

Abstract
The pKas of ferric aquo–heme and aquo–heme electrochemical midpoints (Ems) at pH 7 in sperm whale myoglobin, Aplysia myoblogin, hemoglobin I, heme oxygenase 1, horseradish peroxidase and cytochrome c oxidase were calculated with Multi-Conformation Continuum Electrostatics (MCCE). The pKas span 3.3 pH units from 7.6 in heme oxygenase 1 to 10.9 in peroxidase, and the Ems range from −250 mv in peroxidase to 125 mV in Aplysia myoglobin. Proteins with higher in situ ferric aquo–heme pKas tend to have lower Ems. Both changes arise from the protein stabilizing a positively charged heme. However, compared with values in solution, the protein shifts the aquo–heme Ems more than the pKas. Thus, the protein has a larger effective dielectric constant for the protonation reaction, showing that electron and proton transfers are coupled to different conformational changes that are captured in the MCCE analysis. The calculations reveal a breakdown in the classical continuum electrostatic analysis of pairwise interactions. Comparisons with DFT calculations show that Coulomb’s law overestimates the large unfavorable interactions between the ferric water–heme and positively charged groups facing the heme plane by as much as 60%. If interactions with CuB in cytochrome c oxidase and Arg 38 in horseradish peroxidase are not corrected, the pKa calculations are in error by as much as 6 pH units. With DFT corrected interactions calculated pKas and Ems differ from measured values by less than 1 pH unit or 35 mV, respectively. The in situ aquo–heme pKa is important for the function of cytochrome c oxidase since it helps to control the stoichiometry of proton uptake coupled to electron transfer
Heme cofactors are found in many proteins. Cytochromes having two axial ligands contributed by the protein are six-coordinate and function as redox intermediates in electron transfer chains. However, five-coordinate hemes with one His ligand and one open position are the most common heme motif (1). The open site can bind different molecules for transport or catalysis. Hemoglobins and myoglobins are O2 transporters (2). Heme oxygenase is an essential protein in heme metabolism, using three O2s and seven electrons to degrade heme to biliverdin (3). Peroxidases reduce toxic hydrogen peroxide to water and then carry out one electron oxidization of a wide range of substrates (4, 5). Cytochrome c oxidase is the terminal electron acceptor in the respiratory chain (6–9). Here Heme a3 forms a binuclear center with a copper complex (CuB) that reduces O2 to water. The protein uses the released chemical energy to pump protons across the membrane. Other five-coordinate heme proteins carry out NO storage and transport (10), steroid synthesis (11, 12), and O2, CO, and NO sensing (13, 14).
Hemes can carry out many biological functions because the protein, especially the active site residues, modulates the ligand binding specificity and resultant chemistry. The diversity of in situ heme functions highlights important interactions of proteins with their cofactors and substrates. Extensive studies have explored how ligand geometry (15–17) and the protein scaffolding (18) affect in situ heme properties (1). The range of redox free energy and pKas of protein-bound hemes and their ligands show the importance of electrostatic interactions (19, 20). For example, six-coordinate bis-His–hemes have Ems ranging from −410 to +360 mV with the redox differences being predominately due to the intraprotein electrostatic environment (1, 20–22). In these proteins the loss of solvation energy (15, 23, 24), interactions with the protein backbone and with other residues (15, 20–22, 25), and local conformation changes on ionization changes (20, 26) determine the thermodynamic equilibrium measured by pKas and Ems.
Theoretical studies have analyzed hemes in different systems using a number of techniques. The large span of Ems in cytochromes has been subject to analysis by Protein Dipole Langevin Dipole (PDLD) (27, 28), continuum electrostatics (CE)1 (20, 21, 29, 30), and other techniques (31–37). Ems of the two hemes in quinol:fumarate reductase have been determined within 30 mV of experimental values using MCCE, allowing the low- and high-potential heme to be assigned to the distal and proximal positions in the structure (38). The heme propionic acid pKas and their influence on the pH dependence of cytochrome Ems have been considered by CE analysis (20, 38–40). The pKas and Ems of aquo–heme a3 and CuB in cytochrome c oxidase have been analyzed by CE (41–43) and density functional theory (DFT) (44). DFT and QM/MM calculations have been used to explain the unusual low-spin state of ferric aquo–heme (45–47) and to study the hydroxylation mechanism (48–51) of cytochrome P450.
In the work presented here, pKas and Ems of aquo–heme in myoglobins from sperm whale and Aplysia, hemoglobin I, heme oxygenase 1, and horseradish peroxidase are calculated with Multi-Conformation Continuum Electrostatics (MCCE) (20, 38). The calculated pKa is compared with the pH of the acid–alkaline transition, which is attributed to the deprotonation of a water molecule as the sixth heme ligand (52). Calculations are also carried out on cytochrome c oxidase. Here the pKa is compared with the pH dependence of the proton release on oxidation of heme a3 (53). Several of these proteins have positively charged groups in the active site. The interaction of the heme with these charges is compared in CE and DFT methods, showing that CE overestimates the interaction. With the appropriate corrections, MCCE calculations reproduce the experimental pKas and Ems, showing how the protein structures yield the observed in situ reaction energetics. The differences in the shifts of aquo–heme pKas and Ems in different binding sites show that electron and proton transfers can be coupled to different active site conformational changes. The aquo–heme Em is not a factor in the function of myoglobin or hemoglobin, nor are the pKas important for the reaction mechanism of most of the proteins studied here. However, the aquo–heme pKa in cytochrome c oxidase appears to determine whether heme a3 binds a proton during the reduction of the binuclear center (43). Thus, this pKa helps to control the electroneutrality of the oxidase reduction. The comparison of calculated and measured pKas and Ems in the smaller and simpler proteins provides a benchmark for the analysis of the more puzzling oxidase system.
METHODS
Structures of sperm whale myoglobin (1A6G, 1A6K, 1A6M, 1A6N, 1HJT, 1JP6, and 1JP9), Aplysia myoglobin (1MBA and 5MBA), monomeric clam hemoglobin I (1B0B, 1EBT, 1FLP, and 1MOH), rat heme oxygenase 1 (1DVE, 1IVE, 1IX4, 1ULX, and 1VGI), horseradish peroxidase (1ATJ, 1H55, 1H57, 1H58, 1H5A, 1H5C, 1H5D, 1H5E, 6ATJ, and 7ATJ), and Rhodobacter sphaeroides cytochrome c oxidase (1M56) were obtained from the Protein Data Bank (54). All crystal waters are deleted and protein cavities filled with continuum water. Multi-Conformation Continuum Electrostatics (MCCE) samples both residue ionization state and rotamer position as a function of pH and Eh (55–57). The cytochrome c oxidase calculations use the model generated for analysis of proton uptake coupled to anaerobic reduction in MCCE2.0 (43). In this larger protein a rotation angle of 120° is used to make side chain rotamers, reducing the number of conformers. All other calculations use MCCE 2.2, which adds local optimization of side chain positions, pruning to delete energetically indistinguishable conformers and corrections of the CE energy terms for the errors in the dielectric boundary due to extra surface conformations (see www.sci.ccny.cuny.edu/~mcce). Preselected side chain rotamers are generated in 60° increments around each rotatable bond for all amino acids. In each protein, a water or hydroxyl is added to the open heme coordinate position. The aquo–oxygen is placed on the heme iron with square bipyramidal geometry and a 1.95 Å Fe–O bond length. Hydrogens are added in a tetrahedral geometry with a 0.96 Å H–O bond length. Then additional hydroxyl and water conformations are generated in 30° increments around the Fe–O bond, creating 12 conformers for both water and hydroxyl.
