

Abstract
Background. Clinical studies suggest that statins reduce proteinuria and slow the decline in kidney function in chronic kidney disease. Given a rich literature identifying podocyte apoptosis as an early step in the pathophysiological progression to proteinuria and glomerulosclerosis, we hypothesized that rosuvastatin protects podocytes from undergoing apoptosis. Regarding a potential mechanism, our lab has shown that the cell cycle protein, p21, has a prosurvial role in podocytes and there is literature showing statins upregulate p21 in other renal cells. Therefore, we queried whether rosuvastatin is prosurvival in podocytes through a p21-dependent pathway.
Methods. Two independent apoptotic triggers, puromycin aminonucleoside (PA) and adriamycin (ADR), were used to induce apoptosis in p21 +/+ and p21 −/− conditionally immortalized mouse podocytes with or without pre-exposure to rosuvastatin. Apoptosis was measured by two methods: Hoechst 33342 staining and fluorescence-activated cell sorting (FACS). To establish a role for p21, p21 levels were measured by western blotting following rosuvastatin exposure and p21 was stably transduced into p21 −/− mouse podocytes.
Results. Rosuvastatin protects against ADR- and PA-induced apoptosis in podocytes. Further, exposure to rosuvastatin increases p21 levels in podocytes in vitro. ADR induces apoptosis in p21 −/− mouse podocytes, but rosuvastatin's protective effect is not seen in the absence of p21. Reconstituting p21 in p21 −/− podocytes restores rosuvastatin's prosurvival effect.
Conclusion. Rosuvastatin is prosurvival in injured podocytes. Rosuvastatin exerts its protective effect through a p21-dependent antiapoptotic pathway. These findings suggest that statins decrease proteinuria by protecting against podocyte apoptosis and subsequent podocyte depopulation.
Keywords: podocyte, apoptosis, p21, statins
Introduction
Podocytes, or glomerular visceral epithelial cells, are highly specialized, terminally differentiated cells with limited replicative capacity. A large body of literature shows that two potential fates for injured podocytes include apoptosis and detachment from the underlying glomerular basement membrane (GBM) [1]. Each of these deleterious pathways decreases podocyte number. Decrement in podocyte number is a critical pathophysiological precursor to the development of proteinuria and glomerulosclerosis, hallmarks of diabetic and non-diabetic kidney disease [2–7].
In recent decades, research has focused on identifying potential therapies that reduce podocytes’ detrimental responses to injury. Statins, or 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, show promise in the clinical realm. Post hoc analyses of large randomized, controlled trials, as well as meta-analyses of small prospective studies suggest a role for statins in decreasing the rate of decline of kidney function and reducing proteinuria in patients with chronic kidney disease stages I–III [8–14]. These effects are independent of lipid lowering, implicating pleiotropic effects of statins on specific kidney cells [15]. We hypothesize that statins protect podocytes from undergoing apoptosis, which in turn prevents podocyte depopulation and subsequent proteinuria. To elucidate a potential mechanism, we focused on the cell cycle regulator, p21, given our experience with p21 as a prosurvival player in podocyte apoptosis and data in other kidney cells showing statins upregulate p21.
Materials and methods
Conditionally immortalized heat-sensitive mouse podocytes in culture
Female p21 Waf1/Cip1−/− mice were crossed with male H-2Kb -tsA58 transgenic mice (ImmortoMouse; Jackson Laboratory, Bar Harbor, ME, USA) and the F1 generation intercrossed. Conditionally immortalized mouse podocytes were then derived from p21 Waf1/Cip1 −/− and p21 Waf1/Cip1 +/+ littermates as described previously [16–19]. Briefly, a thermosensitive SV40 large T-cell antigen (tsA58 Tag) is controlled by an interferon-γ-inducible H-2Kb promoter. Cells proliferate when grown at 33°C on collagen I- (BD Biosciences, Bedford, MA, USA) coated plastic plates (Primaria tissue culture dish, 100 × 20 mm; Becton Dickinson Labware, Franklin Lakes, NJ, USA) in the presence of 10 U/ml recombinant mouse interferon-γ (Coulter, Hialeah, FL, USA). The quiescent, differentiated or ‘growth-restricted’ phenotype is achieved by transitioning cells to uncoated plastic plates at 37°C without interferon-γ for 12–14 days, at which time cells express podocyte-specific immunohistologic markers, as described previously [20].
