capacity for regeneration (Abi-Hanna et al., 1988; Lampson and Fisher, 1984; LeBouteiller, 1994). MHC class Ia molecules are absolutely required as restricting elements for CTL-mediated lysis of virus-infected cells. Cells lacking MHC class Ia molecules escape CTL-mediated lysis unless they are transfected with transgenes encoding MHC class Ia molecules (Joly et al., 1991). Thus, the absence of MHC class Ia molecules appears to be an adaptation to reduce the likelihood of CTL-mediated killing of corneal endothelial cells and retinal cells, both of which cannot regenerate. Moreover, both of these cell layers are crucial for normal vision. CTL-mediated lysis of corneal endothelial cells or retinal cells would lead to blindness.
Some viruses induce down-regulation of MHC class Iaexpression on the host cells as a means of evading CTL-mediated clearance. To compensate for this, NK cells are programmed to lyse MHC class Ia negative cells (Ljunggren and Karre, 1990). Thus, the absence of MHC class Ia molecules on corneal endothelial cells and retinal cells renders them vulnerable to cytolysis by NK cells. However, corneal endothelial cells and retinal cells express MHC class Ib molecules such as HLA-G and HLA-E in humans and Qa-2 in mice (Ishitani and Geraghty, 1992; Le Discorde et al., 2003; Niederkorn et al., 1999). The nonclassical MHC class Ib molecules engage the NK cell inhibitory receptor CD94-NKG2 and silence NK cell-mediated cytolysis (Kovats et al., 1990; Lee et al., 1998; Rouas-Freiss et al., 1997). Thus, structural features of the eye limit the entry of immune cells into the eye, and if such cells succeed in entering the eye, their cytolytic activity is either silenced or disabled due to the expression of inhibitory molecules (e.g., MHC class Ib) or by the absence of essential restricting elements (e.g., down-regulation of MHC class Ia) (Table 1).
Table 1.
Structural Features of the Eye that Contribute to Immune Privilege.
| Feature |
Effect |
Location |
|---|---|---|
| Blood: Eye Barrier | Restricts entry of macromolecules and inflammatory cells into the eye. | Tight junctions of vascular endothelial cells and non- fenestrated blood vessels. |
| Reduced expression of MHC class I and II molecules | Reduces susceptibility of ocular cells to cytotoxic T lymphocyte- mediated killing. | Corneal endothelium and retina |
| Expression of MHC Class 1b molecules | Sends inhibitor signals that turn NK cells “off” and prevents cytolysis of MHC class 1a negative cells. | Corneal endothelium and retina |
| Avascular and alymphatic cornea | Reduces trafficking of immune cells in the cornea. | Cornea |
3.2 Cell Membrane Molecules Affecting Ocular Immune Privilege
Cells lining the anterior and posterior segments of the eye are “decorated” with cell membrane-bound molecules that inhibit cells of the innate and adaptive immune responses (Table 2). Fas ligand (FasL) is expressed throughout the eye and induces apoptosis of Fas receptor-bearing inflammatory cells that enter the eye in response to viral infections (Griffith et al., 1995). FasL is also crucial for preventing the immune rejection of corneal allografts (Stuart et al., 1997; Yamagami et al., 1997). Ocular cells also express programmed cell death ligand-1 (PD-L1), which is a member of the B7 family of membrane proteins. Interaction of PD-L1 with its receptor PD-1, which is expressed on a variety of cells including T lymphocytes, results in down-regulation of T lymphocyte proliferation and cytokine production, and induction of T cell apoptosis (Ding et al., 2005; Dong et al., 2002; Okazaki and Honjo, 2007; Saunders et al., 2005). PD-L1 is expressed throughout the interior of the mouse and human eye (Hori et al., 2006; Shen et al., 2007; Yang et al., 2009). Like FasL, PD-L1 expression is crucial for corneal allograft survival; corneal allografts from PD-L1 KO mice or corneal allografts transplanted to mice treated with anti-PD-1 antibody invariably undergo immune rejection (Hori et al., 2006; Shen et al., 2007). PD-L1 appears to be an adaptive mechanism for dampening inflammation, as it is upregulated in the eyes of patients with at least one inflammatory eye disease (sympathetic ophthalmia) and inhibits T lymphocyte production of proinflammatory cytokines such as TNF-α and IFN-γ (Yang et al., 2009). Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), like FasL, is a member of the tumor necrosis factor family. TRAIL is expressed on many of the tissues in the eye that also express FasL and is believed to contribute to ocular immune privilege, although the evidence supporting this is presumptive (Lee et al., 2002; Wang S, 2003).
Table 2.
Cell Membrane-Bound Molecules that Contribute to Immune Privilege in the Eye.
| Molecule |
Effect |
Location |
|---|---|---|
| CRP | Inhibits complement cascade and inflammation elicited by anaphylatoxins generated during complement activation. | Cornea and retina |
| FasL | Induces apoptosis of activated T cells and neutrophils. | Cornea, iris, retina |
| TRAIL | Induces apoptosis of macrophages and neutrophils. | Cornea, iris, retina |
| PD-L1 | Inhibits T lymphocyte proliferation; induces apoptosis of T lymphocytes. | Cornea, iris, retina |
CRP = complement regulatory proteins; FasL = CD95L, Fas ligand; TRAIL = tumor necrosis factor-related apoptosis-inducing ligand; PD-L1 = programmed death ligand-1.
The complement system is composed of 11 plasma proteins that are activated in a cascade-like manner and generate a membrane attack complex (MAC) that punctures the cell membranes of both bacterial and mammalian cells, resulting in osmotic lysis. Complement can be activated by the conventional pathway when complement-fixing antibodies bind to their cognate antigen, or in the absence of antibody by the alternative pathway by bacterial products. Thus, the complement system straddles both the adaptive and innate immune systems. Complement activation also generates soluble factors that recruit and activate inflammatory cells including granulocytes. Although the complement system is crucial for protection against bacterial infections, the provocation of granulocytic inflammation can inflict extensive injury to innocent bystander cells through the generation of reactive oxygen species and the elaboration of proteases by the inflammatory cells. However, cells within the anterior and posterior segments of the eye express cell membrane-bound complement regulatory proteins (CRP’s) that inactivate the complement cascade and protect ocular cells from complement-mediated cytolysis and prevents the induction of granulocytic inflammation (Bora et al., 1993; Lass et al., 1990; Sohn et al., 2000a, b).
It is clear that cell membrane-bound molecules that are expressed extensively throughout the eye protect ocular tissues from injury inflicted by elements of both the innate and adaptive immune responses.
3.3 Soluble Factors that Contribute to Ocular Immune Privilege: “There’s Something Funny about Aqueous Humor”
The aqueous humor (AH) that fills the AC of the eye is richly endowed with immunosuppressive and anti-inflammatory factors that affect both the innate and adaptive immune responses (Table 3) (Taylor, 2007). At least four different molecules are present in the AH that suppress delayed-type hypersensitivity (DTH). DTH responses are known to produce extensive collateral damage to juxtaposed tissues and controlling this form of immune-mediated inflammation is crucial for maintaining the integrity of the visual axis and preserving vision. However, the AH contains at least four molecules that inhibit DTH responses: a) transforming growth factor-β (TGF-β); b) α-melanocyte-stimulating hormone (α-MSH); c) vasoactive intestinal peptide (VIP), and d) calcitonin gene-related peptide (CGRP). The AH also contains a variety of CRP that profoundly inhibit the complement cascade (Bora et al., 1993; Lass et al., 1990; Sohn et al., 2000b). In addition to α-MSH, vasoactive intestinal peptide (VIP), and calcitonin gene-related peptide (CGRP), another neuropeptide, somatostatin, is found in the AH (Taylor and Yee, 2003). Somatostatin (SOM) not only suppresses IFN-γ production by activated T cells, but also induces the production of α-MSH, which is involved in the generation of CD4+, CD25+ regulatory T cells (Taylor and Yee, 2003).
Table 3.
Soluble Factors that Contribute to Immune Privilege in the Eye.
| Factor |
Effect |
Location |
|---|---|---|
| TGF-β | Suppresses activation of T lymphocytes, NK cells, macrophages; renders APC tolerogenic and facilitates induction of ACAID. | Aqueous humor; vitreous; secreted by iris and ciliary body cells. |
| VIP | Inhibits T lymphocyte activation and proliferation. | Aqueous humor. |
| CGRP | Inhibits elaboration of proinflammatory factors by macrophages. | Aqueous humor. |
| α-MSH | Inhibits DTH and release of inflammatory factors by macrophages; inhibits activation of neutrophils; induces CD4+CD25+ T regulatory cells. | Aqueous humor. |
| SOM | Suppresses interferon-γ production by activated T lymphocytes; induces production of α-MSH. | Aqueous humor. |
| sFasL | Suppresses neutrophil recruitment and activation. | Aqueous humor. |
| CRP | Inhibits complement cascade. | Aqueous humor, vitreous, and tears. |
| IDO | Depletes tryptophan; inhibits T lymphocyte and NK cell Proliferation; inhibits NK cell-mediated cytolysis. | In cytoplasm of corneal cells, iris/ciliary body cells, and retinal cells. |
TGF-β, transforming growth factor-beta; VIP, vasoactive intestinal peptide; CGRP, calcitonin gene-related peptide; α-MSH, alpha melanocyte stimulating hormone; SOM, somatostatin; sFasL, soluble Fas ligand; CRP, complement regulatory proteins; IDO, indoleamine dioxygenase.
AH-borne factors also inhibit elements of the innate immune apparatus. A <10 KDa peptide present in AH induces apoptosis of NK cells, macrophages, and neutrophils (D’Orazio et al., 1999). Although it is not found as a soluble molecule within the AH, indoleamine dioxygenase (IDO) is produced by corneal cells and catabolizes tryptophan, which is a key amino acid that is vital for T cell survival. IDO in the cornea abrogates clonal expansion of allospecific T lymphocytes (Beutelspacher et al., 2006; Ryu and Kim, 2007) and inhibits the proliferative and cytolytic activity of NK cells (Della Chiesa et al., 2006; Frumento et al., 2002). As mentioned earlier, NK cells recognize cells failing to express MHC class Ia molecules and as a result, these cells are targeted for cytolysis by NK cells. However, the AH contains at least two factors that inhibit NK cell-mediated cytolytic activity: macrophage migration inhibitory factor (MIF) and TGF-β (Apte et al., 1997; Apte and Niederkorn, 1996; Apte et al., 1998). Moreover, corneal endothelial cells produce IDO, which inhibits the proliferation and cytolytic activity of NK cells (Beutelspacher et al., 2006).
It has been suggested that a low level of spontaneous activation of the complement system is necessary for protecting against occasional bacterial infections and that CRP’s prevent unbridled complement activation as part of maintaining immunological homeostasis. Evidence supporting this was reported by Sohn et al. who found that the administration of neutralizing antibody to one of the CRP, Crry, resulted in severe intraocular inflammation in rats (Sohn et al., 2000a).
3.4 Anterior Chamber-Associated Immune Deviation: Evidence that Ocular Immune Privilege Reaches Beyond the Eye
Medawar’s original explanation that immune privilege of the eye was simply due to sequestration of antigens due to the absence of patent lymphatic drainage was tacitly accepted for over a quarter of a century until seminal studies by Streilein and co-workers demonstrated that alloantigenic cells introduced into the AC elicited a unique systemic immune response that was characterized by the down-regulation of systemic cell-mediated immune responses, but the preservation of antibody production (Kaplan and Stevens, 1975; Kaplan and Streilein, 1977, 1978; Kaplan et al., 1975). Subsequent studies indicated that allogeneic tumors introduced into the AC grew progressively while similar allogeneic tumor cell inocula were routinely rejected if they were injected to sites outside of the eye (Niederkorn et al., 1981). Moreover, AC injection of allogeneic DBA/2 tumor cells not only resulted in the progressive intraocular tumor growth, but also the suppression of the host’s capacity to reject DBA/2 skin allografts, yet the rejection of unrelated skin allografts was preserved. This unique spectrum of systemic immunity was termed anterior chamber-associated immune deviation (ACAID)(Streilein and Niederkorn, 1981) and has been demonstrated with a wide range of antigens including viruses, tumor antigens, soluble proteins, and histocompatibility antigens (Niederkorn, 2006a, b, 2007; Streilein, 2003). The hallmarks of ACAID are the antigen-specific down-regulation of DTH and CTL responses, with the preservation of antibody production (Niederkorn, 2006b, 2007; Streilein, 2003). ACAID is a remarkably complicated immunoregulatory process that involves at least four separate organ systems: a) eye; b) thymus; c) spleen; and d) sympathetic nervous system (Figure 1).
Figure 1.
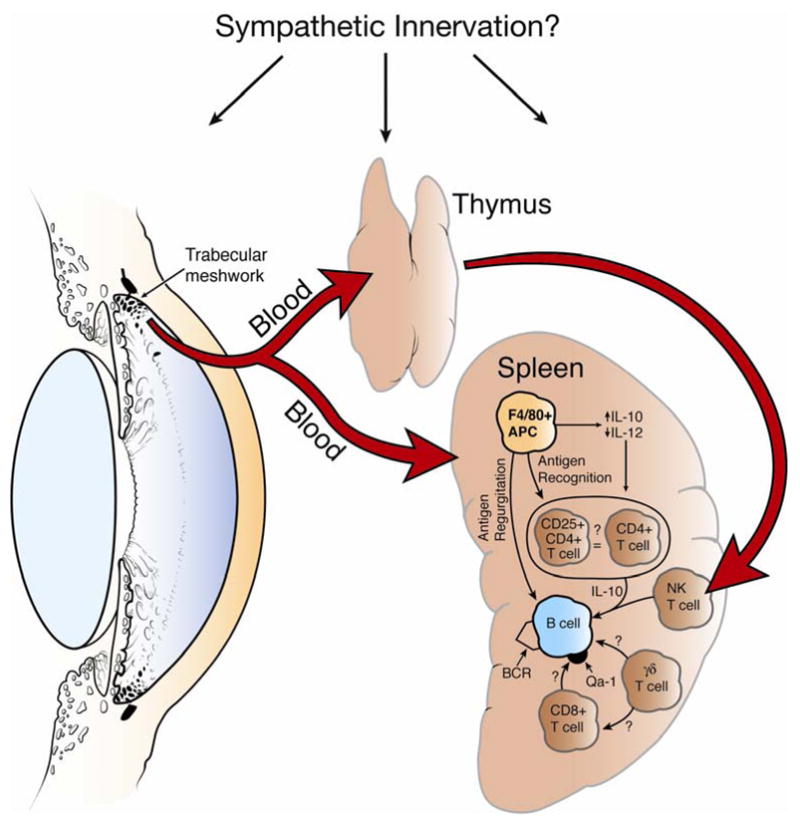
Organ systems and immune cells involved in the induction of ACAID. Removal of the eye, thymus, or spleen within 72 hours of anterior chamber injection prevents the induction of ACAID. Chemical sympathectomy prior to anterior chamber injection of antigen also prevents the induction of ACAID. BCR = B cell receptor. Reproduced with permission (Niederkorn, 2006b).
The induction of ACAID requires the presence of an intact eye, thymus, and spleen, as removal of any of these organs with 72 hr of injecting antigen into the AC prevents the generation of ACAID and instead, elicits DTH responses similar to those induced by injecting antigen into conventional sites (Streilein and Niederkorn, 1981; Wang et al., 2001; Whittum et al., 1982). The sympathetic nervous system is necessary for the induction of ACAID. Chemical sympathectomy prevents ACAID, possibly by disrupting the extensive sympathetic innervations of the eye, thymus, and spleen (Li et al., 2004). The induction of ACAID begins when antigens introduced into the AC are captured by resident F4/80+ macrophages. Under the influence TGF-β and thrombospondin, which are present in the AH, ocular macrophages are imprinted to simultaneously up-regulate IL-10 and down-regulate IL-12 (D’Orazio and Niederkorn, 1998b; Wilbanks et al., 1992; Wilbanks and Streilein, 1992; Zamiri et al., 2005). Within 48 hr of AC injection of antigen, F4/80+ macrophages enter the peripheral blood and migrate to the spleen and thymus (Wang et al., 2001; Wilbanks and Streilein, 1991). The blood-borne F4/80+ macrophages are potent amplifying antigen presenting cells (APC) – as few as 20 of these cells can induce ACAID if adoptively transferred to naïve recipients (Wilbanks et al., 1992). The blood-borne ocular APC migrate to the thymus and spleen within 72 hr of AC injection. Within the thymus the APC elicit the development of CD4−, CD8−, NK1.1+ T cells (NKT cells), which then emerge from the thymus and enter the bloodstream where they migrate to the spleen. In addition to migrating to the thymus, ocular APC migrate to the spleen where they, in concert with thymic NKT cells, initiate a series of cellular interactions involving marginal zone B cells, CD4+ T cells, CD8+ T cells, γδ T cells and the third component of complement. These interactions culminate in the generation of CD8+ T regulatory cells (Tregs) that inhibit both Th1- and Th2-based immune inflammation (Niederkorn, 2006b). All three organs involved in the induction of ACAID – eye, spleen, and thymus – possess dense sympathetic innervations. The sympathetic nervous system is important in the maintenance of ocular immune privilege as chemical sympathectomy prior to AC injection of antigen prevents the development of ACAID (Li et al., 2004). It is not clear how the sympathetic nervous system exerts its effect, but it appears that it is not at the level of the AC, as chemical sympathectomy does not affect the generation of F4/80+ ocular APC’s (Li et al., 2004). Thus, the induction and expression of ACAID is remarkably complex and requires the active participation of multiple organs and organ systems including the circulatory system, sympathetic nervous system, thymus, spleen, and eye.