Look-up tables are calculated for electrostatic and non-electrostatic conformer self and conformer–conformer pairwise interactions. The electrostatic pairwise interactions and reaction field energies are determined with a finite-difference technique to solve the Poisson–Boltzmann equation using the program DelPhi (58–60). Focusing (61) is used to ensure a final grid spacing of >2 grids/Å. The protein residues are given PARSE charges and radii (62). The protein has a dielectric constant, ε, of 4 while the surrounding water has an ε of 80 with a salt concentration of 150 mM. Cytochrome c oxidase is surrounded by a 32 Å slab with ε = 4 placed with IPECE to bury the fewest ionizable residues to simulate the membrane (63). The Lennard-Jones interactions are calculated with AMBER parameters (64), rescaled by 0.25 (65). A metal-centered charge set is used for hemes with a +2 or +3 charge on the ferrous and ferric Fe and −0.5 on each N atom of the porphyrin as used previously in cytochrome benchmark calculations (20, 21). The water–heme ligand uses TIP3 charges, and the hydroxyl charge, calculated with B3LYP (66) in Gaussian98 (67), has +0.2 on H and −1.2 on O. For the a-type heme in cytochrome c oxidase, a +0.3 charge is placed on the formyl group C and −0.3 on O.
MCCE calculates the shift in pKa or Em when a group is moved from aqueous solution to the protein. The solution pKa (pKa,sol) and Em (Em,sol) are obtained from measurements in water. pKa,sol for amino acids are taken from studies of peptides (68, 69). The heme propionic acids are treated as described previously with a pKa,sol of 4.9 (20). The five-coordinate hemes are assumed to bind a water or hydroxide as the sixth ligand. Heme and its His and water or hydroxide ligands are treated as a complex. A pKa,sol of 9.6 for the hydroxyl in the ferric (70, 71) and 10.9 in the ferrous (71) His–aquo–heme has been measured in microperoxidase 8 (MP8) which has a heme c. The water pKa,sols are assumed to be the same for hemes a and c. The Em,sol for the His–water heme is −120 mV, and −200 mV (72) for the His–hydroxyl heme. The value of −120 mV is slightly higher than the measured value of −140 mV (72, 73), giving an 80 mV shift from His–hydroxyl, consistent with the 1.3 pKa shift between the pKas of ferric and ferrous aquo–heme (70,71). With the same ligands, the Em of heme a is 100 (1) to 160 (74, 75) mV more positive than that of heme c. An Em,sol of − 20 mV for His–water heme a3 and −100 mV for His–hydroxyl heme a3 is used here. No explicit pairwise interactions of water or hydroxide ligands with the His ligand or heme to which they are bound are considered since these interactions are included in Em,sol and pKa,sol. The reaction field energies in both solution and the protein were calculated for the aquo–heme complexes. A systematic shift of −0.74 ΔpK unit (−1 kcal/mol) is added to the reference reaction field energy of ferric hydroxyl–heme in the solution (ΔGRX-N,sol) and 0.5 ΔpK unit (0.67 kcal/mol) to that of ferrous water–heme. This raises all ferric heme pKas by 0.74 pH unit and raises the aquo–heme Ems by 30 mV. Systematic shifts are also applied to the ionized form of ionizable amino acids on the basis of pKa benchmark studies (43, 56, 65).
Possible microstates with different ligand and side chain position and protonation states are subjected to Monte Carlo sampling. A microstate is made up of one conformer for each residue, cofactor, and water. The energy of microstate n (ΔGn) is the sum of the electrostatic and nonelectrostatic energies (20):
| (1) |
where the summation is over the total M conformers of all residues in the protein; for each conformer i, δi) 1 if it is present in the current microstate n and 0 if it is not. Each residue has one conformer with δi) 1 and the rest δi) 0. In the double summation δi and δj are both 1 only when conformer i and j are from two different residues. kbT is 0.59 kcal/mol (25.8 meV), and mi is 1 for bases, −1 for acids, and 0 for polar groups and waters. ni is the number of electrons gained or lost compared to the ground state conformer. For example, if an oxidized conformer is defined as the ground state, it has ni = 0 and the reduced conformer has ni = 1; F is the Faraday constant. pKsol,i is the pKa and Em,sol,i the midpoint potential of the ith cofactor in solution. ΔΔGrxn,i is the difference between the conformer reaction field energy in solution and in the protein (desolvation energy). ΔGpol,i is the pairwise electrostatic and nonelectro-static interaction of the conformer with the backbone and with side chains that have no conformational degrees of freedom. The torsion energy for each conformer is added to ΔGpol,i. ΔGij is the electrostatic and Lennard-Jones pairwise interaction between each pair of conformers in the microstate. The limits on the summation of the interconformer terms ensure that each interaction is counted once. Monte Carlo sampling establishes the Boltzmann distribution of the different conformers of each residue at 25 °C. Residue pKas and Ems are determined from the fraction group ionization in a series of Monte Carlo simulations at different pHs or Ehs (eq 1). Multiflip (76) between closely coupled residues is implemented (63). The SOFT function is not used (56). For pKa calculations the heme is fixed in its oxidized state, and the probability of residue protonation, including that of the aquo–heme, is determined as a function of pH (56). For Em calculations the pH is fixed at 7 and the Eh varied. All groups are allowed to sample different protonation states throughout the heme titration.
Analysis of the MCCE pKas and Ems
The Em or pKa changes on moving a group from solution to its position in the protein. Thus
| (2) |
where pKa is the group pKa in the protein, m is −1 for an acid and +1 for a base, and
| (3) |
where Em is the heme in situ Em and n is the number of electrons. The shift in energy of ionization (ΔΔGprotein) is due to different changes of the reaction reactant and product energies when the reaction is moved into the protein. This term can be broken down in MCCE into the sum:
| (4) |
including the change in reaction field energy from solution (ΔΔGrxn), the added interaction with the rigid backbone (ΔGpol), and the side chains in their ionization and conformation states equilibrated at the given pH and Eh (ΔGres) (20, 43, 77).
MCCE uses continuum electrostatics to calculate the interaction between the aquo–heme and charges and dipoles in the protein. Selected interactions with the ferric heme porphyrin together with its imidazole and water or hydroxyl ligand were also calculated using Gaussian 98 (67). The heme and imidazole are taken from the crystal structure of sperm whale myoglobin (1A6K), with water or hydroxyl added in GaussView (67). Each structure is then optimized using the B3LYP method (66) with the LANL2DZ basis set (78). Hydroxyl–hemes are expected to be low spin (53, 79) while water–hemes are generally high spin (53, 80). The water–heme is given a net charge +1 and spin 5/2, while the hydroxyl–heme has a net charge 0 and spin 1/2. The change in system energy is then calculated with an external charge of +1 or −1 placed next to the aquo–heme complex in positions shown in Figure 1. All atoms are fixed in the structure optimized before the charge is added.
Figure 1.
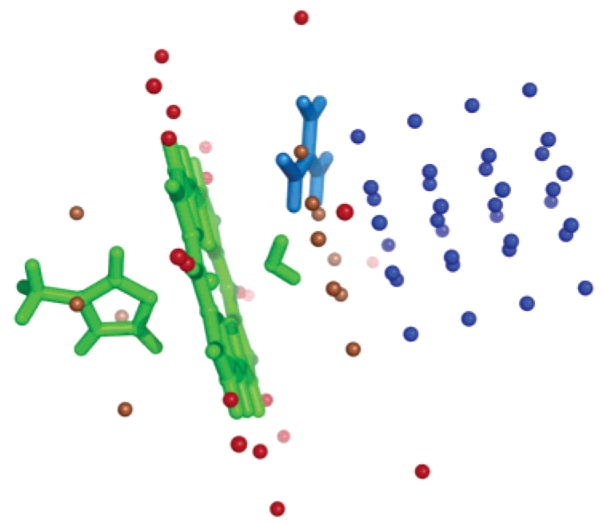
Position of external charges for comparison of CE and DFT interactions. Blue points are near the heme face; red points are in the plane of the heme and near the heme edge. Those within 4 Å of the heme plane are in brown. His–aquo–heme is in green. The position of Arg 38 in horseradish peroxidase is shown in blue.
