

Abstract
Objective:
To evaluate the spatial pattern and regional rates of neocortical atrophy from normal aging to early Alzheimer disease (AD).
Methods:
Longitudinal MRI data were analyzed using high-throughput image analysis procedures for 472 individuals diagnosed as normal, mild cognitive impairment (MCI), or AD. Participants were divided into 4 groups based on Clinical Dementia Rating Sum of Boxes score (CDR-SB). Annual atrophy rates were derived by calculating percent cortical volume loss between baseline and 12-month scans. Repeated-measures analyses of covariance were used to evaluate group differences in atrophy rates across regions as a function of impairment. Planned comparisons were used to evaluate the change in atrophy rates across levels of disease severity.
Results:
In patients with MCI–CDR-SB 0.5–1, annual atrophy rates were greatest in medial temporal, middle and inferior lateral temporal, inferior parietal, and posterior cingulate. With increased impairment (MCI–CDR-SB 1.5–2.5), atrophy spread to parietal, frontal, and lateral occipital cortex, followed by anterior cingulate cortex. Analysis of regional trajectories revealed increasing rates of atrophy across all neocortical regions with clinical impairment. However, increases in atrophy rates were greater in early disease within medial temporal cortex, whereas increases in atrophy rates were greater at later stages in prefrontal, parietal, posterior temporal, parietal, and cingulate cortex.
Conclusions:
Atrophy is not uniform across regions, nor does it follow a linear trajectory. Knowledge of the spatial pattern and rate of decline across the spectrum from normal aging to Alzheimer disease can provide valuable information for detecting early disease and monitoring treatment effects at different stages of disease progression.
GLOSSARY
- AD
= Alzheimer disease;
- ADNI
= Alzheimer’s Disease Neuroimaging Initiative;
- ANCOVA
= analysis of covariance;
- CDR
= Clinical Dementia Rating;
- CDR-SB
= Clinical Dementia Rating Sum of Boxes score;
- MCI
= mild cognitive impairment;
- MMSE
= Mini-Mental State Examination;
- PI
= Principal Investigator;
- RM
= repeated-measures;
- ROI
= region of interest;
- TIV
= total intracranial volume;
- UCSD
= University of California, San Diego.
Whole brain atrophy rates differ among normal aging individuals and those diagnosed with mild cognitive impairment (MCI) and Alzheimer disease (AD).1–3 Recent studies have demonstrated that the spatial pattern of neocortical atrophy associated with normal vs pathologic aging is not uniform and may depend on the degree of disease severity.4 However, few longitudinal studies are available that describe regional atrophy rates and changes in atrophy rates across the disease spectrum from normal aging to early AD.5–7
We examined longitudinal rates of regional neocortical atrophy over 1 year in 137 healthy controls and 335 individuals at different stages of clinical impairment. Annual atrophy rates were of interest because they have demonstrated high sensitivity to subtle brain changes in AD and may discriminate between diagnostic groups better than baseline brain measures.1 Furthermore, we used cross-sectional data to estimate the change in atrophy rates with increasing levels of disease. Change in atrophy rates may be particularly useful for evaluating the efficacy of disease-modifying therapeutics.5,8 We hypothesized that in normal aging, mild atrophy would be observed in prefrontal and parietal regions with relative sparing of posterior neocortex. Conversely, we predicted that in patients with mild MCI, atrophy rates would be greatest in medial temporal lobe regions and would spread to other cortical regions along a posterior-to-anterior gradient with increasing levels of clinical impairment. The longitudinal and cross-sectional MRI data reported in this study provide a qualitative and quantitative depiction of regional brain changes that accompany normal aging and progression from prodromal to early AD.
METHODS
Alzheimer’s Disease Neuroimaging Initiative.
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (www.loni.ucla.edu/ADNI). The ADNI was launched in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, private pharmaceutical companies and nonprofit organizations, as a $60 million, 5-year public–private partnership. ADNI’s goal is to test whether serial MRI, PET, other biologic markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials.