3.5 In Situ Induction of Ocular Regulatory Cells
The pigmented epithelial cells of the iris and ciliary body (I/CB) secrete the constituents of the AH and display important immunomodulatory properties (Helbig et al., 1990; Hooper et al., 1991; Knisely et al., 1991). In addition to secreting soluble factors that exert immunomodulatory and anti-inflammatory effects, I/CB cells directly suppress T lymphocyte proliferation and production of IFN-γ by a contact-dependent mechanism, which does not rely on TGF-β, IL-10, or TNF-α (Yoshida et al., 2000b). Cell to cell contact between pigmented I/CB epithelial cells and T lymphocytes results in the generation of Tregs that inhibit T lymphocyte proliferation and antigen-specific DTH (Yoshida et al., 2000a). The locally-induced Tregs mediate suppression by secreting active and latent forms of TGF-β. These findings suggest that inflammatory cells that enter the eye by way of the I/CB blood vessels encounter I/CB pigmented epithelial cells and are converted from potential inflammatory cells to Tregs. Thus, subsequent waves of inflammatory cells that enter the eye come under the influence of the locally-induced Tregs.
A second category of antigen non-specific T regs can be generated in the eye through the action of α-MSH. α-MSH, like other cytokines in the AH, suppresses IFN-γ production by activated T lymphocytes and enhances T lymphocyte production of anti-inflammatory cytokines, such as TGF-β1(Taylor, 2007). In addition to its immunosuppressive properties, α-MSH converts T lymphocytes that enter the eye into CD4+, CD25+ Tregs that suppress DTH responses and mitigate immune-mediated inflammation such as EAU (Namba et al., 2002; Nishida and Taylor, 1999; Taylor and Namba, 2001). Thus, the neuropeptides in the AH provide yet one more pathway for the generation of ocular regulatory T cells.
The role of I/CB-induced and neuropeptide-induced T regs on the immune surveillance of intraocular tumors has not been explored, but is worthy of further investigation.
3.6 Relevance of ACAID: Why Does the Eye Tolerate an “Immunological Blind Spot”?
ACAID has been used experimentally as a strategy for inducing Tregs that mitigate immune-mediated diseases such as experimental autoimmune uveitis (EAU)(Mizuno et al., 1989; Mizuno et al., 1988) and for preventing the immune rejection of corneal allografts (Niederkorn and Mellon, 1996; She et al., 1990; Sonoda et al., 2002; Sonoda and Streilein, 1993). Orthotopic corneal allografts are placed over the AC of the eye and in fact, form the anterior wall of the AC of the eye. Antigens sloughed from the orthotopic corneal allograft into the AC are believed to induce ACAID. Moreover, rodents with long-term clear corneal allografts display an immunologic phenotype that is similar, if not identical, to ACAID (Sonoda et al., 2002; Sonoda and Streilein, 1993). In addition, maneuvers that prevent the induction of ACAID invariably lead to the immune rejection of corneal allografts (Niederkorn, 2006a). The preservation of a dynamic and complex immunoregulatory phenomenon such as ACAID suggests that this form of immune regulation provides significant benefit to the host. Corneal allografts appear to induce ACAID by virtue of their sloughing of corneal antigens into the AC and the induction of ACAID by AC injection of donor cells conveys considerable benefit to the long-term survival of corneal allografts, however it is unlikely that the immune system anticipated corneal transplantation when it created ACAID. Moreover, the presence of such an “immunological blind spot”, would seem to be counterintuitive, as it would render the host vulnerable to a variety of infectious agents. However, the eye is an extension of the brain, and like the brain, it has a very limited capacity to regenerate. Therefore, immune-mediated inflammation that inflicts considerable collateral damage to juxtaposed normal tissues, such as that produced by DTH responses, would jeopardize the function of the eye. ACAID appears to be an elegant counter measure to control DTH and complement-mediated inflammation elicited by antigens that enter the eye, that either do not pose significant threats to the host’s well-being or can be controlled by other means. For example, non-complement-fixing antibodies that are induced by ACAID would be capable of neutralizing viruses without arousing inflammation. Type I interferons are also capable of controlling virus infections without inflicting collateral injury. However, it should be noted that ACAID is neither universal nor immutable. Not all antigens induce ACAID (D’Orazio and Niederkorn, 1998a; Knisely et al., 1987; Li and Niederkorn, 1997; Niederkorn, 1991; Niederkorn, 1995; Niederkorn, 1997; Niederkorn and Knisely, 1988) and the presence of immune-mediated ocular diseases such as uveitis indicates that immune regulation in the eye is imperfect. Moreover, the weight of evidence suggests that ACAID is not necessarily a means of maintaining tolerance to self antigens that are not expressed in the thymus during the critical clonal deletion phase of tolerance induction. Instead, ACAID appears to be an effective adaptation for regulating immune responses in an organ that cannot tolerate unrestrained inflammation and the elimination of crucial parenchymal cells that are unable to undergo mitosis.
4. Circumvention of Immune Privilege and Intraocular Tumor Rejection
On first blush, one might assume that the redundant processes that provide immune privilege would exclude immune surveillance from functioning against intraocular tumors. However, the development of uveitis and the immune rejection of corneal allografts indicate that ocular immune privilege is not universal and under defined conditions, can be circumvented. There is evidence that human intraocular tumors are potentially susceptible to immune surveillance. Human uveal melanomas express melanoma-specific MAGE antigens and melanoma-associated antigens gp100 and tyrosinase, which are potential targets for immunological attack (Chen et al., 1997; Luyten et al., 1998; Mulcahy et al., 1996). Moreover, CD8+ CTL isolated from either the peripheral blood of uveal melanoma patients or from primary uveal melanomas have been shown to lyse human uveal melanoma cells in vitro (Kan-Mitchell et al., 1991; Ksander et al., 1998; Ksander et al., 1991). Elements of the innate immune system, namely NK cells, are also capable of killing human uveal melanoma cells in vitro (He et al., 2004; Ma and Niederkorn, 1995) and inhibiting the growth of human uveal melanomas transplanted into the eyes of NK-competent athymic nude mice (Apte et al., 1997). The most compelling data indicating that intraocular tumors can undergo immune rejection are derived from prospective studies in animals (Chen and Ksander, 2007; Niederkorn, 1991, 1997; Niederkorn, 2002; Niederkorn and Wang, 2005).
4.1 Allogeneic, Xenogeneic, and Syngeneic Intraocular Tumor Rejection: Lessons from Three Blind Mice
The majority of animal studies on the immunology of intraocular tumors have employed intraocular transplantation of tumor cells into: a) syngeneic, b) allogeneic, or c) xenogeneic mice. Early studies used the Greene hamster melanoma cell line, which was derived from a cutaneous melanoma that arose in an outbred Syrian hamster (Greene, 1958). The Greene hamster melanoma has been transplanted into the eyes of rabbits, rats, and mice for studies on the immunology and therapy of uveal melanomas. This model is of questionable value and has fallen out of favor for several fundamental reasons. The Greene hamster melanoma cell line was derived from outbred Syrian hamsters and therefore, represents a xenograft that elicits a robust xenograft rejection even in the AC of the eye, thereby disqualifying its use for immunology studies. Moreover, this tumor cell line is known to be contaminated with at least one C-type retrovirus that is capable of transforming juxtaposed cells (Russell et al., 1979).
Early studies on ACAID employed DBA/2 (P815) mastocytoma cells transplanted into the eyes of allogeneic Balb/c mice. The P815 tumor cell line originated in DBA/2 mice and expresses both MHC antigens and the full array of multiple minor histocompatibility (H) antigens of the DBA/2 mouse. Minor H antigens have been used by many investigators as surrogate tumor antigens. P815 cells undergo conventional immune rejection following subcutaneous transplantation in allogeneic Balb/c mice. However, P815 cells transplanted into the AC induce ACAID and grow progressively (Niederkorn et al., 1981). The progressive intraocular tumor growth coincides with the antigen-specific suppression of systemic DTH responses. As stated earlier, the induction of ACAID requires an intact spleen, as splenectomized Balb/c mice fail to develop ACAID, and instead mount robust DTH responses to the P815 tumors. In splenectomized Balb/c mice the intraocular tumors undergo immune rejection that is characterized by ischemic necrosis that culminates in phthisis of the affected eye (Streilein and Niederkorn, 1981). The histopathological sequelae of the rejected intraocular P815 tumors are reminiscent of DTH lesions with infarction, perivascular cuffing of lymphocytes, and ischemic necrosis of innocent bystander cells (Niederkorn, 1991; Niederkorn, 1995; Niederkorn, 1997). Thus, circumventing immune privilege by splenectomy resulted in the emergence of allodestructive DTH responses that promote the rejection of the intraocular tumor allograft, but at the expense of the eye.
This phthisical form of intraocular tumor rejection is not unique to allogeneic tumor allografts, but also occurs with highly immunogenic syngeneic tumors. A mutant of P815 mastocytoma (P91) was created by chemical mutagenesis and expresses potent tumor-specific antigens that elicit strong DTH responses following intraocular transplantation in syngeneic DBA/2 mice, even in the presence of an intact spleen (Niederkorn and Meunier, 1985). The development of DTH responses to the tumor-specific antigens on the P91 tumor cells coincides with the onset of intraocular tumor rejection. Like the P815 tumor allograft model, the P91 syngraft tumor undergoes immune rejection by a process that produces extensive ischemic necrosis and culminates in phthisis.
Circumvention of immune privilege and intraocular tumor rejection does not always culminate in phthisis and blindness. A variety of transplantable murine tumors undergo immune rejection in the eyes of syngeneic hosts by immune responses directed against tumor antigens without inflicting collateral injury to normal ocular tissues. Ultraviolet light (UV)-induced tumors express potent tumor-specific antigens and undergo immune rejection following intraocular transplantation in syngeneic mice (Knisely et al., 1987; Knisely and Niederkorn, 1990). Tumor resolution does not culminate in phthisis, but instead is characterized by piecemeal necrosis of the intraocular tumors, which is consistent with a CTL-mediated form of tumor rejection. Moreover, the resolving tumors were infiltrated with CD8+ T cells, which produced extensive cytolysis of the tumor cells in vitro (Knisely and Niederkorn, 1990). Experiments using a “Koch’s postulate-like” design demonstrated that the CD8+ tumor-infiltrating lymphocytes (TIL) produced non-necrotizing rejection when adoptively transferred to naïve immunoincompetent nude mice that were subsequently challenged intraocularly with the original UV-induced tumor (Knisely and Niederkorn, 1990). Thus, CD8+ T cells are found in rejecting intraocular tumors, can be isolated and shown to produce anti-tumor immunity in vitro, and can be transferred into second generation hosts where they recapitulate their original function.
Two other murine syngeneic intraocular models lend support to the hypothesis that immune privilege can be circumvented without tumor rejection leading to destruction of the eye and blindness (Ma et al., 1994a; Schurmans et al., 1999; Schurmans et al., 2001). The Ad5E1 tumor was induced in C57BL/6 mouse cells using the adenovirus type 5 early region oncogenes and expresses tumor-specific antigens that elicit potent CD4+ T cell responses that lead to a non-phthisical form of intraocular tumor rejection (Schurmans et al., 1999; Schurmans et al., 2001). The rejection of intraocular Ad5E1 tumors is not characterized by piecemeal necrosis and the presence of a moth-eaten appearance, and occurs in mice depleted of CD8+ T cells and in perforin KO mice, suggesting that CD8+ CTL are not involved in the non-phthisical rejection in this tumor model (Schurmans et al., 2001). Further studies indicated that CD4+ T cells, IFN-γ, and macrophages were absolutely necessary for the rejection of Ad5E1 tumors (Boonman et al., 2006; Schurmans et al., 2001; Wang et al., 2003). Adoptive transfer studies demonstrated that CD4+ T cells collected from mice that had rejected their intraocular tumors could produce non-phthisical intraocular tumor rejection when transferred to T cell-deficient mice (Dace et al., 2008). However, there were two prerequisites for CD4+ T cells to produce tumor rejection: a) the CD4+ T cells must be capable of producing IFN-γ, and b) the host must have a functional, intact ocular macrophage repertoire. The results of these studies suggest that IFN-γ serves two important roles in the rejection of the intraocular Ad5E1 tumors: a) it exerts an anti-angiogenic effect and b) it activates macrophages to become cytotoxic to the tumor cells. A series of experiments with bone marrow chimeric mice demonstrated that although CD4+ T cells needed to produce IFN-γ, the bone marrow-derived cells (including T lymphocytes and macrophages) did not need to respond to IFN-γ (Dace et al., 2007a). That is, tumor rejection occurred in IFN-γ receptor KO mice, which were capable of producing IFN-γ, but could not respond to this cytokine. Microarray analysis of Ad5E1 tumor cells revealed that IFN-γ acted directly on Ad5E1 tumor cells and induced the down-regulation of five proangiogenic genes and the up-regulation of four anti-angiogenic genes in the tumor cells (Dace et al., 2007a). Immunohistochemical studies have confirmed that macrophages infiltrate intraocular Ad5E1 tumors (Boonman et al., 2006) and recent in vitro studies have demonstrated that activated macrophages are capable of directly killing Ad5E1 tumor cells (Niederkorn et al. unpublished findings). Thus, non-phthisical rejection of syngeneic intraocular Ad5E1 tumors can occur in the absence of CD8+ CTL by a process that requires CD4+ T cells, yet tumor rejection is not directly mediated by CD4+ T cells themselves. These results suggest that when confronted with Ad5E1 tumor antigens, CD4+ T cells elaborate IFN-γ, which exerts an anti-angiogenic effect and thereby inhibits tumor growth and coincidentally activates macrophages that are capable of directly killing tumor cells.
Although the non-phthisical form of Ad5E1 tumor rejection can occur in the absence of CD8+ T cells and perforin, CD8+ T cells have the capacity to independently mediate the non-phthisical form of immune rejection (Dace et al., 2007b). CD8+ cells collected from Ad5E1 tumor-rejector mice and adoptively transferred to immunoincompetent severe combined immune deficient (SCID) mice induce non-phthisical rejection of intraocular Ad5E1 tumors. Interestingly, CD8+ T cells from perforin KO mice produce the same pattern of intraocular tumor rejection, which indicates that under these conditions, non-phthisical tumor rejection is not mediated by conventional CTL. However, the CD8+ T cell-mediated rejection does require TNF-α, as intraocular tumors grow progressively in the eyes of SCID mice that receive CD8+ T cells from TNF-α KO donor mice (Dace et al., 2007b).
A second tumor model involving the in situ transformation of retinal cells has been used to examine the pathophysiology of intraocular tumor rejection. Transgenic FVB/n mice bearing the simian virus 40 (SV40) oncogene under the influence of a tyrosinase promoter develop retinal pigment epithelial (RPE) carcinomas by in situ transformation (Anand et al., 1994). The transgenic mice develop bilateral intraocular tumors that grow progressively and fail to elicit immune responses to the intraocular tumors due to the germline expression of SV40 large T antigen, which induces classical immunological self-tolerance (Ma et al., 1994b). However, RPE carcinomas from the SV40 transgenic mice can be transplanted into the AC of syngeneic FVB/n mice or athymic nude mice. The RPE carcinomas grow progressively in the eyes of nude mice, but are rejected in the eyes of syngeneic immunocompetent, non-transgenic FVB/n mice (Ma et al., 1994a). Intraocular rejection of the RPE carcinomas is characterized by a heavy infiltration of CD8+ TIL that display antigen-specific in vitro tumor cell cytolysis. Moreover, adoptive transfer of the TIL to third-party athymic nude mice that are challenged in the AC with the RPE carcinoma cells results in non-phthisical tumor rejection that is characterized by piecemeal necrosis of the intraocular tumors. Unfortunately, these studies did not determine if the CD8+ T cell-mediated tumor rejection required perforin.