Under both conditions, cells were grown in RPMI 1640 media (Life Technologies, Inc., Gaithersburg, MD, USA) to which the following was added: 9% fetal bovine serum (Summit Biotechnology, Fort Collins, CO, USA), 2 mmol/L glutamine, 10 mmol/L HEPES, 0.075% sodium bicarbonate (all from Sigma, St. Louis, MO, USA) and 1 mmol/L sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin (all from Irvine Scientific, Santa Ana, CA, USA).
In all studies, early passage (12–22) conditionally immortalized heat-sensitive mouse podocytes were utilized. Two independent wildtype podocyte clones were used.
Measuring cell viability with different concentrations of rosuvastatin
To ensure that statins were not toxic to podocytes, podocyte viability was measured after 24, 48 and 72 h of exposure to a range of rosuvastatin concentrations: 1 μM, 20 μM and 100 μM. A viable cell number was assessed by methylthiazoletetrazolium (MTT) assay using a CellTiter 96 non-radioactive cell proliferation assay kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. This assay identifies living cells by measuring the cellular conversion of a tetrazolium salt into a chromofore that is quantified by absorbance at 560 nm.
Retroviral transduction of p21 −/− podocytes
Reconstitution of p21 protein expression in p21 −/− podocytes was achieved using a retroviral transduction system. The pBabe vectors encoding full-length human p21 +/+ or GFP were transfected into Phoenix packaging cells to generate retrovirus. The retrovirus-containing media were harvested and filtered onto 50% confluent proliferating, undifferentiated p21 −/− podocytes. Transfected cells were identified by puromycin selection or fluorescence-activated cell sorting (FACS). Transfected podocytes were further subcultured, passaged and transferred to 37°C to initiate growth arrest and differentiation.
Induction of apoptosis in vitro
Podocyte apoptosis was induced by exposing cells to either adriamycin (ADR) 0.25 μg/ml or puromycin aminonucleoside (PA) 30 μg/ml for 48 h. Cells were pre-incubated with rosuvastatin 20 μM or DMSO (vehicle) for 24 h prior to application of the apoptotic trigger. Each experiment was performed a minimum of three times.
Detection of apoptosis
Percentage of podocyte apoptosis was measured and quantitated by two independent methods
Hoechst staining
Podocytes were grown to 80% confluency on plastic plates (Primaria tissue culture dish, 60 × 15 mm; Becton Dickinson Labware, Franklin Lakes, NJ, USA) under growth-restricted conditions and treated as described above. Hoechst 33342 (Sigma-Aldrich) was added to achieve a final concentration of 10 μM and cells analyzed after 5 min. Apoptotic cells, defined by the presence of chromatin condensation, were counted in a minimum of 20 high-powered fields per experimental condition. The number of apoptotic nuclei was expressed as a percentage of the total.
Fluorescence-activated cell sorting (FACS)
The cells were grown to 90% confluency on plastic plates (Primaria tissue culture dish, 100 × 20 mm; BD Labware) under growth-restricted conditions. The media from each experiment were centrifuged at 300 × g for 5 min and the pellets saved. The cells were washed with Hank's buffered salt solution (HBSS, Invitrogen, Carlsbad, CA, USA) three times. Adherent mouse podocytes were then incubated with 5 ml versene with 0.05% collagenase for 10 min at 37°C in 95% air/5% CO2 followed by mechanical disruption. Detached podocytes were added to the saved pellet and centrifuged for 5 min at 300 × g. After two wash cycles with PBS, the pellet was resuspended in 1:10 binding buffer (BD Pharmingens, Franklin Lakes, NJ, USA) in 12 × 75 polystyrene falcon tubes (BD) to which 5 μl of Annexin V-FITC or Annexin V-PE and 5 μl propidium iodide (PI) or 7-aminoactinomycin D (7-AAD) (all from BD Pharmingens) were added. The cells were then analyzed for apoptosis using FACScalibur. Cells positive for Annexin V-FITC (Annexin V-PE) and negative for PI (AAD) were deemed apoptotic.