The principal investigator of this initiative is Michael W. Weiner, MD, VA Medical Center and University of California, San Francisco. ADNI is the result of efforts of many coinvestigators from a broad range of academic institutions and private corporations. Subjects have been recruited from more than 50 sites across the United States and Canada (www.adni-info.org).
Standard protocol approvals, registrations, and patient consents.
This study was approved by an ethical standards committee on human experimentation at each institution. Written informed consent was obtained from all patients or authorized representatives participating in the study.
Participants.
ADNI eligibility criteria are described at http://www.adni-info.org/index.php?option=com_content&task=view&id=9&Itemid=43). Briefly, subjects are aged 55–90 years and have a study partner able to provide an independent evaluation of functioning. Control subjects have a Mini-Mental State Examination (MMSE) score between 24 and 30 (inclusive) and a global Clinical Dementia Rating (CDR) of 0. Subjects with MCI have MMSE scores between 24 and 30, a subjective memory symptom, objective memory loss measured by education-adjusted scores on Wechsler Memory Scale Logical Memory II, a global CDR of 0.5, preserved activities of daily living, and an absence of dementia. Subjects with mild AD have MMSE scores between 20 and 26, have a global CDR of 0.5 or 1.0, and meet National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association criteria for probable AD.9
In this study, MRI data were included on all ADNI subjects for whom baseline and 12-month MRI scans were available and had passed local quality inspection by December 2008. To estimate the level of clinical impairment, the CDR Sum of Boxes score (CDR-SB) was calculated at baseline for all participants. This score has been described as a useful and reliable measure for detecting subtle clinical change, ideal for use in longitudinal assessments of dementia.10 The CDR-SB score was used to divide the study sample into groups reflecting degree of impairment. From highest to lowest, our initial sample included 151 individuals with a CDR-SB = 0 (normal controls), 105 with a CDR-SB 0.5–1.0, 126 with a CDR-SB 1.5–2.5, and 104 with a CDR-SB >2.5 (mild AD). Because we wished to compare atrophy rates in normal aging with those across different patient groups, individuals in the normal group who progressed to a CDR-SB of 0.5 or greater at follow-up (n = 14) were removed from the analysis. Final group demographics are presented in table 1. Information on CDR-SB progression of individual participants over the course of 1 year is included in table e-1 on the Neurology® Web site at www.neurology.org. Groups did not differ in age [F(3,471) = 1.9, p > 0.05] or sex distribution [κ2(3) = 0.28, p > 0.05]. The groups did differ in level of education [F(3, 471 = 2.8, p < 0.05]. Those with a CDR-SB = 0 obtained a higher level of education relative to those with a CDR-SB >2.5.
Table 1 Demographic characteristics of the control and patient groups
Procedures.
Dual 3-dimensional T1-weighted volumes were downloaded from the public ADNI database (http://www.loni.ucla.edu/ADNI/Data/index.shtml). All image processing and analyses occurred at the Multimodal Imaging Laboratory, University of California, San Diego. Images were corrected for gradient nonlinearities11 and intensity nonuniformity12 using methods developed within the NIH/National Center for Research Resources–sponsored Morphometry Biomedical Informatics Research Network (http://www.nbirn.net/). The T1-weighted images were aligned, averaged to improve signal-to-noise ratio, and resampled to isotropic 1-mm voxels. Methods based on FreeSurfer 3.02 software were used to obtain cortical gray matter volume and thickness measures in distinct regions of interest (ROIs).13–18 Baseline volumetric data were corrected for differences in head size by regressing the estimated total intracranial volume (TIV) from the data.19
Longitudinal analysis.