Collectively, these prospective studies using transplantable murine tumors have revealed an interesting spectrum of immune responses and pathological sequelae (Table 4). Weakly allogeneic tumors, such as P815 mastocytoma, grow progressively in the eyes of allogeneic mice due to the induction of ACAID. However, if ACAID is abrogated the allogeneic tumors undergo immune rejection by a process that appears to be DTH-mediated and culminates in phthisis of the affected eye. Blindness is the consequence of tumor rejection under these conditions. The phthisical form of immune rejection is not restricted to allogeneic tumors, but also occurs in at least one syngeneic tumor model (i.e., P91 mastocytoma). A second pattern of intraocular tumor rejection does not lead to phthisis and instead, leaves the eye anatomically and presumably functionally intact. The non-phthisical rejection is CD4+ T cell-dependent, but is not necessarily CD4+ T cell-mediated. Depending on the experimental conditions, CD4+ T cells can mediate non-phthisical tumor rejection in the absence of CD8+ T cells through the generation of IFN-γ, which concomitantly produces anti-angiogenic effects and activates tumoricidal macrophages. CD8+ T cells can independently mediate non-phthisical tumor rejection by acting as classical CTL and killing tumor cells by a perforin-dependent mechanism or by elaborating TNF-α, which induces tumor cell apoptosis. In each of these cases, the anatomical integrity of the eye is left intact and vision is preserved.
Table 4.
Mechanisms and Patterns of Intraocular Tumor Rejection
| Intraocular Tumor |
Host |
Pathological Sequelae |
Mechanism |
|---|---|---|---|
| P815 | Splenectomized Balb/c (allogeneic) | Phthisis | Delayed-type hypersensitivity |
| P91 | DBA/2 (syngeneic) | Phthisis | Delayed-type hypersensitivity |
| SV40-induced RPE carcinoma | FVB/n (syngeneic) | Non-phthisis | CD8+ cytotoxic T lymphocytes |
| Adenovirus-induced tumors | C57BL/6 (syngeneic) | Non-phthisis | CD4+ T lymphocytes produce IFN-γ and generate tumoricidal macrophages and lymphocytes. |
| Adenovirus-induced tumors | SCID (T cell-deficient) | Non-phthisis | Adoptively transferred CD8+ T lymphocytes mediate rejection via production of TNF-α. |
| UV-induced fibrosarcoma | Balb/c (syngeneic) | Non-phthisis | CD8+ cytotoxic T lymphocytes |
4.2 Immune Surveillance of Uveal Melanomas
In spite of the redundant mechanisms that support immune privilege, there is evidence that immune inflammation can occur within the eye. Anterior uveitis and corneal allograft rejection remind us that the adaptive immune system can be aroused and produce immune-mediated injury within the eye. Although animal studies have provided evidence that tumor-specific antigens expressed on intraocular tumors can provoke T cell-dependent immune responses, there is a dearth of data implicating a role for adaptive immunity to intraocular tumors in humans. Spontaneous resolution of uveal melanomas and retinoblastomas has been reported, although the incidence is exceptionally rare (Ashton, 1964; Jensen, 1974; Khodadoust et al., 1977; Reese, 1966).
Lymphocytes have been found within 7–20% of uveal melanomas, which suggests the presence of an adaptive immune response to the tumor antigens (de la Cruz et al., 1990; Durie et al., 1990; Nitta et al., 1990). One study reported that the TIL in seven of the eight uveal melanomas examined shared the same Vα T cell receptor (TCR) genotype suggesting the presence of a uveal melanoma-specific T cell response (Nitta et al., 1990). Human uveal melanomas express an array of melanoma-specific and melanoma-associated antigens that are potential targets for immunological attack (Chen et al., 1997; Luyten et al., 1998; Mulcahy et al., 1996). Moreover, CD8+ lymphocytes collected from uveal melanoma patients kill uveal melanoma cells in vitro (Kan-Mitchell et al., 1991; Ksander et al., 1998; Ksander et al., 1991). Thus, there is a significant body of evidence supporting the notion that the adaptive immune system can recognize and respond to uveal melanomas. Whether adaptive immunity has a significant impact in controlling primary uveal melanoma and its metastases remains to be established.
Recently, the role of the innate immune response in uveal melanoma has received a large amount of attention. Studies using uveal melanoma cell lines have demonstrated that many uveal melanomas are susceptible to NK cell-mediated cytolysis (He et al., 2004; Ma et al., 1995; Ma and Niederkorn, 1995). However, the susceptibility of uveal melanoma cells to NK cell-mediated cytolysis is inversely correlated with the expression of MHC class I molecules (Ma et al., 1995; Ma and Niederkorn, 1995). This is consistent with the “missing self” hypothesis, which states that MHC class I molecules expressed on target cells engage killer inhibitory receptors (KIR) that are expressed on NK cells and transmit inhibitory signals to the NK cells thereby silencing their cytolytic machinery and protecting the MHC class I positive cells from NK cell-mediated destruction (Ljunggren and Karre, 1990). Uveal melanomas, especially those arising in the iris, are bathed in AH, which contains large quantities of TGF-β (Cousins et al., 1991; Jampel et al., 1990). TGF-β, at the concentration found in normal AH, down regulates MHC class I expression on uveal melanoma cells and increases their susceptibility to NK cell-mediated cytolysis in vitro (Ma and Niederkorn, 1995). Thus, uveal melanomas reside in an environment that would appear to increase their vulnerability to NK cell-mediated clearance. As mentioned earlier, TIL have been detected in uveal melanomas (Anastassiou et al., 2001; de la Cruz et al., 1990; Durie et al., 1990; Nitta et al., 1990; Staibano et al., 2006). NK cells comprise 10% to 40% of the TIL population in uveal melanomas (de Waard-Siebinga et al., 1996; Ksander et al., 1991; Meecham et al., 1992) and NK cells isolated from uveal melanoma-containing eyes mediate cytolysis of NK sensitive tumor cells in vitro (Ksander et al., 1991). Thus, four observations suggest that NK cells might have a role in the immune surveillance of uveal melanomas: a) uveal melanoma cells are susceptible to NK cell-mediated cytolysis in vitro; b) NK cells enter melanoma-containing eyes; c) NK cells isolated from uveal melanomas kill tumor cells in vitro; and d) uveal melanomas reside in an environment that down-regulates their MHC class I expression and accordingly, would increase their susceptibility to NK cell-mediated cytolysis. However, a growing body of evidence suggests that NK cells evade NK cell-mediated immune surveillance in the eye.
One of the first observations suggesting the immune privilege in the eye might be extended to NK cell activity came from studies demonstrating that corneal endothelial cells, which lack MHC class I expression, were highly susceptible to NK cell-mediated cytolysis in vitro, yet there is no evidence to indicate that the corneal endothelium is damaged in eyes containing NK cells (Apte and Niederkorn, 1996). An important difference between corneal endothelial cells tested in vitro and those residing in the AC of the eye is the presence of AH that bathes the corneal endothelium in situ. Further studies revealed that NK cells exposed to AH, were no longer capable of killing corneal endothelial cells or NK-sensitive tumor cells (Apte and Niederkorn, 1996; Apte et al., 1998). Examination of AH reveals that it contains two important inhibitors of NK cells, TGF-β and macrophage migration inhibitory factor (MIF), both of which occur in concentrations that profoundly inhibited NK cell activity (Apte and Niederkorn, 1996; Apte et al., 1998; Rook et al., 1986). Interestingly, MIF produces an immediate inhibition of NK cell activity (Apte and Niederkorn, 1996; Apte et al., 1998), while the peak inhibitory effects of TGF-β requires18–22 hr (Rook et al., 1986). Thus, the combined effect of immediate-acting (MIF) and delayed-acting (TGF-β) inhibitors on NK cell activity provides uveal melanomas of the anterior segment of the eye with sustained protection from NK cell-mediated surveillance (Figure 2). In vivo studies in SCID mice provided further support for this hypothesis. Although SCID mice lack functional T cells, they display robust NK cell activity. Accordingly, NK-sensitive uveal melanoma cells underwent NK cell-mediated rejection following subcutaneous transplantation in nude mice, yet grew progressively when transplanted into the AC of SCID mice (Apte et al., 1997). The subcutaneous tumor rejection occurred even when the mice were challenged with tumor cell suspensions that were 50 times higher than the tumor cell suspensions that grew progressively when transplanted into the AC of the eye. Subcutaneous tumors did grow progressively if the SCID mice were depleted of NK cells by in vivo administration of anti-asialo GM1 antibody (Apte et al., 1997). Thus, uveal melanomas reside in an environment that provides them sanctuary from NK cell-mediated elimination.
Figure 2.
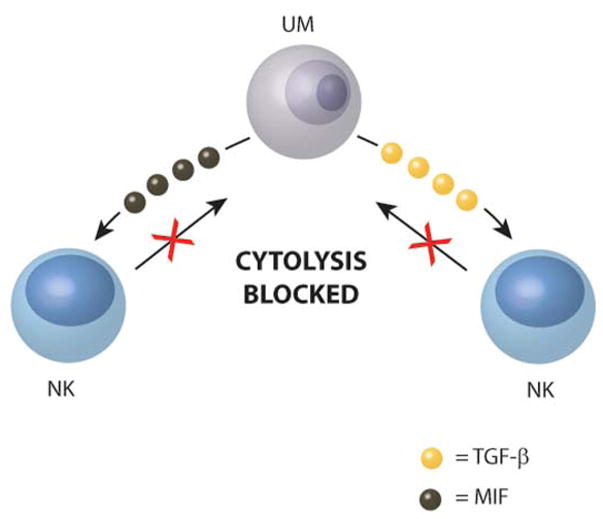
Uveal melanoma cells secrete factors that produce immediate (MIF) and delayed (TGF-β) suppression of NK cytolysis of tumor cells. UM = uveal melanoma cell.
Although cutaneous and uveal melanomas arise from neural crest progenitors, they differ significantly in their epidemiological (Mahoney et al., 1990), cytogenetic (McCarthy et al., 1993), metastatic (Augsburger et al., 1993; Char, 1978; Niederkorn, 1995; Vijayasaradhi and Houghton, 1995), and immunological (Niederkorn, 1995) characteristics. Uveal melanomas and cutaneous melanomas display remarkably different metastatic behavior. While cutaneous melanomas can metastasize to a wide variety of organs, uveal melanomas display a propensity to metastasize to the liver. In fact, liver metastases are present in up to 95% of the patients who die with uveal melanoma (Albert, 1997; Augsburger et al., 1993; COMS, 2001; Kath et al., 1993). An interesting dichotomy between cutaneous melanoma and uveal melanoma is found in the correlation between MHC class I expression and malignancy. Low expression of MHC class I molecules on cutaneous melanomas is associated with increased tumor thickness, progressive tumor growth, and poor prognosis (Brocker et al., 1985). Low expression of MHC class I molecules has been noted on metastases arising from a variety of non-ocular tumors including head and neck squamous cell carcinomas, as well as lung, colon, and cervical cancers (Hicklin et al., 1999). Metastases of cutaneous melanomas express reduced levels of MHC class I molecules compared to the primary tumors, which correlates with a poor prognosis (Ruiter et al., 1984; Ruiter et al., 1982; Ruiter et al., 1991; van Duinen et al., 1988). By contrast, the opposite occurs with uveal melanoma metastases (Jager et al., 2002b). High expression of HLA-B on primary uveal melanomas is correlated with increased malignancy and reduced survival (Blom et al., 1997d; Ericsson et al., 2001; Jager et al., 2002a). Moreover, elevated HLA-B expression is correlated with uveal melanomas that are predominantly composed of an epithelioid morphology, a cell type that has been consistently associated with increased malignancy (Blom et al., 1997c). Likewise, reduced expression of MHC class I molecules on primary uveal melanomas is associated with improved survival (Blom et al., 1997b; Blom et al., 1997d; Ericsson et al., 2001; Jager et al., 2002a). These findings are consistent with previous in vitro studies demonstrating a correlation between high expression of MHC class I on uveal melanoma cells and reduced susceptibility to NK cell-mediated cytolysis (Ma et al., 1995; Ma and Niederkorn, 1995).
5. Liver Metastasis of Uveal Melanoma: Immunoediting of NK Cell-Mediated Immune Responses
Mouse studies have demonstrated that NK cells can control the development of liver metastases arising from intraocular melanomas. Disabling the host’s NK cell repertoire results in a sharp increase in metastases (Dithmar et al., 1999), while augmenting NK cell activity with type I interferons reduces liver metastases (Alizadeh et al., 2003; Dithmar et al., 2000). Collectively, these findings indicate that uveal melanomas enjoy immune privilege from NK cell-mediated surveillance in the eye, but are vulnerable to NK cell-mediated elimination when they reach the liver. Since liver metastasis is the leading cause of death in uveal melanoma patients, the correlation between high MHC class I expression and poor prognosis is most likely the result of the uveal melanoma cells’ resistance to NK cell-mediated cytolysis in the liver. The weight of evidence supports the following scenario. Primary uveal melanomas contain high and low MHC class I expressing populations. Those populations of uveal melanoma cells expressing low amounts of MHC class I molecules are highly susceptible to NK cell-mediated cytolysis, but are shielded from NK cells by the MIF and TGF-β that are present in the AH and are secreted by cells in the uveal tract. However, once the uveal melanoma cells leave the eye and enter the liver, they find themselves in an organ that has one of the highest concentrations of NK cells of any organ in the body. Uveal melanoma cells expressing low MHC class I molecules would be promptly eliminated by NK cells in the liver, while melanoma cells expressing high quantities of MHC class I would be preserved and would become the dominant phenotype in the progressively growing liver metastases. This prediction is supported by a novel study by Verbick and coworkers who had the unusual opportunity to obtain primary uveal melanoma cells and cells from liver metastases in the same patient (Verbik et al., 1997). These investigators found that only 4% of the primary uveal melanoma cells expressed detectable MHC class I molecules, while tumor cells isolated from liver metastases expressed nine times as much MHC class I as the primary uveal melanoma cells.
NK cells can also escape NK cell-mediated cytolysis by elaborating the same inhibitory factors that protect them in the eye, (i.e., MIF and TGF-β). It is noteworthy that liver metastases of uveal melanomas produce approximately twice as much biologically active MIF as primary uveal melanoma cells (Repp et al., 2000). A recent study reported that all 13 of the uveal melanoma-containing eyes examined expressed TGF-β2, the isoform of TGF-β that is known to suppress NK cell activity (Esser et al., 2001). Thus, uveal melanoma cells have the capacity to secrete soluble factors that suppress NK cell activity in the tumor nidus and in a sense, recreate a version of the anterior chamber.
In addition to expressing killer inhibitory receptors (KIR), NK cells also express receptors that activate the cytolytic machinery of NK cells. NKG2D is an activating receptor that is expressed on the cell membranes of NK cells and engages its ligand, MIC-A/B, which is expressed on various tumor cells, including uveal melanomas. A recent study found that MICA/B was expressed on 50% of the primary uveal melanoma-containing eyes examined, but was absent on all 11 metastases specimens tested (Vetter et al., 2004). This finding is further evidence that a selection process favors the emergence of liver metastases that either resist NK cell-mediated cytolysis or in the case of MIC-A/B, fail to activate NK cells.
Collectively, these results suggest that a form of “immunoediting” occurs in the development of liver metastases of uveal melanomas. A mixed population of uveal melanoma cells leaves the eye and enters the bloodstream. NK cells within the bloodstream and in the liver eliminate NK cell-susceptible uveal melanoma cells, leaving behind a population of NK cell-resistant melanoma cells that form liver metastases. The emergence of liver metastases with a predominantly NK cell-resistant phenotype represents a form of “immunoediting” by NK cells and as such, is an important barrier for the immunotherapy of uveal melanoma.
6. Immune Escape Mechanisms of Uveal Melanomas: Creating Ad Hoc Immune Privilege
There is ample experimental and anecdotal data indicating that intraocular tumors in general and uveal melanomas in specific express tumor-specific antigens and can arouse both adaptive and innate immune responses. However, there is mounting evidence that uveal melanoma cells employ an impressive array of escape mechanisms to elude both adaptive and innate immune effector mechanisms. These escape mechanisms share a curious similarity to the aforementioned mechanisms that contribute to immune privilege in the eye.
