

Abstract
Polymorphisms in NQO1, a gene coding for the phase II enzyme involved in the detoxification of quinone carcinogens, have been associated with childhood leukemia in some studies, although the observed direction and magnitude of effects have been inconsistent. Therefore, the authors systematically reviewed all published reports describing the effect of NQO1 in de novo childhood leukemia and conducted a meta-analysis of 7 case-control studies that examined the association between NQO1*2 and childhood leukemia. Although a family-based study previously demonstrated overtransmission of this allele among childhood acute lymphoblastic leukemia cases, the meta-analysis showed that the presence of a NQO1*2 variant allele, which reduces the activity of the enzyme NAD(P)H:quinone oxidoreductase 1 (NQO1), had no significant effect on childhood leukemia. However, there was an increased risk associated with having at least 1 copy of the NQO1*2 allele in a subset of cases with MLL translocations (summary odds ratio = 1.39, 95% confidence interval: 0.98, 1.97). Heterogeneity between studies may be due to differences in population exposures to NQO1 substrates and small sample sizes, as well as potential population stratification in non-family-based studies. Therefore, further research is warranted on the role of NQO1 polymorphisms in the etiology of childhood leukemia, especially among MLL-positive leukemias.
Keywords: epidemiology; leukemia; meta-analysis; NAD(P)H dehydrogenase (quinone); NQO1; polymorphism, single nucleotide; systematic review
GENE AND ENZYME
The human NAD(P)H:quinone oxidoreductase 1 gene (NQO1; DT-diaphorase, Enzyme Commission (EC) number 1.6.99.2) occupies 17 kilobase pairs (kb) within a gene-rich region on chromosome 16 at 16q22.1 (1). This cytosolic flavoenzyme detoxifies quinones (a large class of aromatic compounds found commonly in plants, benzene metabolites, and chemotherapies) to hydroquinones or catechols (2–4). The enzyme NAD(P)H:quinone oxidoreductase 1 (NQO1) acts as an antioxidant by catalyzing a 2-electron reduction that bypasses the need for two 1-electron reductions that can result in the production of DNA and protein-damaging reactive oxygen species. In certain conditions (e.g., the presence of myeloperoxidase or autooxidants), NQO1 can contribute to the formation of reactive oxidation species via oxidative cycling and therefore can act as a prooxidant (2, 5). NQO1 is constitutively expressed in most tissues including the bone marrow, where expression is thought to be highly inducible by xenobiotics with quinone moieties, and is upregulated during times of oxidative or electrophilic stress (6). NQO1 expression is also upregulated in tumor tissues as the result of hypoxia (7): The success of quinone-based chemotherapies to exert their cytotoxic effects depends on their bioactivation by elevated levels of NQO1.
GENE VARIANTS
There have been more than 93 single nucleotide polymorphisms (SNPs) identified in the NQO1 gene. The most widely studied SNP of NQO1 is a C to T change at nucleotide position 609 (rs1800566), also known as NQO1*2. This results in a proline to serine amino acid change at codon 187 that is associated with a loss of enzyme activity due to instability of the protein product (8). Thus, the enzyme activity of the homozygous variant genotype (NQO1*2/*2) is almost undetectable, and the enzyme activity of the heterozygous genotype (NQO1*1/*2) is intermediate between the homozygous variant genotype and wild type (NQO1*1/*1) (6). The NQO1*2 allele frequency varies between different ethnic groups, ranging from 16% in Caucasians to 49% in Chinese populations. Prevalence of the NQO1*2/*2 genotype is 4.4% in non-Hispanic whites, 5.2% in African Americans, 12.2% in Japanese, 15.5% in Mexican Hispanics, 17.9% in Native Americans, 18.8% in Koreans, and 22.4% in Chinese (6, 9–11).
Another genetic polymorphism of NQO1 is a single nucleotide change from C to T at nucleotide position 465 (rs4986998), also known as NQO1*3, which changes the amino acid at codon 139 from arginine to tryptophan. This SNP results in alternative messenger RNA splice sites that can lead to a deletion of exon 4 and create a protein lacking the quinone binding site (12–15) for which enzyme activity differs according to the substrate (14). The frequency of the NQO1*3 polymorphism is generally low and ranges from 0% to 5% among different ethnic populations (9).
LABORATORY TESTS
Most publications describing NQO1 genotyping utilized polymerase chain reaction followed by restriction fragment length polymorphism analysis or allele-specific oligonucleotide hybridization assays, as described previously (16, 17). The SNPs on NQO1 are also amenable to genotyping by using more modern array-based methods.
DISEASE
Leukemia represents one-third of all cancer cases under the age of 15 years and is the most common pediatric cancer (18). A heterogeneous disease that disrupts normal hematopoiesis, it can present as either acute or chronic: Approximately 80% of childhood leukemias (<15 years of age) are acute lymphoblastic leukemia (ALL), less than 20% are acute myeloid leukemia (AML), and 2% are chronic myeloid leukemia, while chronic lymphocytic leukemia rarely occurs in children (19).
The incidence of childhood ALL varies by and within countries, with the lowest rates observed by the African countries (from 4.5 per million to 14.3 per million), followed by Asian countries (from 9.2 per million to 40.6 per million), with the highest rates observed by European, American (North, Central, and South), and Oceania (Australia and New Zealand) countries (from 16.1 per million to 48 per million) (20). In contrast, the differences in the incidence of childhood AML among different countries are not as distinct (20). In addition, the ratio of ALL to AML also varies by region, with the lower ratios observed in most African and Asian countries (i.e., more AML than ALL cases) and higher ratios observed in the European, American, and Oceania countries (20).
The most common cytogenetic changes in childhood leukemias include high hyperdiploidy (>50 chromosomes in karyotype) and specific recurrent chromosomal translocations; these are both believed to occur in utero after prenatal exposure to toxic agents. About 80% of infant leukemia cases (acute leukemia in children <12 months of age) harbor the MLL gene rearrangement at 11q23, and the most common translocation among children aged 2–5 years is the TEL-AML1. Despite our knowledge of tumor genetics, the causes of childhood leukemia are largely unknown: Currently, the only established risk factors are ionizing radiation and genetic syndromes such as Down syndrome (21). Accordingly, exogenous factors including history of infections, maternal diet and medication use during pregnancy, and benzene exposures from pesticides, parental smoking, parental occupation, and traffic emissions have been the subject of investigation. Several compounds from these exposures can be oxidized to semiquinones and quinones by peroxidases (e.g., myeloperoxidase), which then are substrates for NQO1 (5, 22).
The NQO1*2 variant allele has been associated with infants' leukemias harboring the MLL rearrangement (17, 23), childhood ALL (16), and therapy-related leukemias (24, 25). These observations are supported by a large epidemiologic study that showed that presence of the NQO1*2 allele increased the risk of hematotoxicity more than 2-fold among a large cohort of Chinese workers exposed to benzene, an established risk factor for adult AML (26). The NQO1*3 allele has been less studied, although Eguchi-Ishimae et al. (27) showed an association with Japanese infant leukemia cases that harbored the MLL-AF4 [t(4;11)] translocation. In contrast, in a comparison of childhood leukemia cases with controls in a British population, there was no evidence of association of the NQO1*3 allele in MLL-positive leukemias in infants (odds ratio (OR) = 1.12) or in TEL-AML1 or hyperdiploid leukemias compared with controls (17).