Nine external charges are placed on a plane parallel to the porphyrin plane located 4, 6, 8, 10, or 12 Å from the heme (Figure 1). Points that overlap the water or imidazole ligand are removed. The interactions are also calculated with Coulomb’s law with the metal-centered charges used in the MCCE calculations with ε = 1. For the ferric hydroxyl–heme, Coulomb’s law reproduces the DFT interactions with an error of <10% and a slope of 0.94 with the exception of points within 4 Å of the heme plane (Figure 2). However, for the water–heme, the Coulomb’s law interactions with the points above the heme plane are significantly larger than those obtained with DFT. In contrast, for charges on the heme edge (Figure 1) the Coulomb’s law interactions reproduce the DFT interactions with <5% error and a slope of 1.0 for both water and hydroxyl ligated hemes (Figure 2). The source of the error in the CE calculation is not known. However, the large unfavorable interactions with water–heme are relaxed in the DFT calculations, while the smaller, favorable interactions with hydroxyl–heme are not. This suggests that there is significantly more polarization of the ferric water–heme by the external charge. In addition, the correction could be needed if there is significant charge at positions that are not well described by the atom-centered charges used here. Further studies are in progress to develop a water–heme charge distribution, perhaps including charges out of the heme plane, which can better reproduce the DFT interaction with Coulomb’s law. Preliminary calculations show that in general there is no systematic error in the CE calculations of the interactions of the standard amino acids with external charges.
Figure 2.

Comparison of interactions with a positive charge (blue ●) near the heme face, (red ■) in the plane of the heme and near the heme edge, and (brown ▲) within 4 Å of the heme plane as shown in Figure 1 calculated with DFT or CE Coulomb’s law with 3ε = 1. (A) Neutral, ferric (hydroxyl–heme). The ideal straight line through the origin with a slope of 1 is shown. (B) Cationic, ferric water–heme. The line through the points, y = 0.95x− (3.2 × 10−3)x2, is the best fit to a binomial function for the points near the heme face (blue). (C) (Hydroxyl–heme) – (water–heme). The line through the points is y = 0.89x + (1.1 × 10−3)x2.
The ferric water–heme interaction of a simple external charge at the position of Arg 38 in peroxidase or CuB in oxidase calculated with DFT is only 60% of that found with Coulomb’s law. The interaction with a charge at the position of Arg 99 in hemoglobin I is 65% as large. The influence of the charge distribution for the external group on the correction was examined by using a PARSE Arg charge distribution, rather than a single +1 charge. The DFT interaction is now 63% of that found with Coulomb’s law, very similar to that found with a unitary charge model. The influence of the charge distribution of CuB is more complex and cannot be described by a simple scaling factor. The details are discussed in ref 43.
Corrections are applied to interactions of the water–heme with Arg 99 in hemoglobin, Arg 38 in peroxidase, and CuB in oxidase. However, pairwise electrostatic interactions in MCCE are calculated with the Poisson–Boltzmann equation (PB), a CE analysis which takes into account the different dielectric constants of the protein and water (30, 81). The scaling factor derived for particular heme–residue geometry from the comparison of Coulomb’s law and DFT interactions in a vacuum (Figure 2) is used to scale the individual PB interactions in MCCE. Corrections used for CuB can be found in ref 43.
Interactions with residues that are on the heme plane or far away from the heme are shown to need no correction. Residues that have significant interactions with the aquo–heme which are not corrected because of their position include the following: all of the heme propionates; Arg 45 which is hydrogen bonded to a propionic acid in myoglobin; Lys 179 which is hydrogen bonded to a propionate in hemoglobin 1; Arg 136 and Asp 140 which form an ion pair on the porphyrin edge furthest from the propionates in hemoglobin 1 (Figure 4).
Figure 4.

Structures of (A) sperm whale myoglobin, (B) Aplysia myoglobin, (C) hemoglobin I, (D) heme oxygenase, and (E) horseradish peroxidase. Residues and backbone segments that interact with the cationic ferric aquo–heme by more than 0.5 ΔpK unit (0.68 kcal/mol) are shown.
RESULTS AND DISCUSSION
The ferric aquo–heme pKas were calculated with MCCE in myoglobins from sperm whale and Aplysia, hemoglobin I, heme oxygenase 1, horseradish peroxidase, and cytochrome c oxidase and compared to measured values. These pKas span 3.3 pH units from 7.6 in heme oxygenase 1 to 10.9 in the peroxidase. Two sets of calculations are presented. In the first set, standard Poisson–Boltzmann continuum electrostatics (CE) calculations are used for all pairwise interactions between the aquo–heme and the surrounding protein residues. In the second, an additional correction derived from DFT calculations is applied. Calculated pKas are compared to the experimental values. Calculations of the heme Ems are discussed below.
The pKas calculated using the standard CE-based pairwise interactions in MCCE (20) do not provide consistent agreement with the experimental data (Figure 3). Calculated pKas for the myoglobins and heme oxygenase are within 1 pH unit of the measured values. However, errors as large as 6 pH units are found in horseradish peroxidase and cytochrome c oxidase. In these proteins a nearby positive charge in the heme distal pocket lowers the calculated pKa, while the myoglobins have no charges in similar positions. As described in the Methods section CE and DFT calculations provide very similar values for pairwise interactions between a charge and a neutral hydroxyl–heme, probably because the electronic polarization does not contribute much to this smaller, favorable interaction. For the cationic water–heme, both methods also yield similar results when the charge is on the heme edge. However, when the charge is above the heme plane near the iron, the interactions derived with a CE analysis are significantly larger than those obtained with DFT calculations. The closer the charge is to the heme plane, the larger the discrepancy (Figure 2). The large unfavorable interactions are relaxed when electronic polarization is considered in the DFT calculations. The DFT corrections are applied to the pairwise electrostatic interaction of the water–heme with Arg 99 in hemoglobin I, Arg 38 in horseradish peroxidase, and CuB in cytochrome c oxidase to recalculate the pKas. Now calculated pKas in the six proteins agree with the measured values within 1 pH unit. Analysis of the MCCE calculations can then show how interactions and conformational changes shift the pKa,sol of 9.6 to different in situ values in these proteins.
Figure 3.

Comparison of calculated and experimental pKas (■) with and (○) without the DFT correction for (a) sperm whale myoglobin, (b) Aplysia myoglobin, (c) hemoglobin I, (d) heme oxygenase, (e) horseradish peroxidase, and (f) cytochrome c oxidase. The ideal line of slope 1 passing through the origin and lines with differences between experimental and calculated of (1 pH unit (dashed lines) are shown. References for experimental values are in Table 1.