6.1 Indoleamine 2,3 Dioxygenase
T lymphocytes require the amino acid tryptophan for clonal expansion, proliferation, and survival. In the absence of tryptophan, T lymphocytes perish. The enzyme indoleamine 2,3 dioxygenase (IDO) catalyzes the rate-limiting step in tryptophan catabolism. Tryptophan depletion by IDO curtails T lymphocyte-mediated immune responses (Frumento et al., 2002; Mellor et al., 2002; Munn et al., 1999; Terness et al., 2002). Proinflammatory cytokines, such as IFN-γ, upregulate IDO expression in macrophages and antigen presenting cells, presumably as a feed back mechanism for regulating T lymphocyte responses (Takikawa et al., 1991). Although IDO is not vital for NK cell survival, it inhibits NK cell proliferative and cytolytic activity (Della Chiesa et al., 2006; Frumento et al., 2002). Thus, IDO affects both the innate and adaptive immune responses. IDO plays a role in immune regulation and immune suppression in several conditions: a) controlling microbial infections (Daubener and MacKenzie, 1999); b) promoting allograft survival (Hainz et al., 2007); c) controlling autoimmune inflammation (Alexander et al., 2002); and d) maintaining the survival of the allogeneic fetus (Munn et al., 1998).
IDO is expressed in a variety of ocular tissues including the retina, iris/ciliary body, lens, and cornea (Beutelspacher et al., 2006; Malina and Martin, 1993, 1996; Ryu and Kim, 2007). IDO functions as an antioxidant and is believed to protect the eye against ultraviolet light-induced injury (Bodaghi et al., 1999; Malina and Martin, 1996; Takikawa et al., 1999). It was proposed that the IDO expressed by corneal cells was instrumental in providing immune privilege to corneal allografts (Beutelspacher et al., 2006). IDO produced by corneal cells was found to be biologically and enzymatically active in vitro (Beutelspacher et al., 2006); however, in vivo administration of an IDO inhibitor, 1-methyl-L-tryptophan, did not affect corneal allograft survival (Beutelspacher et al., 2006) Interestingly, over-expression of the IDO gene in corneal cells resulted in the suppression of T lymphocyte proliferation in mixed lymphocyte responses in vitro and the significant prolongation of corneal allografts in vivo (Beutelspacher et al., 2006).
IDO is believed to be an escape mechanism employed by tumors to avoid immune surveillance (Uyttenhove et al., 2003) and has been implicated in human malignancies including lung (Astigiano et al., 2005), colon (Brandacher et al., 2006), liver (Ishio et al., 2004), and endometrial (Ino et al., 2006) carcinomas as well as skin melanoma (Weinlich et al., 2007). Chen and co-workers examined uveal melanoma-bearing eyes and livers containing uveal melanoma metastases by immunohistochemistry and found that IDO was not expressed in situ in either primary uveal melanoma cells or liver metastases (Chen et al., 2007). However, in vitro studies on uveal melanoma cell lines determined that exposure to IFN-γ, a cytokine produced by both T lymphocytes and NK cells, resulted in an upregulation of IDO mRNA and protein in all five of the primary uveal melanoma cell lines and both of the metastases cell lines tested (Chen et al., 2007). Importantly, the IDO induced by IFN-γ was enzymatically and biologically active. These findings add to a growing list of tumors that appear to utilize IDO as an immune escape mechanism. It is conceivable that the induction of IDO by IFN-γ is a defensive mechanism on the part of uveal melanoma cells in response to the presence of T lymphocytes and NK cells, both of which are capable of producing this cytokine and killing uveal melanoma cells.
6.2 Programmed Death Ligand-1
Programmed cell death-1 (PD-1) is a receptor belonging to the CD28/CTLA-4 family and is expressed on a subset of thymocytes (Nishimura and Honjo, 2001), activated T and B cells (Agata et al., 1996; Blank et al., 2004; Carreno and Collins, 2002), and myeloid cells (Nishimura and Honjo, 2001). PD-1 has two ligands: PD-L1 and PD-L2. Both ligands are cell membrane-bound molecules and are members of the B7 family (Dong et al., 1999; Freeman et al., 2000; Latchman et al., 2001). PD-L1 is expressed on a wide variety of tissues and its expression is upregulated by the proinflammatory cytokine IFN-γ (Okazaki and Honjo, 2007; Yang et al., 2008; Yang et al., 2009). PD-L1 is also expressed on cells in the cornea and retina of human eyes and its expression is elevated in at least one immune-mediated ocular disease, sympathetic ophthalmia (Yang et al., 2009). PD-L2 expression appears to be limited to antigen presenting cells such as macrophages and dendritic cells (Latchman et al., 2001; Okazaki and Honjo, 2007). Engagement of PD-1 with either PD-L1 or PD-L2 results in the downregulation of T lymphocyte proliferation and cytokine production and culminates in apoptosis (Ding et al., 2005; Dong et al., 2002; Latchman et al., 2001; Okazaki and Honjo, 2007; Saunders et al., 2005). PD-L1 is constitutively expressed on human (Sugita et al., 2009; Yang et al., 2009) and murine (Hori et al., 2006; Shen et al., 2007) corneal cells and is crucial for the long-term survival of corneal allografts (Hori et al., 2006; Shen et al., 2007). In vivo treatment with blocking anti-PD-1 antibody results in a steep increase in the tempo and incidence of corneal allograft rejection(Hori et al., 2006; Shen et al., 2007). Likewise, corneal allografts prepared from PD-L1−/− mice invariably undergo immune rejection (Shen et al., 2007).
A recent investigation has reported that five of nine primary uveal melanoma cell lines and one of five metastatic cell lines from uveal melanoma patients constitutively expressed PD-L1 protein (Yang et al., 2008). However, exposure to the proinflammatory cytokine, IFN-γ, resulted in the expression of PD-L1 on all of the uveal melanoma and metastases cell lines. Moreover, IL-2 production by activated T lymphocytes was significantly reduced when the lymphocytes were co-cultured with IFN-γ-treated uveal melanoma cells. Addition of either anti-PD-L1 or anti-PD-1 blocking antibodies restored IL-2 production by the activated T lymphocytes, confirming that the impaired T lymphocyte function induced by the uveal melanoma cells was PD-L1-dependent. These results, like those related to IDO production, imply that uveal melanoma cells and metastases of uveal melanoma cells have the capacity to respond to the presence of cells of the adaptive (T lymphocytes) and innate (NK cells) immune systems by detecting the production of IFN-γ, which triggers the tumor cells to upregulate soluble and cell membrane-bound molecules that disable immune cells.
6.3 Complement Regulatory Proteins
The complement system is a group of plasma and membrane-bound proteins that are activated in a cascade-like manner. The complement cascade can be activated by three pathways: a) classical, b) alternative, and c) lectin pathways. The classical pathway is activated when complement-fixing antibodies - typically IgG and IgM isotypes - bind to their cognate antigen. The ensuing enzymatic reactions culminate in the generation of two important categories of biologically active molecules: a) peptide-like anaphylatoxins (e.g., C3a, C4a, and C5a), and b) the membrane attack complex (MAC). The anaphylatoxins act as potent chemoattractants that recruit and activate granulocytes. The complement cascade can also be activated by the lectin and alternative pathways by products produced by microorganisms. The lectin and alternative pathways act as a “first-responder” mechanism for rapidly activating the complement system before the adaptive immune system can be aroused to generate complement-fixing antibodies. Although anaphylatoxins serve an important function in focusing granulocytes to the site of an infection, the inflammation that they provoke can inflict injury to neighboring innocent bystander cells of the host through the release of reactive oxygen species (ROS) and proteases. Since the complement cascade can be activated by either microorganisms or by specific antibody, it straddles both the adaptive and innate immune systems. However, the complement system is controlled by CRP, which inactivate various stages of the complement cascade. CRP occur in soluble and cell membrane forms and are strategically located in organs such as the eye and placenta that are vulnerable to the collateral injury that is inflicted by granulocytic inflammation and the generation of MAC. Decay accelerating factor (DAF; CD55) and membrane cofactor protein (MCP; CD46) are expressed in both the eye and the trophoblast and play a crucial role in preserving vision and preventing immune-mediated abortion respectively (Bora et al., 1993; Goslings et al., 1998; Holmes et al., 1992; Holmes et al., 1990; Lass et al., 1990; Rooney et al., 1993; Sohn et al., 2000a, b). Crry is a functional homolog of DAF and MCP and is found only in rodents. Crry is crucial for protecting the rodent fetus from immune-mediated abortion. Mouse embryos with a deleted Crry gene perish in utero as a result of complement-activation and granulocytic inflammation (Xu et al., 2000). Crry is also expressed throughout the rodent eye and is important in buffering the effects of complement activation. Administration of anti-Crry antibody results in extensive intraocular inflammation and injury to ocular tissues (Sohn et al., 2000a). CRP are expressed on the cell membranes of numerous ocular tissues and in soluble forms in the AH and protect corneal endothelial cells, which do not express CRP, from complement-mediated cytolysis (Hargrave et al., 2003; Hegde et al., 2002).
Immunohistochemical analysis of uveal melanoma-containing eyes revealed the presence of all three CRP (CD46, CD55, and CD59) on uveal melanoma cells (Goslings et al., 1996). Studies on human uveal melanoma cell lines confirmed this observation and also demonstrated that the CRP protected uveal melanoma cells from complement-mediated cytolysis in vitro (Goslings et al., 1996). However, enzymatic removal of CRP rendered the uveal melanoma cells susceptible to complement-mediated lysis. Other investigations noted that the proinflammatory cytokine TNF-α upregulated CRP expression on some, but not all, uveal melanoma cells (Blom et al., 1997a). The latter finding is reminiscent of the upregulation of IDO and PD-L1 in uveal melanoma cells by proinflammatory cytokines elaborated by elements of the adaptive and innate immune systems. Although there is no compelling evidence to date suggesting that uveal melanomas elicit the production of complement-fixing antibodies or that antibodies play a role in the immune surveillance of uveal melanoma, it is intriguing that the expression of CRP represents yet one more modality that uveal melanomas have adopted from the repertoire of strategies that sustain immune privilege in the eye.
6.4 Resistance to Perforin-Mediated Cytolysis
The leukocytes of patients in the late stages of uveal melanoma produce increased amounts of IFN-γ. Increased serum levels of IFN-γ are associated with the development of metastases in uveal melanoma patients (Likhvantseva et al., 1999). Moreover, IFN-γ is frequently detected in uveal melanoma specimens (Filer et al., 1993). IFN-γ is a potent upregulator of MHC class I and II molecules and the increased presence of IFN-γ in uveal melanomas as well as in the circulating blood would be expected to increase the susceptibility of uveal melanoma cells to CTL-mediated cytolysis. However, high expression of MHC class I molecules on primary uveal melanomas is correlated with a poor prognosis (Blom et al., 1997b; Blom et al., 1997c; Blom et al., 1997d; Ericsson et al., 2001). The paradoxical effect of IFN-γ in the pathophysiology of uveal melanoma has several potential explanations. As stated earlier, IFN-γ upregulates the expression of two immunoinhibitory molecules, IDO and PD-L1, in uveal melanoma cells. Either or both of these molecules could disarm CTL. However, a recent study has uncovered yet one more effect of IFN-γ in promoting the escape of uveal melanoma cells from immune surveillance. Uveal melanoma cells treated with IFN-γ display a five-fold increase in the expression of MHC class I molecules, yet experience a decreased susceptibility to MHC class I-restricted cytolysis by CD8+ CTL (Hallermalm et al., 2008). IFN-γ does not affect the capacity of CD8+ CTL to become activated or to release cytolytic perforin granules. Instead, the inhibitory effect of IFN-γ is focused on the uveal melanoma cells themselves. Untreated uveal melanoma cells are susceptible to cytolysis when incubated with perforin, but become highly resistant to perforin-mediated cytolysis (>70% inhibition) following incubation with IFN-γ. The current paradigm of CTL-mediated cytolysis proposes that perforin released from CTL binds to the target cell, perforates the cell membrane, and facilitates the entry of granzyme B (grB) into the target cell (Raja et al., 2003). Granzyme B then initiates the apoptotic cell death program of the target cell by activating the caspase pathway. Treatment with IFN-γ significantly decreases the ability of grB to bind to and induce apoptosis of uveal melanoma cells. By contrast, neither type I interferon nor TNF-α affects grB-induced apoptosis or CTL-mediated cytolysis of uveal melanoma cells. Importantly, the inhibitory effect of IFN-γ is limited to the uveal melanoma cells and does not affect the function or viability of CTL. Since NK cells also utilize perforin to mediate cytolysis of tumor cells, it is likely that IFN-γ also protects uveal melanoma cells from NK cell-mediated killing. Moreover, IFN-γ also upregulates MHC class I molecules and IDO, both of which inhibit NK cell-mediated cytolysis. Thus, IFN-γ acts through three independent pathways to disable NK cell-mediated surveillance of uveal melanoma cells. This should raise a warning flag when the use of IFN-γ is suggested as a therapeutic modality for the management of uveal melanoma.
6.5 Blockade of FasL-Induced Apoptosis
In addition to utilizing perforin, CTL and NK cells can mediate tumor cell death by inducing apoptosis via engagement of death receptors on the target cell membrane. Death receptors belonging to the TNF superfamily transmit apoptosis-inducing signals when they engage their ligands that are expressed on NK cells and CTL. The key death-inducing ligands expressed by CTL and NK cells include TNF-α, TRAIL, and FasL. Interestingly, uveal melanoma cells express both Fas and FasL, yet are resistant to FasL-induced apoptosis mediated by either CTL or NK cells (Hallermalm et al., 2004). Exposure to proteases results in the production of a soluble form of FasL due to the cleavage of cell membrane-bound FasL from the uveal melanoma cell surface. The soluble FasL binds to Fas on the uveal melanoma cell membrane. However, apoptosis is not induced, as soluble FasL is over 1,000 times less efficient in the induction of apoptosis compared with the membrane-bound trimeric complex for of FasL (Schneider et al., 1998). This autocrine secretion and binding of soluble FasL to Fas on melanoma cells blocks engagement of Fas with cell membrane FasL that is expressed on CTL and NK cells. Thus, soluble FasL shields uveal melanoma cells from FasL-induced apoptosis and denies CTL and NK cells yet one more avenue for immune surveillance (Figure 3).
Figure 3.

Soluble FasL shed from uveal melanoma cells binds to Fas and blocks FasL-induced apoptosis by membrane-bound FasL that is expressed on NK cells and CTL. CTL = cytotoxic T lymphocyte; NK = natural killer cell.
6.6 Disabling NK Cell-Mediated Immune Surveillance
In vitro studies have demonstrated that uveal melanoma cell populations differ in their susceptibility to NK cell-mediated cytolysis (He et al., 2004; Ma et al., 1995; Ma and Niederkorn, 1995). Several observations support the notion that NK cells can exert a selective pressure on the development of uveal melanoma liver metastases. Uveal melanoma cells displaying a spindle cell morphology are less malignant and coincidentally, express little or no MHC class I molecules (Natali et al., 1989). By inference, one would predict that the absence or low expression of MHC class I molecules on uveal melanomas predominantly comprised of spindle cell melanoma cells should be vulnerable to NK cell-mediated surveillance. This would correlate with reduced malignancy that is typically associated with uveal melanomas comprised predominantly of tumor cells with a spindle cell morphology. By contrast, epithelioid uveal melanoma cells, a phenotype that is characteristically associated with heightened malignancy, typically express high levels of MHC class I (Blom et al., 1997c). Interestingly, those epithelioid uveal melanoma cells that express normal or reduced levels of MHC class I molecules, and presumably normal or enhanced susceptibility to NK cell-mediated cytolysis, display elevated amounts of c-myc, a transcription factor that stimulates tumor cell proliferation. Although, highly speculative, the increased proliferative potential conferred by c-myc might permit low MHC class I-bearing epithelioid uveal melanoma cells to escape immune elimination due to their enhanced proliferative capacities.
The intraocular milieu contains at least three molecules that inhibit NK cell activity: a) MIF; b) TGF-β; and c) IDO. It is noteworthy that uveal melanoma cells elaborate all three of these molecules and thus, in a sense, recapitulate the AH environment in terms of NK cell inhibition (Esser et al., 2001; Repp et al., 2000). In addition to inhibiting NK cell activity, uveal melanoma cells appear to down-regulate NK stimulatory molecules (Vetter et al., 2004).
It is clear that uveal melanoma cells employ a complex array of strategies for escaping immune surveillance by NK cells (Table 5).