Western blot analysis
Tissue culture plates were placed on ice, and the cells were washed three times in ice-cold PBS. Podocytes were lysed using an RIPA lysis buffer [50 mM Tris–HCl, pH 8.0, 5 mM EDTA, 150 mM NaCl, 1% IP-40, 1% Trion X-100, 50 mM NaF, 1 mM Na-orthovanadate (all from Sigma-Aldrich)] and a 40 mM protease inhibitor (Roche Molecular Biochemicals, Indianapolis, IN, USA). After an overnight freeze-thaw cycle, lysates were cleared by centrifugation at 14 000 rpm for 5 min at 4°C. Protein concentrations were determined by a BCA protein assay kit (Pierce, Rockford, IL, USA) according to the manufacturer's protocol. Reducing buffer was added to each protein extract and each sample boiled for 5 min. Reduced protein sample of 10– 20 μg was loaded per lane on a 12% SDS–polyacrylamide gel and subsequently transferred by electroblotting to a PVDF membrane (Sigma-Aldrich) at 350 mA for 75 min.
After blocking the membrane in 5% dried milk in a TBST solution (10 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.1% Tween) for 1 h at room temperature, the blot was incubated overnight at 4°C with anti-p21WAF1/CIP1 +/+ (BD Biosciences, San Diego, CA, USA) and anti-actin (Chemicon International, Temecula, CA, USA) antibodies. After washing three times in TBST, membranes were incubated with the appropriate horseradish peroxidase secondary antibody (Sigma-Aldrich) for 1 h. After additional washings in TBST, proteins were visualized using enhanced chemiluminescence technology according to the manufacturer's instructions (GE Healthcare, Piscataway, NJ, USA).
Statistical analysis
Unless otherwise noted, all experiments were performed a minimum of three times. All results are expressed as mean ± SD. For statistical comparisons of two groups, unpaired t-tests were applied. One-way ANOVA was used to determine the significance of differences among multiple experimental groups. A P-value of <0.05 was considered to be significant.
Results
Rosuvastatin protects against PA-induced podocyte apoptosis
Clinical studies show that statins may reduce proteinuria and slow the decline in kidney function in chronic kidney disease. Given an abundance of literature showing that podocyte apoptosis is an early step in the pathophysiological progression to proteinuria and glomerulosclerosis, we hypothesized that rosuvastatin protects podocytes from undergoing apoptosis. We examined two independent podocyte apoptotic triggers: PA 30 μg/ml and ADR 0.25 μg/ml.
To determine an appropriate concentration of rosuvastatin, dose titration studies were performed. Growth-restricted conditionally immortalized p21 +/+ mouse podocytes were incubated in media containing escalating doses of rosuvastatin. Cell viability was measured by MTT assay at 24, 48 and 72 h (Figure 1). Rosuvastatin 100 μM proved toxic to podocytes compared to DMSO (vehicle) as evidenced by decreased cell viability at all timepoints. However, lower rosuvastatin doses of 1 μM and 20 μM were well tolerated with preserved cell number relative to control and DMSO. Consistent with in vitro studies reported elsewhere in the literature and reports of systemic, therapeutic concentrations in humans, a working concentration of 20 μM was chosen for all subsequent studies [21,22].
Fig. 1.

Low-dose rosuvastatin does not affect podocyte viability in vitro. Growth-restricted conditionally immortalized mouse podocytes were incubated with escalating doses of rosuvastatin or DMSO as a vehicle control. Cell viability was determined by MTT assay at 24, 48 and 72 h time points (*P < 0.05).