REVIEW AND METHODS—IDENTIFICATION AND ELIGIBILITY OF RELEVANT STUDIES
Our objective was to identify all published articles examining the association between NQO1 polymorphisms and childhood leukemia. We searched PubMed for all articles published up to September 24, 2007. We identified 56 articles with the search terms (“NAD(P)H Dehydrogenase (Quinone)” [MeSH] or “NQO1”) and “leukemia” and limiting the search to studies in human populations. Articles with the following characteristics were excluded from the review: 1) non-English articles; 2) review articles; 3) nonepidemiologic studies (e.g., studies on animals or cell culture); 4) treatment outcome studies; 5) studies with therapy-related leukemia; 6) studies that included nonleukemia cases (e.g., non-Hodgkin's lymphoma) without reporting separate results for leukemia; and 7) studies with only adults or studies that combined adult and childhood leukemia but did not report results separately by age group (Figure 1). As of September 24, 2007, we had identified 9 published studies describing the association between NQO1 polymorphisms and de novo childhood leukemia: 1 family-based study (28) and 8 “case-control” studies (16, 17, 23, 27, 29–32) for which 2 of these studies (23, 31) included case-only analyses.
Figure 1.
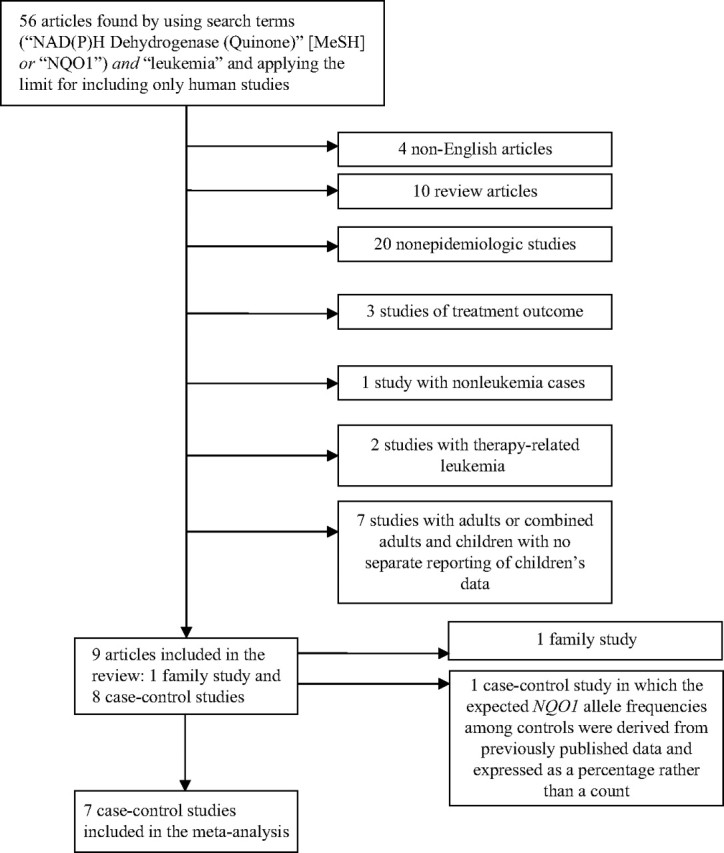
Process of article exclusion. MeSH, Medical Subject Headings; NQO1, NAD(P)H:quinone oxidoreductase 1.
ASSOCIATIONS
Family-based studies
One study with a family-based design of 657 case-parent trios and case-parent-grandparent clusters found that the NQO1*2 allele was transmitted to childhood ALL cases more frequently than expected, with a relative risk (dominant model) of 1.39 (95% confidence interval (CI): 1.07, 1.79) (28). This study also examined transmission of the NQO1*2 allele among 21 trios of infants (aged ≤12 months) with leukemia and found that there was a large, nonstatistically significant increased risk with carrying the variant allele (relative risk = 4.29, 95% CI: 0.58, 31.68). The family-based design is a powerful approach to study the effects of genetic variation in disease susceptibility, because it is immune to the effects of population stratification that could bias the results of case-case and case-control studies.
Case-case studies
Two studies used a case-only approach to examine the association between NQO1*2 and MLL-positive leukemias in infants (23, 31). Both studies used patients with de novo B-lineage ALL without the MLL gene rearrangement as a comparison group. The NQO1*2 allele significantly increased the risk of MLL-negative leukemia in infants in both the Italian (OR = 5.55, 95% CI: 1.81, 16.98) (31) and American (OR = 2.47, 95% CI: 1.08, 5.68) populations (23).
Case-control studies
Among the 5 traditional case-control studies that examined the association between NQO1*2 and acute childhood leukemia, the results were inconclusive (Table 1) (16, 27, 29, 31, 32). In a Canadian population, the presence of at least 1 variant NQO1*2 allele increased the risk of childhood ALL in cases compared with controls by approximately 50% (OR = 1.49, 95% CI: 1.02, 2.19) (Figure 2) (16). In contrast, a decreased risk of childhood ALL associated with the NQO1*2 allele was observed in a Turkish population (OR = 0.79, 95% CI: 0.58,1.08) (32). The other case-control studies showed essentially null results (27, 29, 31). In addition, Wiemels et al. (17) did not detect any association of the NQO1*2 allele in a subset of TEL-AML1-positive and hyperdiploid childhood ALL cases. There was no evidence of association between NQO1*2 and childhood AML among the 3 case-control studies that examined this outcome (27, 29, 32).
Table 1.