Table 5.
Uveal Melanoma Escape Mechanism for Eluding NK Cell-Mediated Destruction
| Molecule |
Mechanism |
Location |
|---|---|---|
| MIF | Inhibits release of perforin by NK cells. | Aqueous humor; secreted by tumor cells. |
| TGF-β | Inhibits NK cell-mediated cytolysis. | Aqueous humor; produced by tumor cells. |
| IDO | Inhibits NK cell proliferation and cytolytic activity. | Produced by tumor cells and cells in iris, choroid, and retina |
| MHC class I molecules | Sends “off” signal to NK cells. | Expressed on epithelioid uveal melanoma cells and liver metastases. |
| Absence of MIC-A/B | Reduced activation of NK cells | Uveal melanoma metastases. |
7. Does the Immune System Promote Tumorigenesis?
7.1 The riddle of tumor-infiltrating lymphocytes and poor prognosis
The immune privilege of the eye provides intraocular tumors, including uveal melanoma, protection from both the innate and adaptive immune systems. This privilege is due at least in part to the action of soluble and cell membrane-bound molecules that disarm, suppress, or eliminate immune effector elements. Interestingly, uveal melanomas express many of these molecules, and in a sense, recapitulate ocular immune privilege. On the surface, one might expect the combination of ocular immune privilege and the acquisition of immunosuppressive capabilities of uveal melanomas would create a condition in which the incidence and malignancy of intraocular tumors would far exceed other body sites. However, uveal melanomas occur at a frequency that is over 25 times less than cutaneous melanomas (see American Cancer Society 2008 statistics; http://www.cancer.gov/cancertopics/types/melanoma/). It is also curious to note that the lens of the eye grows throughout life (although at a reduced rate as we age) and resides within an immunologically privileged milieu, yet spontaneous tumors of the lens (other than experimentally induced neoplasms) have not been described in any species except the cat (Zeiss et al., 2003). Thus, ocular immune privilege does not eventuate in an increased frequency or severity of intraocular tumors; in fact, one could argue that the opposite occurs. However, uveal melanomas that arise in the eye undoubtedly benefit from ocular immune privilege. The capacity of uveal melanoma cells to express MHC class I molecules, IDO, CRP, MIF, and PD-L1 after leaving the eye recapitulates the immune privilege that they left behind.
The past 20 years has witnessed a rebirth of interest in the immune surveillance hypothesis. The research that emerges in the years ahead will undoubtedly reshape the immune surveillance hypothesis to incorporate the immunoediting concept and to possibly consider the notion that in some conditions, the immune response might in fact contribute to tumor development. The latter hypothesis was introduced by Prehn almost 40 years ago (Prehn and Lappe, 1971) and there are observations in the past decade that lend credence to this iconoclastic concept. The presence of either TIL or tumor-associated macrophages in uveal melanomas carries a poor prognosis (Anastassiou et al., 2001; de la Cruz et al., 1990; Makitie et al., 2001). The association between TIL and a poor prognosis seems counterintuitive. However, two possible explanations may account for increased malignancy in uveal melanomas infiltrated with TIL. The first possibility is that T lymphocytes or NK cells in the TIL population might produce IFN-γ, which has multiple effects on uveal melanoma cells including: a) upregulation of MHC class I, which increases their resistance to NK cell-mediated cytolysis; b) increased resistance to perforin-induced cytolysis by CTL and NK cells; c) increased expression of IDO; and d) increased expression of PD-L1 (Figures 4 and 5). A second explanation, although not mutually exclusive with the first, is that T lymphocytes in the TIL population produce IL-2, which serves as a growth factor for uveal melanoma cells (Figure 6). It bears noting that some human uveal melanoma cells express IL-2 and IL-15 receptors and exposure to human recombinant IL-2 and IL-15 stimulates their growth and reduces their susceptibility to NK cell-mediated cytolysis in vitro (He et al., 2004)(Figure 6).
Figure 4.

Interferon-γ secreted by tumor-infiltrating lymphocytes upregulate MHC class I on uveal melanoma cells and stimulates their production of IDO. MHC class I blocks NK cell-mediated cytolysis while IDO inhibits NK cell proliferation. IFN-γ = interferon-γ; IDO = indoleamine dioxygenase; KIR = killer inhibitory receptor; TIL = tumor-infiltrating lymphocytes;
Figure 5.

TIL elaborate interferon-γ, which induces multiple protective mechanisms to prevent T cell-mediated immune surveillance.
Figure 6.
TIL produce IL-2, which stimulates uveal melanoma cell proliferation and reduces the susceptibility of uveal melanoma cells to NK cell-mediated cytolysis.
Experimental evidence suggesting that a weak immune response promotes, rather than inhibits carcinogenesis, relates to the pleiotropic effects of IFN-γ in tumor progression. Low-level expression of IFN-γ promoted the growth of various transplanted murine tumors and enhanced the outgrowth of chemically induced tumors in mice (He et al., 2005; Roberts et al., 2007). Other investigations have shown that IFN-γ can promote tumor growth by down regulating tumor antigens (Beatty and Paterson, 2000; Le Poole et al., 2002) and enhancing the survival to tumor cells and their metastatic behavior (Kelly et al., 1991; Taniguchi et al., 1987). In a chemically induced cancer model, Roberts and co-workers demonstrated that CD8+ T lymphocytes act as tumor-promoting cells rather than Tregs (Roberts et al., 2007). That is, the tumor burden in CD8−/− mice was half that found in wild-type mice and CD4−/− mice indicating that the CD8+ T lymphocytes acted as tumor promoting cells, rather than as regulatory cells. It remains to be determined if these observations are idiosyncracies unique to these specific tumor models or if they are forecasting a new paradigm in tumor immunology.
7.2 Myeloid-derived suppressor cells: “Immunological betrayal of the worst kind”
The notion that chronic inflammation contributes to tumorigenesis is not a new concept and in fact, was proposed over 140 years ago by Virchow (Balkwill and Mantovani, 2001). A wide range of neoplasms are associated with chronic inflammation including mesothelioma, lung, prostate, bladder, pancreatic, cervical, and esophageal cancers, as well as cutaneous melanoma (Coussens and Werb, 2002; Mantovani et al., 2008; Shacter and Weitzman, 2002). Myeloid-derived cells are common inflammatory cells present in many, if not most tumors and it is widely recognized that the presence of TAM carries a poor prognosis for multiple tumors, including uveal melanoma (Lin and Pollard, 2004; Makitie et al., 2001). TAM produce a wide range of factors that promote malignancy and enhance metastasis including: a) vascular endothelial growth factor (VEGF); b) epidermal growth factor (EGF); c) matrix metalloproteinases (MMP); d) tissue-type plasminogen activator (t-PA); e) IL-6; and f) TGF-β (Lin and Pollard, 2004). There is a growing awareness that myeloid-derived suppressor cells (MDSC) are an immature population of myeloid cells that are consistently present in most cancers and are believed to be the primary cause of immune suppression that occurs in patients with progressive tumors (Gabrilovich and Nagaraj, 2009; Ostrand-Rosenberg and Sinha, 2009). MDSC suppress elements of the innate and adaptive immune responses using a wide range of mechanisms. MDSC deplete intratumoral levels of arginine and cysteine, which are two key amino acids that are necessary for the survival of T lymphocytes (Bronte et al., 2003; Bronte and Zanovello, 2005; Kusmartsev et al., 2004; Ostrand-Rosenberg and Sinha, 2009). MDSC produce arginase, which in addition to depleting arginine also suppress adaptive immunity by down-regulating the T cell receptor (TCR)-associated CD3zeta-chain (ζ-chain) (Zea et al., 2004). CD3 ζ-chain expression is necessary for T cell activation (Irving and Weiss, 1991; Lai et al., 1996) and reduced expression inhibits T cell function (Baniyash, 2004). Down-regulation of CD3 ζ-chain has been observed in several types of cancers(Baniyash, 2004), including uveal melanomas (McKenna et al., 2009). The percentage of putative MDSC in the peripheral blood increased 1.8-fold in uveal melanoma patients in comparison with healthy controls and was accompanied by a 2.7-fold decrease in the expression of CD3 ζ-chain on T cells in the peripheral blood (McKenna et al., 2009). An immunohistological study on 61 uveal melanoma-containing eyes reported reduced expression of CD3 ζ-chain on the CD8+ TIL in a subgroup of uveal melanomas that were predominantly epithelioid, which coincided with poor survival (Staibano et al., 2006). Animal studies have demonstrated that CD11b+Gr1+ MDSC are present in intraocular tumors and suppress tumor-specific CD8+ CTL responses in vitro and presumably in vivo (McKenna and Kapp, 2006). The central role for MDSC in the evasion of immune surveillance is emerging as one of the most provocative topics in tumor immunology and will undoubtedly be a target for future immunotherapeutic modalities for many neoplasms, including uveal melanoma.
8. Future Directions
Although uveal melanomas arise in an immune privileged environment and have adopted many of the mechanisms and express many of the molecules that contribute to ocular immune privilege, there are glimmers of hope for counteracting the immune privilege of uveal melanomas. Bundling immunotherapeutic modalities that simultaneously disarm MSDC and enhance innate and adaptive immune responses offers an opportunity to target multiple weak spots in uveal melanoma’s evasive repertoire. Augmenting the host’s NK cell repertoire using biological response modifiers other than IFN-γ (e.g., IL-2) would activate innate immunity without eliciting the untoward effects of IFN-γ (e.g., upregulating IDO, MHC class 1a molecules, and PD-L1). Broad-based activation of the adaptive immune response, especially CD4+ T cell-mediated effector elements, could be combined with the aforementioned stimulation of the innate immune apparatus. One example of a promising new approach for initiating CD4+ T cell-mediated tumor immunity is the development of “designer” tumor vaccines using engineered MHC class II-matched uveal melanoma cells selected from a bank of uveal melanoma cell lines that express different MHC class II haplotypes (Bosch et al., 2007). The MHC class II melanoma cell lines can be matched with the MHC class II haplotypes of the uveal melanoma patients. The MHC class II melanoma cell lines can be engineered to express co-stimulatory molecules so that they act as surrogate APC’s to stimulate melanoma-specific, MHC class II-dependent, CD4+ T cell responses. MHC class II alleles are not as polymorphic as MHC class I alleles, and thus, a relatively small set of uveal melanoma cell lines with the most common MHC class II phenotype would in theory be sufficient for use in tailoring vaccines for individual patients. These vaccines could be applied shortly after diagnosing patients who are at high risk for metastatic disease. The potential of such “designer” vaccines is risky, but considering that the 5-year survival of uveal melanoma patients has not improved in the past 30 years (Singh and Topham, 2003) and there is no currently available therapeutic agent that has been shown to significantly prolong the survival of uveal melanoma patients with liver metastases (Gombos and Mieler, 2003), it is a risk worth taking.
Acknowledgments
The preparation of this manuscript was supported in part by EY005631 and CA30276 and an unrestricted grant from Research to Prevent Blindness. The author thanks Dr. Peter Chen for critically reviewing the manuscript.
Abbreviations
- AC
anterior chamber
- ACAID
anterior chamber-associated immune deviation
- CGRP
calcitonin gene-related peptide
- CRP
complement regulatory proteins
- MHC
major histocompatibility complex
- MIF
macrophage migration inhibitory factor
- SOM
somatostatin
- TGF-β
transforming growth factor beta
- TIL
tumor-infiltrating lymphocytes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abi-Hanna D, Wakefield D, et al. HLA antigens in ocular tissues. I. In vivo expression in human eyes. Transplantation. 1988;45:610–613. doi: 10.1097/00007890-198803000-00021. [DOI] [PubMed] [Google Scholar]
- Agata Y, Kawasaki A, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- Albert DM. The ocular melanoma story. LIII Edward Jackson Memorial Lecture: Part II. Am J Ophthalmol. 1997;123:729–741. doi: 10.1016/s0002-9394(14)71119-5. [DOI] [PubMed] [Google Scholar]
- Alexander AM, Crawford M, et al. Indoleamine 2,3-dioxygenase expression in transplanted NOD Islets prolongs graft survival after adoptive transfer of diabetogenic splenocytes. Diabetes. 2002;51:356–365. doi: 10.2337/diabetes.51.2.356. [DOI] [PubMed] [Google Scholar]
- Algarra I, Cabrera T, et al. The HLA crossroad in tumor immunology. Hum Immunol. 2000;61:65–73. doi: 10.1016/s0198-8859(99)00156-1. [DOI] [PubMed] [Google Scholar]
- Alizadeh H, Howard K, et al. Reduction of liver metastasis of intraocular melanoma by interferon-beta gene transfer. Invest Ophthalmol Vis Sci. 2003;44:3042–3051. doi: 10.1167/iovs.02-1147. [DOI] [PubMed] [Google Scholar]
- Anand R, Ma D, et al. Characterization of intraocular tumors arising in transgenic mice. Invest Ophthalmol Vis Sci. 1994;35:3533–3539. [PubMed] [Google Scholar]
- Anastassiou G, Coupland SE, et al. Expression of Fas and Fas ligand in uveal melanoma: biological implication and prognostic value. J Pathol. 2001;194:466–472. doi: 10.1002/path.926. [DOI] [PubMed] [Google Scholar]
- Apte RS, Mayhew E, et al. Local inhibition of natural killer cell activity promotes the progressive growth of intraocular tumors. Invest Ophthalmol Vis Sci. 1997;38:1277–1282. [PubMed] [Google Scholar]
- Apte RS, Niederkorn JY. Isolation and characterization of a unique natural killer cell inhibitory factor present in the anterior chamber of the eye. J Immunol. 1996;156:2667–2673. [PubMed] [Google Scholar]
- Apte RS, Sinha D, et al. Cutting edge: role of macrophage migration inhibitory factor in inhibiting NK cell activity and preserving immune privilege. J Immunol. 1998;160:5693–5696. [PubMed] [Google Scholar]
- Ashton N. Primary tumors of the iris. Br J Ophthalmol. 1964;48:650–668. doi: 10.1136/bjo.48.12.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astigiano S, Morandi B, et al. Eosinophil granulocytes account for indoleamine 2,3-dioxygenase-mediated immune escape in human non-small cell lung cancer. Neoplasia. 2005;7:390–396. doi: 10.1593/neo.04658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augsburger JJ, Mullen D, et al. Planned combined I-125 plaque irradiation and indirect ophthalmoscope laser therapy for choroidal malignant melanoma. Ophthalmic Surg. 1993;24:76–81. [PubMed] [Google Scholar]
- Balkwill F, Charles KA, et al. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- Baniyash M. TCR zeta-chain downregulation: curtailing an excessive inflammatory immune response. Nat Rev Immunol. 2004;4:675–687. doi: 10.1038/nri1434. [DOI] [PubMed] [Google Scholar]
- Beatty GL, Paterson Y. IFN-gamma can promote tumor evasion of the immune system in vivo by down-regulating cellular levels of an endogenous tumor antigen. J Immunol. 2000;165:5502–5508. doi: 10.4049/jimmunol.165.10.5502. [DOI] [PubMed] [Google Scholar]