To determine whether rosuvastatin protects podocytes from apoptosis, podocytes were exposed to rosuvastatin 20 μM for 24 h prior to administering PA. The percentage of apoptosis was measured at 48 h by Hoechst staining (Figure 2A). As shown in Figure 2B, exposure of growth-restricted p21 +/+ podocytes to PA resulted in an 11-fold increase in apoptosis compared to cells grown under normal conditions (P < 0.0001). In contrast, pre-exposure to rosuvastatin markedly attenuated PA-induced apoptosis, resulting in a 3.5-fold reduction in apoptosis relative to PA-only treated cells (P < 0.0001). There were no statistically significant differences in apoptosis among control podocytes and those exposed to vehicle (DMSO) or rosuvastatin alone.
Fig. 2.
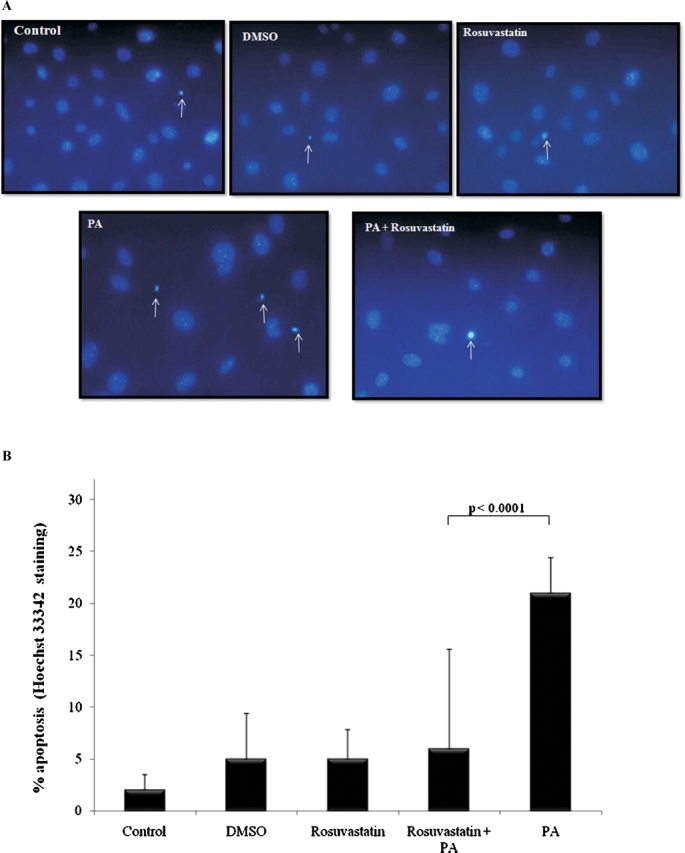
Rosuvastatin protects podocytes from puromycin aminonucleoside-induced apoptosis. Growth-restricted mouse podocytes pre-incubated with rosuvastatin (20 μM) or DMSO (vehicle) for 24 h were exposed to puromycin aminonucleoside (PA, 30 μg/ml) for 48 h. Hoechst staining was performed (A) and the percentage of apoptosis was measured (B).
Rosuvastatin protects against ADR-induced podocyte apoptosis
To determine whether the prosurvival benefits of rosuvastatin extend to other forms of podocyte injury, ADR was used as a second independent method of inducing apoptosis. Representative images of apoptosis measured by Hoechst staining are shown in Figure 3A. Figure 3B shows exposure of conditionally immortalized mouse podocytes to ADR 0.25 μg/ml resulted in an 8-fold increase in apoptosis at 48 h compared to control cells (P < 0.0001). Similar to the results with PA, pre-incubation with rosuvastatin conferred a dramatic survival advantage with a 54% decrease in apoptosis compared to cells exposed to ADR only (P < 0.005).
Fig. 3.