Case-Control Studies Reporting the Association Between NQO1 and Incident Childhood Leukemia That Were Included in the Meta-Analysis, 1999–2005
| First Author, Year, Location (Reference No.) | Study Population | Case Detailsa | Odds Ratiob | 95% Confidence Interval | MAF and Genotyping Method | Comments, Concerns, and Bias |
| Clavel, 2005, France (29) | Cases: 219 incident acute leukemia (ALL, n = 191; ANLL, n = 28) cases diagnosed in Paris, Lyon, Lille, and Nancy from January 1995 to December 1999; aged <15 years; 129 males, 90 females. Controls: 105 frequency matched to cases by age, gender, hospital, and ethnicity and hospitalized for diseases other than cancer or birth defects (mainly from orthopedic department); aged <15 years; 57 males, 48 females. | Acute leukemia (n = 219) | *2: 19%; DNA extracted from lymphocytes, and PCR-RFLP was used for genotyping. | Genotypes were tested and found to be in HWE. Population stratification possible but unlikely in this 92% white population. Odds ratios should not have been adjusted for parental socioprofession because it does not affect the NQO1 genotype. Selection bias minimized by including different diagnostic categories for controls in case a particular condition was related to the exposures of interest. | ||
| *1/*2 | 0.9 | 0.5, 1.6 | ||||
| *2/*2 | 1.7 | 0.4, 6.6 | ||||
| ALL (n = 191) | ||||||
| *1/*2 | 0.9 | 0.5, 1.6 | ||||
| *2/*2 | 2.1 | 0.5, 8.3 | ||||
| ANLL (n = 28) | ||||||
| *1/*2 | 0.9 | 0.3, 2.3 | ||||
| Eguchi-Ishimae, 2005, Japan (27) | Cases: 103 infants diagnosed with acute leukemia according to FAB classification, Southern blotting, cytogenetics, and RT-PCR and registered by the Japan Infant Leukemia Study Group between December 1995 and December 1998; aged <18 months; gender distribution not reported. Controls: 185 healthy newborn Japanese infants’ umbilical cord samples; gender distribution not reported. | MLL-positive total (n = 64) | 1.44 | 0.80, 2.58 | *2: 34%; *3: 1.9%; DNA extracted from blood, and PCR-RFLP was used for genotyping. | Authors did not report HWE testing, but our calculations show that all genotype frequencies were in HWE among controls. Population stratification unlikely in this Japanese population. Unknown whether controls arose from the same source population as the cases and thus could be prone to selection bias. No replication of genotyping, and thus bias could occur from genotyping error; however, control genotypes were in HWE. Not reported if genotyping calls were blinded to case-control status and could be prone to observer bias. |
| ALL (n = 49) | 1.17 | 0.62, 2.21 | ||||
| MLL-AF4 (n = 25) | 1.03 | 0.44, 2.38 | ||||
| AML (n = 15) | 3.23 | 0.88, 11.80 | ||||
| MLL-negative total (n = 39) | 1.16 | 0.58, 2.33 | ||||
| ALL (n = 23) | 1.26 | 0.52, 3.04 | ||||
| AML (n = 16) | 1.04 | 0.37, 2.90 | ||||
| MLL-positive totalc (n = 64) | 2.63 | 0.85, 8.14 | ||||
| ALLc (n = 49) | 3.55 | 1.13, 11.10 | ||||
| MLL-AF4c (n = 25) | 6.36 | 1.84, 21.90 | ||||
| MLL-negative totalc (n = 37) | 0.71 | 0.08, 5.92 | ||||
| ALLc (n = 21) | 1.27 | 0.15, 10.87 | ||||
| Kracht, 2004, Austria, Germany, Czechoslovakia (30) | Cases: 138 incident childhood ALL cases diagnosed by FAB criteria, cytochemistry, and multiplex PCR from October 1986 to December 2000 through German, Austrian, and Czech study centers; aged <19 years; 78 males, 60 females. Controls: 190 healthy blood donors with no history of cancer from a German center; 18–68 years; 130 males, 60 females. Restricted to individuals of Caucasian descent. | MLL-AF4d (n = 35) | 0.79 | 0.36, 1.74 | *2: 17.6%; DNA extracted from blood (controls) or leukemic bone marrow (cases), and SNPs identified by PCR-RFLP. | Genotypes were tested and found to be in HWE. Population stratification unlikely as the subjects were restricted to Caucasians. Controls and cases don't have the same age distribution; thus, this could introduce bias. Controls are adults and composed of volunteer blood donors; therefore, they may not be representative of the source population. Controls were recruited from one medical center in Germany, but the case children were from sites from 3 countries. |
| MLL-AF4, <18 monthsd (n = 22) | 0.44 | 0.14, 1.35 | ||||
| TEL-AML1d (n = 72) | 0.92 | 0.52, 1.65 | ||||
| BCR-ABLd (n = 31) | 1.42 | 0.38, 3.78 | ||||
| Krajinovic, 2002, Quebec, Canada (16) | Cases: 174 incident ALL cases (134 pre-B, 23 T-cell, 17 undetermined lineage) diagnosed in a single Montreal hospital from August 1988 to September 1998; 97 males, 77 females; age range, 1–21 years; median age, 5.2 years. Controls: 323 anonymous, healthy, unrelated white individuals recruited from a large, institutional DNA bank serving the same hospital as cases successfully genotyped for NQO1; not reported if controls were recruited during the same time period as cases; age and gender distribution not reported. Subjects restricted to white, French-Canadian origin and residing in the Province of Quebec as judged by their names, languages, and places of birth. | ALL (n = 174) | 1.49e | 1.02, 2.19e | *2: 17.8%; *3: 5%; DNA from buccal cells, peripheral blood, or bone marrow was used to investigate SNPs by PCR-ASO hybridization assays. | Authors did not report HWE testing, but our calculations show that all genotype frequencies were in HWE among controls. Age and health status for controls unknown; this could introduce bias. Population stratification unlikely because there is a founder effect in Quebec that indicates a relative genetic homogeneity. Not reported if genotyping calls were blinded to case-control status and could be prone to observer bias. |
| *1/*2 | 1.54e | 1.04, 2.29e | ||||
| *2/*2 | 1.34e | 0.53, 3.37e | ||||
| Lanciotti, 2005, Italy (31) | Cases: 50 infant ALL cases diagnosed by banded karyotyping, RT-PCR, and FISH and enrolled in the AIEOP protocol between 1995 and 2003; aged ≤12 months; gender distribution not reported. Healthy controls: 147 healthy control children admitted for traumas, acute infections, or minor surgical procedures; gender distribution not reported. | ALL total (n = 50) | 1.91 | 1.00, 3.65 | *2f; DNA was extracted from blood (controls) or bone marrow (cases), and SNPs were identified by PCR-RFLP. | Genotypes were tested and found to be in HWE. Population stratification unlikely in this Italian population. Selection bias possible because it is not reported if cases and controls were admitted to the same hospital or came from the same source population. |
| MLL-negative (n = 18) | 4.22 | 1.43, 12.49 | ||||
| MLL-positive (n = 32) | 1.26 | 0.58, 2.74 | ||||
| Sirma, 2004, Turkey (32) | Cases: 273 (189 ALL, 84 AML) cases diagnosed between 1996 and 2001 in 2 different medical faculties in Istanbul, Turkey, according to standard morphologic, cytochemical, and immunophenotypic criteria; aged 1–16 years (median ages, 8 (ALL) and 9 (AML) years); 128 females, 145 males. Controls: 286 healthy volunteers from different geographic areas of Turkey; aged 2–60 years (median, 19 years); 141 females, 145 males. | ALL | 0.75 | 0.25, 2.23 | *2: 24.8%; DNA was extracted from blood, and SNPs were identified by using PCR-RFLP genotyping. | Genotypes were tested and found to be in HWE. Controls and cases don't have the same age distribution; thus, this could introduce bias. Cases and controls are drawn from different source populations and could have very different exposure histories. Population stratification unlikely in this Turkish population. |
| ALL *2 allele | 0.79 | 0.58, 1.08 | ||||
| AML | 0.67 | 0.15, 3.13 | ||||
| AML *2 allele | 0.71 | 0.46, 1.09 | ||||
| ALL + AML | 0.73 | 0.27, 1.94 | ||||
| ALL + AML *2 allele | 0.76 | 0.58, 1.01 | ||||
| Wiemels, 1999, United Kingdom (17) | Cases: 115 pediatric leukemia cases diagnosed by banded karyotyping, FISH, and RT-PCR and enrolled in the UKCCS between 1991 and 1996; aged <15 years; gender distribution not reported. Controls: 100 umbilical cord samples from healthy newborn infants; gender distribution not reported. Subjects were >90% white and <10% from minority groups (Asian, Oriental, Afro-Carribean, black). | MLL-positive total (n = 36) | 2.54 | 1.08, 5.96 | *2: 17%; *3: 5.5%; DNA was extracted from blood, and SNPs were identified by using PCR-RFLP genotyping. | Authors did not report HWE testing, but our calculations show that all genotype frequencies were in HWE among controls. Unknown whether controls arose from the same source population as the cases and thus could be prone to selection bias. No replication of genotyping, and bias could occur from genotyping error; however, control genotypes were in HWE. Not reported if genotyping calls were blinded to case-control status and could be prone to observer bias. |
| MLL-AF4 (n = 21) | 8.63 | 2.45, 33.22 | ||||
| TEL-AML1 (n = 50) | 1.52 | 0.71, 3.25 | ||||
| Hyperdiploid (n = 29) | 0.91 | 0.33, 2.38 |
Abbreviations: AIEOP, Italian Pediatric Hematology and Oncology Group; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; ANLL, acute nonlymphocytic leukemia; ASO, allele-specific oligoprimer; FAB, French-American-British; FISH, fluorescence in situ hybridization; HWE, Hardy-Weinberg equilibrium (calculated among controls); MAF, minor allele frequency (among controls); PCR, polymerase chain reaction; RFLP, restriction fragment length polymorphism; RT, reverse transcription; SNP, single nucleotide polymorphism; UKCCS, United Kingdom Childhood Cancer Study.