For each subject, dual 3-dimensional follow-up structural scans were rigid-body aligned, averaged, and affine aligned to the subject’s baseline. A deformation field was calculated from nonlinear registration according to Holland et al.20 in a manner related to that of Ashburner and Friston21 and Christensen et al.22 and then used to align scans at the subvoxel level. The follow-up aligned image undergoes skull stripping and subcortical segmentation, with labels applied from the baseline scan. For cortical reconstruction, surface coordinates for the white and pial boundaries were derived from the baseline images and mapped onto the follow-up images using the deformation field. Parcellation and labeling17 from the baseline image was then applied to the follow-up image. This results in a 1:1 correspondence between each vertex in the base image and in the 12-month image. This method produces an estimate of the annual percent cortical volume loss at each vertex and within each ROI. Atrophy rates in this study were defined as the percent cortical volume loss over the course of 1 year. These atrophy rates represent the within-subject change over time and are independent of group differences in baseline measurements (e.g., TIV).
Statistical analyses.
To examine whether the rate of neocortical loss varies across regions as a function of degree of impairment, a repeated-measures (RM) analysis of covariance (ANCOVA) was performed with CDR-SB group as a between-subject variable and hemisphere (left vs right) and rate of lobar atrophy (lateral temporal, medial temporal, parietal, frontal, cingulate, occipital, and primary sensory) as within-subjects variables. Age and sex were entered as covariates in each model. The primary analyses of interest were 1) whether there was a main effect of CDR-SB group, indicating that atrophy rate varies with level of impairment, and 2) whether there was an interaction between CDR-SB group and region or hemisphere, indicating different spatial patterns of atrophy across levels of impairment. When significant group effects were observed for a lobar region, univariate analyses were performed within smaller ROIs using a threshold of p < 0.001. Because of evidence that whole brain atrophy rates do not appear constant over the course of disease,5 changes in regional atrophy rates were evaluated by performing independent t tests on the difference in atrophy rates (i.e., slope of the lines) between the normal control group and MCI patients with CDR-SB 0.5–1 vs the difference in atrophy rates between MCI patients with CDR-SB 1.5–2.5 and early AD. Greater atrophy rates early relative to late in disease were defined as a steeper slope between the normal and MCI–CDR-SB 0.5–1 groups, whereas greater atrophy rates later relative to early in disease defined as a steeper slope between the MCI–CDR-SB 1.5–2.5 and early AD groups.
RESULTS
Continuous surface maps of cortical volume loss as a function of disease severity demonstrate that cortical atrophy over 12 months is minimal in the normal control group (figure 1). In MCI patients with CDR-SB 0.5–1, cortical loss appears most prominent in medial and lateral temporal and inferior parietal cortex, spreading to widespread frontal regions as impairment increases to early AD.

Figure 1 Annual atrophy rates as a function of degree of clinical impairment
Annual atrophy rates as a function of degree of clinical impairment (i.e., baseline Clinical Dementia Rating Sum of Boxes score [CDR-SB]). Mean atrophy rates are represented as a percent change in neocortical volume and mapped onto the lateral (left), ventral (middle), and medial (right) pial surface of the left hemisphere. These data demonstrate that atrophy rates are most prominent in posterior brain regions early in the course of disease, spreading to anterior regions as the level of impairment increases, with relative sparing of sensorimotor regions. MCI = mild cognitive impairment; AD = Alzheimer disease.
ROI analysis.
The RM ANCOVA for lobar regions revealed a main effect of region [F(6, 1,892) = 15.4, p < 0.001], a main effect of group [F(3,466) = 45.4, p < 0.001], and a region-by-group interaction [F(18, 1,892) = 23.4, p < 0.001]. There was no main effect of hemisphere [F(1,466) = 3.0, p = 0.067], nor was there a group-by-hemisphere interaction [F(3,466) = 1.6, p = 0.181]. Therefore, follow-up analyses were collapsed across right and left hemisphere values. Univariate ANCOVAs revealed group differences across all lobar regions. Follow-up comparisons were performed within each ROI between normal controls and each patient group (table 2). Atrophy rates in all ROIs for the 4 groups can be seen in figure 2.
Table 2 One-year atrophy rates in the control and patient groups

Figure 2 Annual neocortical atrophy rates in regions of interest
Graphs depicting annual neocortical atrophy rates (percent volume change) in regions of interest in the normal control, mild cognitive impairment (MCI)–Clinical Dementia Rating Sum of Boxes score (CDR-SB) 0.5–1, MCI–CDR-SB 1.5–2.5, and early Alzheimer disease (AD) groups.