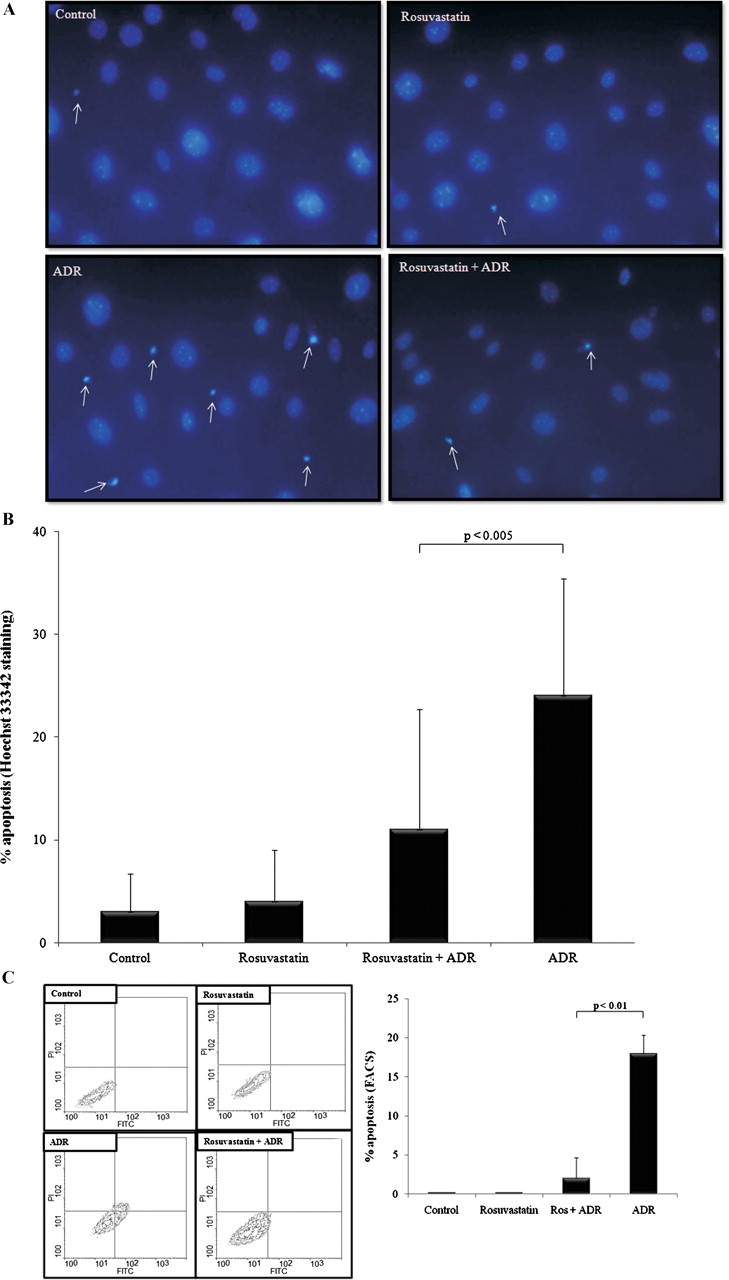
Rosuvastatin protects podocytes from adriamycin-induced apoptosis. Podocytes pre-incubated for 24 h with rosuvastatin or DMSO were exposed to adriamycin (ADR, 0.25 μg/ml) for 48 h. Hoechst staining was performed (A) and the percentage of apoptosis was measured (B). Additionally, cells were dual-labeled with FITC-Annexin V and propidium iodide and fluorescent-activated cell sorting (FACS) performed. Representative contour plots (C) show virtually all of the cells in control and rosuvastatin-treated groups were viable, as detected in the lower-left quadrant (FITC−, PI−). ADR exposure significantly increased the percentage of apoptotic (FITC+, PI−) cells, as detected in the lower-right quadrant. Pre-incubation with rosuvastatin largely abrogated the rightward shift induced by ADR, consistent with a prosurvival effect. The results of three separate FACS experiments were pooled and averaged results are shown graphically.
In order to validate the results obtained by Hoechst staining, FACS was performed as a second independent method to measure apoptosis. The advantages of FACS over measuring Hoechst-stained apoptotic cells using fluorescence microscopy are many-fold. First, while fluorescence microscopy is a subjective morphologic assessment of a sample of cells, the flow cytometer analyzes each cell individually giving information about the heterogeneity of a sample of cells. Further, FACS is rapid and precise, allowing for the evaluation of thousands of cells in seconds. Finally, cells are quantified into subpopulations of interest based on their fluorescent intensity and specific wavelength.