The referent is *1/*1. The odds ratio is for *1/*2 + *2/*2 genotypes combined unless otherwise specified.
All odds ratios were unadjusted except those for the study by Clavel et al. (29) that were adjusted for parental socioprofessional category and matching factors (age, gender, hospital, ethnicity).
Odds ratio for *1/*3 + *3/*3 genotypes combined.
There were insufficient numbers for the *2/*2 genotype, and therefore it was not included in calculations.
Odds ratios and 95% confidence intervals were calculated by using genotype frequencies extracted from haplotype frequencies presented in Table III of Krajinovic et al. (16).
Not able to calculate the frequency of the NQO1*2 allele as the authors did not present separate genotype frequencies for heterozygote and homozygote variants.
Figure 2.

Forest plot of NQO1*2 and childhood acute lymphoblastic leukemia, random effects model. Test for heterogeneity: χ2 = 6.98, df = 4 (P = 0.14), I2 = 42.7%; test for overall effect: Z = 0.54 (P = 0.59). The odds ratio for each study is represented by a black square that is proportional to the sample size; the horizontal line shows the corresponding 95% confidence interval (CI). A dashed line marks the combined estimate, while the vertical solid line represents the null result.
Four case-control studies examined the association between NQO1*2 and MLL-positive childhood leukemia (17, 27, 30, 31). A significantly increased risk of MLL-positive leukemia associated with the NQO1*2 allele was observed in a British population (OR = 2.5, 95% CI: 1.08, 5.96) (17), while results from 2 other studies were also suggestive of an increased risk (27, 31). Additionally, in a case-control analysis comparing allele frequencies in cases with the expected population distribution of NQO1 allele frequencies at nucleotide position 609 derived from the published literature, Smith et al. (23) observed that MLL-positive leukemia cases were significantly more likely to have at least 1 copy of the NQO1*2 allele compared with ethnically matched, disease-free subjects (OR = 2.50; P = 0.02). Three of these studies further classified MLL-positive cases and identified a small subset (n ≤ 35) of cases harboring the MLL-AF4 translocation [t(4;11)] (17, 27, 30). Wiemels et al. (17) authored the only case-control study to show a significantly increased risk of MLL-AF4-positive leukemias associated with the NQO1*2 allele (OR = 8.63, 95% CI: 2.45, 33.22). It should be noted that this study (17) had the smallest sample size (21 MLL-AF4-positive cases) among all 3 studies. In summary, the results of 4 case-control studies suggest that the NQO1*2 allele may be associated with MLL-positive leukemia (17, 27, 30, 31) and possibly with the MLL-AF4-positive subtype (17).
INTERACTIONS
The presence of a functional polymorphism in NQO1 does not appear to confer a strong increased risk of childhood leukemia in general but may do so in the presence of an environmental exposure to quinones. Furthermore, this effect may be modified by polymorphisms in other carcinogen-metabolizing genes such as the GSTs, CYP2E1, and MPO. Therefore, it is important to study gene-gene and gene-environment interactions. In a French study, Clavel et al. (29) examined interactions among the NQO1*2 polymorphism, maternal tobacco smoking, and alcohol and coffee consumption during pregnancy. Using a case-control analysis, they observed that maternal coffee consumption negatively interacted with the NQO1*2 allele to reduce the risk of childhood acute leukemia (OR = 0.3, 95% CI: 0.1, 1.0). However, they found no significant interactions between the NQO1*2 polymorphism and either maternal smoking or alcohol consumption during pregnancy. A case-only analysis produced similar results.
Using a trio design (biologic parents and the affected child) to study a French-Canadian population, Infante-Rivard et al. (28) did not observe a multiplicative interaction between prenatal exposure to mononuclear aromatic hydrocarbons and the child's (NQO1) genotype (likelihood ratio test: P = 0.33) but did find weak evidence for a “supermultiplicative” interaction between maternal smoking during pregnancy and the child's (NQO1) genotype (likelihood ratio test: P = 0.15). In a small sample of study subjects, Krajinovic et al. (16) found a suggestion of a combined 3-way interaction between NQO1 and CYP2E1 risk-elevating genotypes and the wild-type MPO allele in increasing the risk of childhood ALL (OR = 5.4, 95% CI: 1.2, 23.4).
META-ANALYSIS
Identification and eligibility of relevant studies for meta-analysis
We decided that a quantitative summary of the role of NQO1 in childhood leukemia would be useful and therefore conducted a meta-analysis using the results of published studies. It was only possible to include case-control studies from the review into the meta-analysis because of the need for exposed and unexposed groups and diseased and nondiseased groups. Therefore, 2 of the studies included in the review were excluded from the meta-analysis because of 1) a family-based design (28) and 2) a case-only analysis of MLL-positive versus MLL-negative cases (23) (Figure 1). Although the latter study (23) included a second comparison group of disease-free, ethnically matched controls, the expected NQO1 allele frequencies were derived from previously published data and expressed as a percentage, rather than a count, and therefore could not be included in our meta-analysis. Table 1 describes the 7 case-control studies included in the meta-analysis.