Normal vs MCI–CDR-SB 0.5–1.
Relative to normal controls, MCI patients with CDR-SB 0.5–1 showed a greater rate of atrophy across all medial temporal lobe regions, inferior and middle temporal gyri, inferior parietal lobule, precuneus, and posterior cingulate. Atrophy rates were not increased in frontal, occipital, or sensory regions relative to controls.
Normal vs MCI–CDR-SB 1.5–2.5.
In addition to the regions described above, MCI patients with CDR-SB 1.5–2.5 showed an increased rate of atrophy within the superior temporal gyrus, as well as lateral orbitofrontal, rostral and caudal middle frontal, frontal operculum, and superior frontal gyrus compared with controls. In addition, atrophy in all parietal regions and the lingual and lateral occipital regions progressed at a faster rate.
Normal vs early AD.
Relative to normal controls, those with mild AD showed a faster rate of atrophy across all temporal, frontal, cingulate, and occipital regions. Greater atrophy rates were also seen within precentral and postcentral gyri with relative sparing of pericalcarine regions.
Changes in regional atrophy rates across levels of disease severity.
Although atrophy rates increased throughout the neocortex with increasing disease severity (table 2), the rate of increase across levels of impairment was not constant within all regions. As shown in figure 3, increases in atrophy rates were greater between controls and those with MCI–CDR-SB 0.5–1 within medial temporal lobe regions (t 471 = 2.01, p < 0.05), including entorhinal (t 471 = 4.69, p < 0.01) and fusiform (t 471 = 2.67, p < 0.01) cortex. This effect did not reach significance in parahippocampal cortex (t 471 = 0.95, p > 0.05), hippocampus (t 471 = 0.96, p > 0.05), or the temporal pole (t 471 = 1.17, p > 0.05). Conversely, increases in atrophy rates were greater between those with MCI–CDR-SB 1.5–2.5 and early AD within frontal (t 471 = 2.17, p < 0.05), parietal (t 471 = 3.34, p < 0.01), anterior cingulate (t 471 = 3.5, p < 0.01), and posterior cingulate (t 473 = 3.10, p < 0.01) cortex. No differences in early vs late changes in atrophy rates were observed in the occipital (t 471 = 0.87, p > 0.05), lateral temporal (t 471 = 1.69, p > 0.05), or hippocampal (t 471 = 0.229, p > 0.05) regions. Whereas occipital regions showed minimal change across groups, atrophy rates within the lateral temporal lobe and hippocampus appeared constant across levels of disease severity.
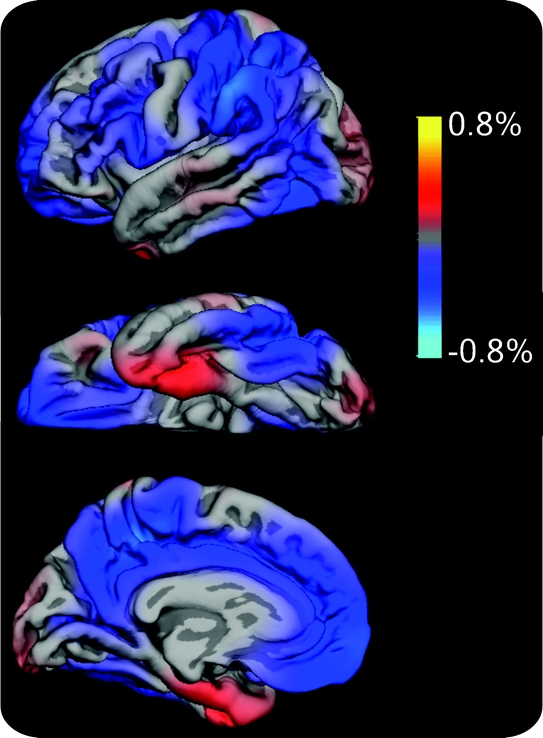
Figure 3 Cortical surface maps representing the mean difference in atrophy rates early vs later in the course of disease
Cortical surface maps representing the mean difference in atrophy rates early (mild cognitive impairment [MCI]–Clinical Dementia Rating Sum of Boxes score [CDR-SB] 0.5–1 − normal) vs later (early Alzheimer disease [AD] − MCI–CDR-SB 1.5–2.5) in the course of disease projected onto the left pial surface. Positive values (red) represent cortical regions showing early > late increases in atrophy rates (i.e., areas in which the difference in atrophy rates between controls and patients with MCI–CDR-SB 0.5–1 