Sperm Whale Myoglobin
Sperm whale myoglobin has a folded six-helix core (Figure 4). The pKa of the ferric aquo–heme is 8.95 (52), little different from the pKa,sol of 9.6. MCCE calculations with seven different crystal structures give an average aquo–heme pKa of 9.1 ( 1.0, in good agreement with experiment. This heme is close to the protein surface. However, pKa ≈ pKa,sol not because the heme does not interact with the protein but rather because a number of effects cancel (Table 1). The water–heme, with a net +1 charge, has 7 ΔpK units (9.52 kcal/mol) less solvation energy in the protein than it does in water, but the hydroxyl–heme, which has a large dipole moment, has a 5 ΔpK unit desolvation energy. Thus, the resultant desolvation penalty (ΔΔGrxn) favors the water–heme complex raising the pKa by about 2 pH units. The backbone amide dipoles tend to raise the electrostatic potential within all proteins, shifting residue pKas (25, 82) and cytochrome Ems (20). Here they favor the hydroxyl–heme, lowering the pKa by 1 pH unit. The E helix, which contains His 64 in the distal pocket, is the largest contributor.
Table 1.
Experimental and Calculated pKas and Ems and Energy Terms Contributing to pKa and Em Shifts in the Protein
| (a) pKa of Ferric Aquo–heme | |||||
|---|---|---|---|---|---|
| exp pKa | MCCE pKa | ΔΔGrxn (ΔpK unit) | ΔGpol (ΔpK unit) | ΔGres (ΔpK unit) | |
| sperm whale myoglobin (7)a | 8.95 (52) | 9.1 ± 1.0 | 2.07 ± 0.29 | 0.91 ± 0.14 | −2.45 ± 0.83 |
| Aplysia myoglobin (2) | 7.6 (52) | 7.7 ± 1.0 | 1.70 ± 0.34 | 0.88 ± 0.23 | −0.75 ± 0.49 |
| hemoglobin I (4) | 9.6 (83) | 9.8 ± 0.3 | 1.30 ± 0.18 | 1.02 ± 0.09 | −2.57 ± 0.24 |
| heme oxygenase 1 (5) | 7.6 (84, 85) | 7.5 ± 1.5 | 0.16 ± 0.37 | 3.34 ± 0.69 | −1.44 ± 1.10 |
| horseradish peroxidase (10) | 10.9 (52) | 10.1 ± 0.7 | 1.58 ± 0.39 | −0.61 ± 0.07 | −1.49 ± 0.66 |
| cyt c oxidase (1) | 9.0 (53) | 8.6 | 3.9 | 0.4 | −3.3 |
| (b) Em of Aquo–heme at pH 7 | |||||
| exp Em (mV) | MCCE Em (mV) | ΔΔGrxn (ΔpK unit) | ΔGpol (ΔpK unit) | ΔGres (ΔpK unit) | |
|
| |||||
| sperm whale myoglobin (7) | 50 (86) | 25 ± 33 | 4.50 ± 0.56 | 0.98 ± 0.15 | −2.97 ± 0.66 |
| Aplysia myoglobin (2) | 125 (86) | 126 ± 64 | 4.25 ± 1.24 | 0.89 ± 0.11 | −0.89 ± 0.24 |
| hemoglobin I (4) | 103 (87) | 83 ± 15 | 4.33 ± 0.17 | 0.85 ± 0.12 | −1.68 ± 0.09 |
| heme oxygenase 1 (5) | NA | 37 ± 13 | 2.47 ± 0.36 | 1.84 ± 0.44 | −1.61 ± 0.30 |
| horseradish peroxidase (10) | −250 (88, 89) | −215 ± 27 | 3.80 ± 0.51 | −0.62 ± 0.06 | −4.79 ± 0.49 |
| cyt c oxidaseb (1) | NA | 9 | 5.77 | 0.77 | −5.70 |
The value in parentheses is the number of PDB files analyzed. NA, not available. Negative free energy terms favor the cationic ferric water–heme raising the aquo–heme pKa and favor the oxidized heme lowering the Em. The ferric aquo–heme pKa,sol is 9.6 (70, 71). The b-type water–hemes have an Em,sol of −120 mV, while the a-type heme in cytochrome c oxidase has an Em,sol of −20 mV. Hydroxyl–hemes have Em,sols 80 mV lower than the water–hemes (72, 73), consistent with the 1.3 pKa unit shift between the pKas of ferric and ferrous aquo–heme (70, 71).
The pKa and Em of the aquo–heme a3 is calculated with CuA oxidized and heme a and CuB reduced to mimic the experimental pKa measurements in the CO photolyzed mixed-valence complex (53). 1 ΔpK unit ) 58 meV ) 1.36 kcal/mol. A systematic shift of −0.74 ΔpK unit (−1 kcal/mol) is added to the reference reaction field energy (ΔΔGRXN,sol) of ferric hydroxyl–heme and 0.5 ΔpK unit (0.67 kcal/mol) to ferrous water–heme in deriving the in situ Ems and pKas using eqs 2–4.
There are 4 Arg, 14 Glu, 7 Asp, and 19 Lys in the protein and all of them are >95% ionized between pH 7 and pH 9. However, most of these charges have little effect on the aquo–heme pKa because they are solvent exposed. Arg 45, which is hydrogen-bonded to the D-ring propionic acid, does shift the pKa down by 0.6 pK unit (propionate A and D designated as assigned in the PDB structure files). The two heme propionic acids are both fully ionized and together shift the pKa up by 2.3 units. As these are all near the heme edge, the CE interaction agrees with the DFT values (Figure 2). His 64 in the distal heme pocket near the aquo–ligand undergoes a conformational change with the change in the aquo–heme protonation state (Figure 4). The His is always neutral, but the δ proton tautomer stabilizes the positive water–heme by 3–4 ΔpK units, while the protonated ε tautomer stabilizes the hydroxyl–heme by 1.6–2.5 ΔpK units. In different PDB structures the His tautomer is restricted by the rigid backbone to different extents, yielding the dependence of the calculated pKa on the starting structure. Arg 45 not only lowers the aquo–heme pKa by direct electrostatic interaction but it also lowers the pKa indirectly by favoring the εHis 64 conformer.
Aplysia Myoglobin
Myoglobin from the sea hare Aplysia limacine has a folded six-helix structure (Figure 4). It is calculated to have a ferric aquo–heme pKa of 7.7, 2 pK units lower than that of sperm whale myoglobin, in agreement with the experimental value of 7.6 (52). The Aplysia and sperm whale proteins have the same protein folds and are 27% identical. The analogue of myoglobin His 64 is Val 64 in the Aplysia distal pocket, providing a bigger cavity, so the heme is slightly better solvated, shifting the pKa up. However, the biggest difference between the two proteins is in ΔGres. The loss of His 64 and protonation of the ring D propionic acid lowers the aquo–heme pKa. The propionate pKa is raised to 8.6 by the nearby Asp 45, while in myoglobin Arg 45 lowers the propionate pKa to 4.6.
Hemoglobin I
Monomeric clam hemoglobin I also has a folded six-helix structure (Figure 4). The aquo–heme pKa is calculated to be 9.8 and measured to be 9.6 (83), close to that of sperm whale myoglobin. The hemoglobin I and myoglobin have similar structures with 23% identity. A larger cavity in the heme distal pocket leads to a smaller desolvation energy. As His 64 does in myoglobin, Gln 64 changes conformation, keeping its oxygen pointing toward the water–heme and nitrogen pointing toward the hydroxyl–heme. Arg 99, on the proximal side of the heme, shifts the pKa down by 1.5 pH units, while the two propionic acids, both of which are ionized, shift the pKa up by 2.2 pH units.
Heme Oxygenase 1
Rat heme oxygenase 1 is a multihelical bundle with two three-helix motifs (Figure 4). The aquo–heme pKa is calculated to be 7.5, in agreement with the experimental value of 7.6 (84, 85). In this protein heme is an exchangeable substrate and is significantly more exposed than in the other proteins considered here and so has a very small ΔΔGrxn. The large positive potential from the backbone lowers the pKa by 3.3 pH units. The distal pocket helix is kinked at Gly 143 and Gly 144 so the backbone amides of Ser 142, Gly 143, and Gly 144 all point toward the heme iron, stabilizing the hydroxyl–heme. Arg 136 and Asp 140 are in a salt bridge near the porphyrin edge. While each interacts with the aquo–heme by over 3 ΔpK units, their combined impact is small.