In this study, wildtype, p21 −/− and p21-transduced p21 −/− podocytes were stained with Annexin V-FITC and propidium iodide (PI) to distinguish among viable, apoptotic and necrotic podocytes. Because GFP is a fluorochrome, p21 −/− podocytes transduced with GFP were stained with Annexin V-PE and 7-aminoactinomycin (7-AAD). Apoptotic cells undergo a perturbation of their plasma membranes that exposes a cell surface marker, phosphatidylserine, to which Annexin V-FITC and Annexin V-PE bind. Necrotic cells bind both PI (7-AAD) and Annexin V-FITC (Annexin V-PE), whereas viable cells bind neither.
Figure 3C shows representative contour plots of wildtype podocytes exposed to ADR with or without pre-exposure to rosuvastatin. Apoptotic cells binding Annexin V-FITC, but not PI, localize to the right lower quadrant. Also shown in Figure 3C are pooled and averaged percentages of apoptosis from three separate experiments of FACS in p21 +/+ podocytes. While apoptosis was negligible in control cells and rosuvastatin-exposed podocytes, ADR induced a dramatic increase in apoptosis relative to control (P < 0.01). Pre-incubation with rosuvastatin imparted a survival benefit producing an 89% reduction in the percentage of apoptosis when compared to podocytes exposed to ADR only (P < 0.01). Taken together, these data convincingly demonstrate that rosuvastatin is prosurvival for podocytes in vitro.
Rosuvastatin increases p21 levels in podocytes in vitro
Given recent reports that statins may regulate p21 in other cells, we hypothesized that a mechanism by which rosuvastatin protects podocytes from apoptosis is through a p21-dependent pathway. To determine the relationship between rosuvastatin and p21 in podocytes in vitro, Western blotting was performed on extracts from cultured podocytes (Figure 4A). At 24 h after rosuvastatin exposure, protein expression of p21 Waf1/Cip1 +/+ was increased 2.4-fold over control (Figure 4B). This increase persisted at 48 h (data not shown). This finding of p21 upregulation after rosuvastatin exposure prompted further investigation of p21's role in rosuvastatin's antiapoptotic effect on podocytes in vitro.
Fig. 4.

Rosuvastatin increases protein levels of p21 in podocytes in vitro. Growth-restricted mouse podocytes were pre-incubated for 24 h with rosuvastatin or DMSO and protein levels of p21WAF1/CIP1 +/+ measured by Western blot analysis. A representative blot is shown (A). β-actin was used as a housekeeping protein to ensure equal protein loading and densitometric analysis performed (B). Values are averages of three separate experiments and normalized for differences in protein loading.
Rosuvastatin's protective effect is negated in the absence of p21
Previous work from our lab has shown a prosurvival role for p21 in safeguarding against podocyte apoptosis. Since exposing podocytes to rosuvastatin led to increased p21 protein levels, we hypothesized that p21 is necessary for rosuvastatin to exert its prosurvival effect. Accordingly, p21 −/− podocytes were exposed to ADR for 48 h and Hoechst staining was performed. As shown in Figure 5A, ADR increased apoptosis in p21 −/− podocytes by 23-fold relative to control (P < 0.0001). However, in stark contrast to p21 +/+ podocytes, in the absence of p21, rosuvastatin did not protect against ADR-induced apoptosis in p21 −/− podocytes (NS). Similarly, when FACS was used to measure apoptosis in p21 −/− podocytes (Figure 5B), ADR exposure resulted in significant apoptosis compared to control (P < 0.05) that was not mitigated by rosuvastatin pre-incubation (NS). Taken together, these results suggest that rosuvastatin protects podocytes from ADR-induced apoptosis in part through a p21-dependent pathway.
Fig. 5.