was greater than that between patients with MCI–CDR-SB 1.5–2.5 and early AD), whereas negative values (blue) represent cortical areas showing late > early increases in atrophy rates (i.e., areas in which the difference in atrophy rates between patients with MCI–CDR-SB 1.5–2.5 and early AD exceeded that between controls and patients with MCI–CDR-SB 0.5–1). Areas showing no difference in atrophy rates at the early vs later stage of disease, whether due to minimal atrophy or to a constant rate of atrophy across disease stages, do not appear in color on the maps.
DISCUSSION
Longitudinal MRI analysis from our large cohort of normal controls, individuals with MCI, and individuals with AD provides strong evidence that neocortical atrophy rates are not uniform across the cortical mantle, nor are changes in atrophy rates constant in many regions with increased level of clinical impairment. We demonstrate a pattern of atrophy in MCI–CDR-SB 0.5–1 that begins in medial and inferolateral temporal, inferior parietal, and posterior cingulate and spreads to encompass superior parietal, widespread prefrontal, and lateral occipital cortex with progression to MCI–CDR-SB 1.5–2.5. In patients with mild AD, atrophy rates are greater in all cortical regions with the exception of primary visual and auditory cortex relative to age-matched controls. These data support our primary hypothesis and are consistent with previous findings that atrophy in MCI and AD progresses along a posterior-to-anterior gradient with relative sparing of sensory regions.4,23,24
However, regional atrophy rates do not always progress in a linear manner. Whereas increases in atrophy rates were greatest between controls and those with MCI–CDR-SB 0.5–1 within most medial temporal regions and plateaued slightly between those with MCI–CDR-SB 1.5–2.5 and early AD, increases in atrophy rates within the hippocampus and lateral temporal neocortex did not differ across levels of impairment. Conversely, increases in atrophy rates within most prefrontal, parietal, and anterior cingulate regions were modest between controls and patients with MCI–CDR-SB 0.5–1, but the degree of increase was greater as disease severity increased. Therefore, although atrophy rates increased throughout the neocortex with disease severity, the rate of increase differed across regions and level of clinical impairment. A few longitudinal studies have shown acceleration in whole brain, ventricular, and hippocampal atrophy rates with disease progression.5–7 Our cross-sectional findings are in line with other studies suggesting that hippocampal atrophy rates increase linearly with level of disease severity.25 Such discrepancies in the literature may reflect methodologic differences in hippocampal measurements, variability in MRI scanning intervals, and/or differences in study design and patient cohorts (i.e., acceleration of hippocampal atrophy rates has been shown in familial AD).7 Nevertheless, our findings extend the literature by revealing that observed increases in atrophy rates vary by neocortical region and level of clinical impairment.