Horseradish Peroxidase
The multihelical peroxidase uses heme to reduce hydrogen peroxide to water. The aquo–heme pKa is calculated to be 10.1, in reasonable agreement with the measured 10.9 (52). The aquo–heme has a similar reaction field energy loss as the globins. Unlike the globins, interactions with the backbone amides near the metal center are small and somewhat negative, raising the pKa by 0.6 pH unit. Interactions with other residues raise the pKa by 1.5 ΔpK units. His 42 and Asp 43 raise the pKa by 6 pH units while Arg 38 shifts the pKa down by 4.5 pH units (assuming a 60% DFT correction). His 42 sits in the distal pocket in a position similar to His 42 in myoglobin. However, this His is fixed in the δ tautomer by Arg 38, Glu 64, and Asn 70, stabilizing the protonated water–heme.
Cytochrome c Oxidase
Heme a3 in cytochrome c oxidase is deeply buried in the transmembrane 12-helix subunit I of the protein. In the mixed-valence state, with heme a and CuB reduced, the ferric aquo–heme pKa has been measured to be 9 (53). Using the full CE interaction between CuB and the aquo–heme, the calculated pKa is 3. The DFT correction reduces the interaction from 6.4 to 3.2 ΔpK units. Since the aquo–heme titration now occurs at higher pH, the protein net charge is more negative than when the titration is at pH 3. This raises the pKa further to 8.6, in good agreement with experiment (43). The cytochrome c oxidase heme is more deeply buried than in the other proteins, losing more reaction field energy, which shifts the pKa down. Within the membrane-embedded protein, pairwise electrostatic interactions with other residues are quite long range. However, the net interaction of the fully ionized propionic acids and the rest of the protein shifts the pKa up by only 2 pH units.
Em Calculations
The aquo–heme Ems were calculated in each protein at pH 7. Ems previously measured in four of the proteins range from −250 mV in peroxidase to 125 mV in Aplysia myoglobin (86) (Table 1b). MCCE calculations agree with the experimental values with errors of e35 mV. MCCE allows protonation of the aquo–heme to be coupled to heme reduction (20). The relevant Em,sol for water–heme of −120 mV (72, 73) and for hydroxyl–heme of −200 mV (72) is included in the microstate energy (eq 1) so the pH dependence of the Em is properly treated in the redox titration. This degree of freedom could be important for Aplysia myoglobin and heme oxygenase, which have pKas near 7. However, since their ferric pKas are above 7 and ferrous pKas are higher, the aquo–heme ligand remains predominately water at pH 7.
Electrostatic interactions that favor the ferric heme with its net charge of +1 over the neutral ferrous heme shift the Em down. The same electrostatic interactions favor the cationic ferric water–heme over the neutral hydroxyl–heme, raising the pKa. While a higher pKa is correlated with a lower Em, the connection is by no means exact (Figure 5). In most proteins, the ΔΔGprotein for the redox reaction is larger than for the protonation reaction so the Em is shifted from Em,sol by more than the pKa is shifted from pKa,sol (Figure 5, eqs 2 and 3). One difference is the desolvation energy is on average 2.4 ± 0.3 ΔpK units larger for the redox reaction than for the protonation reaction (Table 1). Because of its significant dipole the solvated neutral ferric hydroxyl–heme has a solvation energy that is not much smaller than the positively charged ferric water–heme. Thus, both reactant and product of the protonation reaction have similar ΔΔGrxn. With the exception of cytochrome c oxidase, the desolvation penalty, which always favors the neutral species, shifts the pKa down by at most 2 pH units. In contrast, when compared to the neutral, ferrous water–heme, the cationic, ferric water–heme has much larger interactions with water. The impact of the desolvation energy on the redox reaction when the heme is buried in the protein is thus much larger.
Figure 5.

Em versus pKa at pH 7 for (a) sperm whale myoglobin, (b) Aplysia myoglobin, (c) hemoglobin I, (d) heme oxygenase, (e) horseradish peroxidase, and (f) cytochrome c oxidase. (△) Calculated and (●) available experimental values. The solid line has the same ΔΔGprotein for the in situ Em and pKa (eqs 2 and 3). The dashed line of ΔΔGprotein is 2.4 ΔpK units larger for Em than pKa; 2.4ΔpK units is the average difference in the ΔΔGrxn for the two types of reactions (Table 1).
Hemoglobin I has a pKa which is only 0.2 pH unit higher than the pKa,sol, while the Em is 223 mV more positive than Em,sol (ΔΔGprotein = 3.8 ΔpK units). The difference in desolvation energy for the two reactions contributes 3 ΔpK units. In addition, Gln 64 in the distal pocket changes conformation in the aquo–heme pKa titration but does not in the Em titration. The rigid Gln 64 raises the water–heme Em by 1 ΔpK unit (58 meV) more than it shifts the ferric aquo–heme pKa down. The reorientation of the Gln is part of the protein dielectric response that is captured by the explicit motions available in MCCE (30, 55). The Gln motion coupled to aquo–heme protonation thus diminishes the effective pairwise interaction.
In horseradish peroxidase, ΔΔGprotein shifts the Em down more than it shifts the pKa up. Besides the 2.2 ΔpK unit difference in the desolvation energy, ΔGres favors heme reduction 3.3 ΔpK units more than water deprotonation. Although both ferrous water–heme and ferric hydroxyl–heme have a net charge of 0, they interact differently with nearby residues. The hydroxyl–heme has a large dipole with its negatively charged hydroxide pointing toward the distal pocket. Arg 38 in the distal pocket and Asp 247 on the opposite side form a dipole stabilizing the hydroxyl–heme. The difference in interactions with hydroxyl–heme and water–heme is relatively small, leading to a small ΔGres, contributing to the pKa shift. On the other hand, the neutral ferrous water–heme is not stabilized by the electrostatic dipole generated by these two amino acids while interactions of the ferric water–heme with nearby negative charges shift the Em down.
As in horseradish peroxidase, the favorable interaction of CuB with the hydroxyl–heme in cytochrome c oxidase makes the contribution of ΔGres to the in situ pKa smaller than found for in situ Em. Assuming the Em,sol of heme a is 100 (1) to 160 (74, 75) mV more positive than that of heme c, the Em of heme a3 is predicted to be 9 to 69 mV in cytochrome c oxidase when CuA is oxidized and heme A and CuB are reduced.
There are no experimental data available for the Em in heme oxygenase 1. These calculations predict an Em of 40 mV. A run of four backbone dipoles lowers the pKa more than the Em. All of the other proteins studied here have similar values of ΔGpol for the redox and protonation reactions.
CONCLUSIONS
The pKas of ferric aquo–heme at pH 7 in sperm whale myoglobin, Aplysia myoglobin, hemoglobin I, heme oxygenase 1, horseradish peroxidase, and cytochrome c oxidase were calculated with MCCE. The calculations reveal a breakdown in the classic continuum electrostatic analysis of pairwise interactions. Large errors in calculated pKas were found in horseradish peroxidase and cytochrome c oxidase. DFT calculations suggest that CE calculations overestimate pairwise interactions between ferric water–heme and a positive charge above the heme plane. This could be due to significant out-of-plane charge density, which would be ignored in the atom-centered charges used in the CE analysis. Alternatively, these external charges could polarize ferric hydroxyl–heme significantly more than the amount accounted for by the ε = 4 used in the MCCE calculation. Using a correction that brings the DFT and CE interactions into agreement, the calculated pKas in six proteins are within 1 pH unit of the experimental values. The calculated Ems using the correction derived from DFT calculations are within 35 mV of the experimental values.