Data abstraction for meta-analysis
All articles were assessed independently by two reviewers (N. G. and J. S. C.), who extracted data from each article that included authors, year of publication, country of origin, case and control selection, demographics of the study subjects, and the number of cases and controls for the genotypes of each NQO1 polymorphism. Any discrepancies between the two reviewers were resolved by discussion and consultation with a third reviewer (A. P. C.).
Statistical methods for meta-analysis
Four childhood leukemia outcomes were assessed in this meta-analysis: 1) ALL, 2) AML, 3) MLL-positive leukemia (ALL or AML), and 4) MLL-AF4-positive leukemia (ALL or AML). We calculated summary odds ratios for studies that assessed the relation between NQO1*2 and each outcome. Because NQO1*3 was assessed in only 2 studies (16, 27), we did not include this marker in the current meta-analysis. Forest plots were used to display the results from individual studies on a common scale, as well as the summary odds ratios, and further allowed for a visual assessment of the degree of heterogeneity among studies (Figures 2–5) (33). We used both random and fixed effect models with weights equal to the inverse of the variance to calculate a summary odds ratio for the effect of NQO1*2 on the different subtypes of childhood leukemia. Results from random effects models, which account for heterogeneity among studies, are presented when Mantel-Haenszel chi-squared (χ2) tests for heterogeneity showed significant differences across studies (conservatively defined as P < 0.20).
Figure 3.

Forest plot of NQO1*2 and childhood acute myeloid leukemia, random effects model. Test for heterogeneity: χ2 = 4.00, df = 2 (P = 0.14), I2 = 50.0%; test for overall effect: Z = 0.36 (P = 0.72). The odds ratio for each study is represented by a black square that is proportional to the sample size; the horizontal line shows the corresponding 95% confidence interval (CI). A dashed line marks the combined estimate, while the vertical solid line represents the null result.
Figure 4.

Forest plot of NQO1*2 and MLL-positive leukemia (acute lymphoblastic leukemia or acute myeloid leukemia), fixed effects model. Test for heterogeneity: χ2 = 4.34, df = 3 (P = 0.23), I2 = 30.9%; test for overall effect: Z = 1.83 (P = 0.07). The odds ratio for each study is represented by a black square that is proportional to the sample size; the horizontal line shows the corresponding 95% confidence interval (CI). A dashed line marks the combined estimate, while the vertical solid line represents the null result.
Figure 5.

Forest plot of NQO1*2 and MLL-AF4 leukemia (acute lymphoblastic leukemia or acute myeloid leukemia), random effects model. Test for heterogeneity: χ2 = 11.98, df = 2 (P = 0.002), I2 = 83.3%; test for overall effect: Z = 0.88 (P = 0.38). The odds ratio for each study is represented by a black square that is proportional to the sample size; the horizontal line shows the corresponding 95% confidence interval (CI). A dashed line marks the combined estimate, while the vertical solid line represents the null result.
The I2 statistic measured the extent of inconsistency among the studies (34). I2 values of 25%–50% indicate moderate inconsistency, while values larger than 50% reflect large inconsistencies among studies. We assumed a dominant mode of inheritance (the presence of at least 1 variant allele vs. wild-type), because only 1 copy of the variant NQO1*2 allele is needed to reduce enzyme activity. However, to explore the possibility of a “threshold effect” of enzyme activity, we also explored the analyses for the ALL and AML subsets assuming a recessive mode of inheritance (homozygous variant vs. heterozygote + wild-type); the results were similar to those assuming a dominant model (data not shown). It should be noted that only 1 study did not separate heterozygote and homozygote variants and therefore was not amenable to assessing a recessive mode of inheritance (31).
We evaluated publication bias by using funnel plots (35); however, this assessment was uninformative because of the limited number of studies. The results of the meta-analysis were generated using RevMan software (Review Manager, version 4.2 for Windows; The Nordic Cochrane Centre, Copenhagen, Denmark, 2003). Forest plots were generated using the “plot.meta” function in R software (http://www-ice.iarc.fr/∼martyn/software/meta/).
META-ANALYSIS RESULTS
Overall effects
Figure 2 presents the results of the 5 previous studies examining the association between the NQO1*2 polymorphism and risk of childhood ALL by using a random effects model. Only 1 of the 5 studies showed a significant positive association between childhood ALL and the presence of at least one NQO1*2 allele (16). Overall, the summary odds ratio showed no evidence of association of the NQO1*2 variant and risk of childhood ALL (OR = 1.08, 95% CI: 0.83, 1.40).
Results of the 3 studies that examined the association between the NQO1*2 polymorphism and childhood AML are shown in Figure 3. None of the 3 individual studies showed a statistically significant association (27, 29, 32). The combined analysis, using a random effects model showed no effect of the NQO1*2 variant on the development of childhood AML (OR = 0.90, 95% CI: 0.50, 1.62). Of the studies that evaluated the relation between the NQO1*2 polymorphism and MLL-positive leukemia (ALL or AML), 3 of 4 showed a positive association (17, 27, 31) (Figure 4), but only 1 study had statistically significant results (17). The combined odds ratio of these 4 studies suggested an increased risk of MLL-positive childhood leukemia associated with the presence of at least 1 NQO1*2 allele (fixed effects model: OR = 1.39, 95% CI: 0.98, 1.97; χ2 test for heterogeneity: P = 0.23). This result was also supported by that of Smith et al. (23) (not included in this meta-analysis), who showed that the NQO1*2 allele increased risk of MLL-positive leukemia (OR = 2.47, 95% CI: 1.08, 5.68).
Results of the 3 studies that examined the associations between risk of childhood ALL or AML with the MLL-AF4 translocation and the presence of the NQO1*2 allele were inconsistent (Figure 5). The combined odds ratio for these 3 studies (OR = 1.79, 95% CI: 0.49, 6.55) suggested that the presence of at least 1 NQO1*2 allele confers an increased risk of MLL-AF4-positive childhood leukemia; however, this result must be interpreted with caution as the confidence interval of the combined odds ratio is very wide and driven mainly by the results of 1 study (17).
DISCUSSION
Overall, we did not find any statistically significant association of the NQO1*2 polymorphism and the risk of childhood ALL. However, a family-based study (28) using trios (biologic parents and the affected child) found that there was excess transmission of the NQO1*2 variant allele from parents to affected offspring, suggesting that the NQO1*2 variant was associated with developing childhood ALL.
When restricting to a subset of studies with MLL-positive leukemia as the outcome, we observed an elevated risk with borderline statistical significance with the presence of at least 1 variant NQO1*2 allele (17, 27, 30, 31). Smith et al. (23) also reported that infants with de novo MLL-positive leukemias were significantly more likely to be heterozygous for the NQO1*2 allele (OR = 2.77, 95% CI: 1.17, 6.57; P = 0.02) and also significantly more likely to have low/null activity NQO1 genotypes (NQO1*1/*2 or NQO1*2/*2) than patients with de novo B-lineage ALL without MLL translocations (OR = 2.47, 95% CI: 1.08, 5.68; P = 0.03). This subgroup of infants with de novo MLL-positive leukemias were also significantly more likely to have low/null activity NQO1 genotypes (NQO1*1/*2 or NQO1*2/*2) than expected in an ethnically matched population of disease-free subjects (OR = 2.50; P = 0.02) (23). The lack of statistically significant results among the MLL-positive subgroup in our meta-analysis may be due to the rare occurrence of MLL-positive leukemias in infants, but these results are biologically plausible.