- Beutelspacher SC, Pillai R, et al. Function of indoleamine 2,3-dioxygenase in corneal allograft rejection and prolongation of allograft survival by over-expression. Eur J Immunol. 2006;36:690–700. doi: 10.1002/eji.200535238. [DOI] [PubMed] [Google Scholar]
- Bill A. Ocular circulation . In: Moses RACV, editor. Adler’s Physiology of the Eye. Mosby Co; St. Louis: 1970. pp. 278–296. [Google Scholar]
- Billingham RE, Boswell T. Studies on the problem of corneal homografts. Proc R Soc Lond B Biol Sci. 1953;141:392–406. doi: 10.1098/rspb.1953.0049. [DOI] [PubMed] [Google Scholar]
- Billingham RE, Krohn PL, et al. Effect of cortisone on survival of skin homografts in rabbits. Br Med J. 1951;1:1157–1163. doi: 10.1136/bmj.1.4716.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkeland SA, Storm HH, et al. Cancer risk after renal transplantation in the Nordic countries, 1964–1986. Int J Cancer. 1995;60:183–189. doi: 10.1002/ijc.2910600209. [DOI] [PubMed] [Google Scholar]
- Blank C, Brown I, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–1145. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- Blom DJ, Goslings WR, et al. Lack of effect of different cytokines on expression of membrane-bound regulators of complement activity on human uveal melanoma cells. J Interferon Cytokine Res. 1997a;17:695–700. doi: 10.1089/jir.1997.17.695. [DOI] [PubMed] [Google Scholar]
- Blom DJ, Luyten GP, et al. Human leukocyte antigen class I expression. Marker of poor prognosis in uveal melanoma. Invest Ophthalmol Vis Sci. 1997b;38:1865–1872. [PubMed] [Google Scholar]
- Blom DJ, Mooy CM, et al. Inverse correlation between expression of HLA-B and c-myc in uveal melanoma. J Pathol. 1997c;181:75–79. doi: 10.1002/(SICI)1096-9896(199701)181:1<75::AID-PATH724>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Blom DJ, Schurmans LR, et al. HLA expression in a primary uveal melanoma, its cell line, and four of its metastases. Br J Ophthalmol. 1997d;81:989–993. doi: 10.1136/bjo.81.11.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodaghi B, Goureau O, et al. Role of IFN-gamma-induced indoleamine 2,3 dioxygenase and inducible nitric oxide synthase in the replication of human cytomegalovirus in retinal pigment epithelial cells. J Immunol. 1999;162:957–964. [PubMed] [Google Scholar]
- Boonman ZF, Schurmans LR, et al. Macrophages are vital in spontaneous intraocular tumor eradication. Invest Ophthalmol Vis Sci. 2006;47:2959–2965. doi: 10.1167/iovs.05-1427. [DOI] [PubMed] [Google Scholar]
- Bora NS, Gobleman CL, et al. Differential expression of the complement regulatory proteins in the human eye. Invest Ophthalmol Vis Sci. 1993;34:3579–3584. [PubMed] [Google Scholar]
- Bosch JJ, Thompson JA, et al. MHC class II-transduced tumor cells originating in the immune-privileged eye prime and boost CD4(+) T lymphocytes that cross-react with primary and metastatic uveal melanoma cells. Cancer Res. 2007;67:4499–4506. doi: 10.1158/0008-5472.CAN-06-3770. [DOI] [PubMed] [Google Scholar]
- Brandacher G, Perathoner A, et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res. 2006;12:1144–1151. doi: 10.1158/1078-0432.CCR-05-1966. [DOI] [PubMed] [Google Scholar]
- Brocker EB, Suter L, et al. Phenotypic dynamics of tumor progression in human malignant melanoma. Int J Cancer. 1985;36:29–35. doi: 10.1002/ijc.2910360106. [DOI] [PubMed] [Google Scholar]
- Bronte V, Serafini P, et al. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 2003;24:302–306. doi: 10.1016/s1471-4906(03)00132-7. [DOI] [PubMed] [Google Scholar]
- Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- Burnet M. Cancer: a biological approach. III. Viruses associated with neoplastic conditions. IV. Practical applications. Br Med J. 1957;1:841–847. doi: 10.1136/bmj.1.5023.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway MP, Briggs JC. The incidence of late recurrence (greater than 10 years); an analysis of 536 consecutive cases of cutaneous melanoma. Br J Plast Surg. 1989;42:46–49. doi: 10.1016/s0007-1226(89)90111-2. [DOI] [PubMed] [Google Scholar]
- Camelo S, Kezic J, et al. Antigen from the anterior chamber of the eye travels in a soluble form to secondary lymphoid organs via lymphatic and vascular routes. Invest Ophthalmol Vis Sci. 2006;47:1039–1046. doi: 10.1167/iovs.05-1041. [DOI] [PubMed] [Google Scholar]
- Camelo S, Shanley A, et al. The distribution of antigen in lymphoid tissues following its injection into the anterior chamber of the rat eye. J Immunol. 2004;172:5388–5395. doi: 10.4049/jimmunol.172.9.5388. [DOI] [PubMed] [Google Scholar]
- Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol. 2002;20:29–53. doi: 10.1146/annurev.immunol.20.091101.091806. [DOI] [PubMed] [Google Scholar]
- Char DH. Metastatic choroidal melanoma. Am J Ophthalmol. 1978;86:76–80. doi: 10.1016/0002-9394(78)90018-1. [DOI] [PubMed] [Google Scholar]
- Chen PW, Ksander BR. Influence of immune surveillance and immune privilege on formation of intraocular tumors. Chem Immunol Allergy. 2007;92:276–289. doi: 10.1159/000099278. [DOI] [PubMed] [Google Scholar]
- Chen PW, Mellon JK, et al. Uveal melanoma expression of indoleamine 2,3-deoxygenase: establishment of an immune privileged environment by tryptophan depletion. Exp Eye Res. 2007;85:617–625. doi: 10.1016/j.exer.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PW, Murray TG, et al. Expression of MAGE genes in ocular melanoma cell lines. J Immunother. 1997;20:265–275. doi: 10.1097/00002371-199707000-00003. [DOI] [PubMed] [Google Scholar]
- Coley WB. II. Contribution to the Knowledge of Sarcoma. Ann Surg. 1891;14:199–220. doi: 10.1097/00000658-189112000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas: with a report of ten original cases. Am J Med Sci. 1893;105:487–511. [PubMed] [Google Scholar]
- COMS. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS): COMS report no. 15. Arch Ophthalmol. 2001;119:670–676. doi: 10.1001/archopht.119.5.670. [DOI] [PubMed] [Google Scholar]
- Cousins SW, McCabe MM, et al. Identification of transforming growth factor-beta as an immunosuppressive factor in aqueous humor. Invest Ophthalmol Vis Sci. 1991;32:2201–2211. [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orazio TJ, DeMarco BM, et al. Effect of aqueous humor on apoptosis of inflammatory cell types. Invest Ophthalmol Vis Sci. 1999;40:1418–1426. [PubMed] [Google Scholar]
- D’Orazio TJ, Niederkorn JY. The nature of antigen in the eye has a profound effect on the cytokine milieu and resultant immune response. Eur J Immunol. 1998a;28:1544–1553. doi: 10.1002/(SICI)1521-4141(199805)28:05<1544::AID-IMMU1544>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- D’Orazio TJ, Niederkorn JY. A novel role for TGF-beta and IL-10 in the induction of immune privilege. J Immunol. 1998b;160:2089–2098. [PubMed] [Google Scholar]
- Dace DS, Chen PW, et al. Ocular immune privilege is circumvented by CD4+ T cells, leading to the rejection of intraocular tumors in an IFN-{gamma}-dependent manner. J Leukoc Biol. 2007a;81:421–429. doi: 10.1189/jlb.0806489. [DOI] [PubMed] [Google Scholar]
- Dace DS, Chen PW, et al. CD8+ T cells circumvent immune privilege in the eye and mediate intraocular tumor rejection by a TNF-a-dependent mechanism. Jour Immunol. 2007b doi: 10.4049/jimmunol.178.10.6115. [DOI] [PubMed] [Google Scholar]
- Dace DS, Chen PW, et al. CD4+ T-cell-dependent tumour rejection in an immune-privileged environment requires macrophages. Immunology. 2008;123:367–377. doi: 10.1111/j.1365-2567.2007.02700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg AJ, Subbaramaiah K. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell. 2003;4:431–436. doi: 10.1016/s1535-6108(03)00310-6. [DOI] [PubMed] [Google Scholar]
- Daubener W, MacKenzie CR. IFN-gamma activated indoleamine 2,3-dioxygenase activity in human cells is an antiparasitic and an antibacterial effector mechanism. Adv Exp Med Biol. 1999;467:517–524. doi: 10.1007/978-1-4615-4709-9_64. [DOI] [PubMed] [Google Scholar]
- de la Cruz PO, Jr, Specht CS, et al. Lymphocytic infiltration in uveal malignant melanoma. Cancer. 1990;65:112–115. doi: 10.1002/1097-0142(19900101)65:1<112::aid-cncr2820650123>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- de Waard-Siebinga I, Hilders CG, et al. HLA expression and tumor-infiltrating immune cells in uveal melanoma. Graefes Arch Clin Exp Ophthalmol. 1996;234:34–42. doi: 10.1007/BF00186516. [DOI] [PubMed] [Google Scholar]
- Della Chiesa M, Carlomagno S, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood. 2006;108:4118–4125. doi: 10.1182/blood-2006-03-006700. [DOI] [PubMed] [Google Scholar]
- Demicheli R, Abbattista A, et al. Time distribution of the recurrence risk for breast cancer patients undergoing mastectomy: further support about the concept of tumor dormancy. Breast Cancer Res Treat. 1996;41:177–185. doi: 10.1007/BF01807163. [DOI] [PubMed] [Google Scholar]
- Ding H, Wu X, et al. PD-L1 is expressed by human renal tubular epithelial cells and suppresses T cell cytokine synthesis. Clin Immunol. 2005;115:184–191. doi: 10.1016/j.clim.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Dithmar S, Rusciano D, et al. Neoadjuvant interferon alfa-2b treatment in a murine model for metastatic ocular melanoma: a preliminary study. Arch Ophthalmol. 2000;118:1085–1089. doi: 10.1001/archopht.118.8.1085. [DOI] [PubMed] [Google Scholar]
- Dithmar SA, Rusciano DA, et al. Depletion of NK cell activity results in growth of hepatic micrometastases in a murine ocular melanoma model. Curr Eye Res. 1999;19:426–431. doi: 10.1076/ceyr.19.5.426.5294. [DOI] [PubMed] [Google Scholar]
- Dong H, Strome SE, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhu G, et al. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Bruce AT, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Koebel CM, et al. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Old LJ, et al. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004a;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Old LJ, et al. The three Es of cancer immunoediting. Annu Rev Immunol. 2004b;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- Durie FH, Campbell AM, et al. Analysis of lymphocytic infiltration in uveal melanoma. Invest Ophthalmol Vis Sci. 1990;31:2106–2110. [PubMed] [Google Scholar]
- Egan RM, Yorkey C, et al. Peptide-specific T cell clonal expansion in vivo following immunization in the eye, an immune-privileged site. J Immunol. 1996;157:2262–2271. [PubMed] [Google Scholar]
- Ehrlich P. Ueber den jetzigen Stand der Karzinomforschung. Ned Tijdschr Geneeskd. 1909;5:273–290. [Google Scholar]
- Ericsson C, Seregard S, et al. Association of HLA class I and class II antigen expression and mortality in uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42:2153–2156. [PubMed] [Google Scholar]
- Esser P, Grisanti S, et al. TGF-beta in uveal melanoma. Microsc Res Tech. 2001;52:396–400. doi: 10.1002/1097-0029(20010215)52:4<396::AID-JEMT1024>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Filer RS, Song JC, et al. Cytokines in uveal melanoma. Original title: uveal melanoma cytokines. Klin Monatsbl Augenheilkd. 1993;202:174–179. doi: 10.1055/s-2008-1045579. [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frumento G, Rotondo R, et al. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196:459–468. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombos DS, Mieler WF, Albert PADM. Ocular Oncology. Marcel Dekker, Inc; New York: 2003. Therapy of Uveal Melanoma: Methods and Risk Factors Associated with Treatment; pp. 321–352. [Google Scholar]
- Goslings WR, Blom DJ, et al. Membrane-bound regulators of complement activation in uveal melanomas. CD46, CD55, and CD59 in uveal melanomas. Invest Ophthalmol Vis Sci. 1996;37:1884–1891. [PubMed] [Google Scholar]
- Goslings WR, Prodeus AP, et al. A small molecular weight factor in aqueous humor acts on C1q to prevent antibody-dependent complement activation. Invest Ophthalmol Vis Sci. 1998;39:989–995. [PubMed] [Google Scholar]
- Greene HS. A spontaneous melanoma in the hamster with a propensity for amelanotic alteration and sarcomatous transformation during transplantation. Cancer Res. 1958;18:422–425. [PubMed] [Google Scholar]
- Griffith TS, Brunner T, et al. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- Hainz U, Jurgens B, et al. The role of indoleamine 2,3-dioxygenase in transplantation. Transpl Int. 2007;20:118–127. doi: 10.1111/j.1432-2277.2006.00370.x. [DOI] [PubMed] [Google Scholar]
- Hallermalm K, De Geer A, et al. Autocrine secretion of fas ligand shields tumor cells from fas-mediated killing by cytotoxic lymphocytes. Cancer Res. 2004;64:6775–6782. doi: 10.1158/0008-5472.CAN-04-0508. [DOI] [PubMed] [Google Scholar]
- Hallermalm K, Seki K, et al. Modulation of the tumor cell phenotype by IFN-gamma results in resistance of uveal melanoma cells to granule-mediated lysis by cytotoxic lymphocytes. J Immunol. 2008;180:3766–3774. doi: 10.4049/jimmunol.180.6.3766. [DOI] [PubMed] [Google Scholar]
- Hargrave SL, Mayhew E, et al. Are corneal cells susceptible to antibody-mediated killing in corneal allograft rejection? Transpl Immunol. 2003;11:79–89. doi: 10.1016/S0966-3274(02)00082-5. [DOI] [PubMed] [Google Scholar]
- He YF, Wang XH, et al. Sustained low-level expression of interferon-gamma promotes tumor development: potential insights in tumor prevention and tumor immunotherapy. Cancer Immunol Immunother. 2005;54:891–897. doi: 10.1007/s00262-004-0654-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YG, Mayhew E, et al. Expression and possible function of IL-2 and IL-15 receptors on human uveal melanoma cells. Invest Ophthalmol Vis Sci. 2004;45:4240–4246. doi: 10.1167/iovs.04-0599. [DOI] [PubMed] [Google Scholar]
- Hegde S, Mellon JK, et al. Effect of alloantibodies on corneal allograft survival. Invest Ophthalmol Vis Sci. 2002;43:1012–1018. [PubMed] [Google Scholar]
- Helbig H, Gurley RC, et al. Dual effect of ciliary body cells on T lymphocyte proliferation. Eur J Immunol. 1990;20:2457–2463. doi: 10.1002/eji.1830201115. [DOI] [PubMed] [Google Scholar]
- Herrlinger U, Hebart H, et al. Relapse of primary CNS lymphoma after more than 10 years in complete remission. J Neurol. 2005;252:1409–1410. doi: 10.1007/s00415-005-0854-4. [DOI] [PubMed] [Google Scholar]
- Hicklin DJ, Marincola FM, et al. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol Med Today. 1999;5:178–186. doi: 10.1016/s1357-4310(99)01451-3. [DOI] [PubMed] [Google Scholar]
- Holmes CH, Simpson KL, et al. Complement regulatory proteins at the feto-maternal interface during human placental development: distribution of CD59 by comparison with membrane cofactor protein (CD46) and decay accelerating factor (CD55) Eur J Immunol. 1992;22:1579–1585. doi: 10.1002/eji.1830220635. [DOI] [PubMed] [Google Scholar]
- Holmes CH, Simpson KL, et al. Preferential expression of the complement regulatory protein decay accelerating factor at the fetomaternal interface during human pregnancy. J Immunol. 1990;144:3099–3105. [PubMed] [Google Scholar]
- Hooper P, Bora NS, et al. Inhibition of lymphocyte proliferation by resident ocular cells. Curr Eye Res. 1991;10:363–372. doi: 10.3109/02713689108996342. [DOI] [PubMed] [Google Scholar]
- Hori J, Wang M, et al. B7-H1-induced apoptosis as a mechanism of immune privilege of corneal allografts. J Immunol. 2006;177:5928–5935. doi: 10.4049/jimmunol.177.9.5928. [DOI] [PubMed] [Google Scholar]
- Ino K, Yoshida N, et al. Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br J Cancer. 2006;95:1555–1561. doi: 10.1038/sj.bjc.6603477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64:891–901. doi: 10.1016/0092-8674(91)90314-o. [DOI] [PubMed] [Google Scholar]