Proteins with higher ferric aquo–heme pKas have lower Ems since both arise from the protein stabilizing a positively charged heme. However, the proteins shift the free energy of the redox reaction (ΔΔGprotein) by an amount that would shift a pKa by >6 pH units (375 mV), while the pKas span only 3.3 pH units (eqs 2 and 3). The difference in ΔΔGprotein shows that there is a larger effective dielectric response for the protonation than the redox reactions. In sperm whale myoglobin and hemoglobin I protonation is coupled to conformational changes while the redox reaction is not. These conformation changes allow a residue in the distal pocket to stabilize both protonation states, reducing the energy difference between them. In contrast, when the group is rigid as it is for the redox reaction, the pairwise interactions are larger, shifting the Em more. This difference in flexibility for the two reaction types found in MCCE is supported by the good match between calculation and experiment for both pKas and Ems.
Supplementary Material
Acknowledgments
We gratefully acknowledge helpful discussions with Ronald Koder.
Footnotes
This work is supported by NIH Grant RO1-GM64540.
Abbreviations: pKa,sol, group pKa in aqueous solution; Em,sol, electrochemical midpoint potential (Em) of a model for the redox cofactor in aqueous solution; CE, continuum electrostatics; MCCE, Multi-Conformation Continuum Electrostatics; DFT, density functional theory. 1 ΔpK unit = 1.36 kcal/mol = 58 meV. The propionic acids are designated A and D as found in the PDB structure file. The IUPAC nomenclature calls them respectively propionate D and C.
SUPPORTING INFORMATION AVAILABLE
Interactions between heme a3 and CuB in different aquo protonation and cofactor redox states calculated in a vacuum by Coulomb’s law and by DFT using Gaussian 98 and in cytochrome c oxidase embedded in a low dielectric slab surrounded by water using DelPhi. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Reedy CJ, Gibney BR. Heme protein assemblies. Chem Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 2.Antonin E, Brunori M. Hemoglobin and Myoglobin in their Reactions with Ligands. North-Holland, Amsterdam: 1971. [Google Scholar]
- 3.Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase. Characterization of the enzyme. J Biol Chem. 1969;244:6388–6394. [PubMed] [Google Scholar]
- 4.Welinder KG. Superfamily of plant, fungal and bacterial peroxidases. Curr Opin Struct Biol. 1992;2:338–393. [Google Scholar]
- 5.Smith AT, Veitch NC. Substrate binding and catalysis in heme peroxidases. Curr Opin Chem Biol. 1998;2:269–278. doi: 10.1016/s1367-5931(98)80069-0. [DOI] [PubMed] [Google Scholar]
- 6.Brzezinski P. Redox-driven membrane-bound proton pumps. Trends Biochem Sci. 2004;29:380–387. doi: 10.1016/j.tibs.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Gennis RB. Some recent contributions of FTIR difference spectroscopy to the study of cytochrome oxidas. FEBS Lett. 2003;555:2–7. doi: 10.1016/s0014-5793(03)01150-5. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson-Miller S, Babcock GT. Heme/copper terminal oxidases. Chem Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 9.Wikstrom M. Cytochrome c oxidase: 25 years of the elusive proton pump. Biochim Biophys Acta. 2004;1655:241–247. doi: 10.1016/j.bbabio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Montfort WR, Weichsel A, Andersen JF. Nitrophorins and related antihemostatic lipocalins from Rhodnius prolixus and other blood-sucking arthropods. Biochim Biophys Acta. 2000;1482:110–118. doi: 10.1016/s0167-4838(00)00165-5. [DOI] [PubMed] [Google Scholar]
- 11.Privalle CT, Crivello JF, Jefcoate CR. Regulation of intramitochondrial cholesterol transfer to side-chain cleavage cytochrome P-450 in rat adrenal gland. Proc Natl Acad Sci USA. 1983;80:702–706. doi: 10.1073/pnas.80.3.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pikuleva I, Waterman M. Cytochromes P450 in synthesis of steroid hormones, bile acids, vitamin D3 and cholesterol. Mol Aspects Med. 1999;20:33–42. 43–37. [PubMed] [Google Scholar]
- 13.Rodgers KR. Heme-based sensors in biological systems. Curr Opin Chem Biol. 1999;3:158–167. doi: 10.1016/S1367-5931(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 14.Chan MK. Recent advances in heme-protein sensors. Curr Opin Struct Biol. 2001;5:216–222. doi: 10.1016/s1367-5931(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 15.Churg AK, Warshel A. Control of the redox potential of cytochrome c and microscopic dielectric effects in proteins. Biochemistry. 1986;25:1675–1681. doi: 10.1021/bi00355a035. [DOI] [PubMed] [Google Scholar]
- 16.Poulos TL. Heme enzyme crystal structure. Heme Proteins: Struct Funct, Genet. 1988;7:2–36. [Google Scholar]
- 17.Rietjens I. The role of the axial ligand in heme-based catalysis. J Biol Inorg Chem. 1996;1:355. [Google Scholar]
- 18.Martin ACR, Orengo CA, Hutchinson EG, Jones S, Karmirantzou M, Laskowski RA, Mitchell JBO, Taroni C, Thornton JM. Protein folds and function. Structure. 1998;6:875–884. doi: 10.1016/s0969-2126(98)00089-6. [DOI] [PubMed] [Google Scholar]
- 19.Stellwagen E. Haem exposure as the determinate of oxidation–reduction potential of haem proteins. Nature. 1978;275:73–74. doi: 10.1038/275073a0. [DOI] [PubMed] [Google Scholar]
- 20.Mao J, Hauser K, Gunner MR. How cytochromes with different folds control heme redox potentials. Biochemistry. 2003;42:9829–9840. doi: 10.1021/bi027288k. [DOI] [PubMed] [Google Scholar]
- 21.Gunner MR, Honig B. Electrostatic control of midpoint potentials in the cytochrome subunit of the Rhodopseudomonas viridis reaction center. Proc Natl Acad Sci USA. 1991;88:9151–9155. doi: 10.1073/pnas.88.20.9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voigt P, Knapp EW. Tuning heme redox potentials in the cytochrome c subunit of photosynthetic reaction centers. J Biol Chem. 2003;278:51993–52001. doi: 10.1074/jbc.M307560200. [DOI] [PubMed] [Google Scholar]
- 23.Kassner RJ. A theoretical model for the effects of local nonpolar heme environments on the redox potentials in cytochromes. J Am Chem Soc. 1973;95:7959–7975. doi: 10.1021/ja00789a044. [DOI] [PubMed] [Google Scholar]
- 24.Kassner RJ. Effects of nonpolar environments on the redox potentials of heme complexes. Proc Natl Acad Sci USA. 1972;69:2263–2267. doi: 10.1073/pnas.69.8.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J, Mao J, Gunner MR. Are acidic and basic groups in buried proteins predicted to be ionized? J Mol Biol. 2005;348:1283–1298. doi: 10.1016/j.jmb.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 26.Muegge I, Qi PX, Wand AJW, Chu ZT, Warshel A. The reorganization energy of cytochrome c revisted. J Phys Chem. 1997;101:825–836. [Google Scholar]