A rearrangement in the MLL gene at 11q23 is common in leukemias of both infants (<2 years) and adults (as secondary leukemias that arise after chemotherapy with topoisomerase II inhibitors), suggesting that topoisomerase II inhibition may be a common mechanism (36). Many of the compounds that are metabolized by NQO1 also inhibit topoisomerase II, an enzyme involved in maintaining DNA structure through cleavage and subsequent repair of double-stranded DNA. Inhibition of topoisomerase II activity can ultimately result in chromosomal translocations, as has been evidenced by the ability of dietary flavonoids to cleave the MLL gene (37).
However, the mechanism by which MLL breakage and subsequent translocation occur still remains to be discovered: Topoisomerase II inhibition, as well as bioactivation of reactive metabolites, and formation of reactive oxygen species have been suggested. The double-stranded DNA breaks created by topoisomerase II inhibition can stimulate apoptosis, which leads to the activation of endonucleases that cause further DNA breaks. Wild-type NQO1 has been shown to sensitize cells to undergo apoptosis by tumor necrosis factor-alpha and also to stabilize the p53 tumor suppressor protein, whereas NQO1*2 does not (38, 39). Therefore, it is also possible that the NQO1*2 variant can enable cells to survive DNA damage, thereby increasing the likelihood of survival with DNA breaks and translocations.
The overall results of the meta-analysis may be biased because of confounding and population stratification that may be present in the individual case-control studies included. However, many of the case-control studies included in this meta-analysis had relatively homogeneous ethnic/racial populations (16, 17, 27, 30–32) and/or were matched for ethnicity (29).
The largely negative results among childhood leukemias (outside the MLL group) may be a reflection of low bioactivation in precursor B-cells (i.e., low myeloperoxidase) and also low relevant exposures in the pediatric population. There may also be unrecognized gene-gene and gene-environment interactions that have yet to be elucidated. The elevated risk in the MLL group and in particular the MLL-AF4 group (17, 23) suggests that the MLL gene or the AF4 gene is particularly sensitive to reactive quinones in the bone marrow. This result was not upheld in the Japanese population (27), leaving open the suggestion that the NQO1*2 allele may be linked to a second causal allele within this gene-rich region of chromosome 16 in non-Japanese populations.
Furthermore, the differences in worldwide incidence rates of childhood leukemia and the worldwide differences in the observed ratios of ALL to AML could be due to the variation of environmental exposures among regions; for example, differences in exposure profiles to infectious agents have also been suggested (40). As suggested by Kracht et al. (30), the specific exposure histories for leukemia cases may reflect differences in exposure profiles among countries.
BIAS
The results of the meta-analysis were most suggestive of the role of low/null activity NQO1*1/*2 and NQO1*2/*2 genotypes in MLL-positive leukemia. Among the 4 studies included in this subgroup analysis, the study by Wiemels et al. (17) was the most influential. Therefore, we also reassessed the results by dropping this study and found weak evidence of association between NQO1*1/*2 and NQO1*2/*2 variant genotypes and MLL-positive infant leukemia (OR = 1.19, 95% CI: 0.80, 1.77).
The different choice of control groups (Table 1) could bias the results of the individual studies and lead to inconsistencies between studies; however, the direction and magnitude of this bias are uncertain. For example, hospital-based controls may not necessarily be representative of the underlying source population, and therefore population-based controls may be a better comparison group. Use of controls that are older than cases can introduce a bias because older subjects could have a very different exposure history and also a different response to toxic exposures (because of differences in body size) than younger subjects. Controls selected from different geographic areas than those of the cases can also introduce bias because the cases and controls could have different genetic ancestries and exposure histories. Lack of quality control measures used during genotyping could introduce misclassification bias from genotyping error. Furthermore, the genotyping calls could be prone to bias if the observer was not blinded to case-control status. Although we used random effects models to account for heterogeneity between studies, results from fixed effects models were similar.
We also evaluated bias in our results using 3 criteria (amount of evidence, replication, and protection from bias) published in the interim guidelines for the assessment of cumulative evidence on genetic associations (41). For the amount of evidence, all subgroup analyses (Figures 2–5) contained a moderate number of subjects studied with the NQO1*2 allele. Furthermore, there was no obvious bias incurred from phenotype misclassification or population stratification; however, there was insufficient information on quality control procedures for genotyping (Figures 2–5; Table 1). To assess replication of the associations, we used the I2 values that ranged from 30.9% to 83.3%. The totality of evidence for the association of the NQO1*2 allele with childhood leukemia, as suggested by these guidelines, is moderate for acute lymphoblastic and MLL-positive leukemia but weak for acute myeloid and MLL-AF4-positive leukemia.
POPULATION TESTING
Currently there is insufficient evidence to establish the role of NQO1 in childhood leukemia and, thus, population testing for polymorphisms in this gene has not been implemented.
OTHER POTENTIAL PUBLIC HEALTH APPLICATIONS
There are currently not enough data to support any public health applications.
CONCLUSIONS AND RECOMMENDATIONS FOR RESEARCH
Results of studies examining the effect of polymorphisms in NQO1 on childhood leukemia have been inconsistent, and this is the first meta-analysis to systematically summarize the association of the NQO1*2 polymorphism and the risk of childhood leukemia by immunophenotypic and cytogenetic subtype, when possible. A more homogeneous and consistent classification of the outcome would increase statistical power to detect any significant associations. Therefore, future studies should also cytogenetically classify leukemia cases. Such efforts will understandably require large numbers of patients to assess statistically meaningful cytogenetic subgroups, requiring coordinated collaboration among epidemiology study groups.
We conclude that the NQO1*2 variant allele appears to have no strong association with childhood ALL or AML, but it may be associated with MLL-positive childhood leukemias. However, these results should be interpreted with caution, as this meta-analysis included only a few studies with small sample sizes. Furthermore, the suggested increase in risk of MLL-positive childhood leukemia among the NQO1*2 allele carriers was driven mainly by the results of 1 case-control study but was also supported by 1 additional case-only study. This emphasizes the need for further replication of these findings. Because this is a rare subtype, this would be a prime candidate for a consortium-based effort.
Because the presence of a functional polymorphism in NQO1 may not confer an increased risk of childhood leukemia alone, but rather in the presence of an environmental exposure to quinones, in future studies it would also be important to incorporate both the effects of environmental agents and genotypes of other enzymes (e.g., GSTs, CYP2E1, MPO) that metabolize these carcinogenic substances to toxic quinones and reactive oxygen species (22). Such investigations will require larger sample sizes to permit assessment of first- and, possibly, second-order interactions.