- Ishio T, Goto S, et al. Immunoactivative role of indoleamine 2,3-dioxygenase in human hepatocellular carcinoma. J Gastroenterol Hepatol. 2004;19:319–326. doi: 10.1111/j.1440-1746.2003.03259.x. [DOI] [PubMed] [Google Scholar]
- Ishitani A, Geraghty DE. Alternative splicing of HLA-G transcripts yields proteins with primary structures resembling both class I and class II antigens. Proc Natl Acad Sci U S A. 1992;89:3947–3951. doi: 10.1073/pnas.89.9.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager MJ, Hurks HM, et al. HLA expression in uveal melanoma: there is no rule without some exception. Hum Immunol. 2002a;63:444–451. doi: 10.1016/s0198-8859(02)00389-0. [DOI] [PubMed] [Google Scholar]
- Jager MJ, Hurks HMH, et al. HLA expressin in uveal melanoma: there is no rule without some exception. Human Immunology. 2002b;63:444–451. doi: 10.1016/s0198-8859(02)00389-0. [DOI] [PubMed] [Google Scholar]
- Jampel HD, Roche N, et al. Transforming growth factor-beta in human aqueous humor. Curr Eye Res. 1990;9:963–969. doi: 10.3109/02713689009069932. [DOI] [PubMed] [Google Scholar]
- Jensen OA, Anderson SR. Spontaneous regression of malignant melanoma of the choroid. Acta Ophthalmol (Copenh) 1974:173–182. doi: 10.1111/j.1755-3768.1974.tb00365.x. [DOI] [PubMed] [Google Scholar]
- Joly E, Mucke L, et al. Viral persistence in neurons explained by lack of major histocompatibility class I expression. Science. 1991;253:1283–1285. doi: 10.1126/science.1891717. [DOI] [PubMed] [Google Scholar]
- Kan-Mitchell J, Liggett PE, et al. Lymphocytes cytotoxic to uveal and skin melanoma cells from peripheral blood of ocular melanoma patients. Cancer Immunol Immunother. 1991;33:333–340. doi: 10.1007/BF01756599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DH, Shankaran V, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan HJ, Stevens TR. A reconsideration of immunological privilege within the anterior chamber of the eye. Transplantation. 1975;19:203–209. [PubMed] [Google Scholar]
- Kaplan HJ, Streilein JW. Immune response to immunization via the anterior chamber of the eye. I. F. lymphocyte-induced immune deviation. J Immunol. 1977;118:809–814. [PubMed] [Google Scholar]
- Kaplan HJ, Streilein JW. Immune response to immunization via the anterior chamber of the eye. II. An analysis of F1 lymphocyte-induced immune deviation. J Immunol. 1978;120:689–693. [PubMed] [Google Scholar]
- Kaplan HJ, Streilein JW, et al. Transplantation immunology of the anterior chamber of the eye. II. Immune response to allogeneic cells. J Immunol. 1975;115:805–810. [PubMed] [Google Scholar]
- Kath R, Hayungs J, et al. Prognosis and treatment of disseminated uveal melanoma [see comments] Cancer. 1993;72:2219–2223. doi: 10.1002/1097-0142(19931001)72:7<2219::aid-cncr2820720725>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Kelly SA, Gschmeissner S, et al. Enhancement of metastatic potential by gamma-interferon. Cancer Res. 1991;51:4020–4027. [PubMed] [Google Scholar]
- Khodadoust AA, Roozitalab HM, et al. Spontaneous regression of retinoblastoma. Surv Ophthalmol. 1977;21:467–478. doi: 10.1016/s0039-6257(77)80003-9. [DOI] [PubMed] [Google Scholar]
- Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein G. Tumor antigens. Annu Rev Microbiol. 1966;20:223–252. doi: 10.1146/annurev.mi.20.100166.001255. [DOI] [PubMed] [Google Scholar]
- Knisely TL, Bleicher PA, et al. Production of latent transforming growth factor-beta and other inhibitory factors by cultured murine iris and ciliary body cells. Curr Eye Res. 1991;10:761–771. doi: 10.3109/02713689109013870. [DOI] [PubMed] [Google Scholar]
- Knisely TL, Luckenbach MW, et al. Destructive and nondestructive patterns of immune rejection of syngeneic intraocular tumors. J Immunol. 1987;138:4515–4523. [PubMed] [Google Scholar]
- Knisely TL, Niederkorn JY. Emergence of a dominant cytotoxic T lymphocyte antitumor effector from tumor-infiltrating cells in the anterior chamber of the eye. Cancer Immunol Immunother. 1990;30:323–330. doi: 10.1007/BF01786881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N. Malignant neoplasms in registered cases of primary immunodeficiency syndrome. Jpn J Clin Oncol. 1985;15 (Suppl 1):307–312. [PubMed] [Google Scholar]
- Kovats S, Main EK, et al. A class I antigen, HLA-G, expressed in human trophoblasts. Science. 1990;248:220–223. doi: 10.1126/science.2326636. [DOI] [PubMed] [Google Scholar]
- Ksander BR, Geer DC, et al. Uveal melanomas contain antigenically specific and non-specific infiltrating lymphocytes. Curr Eye Res. 1998;17:165–173. doi: 10.1076/ceyr.17.2.165.5607. [DOI] [PubMed] [Google Scholar]
- Ksander BR, Rubsamen PE, et al. Studies of tumor-infiltrating lymphocytes from a human choroidal melanoma. Invest Ophthalmol Vis Sci. 1991;32:3198–3208. [PubMed] [Google Scholar]
- Kusmartsev S, Nefedova Y, et al. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- Lai P, Rabinowich H, et al. Alterations in expression and function of signal-transducing proteins in tumor-associated T and natural killer cells in patients with ovarian carcinoma. Clin Cancer Res. 1996;2:161–173. [PubMed] [Google Scholar]
- Lampson LA, Fisher CA. Weak HLA and beta 2-microglobulin expression of neuronal cell lines can be modulated by interferon. Proc Natl Acad Sci U S A. 1984;81:6476–6480. doi: 10.1073/pnas.81.20.6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lass JH, Walter EI, et al. Expression of two molecular forms of the complement decay-accelerating factor in the eye and lacrimal gland. Invest Ophthalmol Vis Sci. 1990;31:1136–1148. [PubMed] [Google Scholar]
- Latchman Y, Wood CR, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- Le Discorde M, Moreau P, et al. Expression of HLA-G in human cornea, an immune-privileged tissue. Hum Immunol. 2003;64:1039–1044. doi: 10.1016/j.humimm.2003.08.346. [DOI] [PubMed] [Google Scholar]
- Le Poole IC, Riker AI, et al. Interferon-gamma reduces melanosomal antigen expression and recognition of melanoma cells by cytotoxic T cells. Am J Pathol. 2002;160:521–528. doi: 10.1016/s0002-9440(10)64871-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBouteiller P. HLA class I chromosomal region, genes, and products: facts and questions. Crit Rev Immunol. 1994;14:89–129. doi: 10.1615/critrevimmunol.v14.i2.10. [DOI] [PubMed] [Google Scholar]
- Lee HO, Herndon JM, et al. TRAIL: a mechanism of tumor surveillance in an immune privileged site. J Immunol. 2002;169:4739–4744. doi: 10.4049/jimmunol.169.9.4739. [DOI] [PubMed] [Google Scholar]
- Lee N, Llano M, et al. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A. 1998;95:5199–5204. doi: 10.1073/pnas.95.9.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Taylor S, et al. The induction of splenic suppressor T cells through an immune-privileged site requires an intact sympathetic nervous system. J Neuroimmunol. 2004;153:40–49. doi: 10.1016/j.jneuroim.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Li XY, Niederkorn JY. Immune privilege in the anterior chamber of the eye is not extended to intraocular Listeria monocytogenes. Ocul Immunol Inflamm. 1997;5:245–257. doi: 10.3109/09273949709085065. [DOI] [PubMed] [Google Scholar]
- Liao MJ, Zhang XX, et al. No requirement for V(D)J recombination in p53-deficient thymic lymphoma. Mol Cell Biol. 1998;18:3495–3501. doi: 10.1128/mcb.18.6.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhvantseva VG, Slepova OS, et al. Interferon status of patients with uveal melanoma. Vestn Oftalmol. 1999;115:35–37. [PubMed] [Google Scholar]
- Lin EY, Pollard JW. Role of infiltrated leucocytes in tumour growth and spread. Br J Cancer. 2004;90:2053–2058. doi: 10.1038/sj.bjc.6601705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–244. doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- Luyten GP, van der Spek CW, et al. Expression of MAGE, gp100 and tyrosinase genes in uveal melanoma cell lines. Melanoma Res. 1998;8:11–16. doi: 10.1097/00008390-199802000-00003. [DOI] [PubMed] [Google Scholar]
- Ma D, Alizadeh H, et al. Rejection of intraocular tumors from transgenic mice by tumor- infiltrating lymphocytes. Curr Eye Res. 1994a;13:361–369. doi: 10.3109/02713689409167300. [DOI] [PubMed] [Google Scholar]
- Ma D, Comerford S, et al. Capacity of simian virus 40 T antigen to induce self-tolerance but not immunological privilege in the anterior chamber of the eye. Transplantation. 1994b;57:718–725. doi: 10.1097/00007890-199403150-00014. [DOI] [PubMed] [Google Scholar]
- Ma D, Luyten GP, et al. Relationship between natural killer cell susceptibility and metastasis of human uveal melanoma cells in a murine model. Invest Ophthalmol Vis Sci. 1995;36:435–441. [PubMed] [Google Scholar]
- Ma D, Niederkorn JY. Transforming growth factor-beta down-regulates major histocompatibility complex class I antigen expression and increases the susceptibility of uveal melanoma cells to natural killer cell-mediated cytolysis. Immunology. 1995;86:263–269. [PMC free article] [PubMed] [Google Scholar]
- Mahoney MC, Burnett WS, et al. The epidemiology of ophthalmic malignancies in New York State. Ophthalmology. 1990;97:1143–1147. doi: 10.1016/s0161-6420(90)32445-4. [DOI] [PubMed] [Google Scholar]
- Makitie T, Summanen P, et al. Tumor-infiltrating macrophages (CD68(+) cells) and prognosis in malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42:1414–1421. [PubMed] [Google Scholar]
- Malina HZ, Martin XD. Indoleamine 2,3-dioxygenase activity in the aqueous humor, iris/ciliary body, and retina of the bovine eye. Graefes Arch Clin Exp Ophthalmol. 1993;231:482–486. doi: 10.1007/BF02044236. [DOI] [PubMed] [Google Scholar]
- Malina HZ, Martin XD. Indoleamine 2,3-dioxygenase: antioxidant enzyme in the human eye. Graefes Arch Clin Exp Ophthalmol. 1996;234:457–462. doi: 10.1007/BF02539413. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, et al. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Marincola FM, Jaffee EM, et al. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000;74:181–273. doi: 10.1016/s0065-2776(08)60911-6. [DOI] [PubMed] [Google Scholar]
- Matsui K, Sawa T, et al. Relapse of stage I small cell lung cancer ten or more years after the start of treatment. Jpn J Clin Oncol. 2006;36:457–461. doi: 10.1093/jjco/hyl044. [DOI] [PubMed] [Google Scholar]
- McCarthy JM, Rootman J, et al. Conjunctival and uveal melanoma in the dysplastic nevus syndrome. Surv Ophthalmol. 1993;37:377–386. doi: 10.1016/0039-6257(93)90068-i. [DOI] [PubMed] [Google Scholar]
- McKenna KC, Beatty KM, et al. Activated CD11b+ CD15+ granulocytes increase in blood of uveal melanoma patients. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.08-3012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna KC, Kapp JA. Accumulation of immunosuppressive CD11b+ myeloid cells correlates with the failure to prevent tumor growth in the anterior chamber of the eye. J Immunol. 2006;177:1599–1608. doi: 10.4049/jimmunol.177.3.1599. [DOI] [PubMed] [Google Scholar]
- Medawar PB. Immunity to homologous grafted skin. III. The fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol. 1948;29:58–69. [PMC free article] [PubMed] [Google Scholar]
- Meecham WJ, Char DH, et al. Infiltrating lymphocytes and antigen expression in uveal melanoma. Ophthalmic Res. 1992;24:20–26. doi: 10.1159/000267140. [DOI] [PubMed] [Google Scholar]
- Mellor AL, Keskin DB, et al. Cells expressing indoleamine 2,3-dioxygenase inhibit T cell responses. J Immunol. 2002;168:3771–3776. doi: 10.4049/jimmunol.168.8.3771. [DOI] [PubMed] [Google Scholar]
- Meng S, Tripathy D, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10:8152–8162. doi: 10.1158/1078-0432.CCR-04-1110. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Altman NF, et al. Histopathologic analysis of experimental autoimmune uveitis attenuated by intracameral injection of S-antigen. Curr Eye Res. 1989;8:113–121. doi: 10.3109/02713688909013900. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Clark AF, et al. Induction of anterior chamber associated immune deviation in rats receiving intracameral injections of retinal S antigen. Curr Eye Res. 1988;7:627–632. doi: 10.3109/02713688809031820. [DOI] [PubMed] [Google Scholar]
- Mulcahy KA, Rimoldi D, et al. Infrequent expression of the MAGE gene family in uveal melanomas. Int J Cancer. 1996;66:738–742. doi: 10.1002/(SICI)1097-0215(19960611)66:6<738::AID-IJC5>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Munn DH, Shafizadeh E, et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Zhou M, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- Nacht M, Jacks T. V(D)J recombination is not required for the development of lymphoma in p53-deficient mice. Cell Growth Differ. 1998;9:131–138. [PubMed] [Google Scholar]
- Namba K, Kitaichi N, et al. Induction of regulatory T cells by the immunomodulating cytokines alpha-melanocyte-stimulating hormone and transforming growth factor-beta2. J Leukoc Biol. 2002;72:946–952. [PubMed] [Google Scholar]
- Natali PG, Bigotti A, et al. Analysis of the antigenic profile of uveal melanoma lesions with anti- cutaneous melanoma-associated antigen and anti-HLA monoclonal antibodies. Cancer Res. 1989;49:1269–1274. [PubMed] [Google Scholar]
- Niederkorn J, Streilein JW, et al. Deviant immune responses to allogeneic tumors injected intracamerally and subcutaneously in mice. Invest Ophthalmol Vis Sci. 1981;20:355–363. [PubMed] [Google Scholar]
- Niederkorn JY. The immunopathology of intraocular tumour rejection. Eye. 1991;5:186–192. doi: 10.1038/eye.1991.33. [DOI] [PubMed] [Google Scholar]
- Niederkorn JY. Immunopathogenesis of intraocular tumors. Prog Retinal and Eye Res. 1995;14:505–526. doi: 10.1016/j.preteyeres.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederkorn JY. Immunoregulation of intraocular tumours. Eye. 1997;11:249–254. doi: 10.1038/eye.1997.60. [DOI] [PubMed] [Google Scholar]
- Niederkorn JY. Natural Killer Cells and Uveal Melanoma. In: Zierhut M, Jager MJ, Ksander B, editors. Immunology of Intraocular Tumors. Swets and Zeitlinger; Lisse: 2002. pp. 73–82. [Google Scholar]
- Niederkorn JY. Anterior chamber-associated immune deviation and its impact on corneal allograft survival. Current Opinion in Organ Transplantation. 2006a;11:360–365. [Google Scholar]
- Niederkorn JY. See no evil, hear no evil, do no evil: the lessons of immune privilege. Nat Immunol. 2006b;7:354–359. doi: 10.1038/ni1328. [DOI] [PubMed] [Google Scholar]
- Niederkorn JY. The induction of anterior chamber-associated immune deviation. Chem Immunol Allergy. 2007;92:27–35. doi: 10.1159/000099251. [DOI] [PubMed] [Google Scholar]
- Niederkorn JY, Chiang EY, et al. Expression of a nonclassical MHC class Ib molecule in the eye. Transplantation. 1999;68:1790–1799. doi: 10.1097/00007890-199912150-00025. [DOI] [PubMed] [Google Scholar]
- Niederkorn JY, Knisely TL. Immunological analysis of a destructive pattern of intraocular tumor resolution. Curr Eye Res. 1988;7:515–526. doi: 10.3109/02713688809031806. [DOI] [PubMed] [Google Scholar]
- Niederkorn JY, Mellon J. Anterior chamber-associated immune deviation promotes corneal allograft survival. Invest Ophthalmol Vis Sci. 1996;37:2700–2707. [PubMed] [Google Scholar]
- Niederkorn JY, Meunier PC. Spontaneous immune rejection of intraocular tumors in mice. Invest Ophthalmol Vis Sci. 1985;26:877–884. [PubMed] [Google Scholar]
- Niederkorn JY, Wang S. Immunology of intraocular tumors. Ocul Immunol Inflamm. 2005;13:105–110. doi: 10.1080/09273940490518586. [DOI] [PubMed] [Google Scholar]