- 27.Churg AK, Weiss RM, Warshel A, Takano T. On the action of cytochrome c: correlating geometry changes upon oxidation with activation energies of electron transfer. J Phys Chem. 1983;87:1683–1694. [Google Scholar]
- 28.Cutler RL, Davies AM, Creighton S, Warshel A, Moore GR, Smith M, Mauk AG. Role of arginine-38 in regulation of the cytochrome c oxidation–reduction equilibrium. Biochemistry. 1989;28:3188–3197. doi: 10.1021/bi00434a012. [DOI] [PubMed] [Google Scholar]
- 29.Warwicker J, Watson HC. Calculation of the electric potential in the active site cleft due to a α-helix dipoles. J Mol Biol. 1982;157:671–679. doi: 10.1016/0022-2836(82)90505-8. [DOI] [PubMed] [Google Scholar]
- 30.Gunner MR, Alexov E. A pragmatic approach to structure based calculation of coupled proton and electron transfer in proteins. Biochim Biophys Acta. 2000;1458:63–87. doi: 10.1016/s0005-2728(00)00060-8. [DOI] [PubMed] [Google Scholar]
- 31.Zhou HX. Control of reduction potential by protein matrix: lesson from a spherical protein model. J Biol Inorg Chem. 1997;2:109–113. [Google Scholar]
- 32.Warshel A, Papazyan A, Muegge I. Microscopic and semimacroscopic redox calculations: what can and can not be learned from continuum models. J Biol Inorg Chem. 1997;2:143–152. [Google Scholar]
- 33.Baptista AM, Martel PJ, Peterson SB. Simulation of protein conformational freedom as a function of pH: constant-pH molecular dynamics using implicit titration. Proteins: Struct Funct, Genet. 1997;27:523–544. [PubMed] [Google Scholar]
- 34.Soares CM, Martel PJ, Mendes J, Carrondo MA. Molecular dynamics simulation of cytochrome c3: studying the reduction processes using free energy calculations. Biophys J. 1998;74:1708–1721. doi: 10.1016/S0006-3495(98)77882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baptista A, Martel PJ, Soares CM. Simulation of electron-proton coupling with a Monte Carlo method: application to cytochrome c3 using continuum electrostatics. Biophys J. 1999;76:2978–2998. doi: 10.1016/S0006-3495(99)77452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martel PJ, Soares CM, Baptista AM, Fuxreiter M, Naray-Szabo G, Louro RO, Carrondo MA. Comparative redox and pKa calculations on cytochrome c3 from several DesulfoVibrio species using continuum electrostatic methods. J Biol Inorg Chem. 1999;4:73–86. doi: 10.1007/s007750050291. [DOI] [PubMed] [Google Scholar]
- 37.Zaric SD, Popovic DM, Knapp EW. Factors determining the orientation of axially coordinated imidazoles in heme proteins. Biochemistry. 2001;40:7914–7928. doi: 10.1021/bi010428q. [DOI] [PubMed] [Google Scholar]
- 38.Haas AH, Lancaster CR. Calculated coupling of transmembrane electron and proton transfer in dihemic quinol: fumarate reductase. Biophys J. 2004;87:4298–4315. doi: 10.1529/biophysj.104.042945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogers NK, Moore GR, Sternberg MJE. Electrostatic interactions in globular proteins: Calculation of the pH dependence of the redox potential of cytochrome c551. J Mol Biol. 1985;182:613–616. doi: 10.1016/0022-2836(85)90248-7. [DOI] [PubMed] [Google Scholar]
- 40.Rogers NK, Moore GR. On the energetics of conformational changes and pH dependent redox behaviour of electron transfer proteins. FEBS Lett. 1988;228:69–73. doi: 10.1016/0014-5793(88)80587-8. [DOI] [PubMed] [Google Scholar]
- 41.Kannt A, Lancaster CRD, Michel H. The coupling of electron transfer and proton translocation: electrostatic calculations on Paracoccus denitrificans cytochrome c oxidase. Biophys J. 1998;74:708–721. doi: 10.1016/S0006-3495(98)73996-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Popovic DM, Stuchebrukhov AA. Electrostatic study of the proton pumping mechanism in bovine heart cytochrome c oxidase. J Am Chem Soc. 2004;126:1858–1871. doi: 10.1021/ja038267w. [DOI] [PubMed] [Google Scholar]
- 43.Song Y, Michonova-Alexova E, Gunner MR. Calculated proton uptake on anaerobic reduction of cytochrome c oxidase: Is the reaction electroneutral? Biochemistry. 2006;45:XXXX–XXXX. doi: 10.1021/bi052183d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siegbahn PE. The catalytic cycle of tyrosinase: peroxide attack on the phenolate ring followed by O–O cleavage. J Biol Inorg Chem. 2003;8:567–576. doi: 10.1007/s00775-003-0449-4. [DOI] [PubMed] [Google Scholar]
- 45.Green MT. Role of the axial ligand in determining the spin state of resting cytochrome P450. J Am Chem Soc. 1998;120:10772–10773. [Google Scholar]
- 46.Guallar V, Friesner RA. Cytochrome P450CAM enzymatic catalysis cycle: a quantum mechanics/molecular mechanics study. J Am Chem Soc. 2004;126:8501–8508. doi: 10.1021/ja036123b. [DOI] [PubMed] [Google Scholar]
- 47.Schoneboom JC, Thiel W. The resting state of P450cam: A QM/MM study. J Phys Chem B. 2004;108:7468–7478. [Google Scholar]
- 48.Ogliaro F, Harris N, Cohen S, Filatov M, deVisser DP, Shaik S. A model “rebound” mechanism of hydroxylation by cytochrome P450: Stepwise and effectively conderted pathways and their reactivity patterns. J Am Chem Soc. 2000;122:8977–8989. [Google Scholar]
- 49.Kamachi T, Yoshizawa K. A theoretical study on the mechanism of camphor hydroxylation by compound I of cytochrome P450. J Am Chem Soc. 2003;125:4652–4661. doi: 10.1021/ja0208862. [DOI] [PubMed] [Google Scholar]
- 50.Guallar V, Baik MH, Lippard SJ, Friesner RA. Peripheral heme substituents control the hydrogen-atom abstraction chemistry in cytochromes P450. Proc Natl Acad Sci USA. 2003;100:6998–7002. doi: 10.1073/pnas.0732000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schoneboom JC, Cohen S, Lin H, Shaik S, Thiel W. Quantum mechanical/molecular mechanical investigation of the mechanism of C–H hydroxylation of camphor by cyto-chrome P450cam: theory supports a two-state rebound mechanism. J Am Chem Soc. 2004;126:4017–4034. doi: 10.1021/ja039847w. [DOI] [PubMed] [Google Scholar]
- 52.Brunori M, Amiconi G, Antonin E, Wyman J, Zito R, Fanelli AR. The transition between “acid” and “alkaline” ferric heme proteins. Biochim Biophys Acta. 1968;154:315–322. doi: 10.1016/0005-2795(68)90045-7. [DOI] [PubMed] [Google Scholar]
- 53.Branden M, Namslauer A, Hansson O, Aasa R, Brzezinski P. Water-hydroxide exchange reactions at the catalytic site of heme-copper oxidases. Biochemistry. 2003;42:13178–13184. doi: 10.1021/bi0347407. [DOI] [PubMed] [Google Scholar]
- 54.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alexov EG, Gunner MR. Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophys J. 1997;72:2075–2093. doi: 10.1016/S0006-3495(97)78851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Georgescu RE, Alexov EG, Gunner MR. Combining conformational flexibility and continuum electrostatics for calculating pKas in proteins. Biophys J. 2002;83:1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gunner MR, Mao J, Song Y, Kim J. Factors influencing energetics of electron and proton transfers in proteins. What can be learned from calculations. Biochim Biophys Acta. 2006 doi: 10.1016/j.bbabio.2006.06.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicholls A, Honig B. A rapid finite difference algorithm utilizing successive over-relaxation to solve the Poisson–Boltzmann equation. J Comput Chem. 1991;12:435–445. [Google Scholar]