Some of the heterogeneity between the risk ratios of individual studies may simply be a result of unmeasured cofactors that vary among the populations studied here; therefore, inconsistent results between studies warrant detailed exploration. Because NQO1*2 is a highly penetrant marker and metabolizes a number of putative leukemogens, it remains an important candidate risk factor for leukemia and deserves further analysis.
Our meta-analysis of case-control studies did not provide evidence for a statistically significant effect of NQO1*2 in childhood ALL, yet a family-based study (28) did demonstrate an overtransmission of NQO1*2 among ALL cases. These conflicting results underscore the need to incorporate genetic markers of ancestry for addressing the bias that may result from potential population stratification in non-family-based study designs (i.e., case-control studies), especially given the wide population differences in NQO1*2 allele frequencies (42–44). In addition, family-based designs such as case-parent trios should be encouraged (44, 45). Future studies are needed in Asians and Hispanics, populations that have a higher occurrence of the NQO1*2 polymorphism, in order to better understand its role in childhood leukemia in these populations. In addition, other SNPs in NQO1—such as the less-studied C465T functional variant (NQO1*3), other haplotype-tagging SNPs, and SNPs in neighboring genes—should be further evaluated to comprehensively assess the importance of NQO1 in the development of childhood leukemia.
Acknowledgments
Author affiliations: Division of Epidemiology, School of Public Health, University of California, Berkeley, California (Neela Guha, Anand P. Chokkalingam, Patricia A. Buffler); Department of Epidemiology and Biostatistics, University of California, San Francisco, California (Jeffrey S. Chang, Joseph L. Wiemels); and Division of Environmental Health Sciences, School of Public Health, University of California, Berkeley, California (Martyn T. Smith).
A. P. C. and P. A. B. were supported by grant R01 ES09137. J. S. C. was supported by a fellowship from the National Cancer Institute (grant R25 CA 112355). M. T. S. was supported by National Institutes of Health (NIH) Superfund grant P42ES04705. J. L. W. is a Scholar of the Leukemia and Lymphoma Society and was supported by NIH grant CA89032.
The authors thank Sébastien Antoni for assistance with formatting the figures.
Conflict of interest: none declared.
Glossary
Abbreviations
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- CI
confidence interval
- NQO1
NAD(P)H:quinone oxidoreductase 1
- OR
odds ratio
- SNP
single nucleotide polymorphism
References
- 1.Jaiswal AK, McBride OW, Adesnik M, et al. Human dioxin-inducible cytosolic NAD(P)H:menadione oxidoreductase. cDNA sequence and localization of gene to chromosome 16. J Biol Chem. 1988;263(27):13572–13578. [PubMed] [Google Scholar]
- 2.Klassen C. Toxicology: The Basic Science of Poisons. New York, NY: McGraw-Hill; 2001. [Google Scholar]
- 3.Nebert DW, Roe AL, Vandale SE, et al. NAD(P)H:quinone oxidoreductase (NQO1) polymorphism, exposure to benzene, and predisposition to disease: a HuGE Review. Genet Med. 2002;4(2):62–70. doi: 10.1097/00125817-200203000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Vasiliou V, Ross D, Nebert DW. Update of the NAD(P)H:quinone oxidoreductase (NQO) gene family. Hum Genomics. 2006;2(5):329–335. doi: 10.1186/1479-7364-2-5-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metodiewa D, Jaiswal AK, Cenas N, et al. Quercetin may act as a cytotoxic prooxidant after its metabolic activation to semiquinone and quinoidal product. Free Radic Biol Med. 1999;26(1–2):107–116. doi: 10.1016/s0891-5849(98)00167-1. [DOI] [PubMed] [Google Scholar]
- 6.Ross D, Siegel D. NAD(P)H:quinone oxidoreductase 1 (NQO1, DT-diaphorase), functions and pharmacogenetics. Methods Enzymol. 2004;382:115–144. doi: 10.1016/S0076-6879(04)82008-1. [DOI] [PubMed] [Google Scholar]
- 7.Waleh NS, Calaoagan J, Murphy BJ, et al. The redox-sensitive human antioxidant responsive element induces gene expression under low oxygen conditions. Carcinogenesis. 1998;19(8):1333–1337. doi: 10.1093/carcin/19.8.1333. [DOI] [PubMed] [Google Scholar]
- 8.Traver RD, Horikoshi T, Danenberg KD, et al. NAD(P)H:quinone oxidoreductase gene expression in human colon carcinoma cells: characterization of a mutation which modulates DT-diaphorase activity and mitomycin sensitivity. Cancer Res. 1992;52(4):797–802. [PubMed] [Google Scholar]
- 9.Gaedigk A, Tyndale RF, Jurima-Romet M, et al. NAD(P)H:quinone oxidoreductase: polymorphisms and allele frequencies in Caucasian, Chinese and Canadian Native Indian and Inuit populations. Pharmacogenetics. 1998;8(4):305–313. doi: 10.1097/00008571-199808000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Kelsey KT, Ross D, Traver RD, et al. Ethnic variation in the prevalence of a common NAD(P)H quinone oxidoreductase polymorphism and its implications for anti-cancer chemotherapy. Br J Cancer. 1997;76(7):852–854. doi: 10.1038/bjc.1997.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiyohara C, Yoshimasu K, Takayama K, et al. NQO1, MPO, and the risk of lung cancer: a HuGE Review. Genet Med. 2005;7(7):463–478. doi: 10.1097/01.gim.0000177530.55043.c1. [DOI] [PubMed] [Google Scholar]
- 12.Gasdaska PY, Fisher H, Powis G. An alternatively spliced form of NQO1 (DT-diaphorase) messenger RNA lacking the putative quinone substrate binding site is present in human normal and tumor tissues. Cancer Res. 1995;55(12):2542–2547. [PubMed] [Google Scholar]
- 13.Hu LT, Stamberg J, Pan S. The NAD(P)H:quinone oxidoreductase locus in human colon carcinoma HCT 116 cells resistant to mitomycin C. Cancer Res. 1996;56(22):5253–5259. [PubMed] [Google Scholar]
- 14.Pan SS, Forrest GL, Akman SA, et al. NAD(P)H:quinone oxidoreductase expression and mitomycin C resistance developed by human colon cancer HCT 116 cells. Cancer Res. 1995;55(2):330–335. [PubMed] [Google Scholar]
- 15.Pan SS, Han Y, Farabaugh P, et al. Implication of alternative splicing for expression of a variant NAD(P)H:quinone oxidoreductase-1 with a single nucleotide polymorphism at 465C>T. Pharmacogenetics. 2002;12(6):479–488. doi: 10.1097/00008571-200208000-00009. [DOI] [PubMed] [Google Scholar]