- Nishida T, Taylor AW. Specific aqueous humor factors induce activation of regulatory T cells. Invest Ophthalmol Vis Sci. 1999;40:2268–2274. [PubMed] [Google Scholar]
- Nishimura H, Honjo T. PD-1: an inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol. 2001;22:265–268. doi: 10.1016/s1471-4906(01)01888-9. [DOI] [PubMed] [Google Scholar]
- Nitta T, Oksenberg JR, et al. Predominant expression of T cell receptor V alpha 7 in tumor- infiltrating lymphocytes of uveal melanoma. Science. 1990;249:672–674. doi: 10.1126/science.2382141. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. 2007;19:813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- Old LJ, Boyse EA. Specific antigens of tumors and leukemias of experimental animals. Med Clin North Am. 1966;50:901–912. doi: 10.1016/s0025-7125(16)33187-x. [DOI] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn I. Malignant melanoma in organ allograft recipients. Transplantation. 1996;61:274–278. doi: 10.1097/00007890-199601270-00019. [DOI] [PubMed] [Google Scholar]
- Prehn RT. The immune reaction as a stimulator of tumor growth. Science. 1972;176:170–171. doi: 10.1126/science.176.4031.170. [DOI] [PubMed] [Google Scholar]
- Prehn RT, Lappe MA. An immunostimulation theory of tumor development. Transplant Rev. 1971;7:26–54. doi: 10.1111/j.1600-065x.1971.tb00462.x. [DOI] [PubMed] [Google Scholar]
- Raja SM, Metkar SS, et al. Cytotoxic granule-mediated apoptosis: unraveling the complex mechanism. Curr Opin Immunol. 2003;15:528–532. doi: 10.1016/s0952-7915(03)00111-0. [DOI] [PubMed] [Google Scholar]
- Reese AB. Precancerous and cancerous melanosis. Am J Ophthalmol. 1966;61:1272–1277. doi: 10.1016/0002-9394(66)90256-x. [DOI] [PubMed] [Google Scholar]
- Reiman JM, Kmieciak M, et al. Tumor immunoediting and immunosculpting pathways to cancer progression. Semin Cancer Biol. 2007;17:275–287. doi: 10.1016/j.semcancer.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repp AC, Mayhew ES, et al. Human uveal melanoma cells produce macrophage migration-inhibitory factor to prevent lysis by NK cells. J Immunol. 2000;165:710–715. doi: 10.4049/jimmunol.165.2.710. [DOI] [PubMed] [Google Scholar]
- Roberts SJ, Ng BY, et al. Characterizing tumor-promoting T cells in chemically induced cutaneous carcinogenesis. Proc Natl Acad Sci U S A. 2007;104:6770–6775. doi: 10.1073/pnas.0604982104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook AH, Kehrl JH, et al. Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness. J Immunol. 1986;136:3916–3920. [PubMed] [Google Scholar]
- Rooney IA, Oglesby TJ, et al. Complement in human reproduction: activation and control. Immunol Res. 1993;12:276–294. doi: 10.1007/BF02918258. [DOI] [PubMed] [Google Scholar]
- Rouas-Freiss N, Goncalves RM, et al. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc Natl Acad Sci U S A. 1997;94:11520–11525. doi: 10.1073/pnas.94.21.11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiter DJ, Bergman W, et al. Immunohistochemical analysis of malignant melanomas and nevocellular nevi with monoclonal antibodies to distinct monomorphic determinants of HLA antigens. Cancer Res. 1984;44:3930–3935. [PubMed] [Google Scholar]
- Ruiter DJ, Bhan AK, et al. Major histocompatibility antigens and mononuclear inflammatory infiltrate in benign nevomelanocytic proliferations and malignant melanoma. J Immunol. 1982;129:2808–2815. [PubMed] [Google Scholar]
- Ruiter DJ, Mattijssen V, et al. MHC antigens in human melanomas. Semin Cancer Biol. 1991;2:35–45. [PubMed] [Google Scholar]
- Russell P, Gregerson DS, et al. Characteristics of a retrovirus associated with a hamster melanoma. J Gen Virol. 1979;43:317–326. doi: 10.1099/0022-1317-43-2-317. [DOI] [PubMed] [Google Scholar]
- Ryu YH, Kim JC. Expression of indoleamine 2,3-dioxygenase in human corneal cells as a local immunosuppressive factor. Invest Ophthalmol Vis Sci. 2007;48:4148–4152. doi: 10.1167/iovs.05-1336. [DOI] [PubMed] [Google Scholar]
- Sagalowsky AI, Molberg K. Solitary metastasis of renal cell carcinoma to the contralateral adrenal gland 22 years after nephrectomy. Urology. 1999;54:162. doi: 10.1016/s0090-4295(98)00661-x. [DOI] [PubMed] [Google Scholar]
- Saunders PA, Hendrycks VR, et al. PD-L2:PD-1 involvement in T cell proliferation, cytokine production, and integrin-mediated adhesion. Eur J Immunol. 2005;35:3561–3569. doi: 10.1002/eji.200526347. [DOI] [PubMed] [Google Scholar]
- Schneider P, Holler N, et al. Conversion of membrane-bound Fas (CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med. 1998;187:1205–1213. doi: 10.1084/jem.187.8.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurmans LR, den Boer AT, et al. Successful immunotherapy of an intraocular tumor in mice. Cancer Res. 1999;59:5250–5254. [PubMed] [Google Scholar]
- Schurmans LR, Diehl L, et al. Rejection of intraocular tumors by CD4(+) T cells without induction of phthisis. J Immunol. 2001;167:5832–5837. doi: 10.4049/jimmunol.167.10.5832. [DOI] [PubMed] [Google Scholar]
- Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park) 2002;16:217–226. discussion 230–212. [PubMed] [Google Scholar]
- Shankaran V, Ikeda H, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- She SC, Steahly LP, et al. Intracameral injection of allogeneic lymphocytes enhances corneal graft survival. Invest Ophthalmol Vis Sci. 1990;31:1950–1956. [PubMed] [Google Scholar]
- Shen L, Jin Y, et al. The function of donor versus recipient programmed death-ligand 1 in corneal allograft survival. J Immunol. 2007;179:3672–3679. doi: 10.4049/jimmunol.179.6.3672. [DOI] [PubMed] [Google Scholar]
- Sherman SH, Green K, et al. The fate of anterior chamber flurescein in the monkey eye. 1. The anterior chamber outflow pathways. Exp Eye Res. 1978;27:159–173. doi: 10.1016/0014-4835(78)90086-6. [DOI] [PubMed] [Google Scholar]
- Singh AD, Topham A. Survival rates with uveal melanoma in the United States: 1973–1997. Ophthalmology. 2003;110:962–965. doi: 10.1016/S0161-6420(03)00077-0. [DOI] [PubMed] [Google Scholar]
- Smyth MJ, Dunn GP, et al. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50. doi: 10.1016/S0065-2776(06)90001-7. [DOI] [PubMed] [Google Scholar]
- Smyth MJ, Thia KY, et al. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000;192:755–760. doi: 10.1084/jem.192.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JH, Kaplan HJ, et al. Chronic low level complement activation within the eye is controlled by intraocular complement regulatory proteins. Invest Ophthalmol Vis Sci. 2000a;41:3492–3502. [PMC free article] [PubMed] [Google Scholar]
- Sohn JH, Kaplan HJ, et al. Complement regulatory activity of normal human intraocular fluid is mediated by MCP, DAF, and CD59. Invest Ophthalmol Vis Sci. 2000b;41:4195–4202. [PMC free article] [PubMed] [Google Scholar]
- Sonoda KH, Taniguchi M, et al. Long-term survival of corneal allografts is dependent on intact CD1d- reactive NKT cells. J Immunol. 2002;168:2028–2034. doi: 10.4049/jimmunol.168.4.2028. [DOI] [PubMed] [Google Scholar]
- Sonoda Y, Streilein JW. Impaired cell-mediated immunity in mice bearing healthy orthotopic corneal allografts. J Immunol. 1993;150:1727–1734. [PubMed] [Google Scholar]
- Staibano S, Mascolo M, et al. Tumor infiltrating lymphocytes in uveal melanoma: a link with clinical behavior? Int J Immunopathol Pharmacol. 2006;19:171–179. [PubMed] [Google Scholar]
- Streilein JW. Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol. 2003;3:879–889. doi: 10.1038/nri1224. [DOI] [PubMed] [Google Scholar]
- Streilein JW, Niederkorn JY. Induction of anterior chamber-associated immune deviation requires an intact, functional spleen. J Exp Med. 1981;153:1058–1067. doi: 10.1084/jem.153.5.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart PM, Griffith TS, et al. CD95 ligand (FasL)-induced apoptosis is necessary for corneal allograft survival. J Clin Invest. 1997;99:396–402. doi: 10.1172/JCI119173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutman O. Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science. 1974;183:534–536. doi: 10.1126/science.183.4124.534. [DOI] [PubMed] [Google Scholar]
- Stutman O. Immunodepression and malignancy. Adv Cancer Res. 1975;22:261–422. doi: 10.1016/s0065-230x(08)60179-7. [DOI] [PubMed] [Google Scholar]
- Sugita S, Usui Y, et al. Human corneal endothelial cells expressing programmed death-ligand 1 (PD-L1) suppress PD-1+ T helper 1 cells by a contact-dependent mechanism. Invest Ophthalmol Vis Sci. 2009;50:263–272. doi: 10.1167/iovs.08-2536. [DOI] [PubMed] [Google Scholar]
- Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117:1137–1146. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takikawa O, Habara-Ohkubo A, et al. Induction of indoleamine 2,3-dioxygenase in tumor cells transplanted into allogeneic mouse: interferon-gamma is the inducer. Adv Exp Med Biol. 1991;294:437–444. doi: 10.1007/978-1-4684-5952-4_40. [DOI] [PubMed] [Google Scholar]
- Takikawa O, Littlejohn T, et al. Regulation of indoleamine 2,3-dioxygenase, the first enzyme in UV filter biosynthesis in the human lens. Relevance for senile nuclear cataract. Adv Exp Med Biol. 1999;467:241–245. doi: 10.1007/978-1-4615-4709-9_31. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Petersson M, et al. Interferon gamma induces lung colonization by intravenously inoculated B16 melanoma cells in parallel with enhanced expression of class I major histocompatibility complex antigens. Proc Natl Acad Sci U S A. 1987;84:3405–3409. doi: 10.1073/pnas.84.10.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A, Namba K. In vitro induction of CD25+ CD4+ regulatory T cells by the neuropeptide alpha-melanocyte stimulating hormone (alpha-MSH) Immunol Cell Biol. 2001;79:358–367. doi: 10.1046/j.1440-1711.2001.01022.x. [DOI] [PubMed] [Google Scholar]
- Taylor AW. Ocular immunosuppressive microenvironment. Chem Immunol. 2007;92:71–85. doi: 10.1159/000099255. [DOI] [PubMed] [Google Scholar]
- Taylor AW, Yee DG. Somatostatin is an immunosuppressive factor in aqueous humor. Invest Ophthalmol Vis Sci. 2003;44:2644–2649. doi: 10.1167/iovs.02-1216. [DOI] [PubMed] [Google Scholar]
- Teng MW, Swann JB, et al. Immune-mediated dormancy: an equilibrium with cancer. J Leukoc Biol. 2008;84:988–993. doi: 10.1189/jlb.1107774. [DOI] [PubMed] [Google Scholar]
- Terness P, Bauer TM, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–457. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas L. Reactions to homologous tissue antigens in relation to hypersensitivity. In: Lawrence HS, editor. Cellular and Humoral Aspects of the Hypersensitive States. Hoebers-Harper; New York: 1959. pp. 529–532. [Google Scholar]
- Uyttenhove C, Pilotte L, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- van Dooremaal JC. Die Entwicklung der in fremden Grund versetzten lebenden Geweba. Albrecht von Graefes Arch Ophthalmol. 1873;19:358–373. [Google Scholar]
- van Duinen SG, Ruiter DJ, et al. Level of HLA antigens in locoregional metastases and clinical course of the disease in patients with melanoma. Cancer Res. 1988;48:1019–1025. [PubMed] [Google Scholar]
- Verbik DJ, Murray TG, et al. Melanomas that develop within the eye inhibit lymphocyte proliferation. Int J Cancer. 1997;73:470–478. doi: 10.1002/(sici)1097-0215(19971114)73:4<470::aid-ijc3>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Vetter CS, Lieb W, et al. Loss of nonclassical MHC molecules MIC-A/B expression during progression of uveal melanoma. Br J Cancer. 2004;91:1495–1499. doi: 10.1038/sj.bjc.6602123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayasaradhi S, Houghton AN. Melanoma and melanocytes: pigmentation, tumor progression, and the immune response to cancer. Adv Pharmacol. 1995;32:343–374. doi: 10.1016/s1054-3589(08)61017-0. [DOI] [PubMed] [Google Scholar]
- Wang S, Boonman ZF, et al. Role of TRAIL and IFN-gamma in CD4+ T cell-dependent tumor rejection in the anterior chamber of the eye. J Immunol. 2003;171:2789–2796. doi: 10.4049/jimmunol.171.6.2789. [DOI] [PubMed] [Google Scholar]
- Wang Y, Goldschneider I, et al. Blood mononuclear cells induce regulatory NK T thymocytes in anterior chamber-associated immune deviation. J Leukoc Biol. 2001;69:741–746. [PubMed] [Google Scholar]
- Weinlich G, Murr C, et al. Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatology. 2007;214:8–14. doi: 10.1159/000096906. [DOI] [PubMed] [Google Scholar]
- Whittum JA, Niederkorn JY, et al. Intracameral inoculation of herpes simplex virus type I induces anterior chamber-associated immune deviation. Curr Eye Res. 1982;2:691–697. doi: 10.3109/02713688209019998. [DOI] [PubMed] [Google Scholar]
- Wilbanks GA, Mammolenti M, et al. Studies on the induction of anterior chamber-associated immune deviation (ACAID). III. Induction of ACAID depends upon intraocular transforming growth factor-beta. Eur J Immunol. 1992;22:165–173. doi: 10.1002/eji.1830220125. [DOI] [PubMed] [Google Scholar]
- Wilbanks GA, Streilein JW. Studies on the induction of anterior chamber-associated immune deviation (ACAID). 1. Evidence that an antigen-specific, ACAID- inducing, cell-associated signal exists in the peripheral blood. J Immunol. 1991;146:2610–2617. [PubMed] [Google Scholar]
- Wilbanks GA, Streilein JW. Fluids from immune privileged sites endow macrophages with the capacity to induce antigen-specific immune deviation via a mechanism involving transforming growth factor-beta. Eur J Immunol. 1992;22:1031–1036. doi: 10.1002/eji.1830220423. [DOI] [PubMed] [Google Scholar]
- Xu C, Mao D, et al. A critical role for murine complement regulator crry in fetomaternal tolerance. Science. 2000;287:498–501. doi: 10.1126/science.287.5452.498. [DOI] [PubMed] [Google Scholar]
- Yamagami S, Kawashima H, et al. Role of Fas-Fas ligand interactions in the immunorejection of allogeneic mouse corneal transplants. Transplantation. 1997;64:1107–1111. doi: 10.1097/00007890-199710270-00004. [DOI] [PubMed] [Google Scholar]
- Yang W, Chen PW, et al. PD-L1: PD-1 interaction contributes to the functional suppression of T-cell responses to human uveal melanoma cells in vitro. Invest Ophthalmol Vis Sci. 2008;49:2518–2525. doi: 10.1167/iovs.07-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Li H, et al. PD-L1 expression on human ocular cells and its possible role in regulating immune-mediated ocular inflammation. Invest Ophthalmol Vis Sci. 2009;50:273–280. doi: 10.1167/iovs.08-2397. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Kezuka T, et al. Participation of pigment epithelium of iris and ciliary body in ocular immune privilege. 2. Generation of TGF-beta-producing regulatory T cells. Invest Ophthalmol Vis Sci. 2000a;41:3862–3870. [PubMed] [Google Scholar]
- Yoshida M, Takeuchi M, et al. Participation of pigment epithelium of iris and ciliary body in ocular immune privilege. 1. Inhibition of T-cell activation in vitro by direct cell-to-cell contact. Invest Ophthalmol Vis Sci. 2000b;41:811–821. [PubMed] [Google Scholar]
- Zamiri P, Masli S, et al. Thrombospondin plays a vital role in the immune privilege of the eye. Invest Ophthalmol Vis Sci. 2005;46:908–919. doi: 10.1167/iovs.04-0362. [DOI] [PubMed] [Google Scholar]
- Zea AH, Rodriguez PC, et al. L-Arginine modulates CD3zeta expression and T cell function in activated human T lymphocytes. Cell Immunol. 2004;232:21–31. doi: 10.1016/j.cellimm.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Zeiss CJ, Johnson EM, et al. Feline intraocular tumors may arise from transformation of lens epithelium. Vet Pathol. 2003;40:355–362. doi: 10.1354/vp.40-4-355. [DOI] [PubMed] [Google Scholar]
- Zirm E. Eine erfolgreiche totale Keratoplastik. Albrecht von Graefes Arch Ophthalmol. 1906;64:580–593. [Google Scholar]