- 59.Bharadwaj R, Windemuth A, Sridharan S, Honig B, Nicholls A. The fast multipole boundary element method for molecular electrostatics: An optimal approach for large systems. J Comput Chem. 1995;16:898–913. [Google Scholar]
- 60.Rocchia W, Alexov E, Honig B. Extending the applicability of the nonlinear Poisson–Boltzmann equation: multiple dielectric constants and multivalent ions. J Phys Chem B. 2001;105:6507–6514. [Google Scholar]
- 61.Gilson MK, Honig B. Calculation of the total electrostatic energy of a macromolecular system: solvation energies, binding energies, and conformational analysis. Proteins: Struct Funct, Genet. 1988;4:7–18. doi: 10.1002/prot.340040104. [DOI] [PubMed] [Google Scholar]
- 62.Sitkoff D, Sharp KA, Honig B. Accurate calculation of hydration free energies using macroscopic solvent models. J Phys Chem. 1994;98:1978–1988. [Google Scholar]
- 63.Song Y, Mao J, Gunner MR. Calculation of proton transfers in bacteriorhodopsin bR and M intermediates. Biochemistry. 2003;42:9875–9888. doi: 10.1021/bi034482d. [DOI] [PubMed] [Google Scholar]
- 64.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz JKM, Ferguson DM, Spellman DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 65.Mao J, Song Y, Gunner MR. Better rotamer packing for continuum electrostatics pKa calculations: MCCE2. J Comput Chem. 2006 in preparation. [Google Scholar]
- 66.Becke AD. Density-Functional Thermochemistry. 3 The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 67.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VGJA, Montgomery J, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA. Gaussian. Vol. 98. Gaussian, Inc; Pittsburgh, PA: 1998. Revision A.9. [Google Scholar]
- 68.Richarz R, Wüthrich K. Carbon-13 NMR chemical shifts of the common amino acid residues measured in aqueous solutions of the linear tetrapeptides H-Gly-Gly-X-L-Ala-OH. Biopolymers. 1975;17:2133–2141. [Google Scholar]
- 69.Matthew JB, Gurd FRN, Garcia-Moreno B, Flanagan MA, March KL, Shire SJ. pH-dependent processes in proteins. CRC Crit Rev Biochem. 1985;18:91–197. doi: 10.3109/10409238509085133. [DOI] [PubMed] [Google Scholar]
- 70.Munro OQ, Marques HM. Heme-peptide models for hemoproteins. 1 solution chemistry of N-acetylmicroperoxi-dase-8. Inorg Chem. 1996;35:3752–3767. doi: 10.1021/ic9502842. [DOI] [PubMed] [Google Scholar]
- 71.Vashi PR, Marques HM. The coordination of imidazole and substituted pyridines by the hemeoctapeptide N-acetyl-ferromicroperoxidase-8 (FeIINAcMP8) J Inorg Biochem. 2004;98:1471–1482. doi: 10.1016/j.jinorgbio.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 72.Marques HM, Cukrowski I, Vashi PR. Coordination of weak field ligands by N-acetylmicroperoxidase-8 (NAcMP8), a ferric haempeptide from cytochrome c, and the influence of the axial ligand on the reduction potential of complexes of NAcMP8. J Chem Soc. 2000;2000:1335–1342. [Google Scholar]
- 73.Santucci R, Reinhard H, Brunori M. Direct electrochemistry of the undacapeptide from cytochrome c (microperoxidase) at a glassy carbon electrode. J Am Chem Soc. 1988;110:8536–8357. [Google Scholar]
- 74.Vanderkooi G, Stotz E. Reductive alteration of heme a hemochromes. J Biol Chem. 1965;240:3418–3424. [PubMed] [Google Scholar]
- 75.Vanderkooi G, Stotz E. Oxidation–reduction potentials of heme a hemochromes. J Biol Chem. 1966;241:3316–3323. [PubMed] [Google Scholar]
- 76.Beroza P, Fredkin DR, Okamura MY, Feher G. Protonation of interacting residues in a protein by a Monte Carlo method: application to Lysozyme and the photosynthetic reaction center of Rhodobacter sphaeroides. Proc Natl Acad Sci USA. 1991;88:5804–5808. doi: 10.1073/pnas.88.13.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu Z, Gunner MR. Energetics of quinone-dependent electron and proton transfers in Rhodobacter sphaeroides photosynthetic reaction centers. Biochemistry. 2005;44:82–96. doi: 10.1021/bi048348k. [DOI] [PubMed] [Google Scholar]
- 78.Hay PJ, Wadt WR. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J Chem Phys. 1985;82:270–283. [Google Scholar]
- 79.Sitter AJ, Shifflett JR, Terner J. Resonance Raman spectroscopic evidence for heme iron-hydroxide ligation in peroxidase alkaline forms. J Biol Chem. 1988;263:13032–13038. [PubMed] [Google Scholar]
- 80.Peisach J, Blumberg WE, Ogawa S, Rachmilewitz EA, Oltzik R. The effects of protein conformation on the heme symmetry in high spin ferric heme proteins as studied by electron paramagnetic resonance. J Biol Chem. 1971;246:3342–3355. [PubMed] [Google Scholar]
- 81.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 82.Gunner MR, Saleh MA, Cross E, ud-Doula A, Wise M. Backbone dipoles generate positive potentials in all proteins: origins and implications of the effect. Biophys J. 2000;78:1126–1144. doi: 10.1016/S0006-3495(00)76671-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kraus DW, Wittenberg JB, Lu JF, Peisach J. Hemoglobins of the Lucina pectinata/bacteria symbiosis. II An electron paramagnetic resonance and optical spectral study of the ferric proteins. J Biol Chem. 1990;265:16054–16059. [PubMed] [Google Scholar]
- 84.Sun J, Wilks A, Ortiz de Montellano PR, Loehr TM. Resonance Raman and EPR spectroscopic studies on heme–heme oxygenase complexes. Biochemistry. 1993;32:14151–14157. doi: 10.1021/bi00214a012. [DOI] [PubMed] [Google Scholar]
- 85.Takahashi S, Wang J, Rousseau DL, Ishikawa K, Yoshida T, Host JR, Ikeda-Saito M. Heme-heme oxygenase complex. Structure of the catalytic site and its implication for oxygen activation. J Biol Chem. 1994;269:1010–1014. [PubMed] [Google Scholar]
- 86.Taylor JF. Measurement of the oxidation-reduction equilibria of hemoglobin and myoglobin. Methods Enzymol. 1981;76:577–582. doi: 10.1016/0076-6879(81)76145-7. [DOI] [PubMed] [Google Scholar]
- 87.Kraus DW, Wittenberg JB. Hemoglobins of the Lucina pectinata/bacteria symbiosis. I Molecular properties, kinetics and equilibria of reactions with ligands. J Biol Chem. 1990;265:16043–16053. [PubMed] [Google Scholar]
- 88.Yamada H, Makino R, Yamazaki I. Effects of 2,4-substituents of deuteropheme upon redox potentials of horseradish peroxidases. Arch Biochem Biophys. 1975;169:344–353. doi: 10.1016/0003-9861(75)90350-1. [DOI] [PubMed] [Google Scholar]
- 89.Harbury HA. Oxidation-reduction potentials of horseradish peroxidase. J Biol Chem. 1957;225:1009–1024. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.