- 16.Krajinovic M, Sinnett H, Richer C, et al. Role of NQO1, MPO and CYP2E1 genetic polymorphisms in the susceptibility to childhood acute lymphoblastic leukemia. Int J Cancer. 2002;97(2):230–236. doi: 10.1002/ijc.1589. [DOI] [PubMed] [Google Scholar]
- 17.Wiemels JL, Pagnamenta A, Taylor GM, et al. A lack of a functional NAD(P)H:quinone oxidoreductase allele is selectively associated with pediatric leukemias that have MLL fusions. United Kingdom Childhood Cancer Study Investigators. Cancer Res. 1999;59(16):4095–4099. [PubMed] [Google Scholar]
- 18.Ries LAG, Smith MA, Gurney JG, et al. Cancer Incidence and Survival Among Children and Adolescents: United States SEER Program 1975–1995. Bethesda, MD: National Cancer Institute, SEER Program; 1999. (NIH publication no. 99-4649) [Google Scholar]
- 19.Pui CH. Childhood Leukemia. New York, NY: Cambridge University Press; 1999. [Google Scholar]
- 20.Parkin DM, Kramarova E, Draper GJ, et al. International Incidence of Childhood Cancer. Vol II. Lyon, France: International Agency for Research on Cancer; 1998. [Google Scholar]
- 21.Greaves MF, Alexander FE. An infectious etiology for common acute lymphoblastic leukemia in childhood? Leukemia. 1993;7(3):349–360. [PubMed] [Google Scholar]
- 22.Ross D. Metabolic basis of benzene toxicity. Eur J Haematol Suppl. 1996;60:111–118. doi: 10.1111/j.1600-0609.1996.tb01656.x. [DOI] [PubMed] [Google Scholar]
- 23.Smith MT, Wang Y, Skibola CF, et al. Low NAD(P)H:quinone oxidoreductase activity is associated with increased risk of leukemia with MLL translocations in infants and children. Blood. 2002;100(13):4590–4593. doi: 10.1182/blood-2001-12-0264. [DOI] [PubMed] [Google Scholar]
- 24.Larson RA, Wang Y, Banerjee M, et al. Prevalence of the inactivating 609C→T polymorphism in the NAD(P)H:quinone oxidoreductase (NQO1) gene in patients with primary and therapy-related myeloid leukemia. Blood. 1999;94(2):803–807. [PubMed] [Google Scholar]
- 25.Naoe T, Takeyama K, Yokozawa T, et al. Analysis of genetic polymorphism in NQO1, GST-M1, GST-T1, and CYP3A4 in 469 Japanese patients with therapy-related leukemia/myelodysplastic syndrome and de novo acute myeloid leukemia. Clin Cancer Res. 2000;6(10):4091–4095. [PubMed] [Google Scholar]
- 26.Rothman N, Smith MT, Hayes RB, et al. Benzene poisoning, a risk factor for hematological malignancy, is associated with the NQO1 609C→T mutation and rapid fractional excretion of chlorzoxazone. Cancer Res. 1997;57(14):2839–2842. [PubMed] [Google Scholar]
- 27.Eguchi-Ishimae M, Eguchi M, Ishii E, et al. The association of a distinctive allele of NAD(P)H:quinone oxidoreductase with pediatric acute lymphoblastic leukemias with MLL fusion genes in Japan. Haematologica. 2005;90(11):1511–1515. [PubMed] [Google Scholar]
- 28.Infante-Rivard C, Vermunt JK, Weinberg CR. Excess transmission of the NAD(P)H:quinone oxidoreductase 1 (NQO1) C609T polymorphism in families of children with acute lymphoblastic leukemia. Am J Epidemiol. 2007;165(11):1248–1254. doi: 10.1093/aje/kwm022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clavel J, Bellec S, Rebouissou S, et al. Childhood leukaemia, polymorphisms of metabolism enzyme genes, and interactions with maternal tobacco, coffee and alcohol consumption during pregnancy. Eur J Cancer Prev. 2005;14(6):531–540. doi: 10.1097/00008469-200512000-00007. [DOI] [PubMed] [Google Scholar]
- 30.Kracht T, Schrappe M, Strehl S, et al. NQO1 C609T polymorphism in distinct entities of pediatric hematologic neoplasms. Haematologica. 2004;89(12):1492–1497. [PubMed] [Google Scholar]
- 31.Lanciotti M, Dufour C, Corral L, et al. Genetic polymorphism of NAD(P)H:quinone oxidoreductase is associated with an increased risk of infant acute lymphoblastic leukemia without MLL gene rearrangements. Leukemia. 2005;19(2):214–216. doi: 10.1038/sj.leu.2403613. [DOI] [PubMed] [Google Scholar]
- 32.Sirma S, Agaoglu L, Yildiz I, et al. NAD(P)H:quinone oxidoreductase 1 null genotype is not associated with pediatric de novo acute leukemia. Pediatr Blood Cancer. 2004;43(5):568–570. doi: 10.1002/pbc.20098. [DOI] [PubMed] [Google Scholar]
- 33.Lewis S, Clarke M. Forest plots: trying to see the wood and the trees. BMJ. 2001;322(7300):1479–1480. doi: 10.1136/bmj.322.7300.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21(11):1539–1558. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 35.Egger M, Davey Smith G, Schneider M, et al. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629–634. doi: 10.1136/bmj.315.7109.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross JA, Potter JD, Robison LL. Infant leukemia, topoisomerase II inhibitors, and the MLL gene. J Natl Cancer Inst. 1994;86(22):1678–1680. doi: 10.1093/jnci/86.22.1678. [DOI] [PubMed] [Google Scholar]
- 37.Strick R, Strissel PL, Borgers S, et al. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc Natl Acad Sci U S A. 2000;97(9):4790–4795. doi: 10.1073/pnas.070061297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asher G, Lotem J, Kama R, et al. NQO1 stabilizes p53 through a distinct pathway. Proc Natl Acad Sci U S A. 2002;99(5):3099–3104. doi: 10.1073/pnas.052706799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siemankowski LM, Morreale J, Butts BD, et al. Increased tumor necrosis factor-alpha sensitivity of MCF-7 cells transfected with NAD(P)H:quinone reductase. Cancer Res. 2000;60(13):3638–3644. [PubMed] [Google Scholar]
- 40.Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006;6(3):193–203. doi: 10.1038/nrc1816. [DOI] [PubMed] [Google Scholar]
- 41.Ioannidis JP, Boffetta P, Little J, et al. Assessment of cumulative evidence on genetic associations: interim guidelines. Int J Epidemiol. 2008;37(1):120–132. doi: 10.1093/ije/dym159. [DOI] [PubMed] [Google Scholar]
- 42.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55(4):997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 43.Pritchard JK, Stephens M, Rosenberg NA, et al. Association mapping in structured populations. Am J Hum Genet. 2000;67(1):170–181. doi: 10.1086/302959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas DC, Witte JS. Point: population stratification: a problem for case-control studies of candidate-gene associations? Cancer Epidemiol Biomarkers Prev. 2002;11(6):505–512. [PubMed] [Google Scholar]
- 45.Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM) Am J Hum Genet. 1993;52(3):506–516. [PMC free article] [PubMed] [Google Scholar]