Understanding the different spatial patterns and regional trajectories of neocortical loss that accompany various stages of disease can provide critical information for early detection of disease, as well as response to treatment. Whereas atrophy rates within medial temporal lobe regions seem to be the earliest indicator of mild disease, atrophy rates within anterior and posterior association cortex may help to determine treatment efficacy with respect to slowing disease progression and cognitive decline. Such longitudinal data will be particularly important for studies of disease-modifying treatments because each subject serves as his or her own control, allowing disease progression to be assessed directly through repeat evaluations. When monitoring response to treatment, our data suggest that the cortical areas targeted will depend on where patients reside along the disease spectrum. Slowing of entorhinal atrophy rates in patients with mild impairment may represent a true treatment effect, whereas later in the disease it may reflect the natural course of disease progression. Similarly, a steady rate of decline in prefrontal regions in patients with greater impairment may reflect a strong response to treatment because increases in atrophy rates would be expected with conversion to early AD. These patterns should be carefully considered in studies that select MRI biomarkers as primary or secondary outcome measures because overlooking the natural history of disease progression could result in overestimation or underestimation of treatment effects. High-throughput image analysis procedures, such as the ones used in this study, would be particularly useful and cost-effective in large-scale clinical trials designed to measure treatment effects in hundreds of individuals, within multiple regions of interest, and over several time points.
Despite support for our hypothesis of the spatial distribution of changes observed in our patient groups, the pattern of atrophy in our control group did not support our hypothesis of greater atrophy rates in prefrontal and parietal regions relative to posterior association cortex. Rather, we found 1-year atrophy rates to be relatively small and of similar magnitude across frontal, parietal, and temporal lobe regions. Although these data initially appear at odds with existing literature, they are commensurate with other studies showing minimal, diffuse cortical atrophy over a 1- to 2-year period in healthy individuals aged 59–85 years.4,26,27 Therefore, it is possible that with a longer time interval, the predicted anterior-to-posterior gradient of atrophy frequently observed in normal aging would have emerged. Additional insights into patterns of normal age-related atrophy will be critical to the interpretation of pathologic patterns of aging.
Despite the potential clinical value of our findings, there are limitations of this study that should be noted. First, longitudinal follow-up was limited to 1 year. Therefore, our longitudinal data were supplemented with cross-sectional analysis to investigate the change in atrophy rates across levels of clinical impairment. Longitudinal studies that follow the same individuals for 5 to 10 years will provide a more definitive analysis of the pattern of atrophy (i.e., regional acceleration) associated with disease progression. Long-term follow-up is particularly important for individuals diagnosed with MCI because of the heterogeneity within MCI and the uncertainty as to what this group truly represents. Second, we do not currently have histologic verification of disease. Therefore, it is possible that some subjects have disorders other than AD, or have comorbid pathology that was not detected with our in vivo measures. Taken together, these data provide additional insight into the complex patterns and rates of neocortical atrophy that characterize different stages of clinical impairment. This information may aid in the selection of biomarkers for future clinical trials designed to evaluate the effect of disease-modifying treatments.
AUTHOR CONTRIBUTIONS
Statistical analysis was performed by Carrie McDonald, PhD. Dr. McDonald is a clinical neuropsychologist and an Assistant Professor of Psychiatry at the University of California, San Diego. She has doctoral level training in multivariate statistics and structural equation modeling.
ACKNOWLEDGMENT
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (www.loni.ucla.edu/ADNI). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. Complete listing of ADNI investigators available at http://www.loni.ucla.edu/ADNI/Data/ADNI_Authorship_List.pdf. The authors thank Robin Jennings, Michele Perry, Chris Pung, and Elaine Wu for downloading and preprocessing the ADNI MRI data. The authors thank Holly Girard for assistance with figures.
DISCLOSURE
Dr. McDonald receives research support from the NIH [NS056091 (Principal Investigator [PI])]. Dr. McEvoy receives research support from the NIH [K01 AG029218 (PI), R01AG031224 (Coinvestigator), and U24 RR021382 (Coinvestigator)], from the HIV Neurobehavioral Research Center, University of California, San Diego (UCSD) (PI), and from the Stein Institute for Research on Aging, UCSD (PI). An immediate family member serves as President of and holds stock and stock options in Cortechs Labs, Inc. and receives research support from the NIH [5R43NS061023 (PI) and 1R43NS061023 (PI)]. Ms. Gharapetian reports no disclosures. Dr. Fennema-Notestine receives research support from the NIH [U24 RR21382 (Coinvestigator), U24 RR021992 (Coinvestigator), U24 RR019701 (Coinvestigator), P30 MH062512 (Coinvestigator), N01 MH22005 (Coinvestigator, PI Imaging Core), R01 MH079752 (Coinvestigator), R01 AG031224 (Coinvestigator), R01 AG024506 (Coinvestigator)], and from the Department of Veterans Affairs (Medical Research Service grant). Dr. Hagler receives research support from the NIH [MH079146-01A2 (PI), 2R01 NS018741-23A1 (Coinvestigator), U54 NS056883-01 (Coinvestigator), and 5K01MH079146-02 (PI)] and has applied for a patent for an automated method for labeling white matter fibers from MRI (2008, details pending). Dr. Holland receives research support from the NIH [7R01AG022381-03 (Programmer Analyst) and 1U01AG024904-02 (Programmer Analyst)]. Mr. Koyama reports no disclosures. Dr. Brewer receives research support from the NIH [K23 NS050305 (PI)]. Dr. Dale receives research support from the NIH [2P50NS022343-21A2 (Coinvestigator), 1R01AG031224 (PI), 1U01AG024904-02 (Subcontract PI), 5 U24 RR021382-04 (Subcontract PI), 1R01MH079752-01 (Coinvestigator), 2 RO1 NS18741-23A1 (Coinvestigator), and 1P50MH081755-01 (Coinvestigator)]; receives funding to his laboratory from General Electric Medical Systems as part of a Master Research Agreement with UCSD; and is a founder of, holds equity in, and serves on the scientific advisory board for CorTechs Labs, Inc. The terms of this arrangement have been reviewed and approved by UCSD in accordance with its conflict of interest policies.
Supplementary Material
Address correspondence and reprint requests to Dr. Carrie R. McDonald, Multimodal Imaging Laboratory, Suite C101, 8950 Villa La Jolla Dr., La Jolla, CA 92037 camcdonald@ucsd.edu
Supplemental data at www.neurology.org
Supported by a grant (U24 RR021382) to the Morphometry Biomedical Informatics Research Network (http://www.nbirn.net) that is funded by the National Center for Research Resources at the National Institutes of Health, USA. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI; Principal Investigator: Michael Weiner; NIH grant U01 AG024904). ADNI is funded by the National Institute on Aging, by the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Pfizer Inc., Wyeth Research, Bristol-Myers Squibb, Eli Lilly and Company, GlaxoSmithKline, Merck & Co. Inc., AstraZeneca AB, Novartis Pharmaceuticals Corporation, Alzheimer’s Association, Eisai Global Clinical Development, Elan Corporation plc, Forest Laboratories, and the Institute for the Study of Aging, with participation from the US Food and Drug Administration. Industry partnerships are coordinated through the Foundation for the National Institutes of Health. The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory of Neuro Imaging at the University of California, Los Angeles.
Disclosure: Author disclosures are provided at the end of the article.
Received February 6, 2009. Accepted in final form May 1, 2009.
REFERENCES
- 1.Sluimer JD, van der Flier WM, Karas GB, et al. Whole-brain atrophy rate and cognitive decline: longitudinal MR study of memory clinic patients. Radiology 2008;248:590–598. [DOI] [PubMed] [Google Scholar]
- 2.Fox NC, Freeborough PA. Brain atrophy progression measured from registered serial MRI: validation and application to Alzheimer’s disease. J Magn Reson Imaging 1997;7:1069–1075. [DOI] [PubMed] [Google Scholar]
- 3.Ezekiel F, Chao L, Kornak J, et al. Comparisons between global and focal brain atrophy rates in normal aging and Alzheimer disease: boundary shift integral versus tracing of the entorhinal cortex and hippocampus. Alzheimer Dis Assoc Disord 2004;18:196–201. [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson PM, Hayashi KM, de Zubicaray G, et al. Dynamics of gray matter loss in Alzheimer’s disease. J Neurosci 2003;23:994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan D, Janssen JC, Whitwell JL, et al. Change in rates of cerebral atrophy over time in early-onset Alzheimer’s disease: longitudinal MRI study. Lancet 2003;362:1121–1122. [DOI] [PubMed] [Google Scholar]
- 6.Jack CR Jr, Weigand SD, Shiung MM, et al. Atrophy rates accelerate in amnestic mild cognitive impairment. Neurology 2008;70:1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ridha BH, Barnes J, Bartlett JW, et al. Tracking atrophy progression in familial Alzheimer’s disease: a serial MRI study. Lancet Neurol 2006;5:828–834. [DOI] [PubMed] [Google Scholar]
- 8.Dickerson BC, Sperling RA. Neuroimaging biomarkers for clinical trials of disease-modifying therapies in Alzheimer’s disease. NeuroRx 2005;2:348–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 10.Lynch CA, Walsh C, Blanco A, et al. The Clinical Dementia Rating sum of box score in mild dementia. Dement Geriatr Cogn Disord 2006;21:40–43. [DOI] [PubMed] [Google Scholar]
- 11.Jovicich J, Czanner S, Greve D, et al. Reliability in multi-site structural MRI studies: effects of gradient non-linearity correction on phantom and human data. Neuroimage 2006;30:436–443. [DOI] [PubMed] [Google Scholar]
- 12.Sled JG, Zijdenbos AP, Evans AC. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging 1998;17:87–97. [DOI] [PubMed] [Google Scholar]
- 13.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–355. [DOI] [PubMed] [Google Scholar]
- 14.Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA 2000;97:11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis, II: inflation, flattening, and a surface-based coordinate system Neuroimage 1999;9:195–207. [DOI] [PubMed] [Google Scholar]
- 16.Fischl B, Sereno MI, Tootell RB, Dale AM. High-resolution intersubject averaging and a coordinate system for the cortical surface. Hum Brain Mapp 1999;8:272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desikan RS, Segonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 2006;31:968–980. [DOI] [PubMed] [Google Scholar]
- 18.Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis, I: segmentation and surface reconstruction. Neuroimage 1999;9:179–194. [DOI] [PubMed] [Google Scholar]
- 19.Buckner RL, Head D, Parker J, et al. A unified approach for morphometric and functional data analysis in young, old, and demented adults using automated atlas-based head size normalization: reliability and validation against manual measurement of total intracranial volume. Neuroimage 2004;23:724–738. [DOI] [PubMed] [Google Scholar]
- 20.Holland D, Hagler DJJ, Fennema-Notestine C, et al. Longitudinal nonlinear registration and quantitative analysis of change in whole brain and regions of interest. Alzheimers Dementia J Alzheimers Assoc 2008;4:T288. [Google Scholar]
- 21.Ashburner J, Friston KJ. Voxel-based morphometry: the methods. Neuroimage 2000;11:805–821. [DOI] [PubMed] [Google Scholar]
- 22.Christensen GE, Rabbitt RD, Miller MI. Deformable templates using large deformation kinematics. IEEE Trans Image Process 1996;5:1435–1447. [DOI] [PubMed] [Google Scholar]
- 23.Braak H, Braak E. Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol Scand Suppl 1996;165:3–12. [DOI] [PubMed] [Google Scholar]
- 24.Whitwell JL, Shiung MM, Przybelski SA, et al. MRI patterns of atrophy associated with progression to AD in amnestic mild cognitive impairment. Neurology 2008;70:512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morra JH, Tu Z, Apostolova LG, et al. Automated mapping of hippocampal atrophy in 1-year repeat MRI data from 490 subjects with Alzheimer’s disease, mild cognitive impairment, and elderly controls. Neuroimage 2009;45:S3–S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith CD, Chebrolu H, Wekstein DR, et al. Age and gender effects on human brain anatomy: a voxel-based morphometric study in healthy elderly. Neurobiol Aging 2007;28:1075–1087. [DOI] [PubMed] [Google Scholar]
- 27.Resnick SM, Goldszal AF, Davatzikos C, et al. One-year age changes in MRI brain volumes in older adults. Cereb Cortex 2000;10:464–472. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.