The prosurvival effect of rosuvastatin is negated in the absence of p21. p21 −/− growth-restricted conditionally immortalized mouse podocytes were pre-incubated with rosuvastatin or DMSO. Cells were exposed to ADR and apoptosis measured 48 h later by Hoechst staining (A) and FACS analysis (B).
Reconstituting p21 in p21 −/− podocytes restores rosuvastatin's protective effects
To confirm that the loss of a prosurvival effect of rosuvastatin observed in p21 −/− podocytes is indeed due to a deficiency in p21, p21 was stably reintroduced into p21 −/− podocytes using a retroviral transduction system, as confirmed by Western blot analysis (Figure 6A). As expected, p21 is not present in p21 −/− podocytes and p21 −/− podocytes transfected with vector (GFP) alone, while it is present in wildtype and p21-reconstituted p21 −/− podocytes.
Fig. 6.
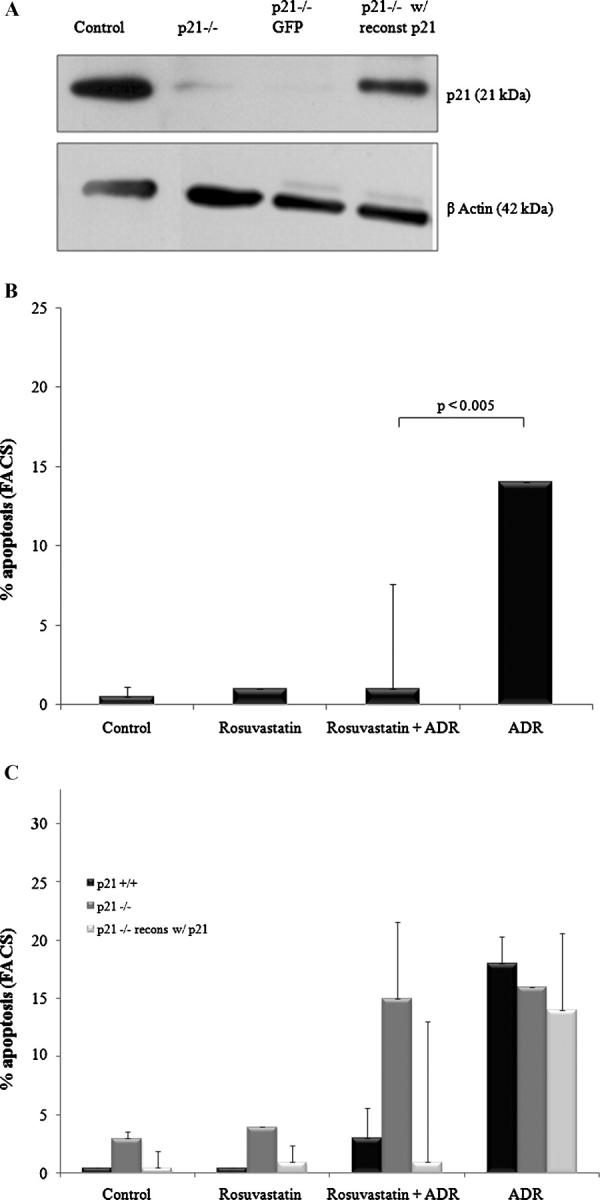
Reconstituting p21 expression in p21 −/− podocytes restores the prosurvival effect of rosuvastatin. Growth-restricted p21 −/− mouse podocytes were stably transfected with a human p21 vector or GFP vector as a control. Western blotting for p21WAF1/CIP1 +/+ showed successful re-expression of p21 in the p21 −/− podocytes (A). Following stable transfection, cells were pre-incubated with rosuvastatin or DMSO and ADR-induced apoptosis was measured by FACS analysis (B). The prosurvival effects of rosuvastatin in podocytes as a function of p21 status are summarized in (C).
The p21 −/− podocytes reconstituted with p21 were exposed to ADR and apoptosis measured by FACS as outlined previously. As shown in Figure 6B, exposure to ADR alone induced a 28-fold increase in the percentage of apoptosis relative to control (P < 0.01). However, reconstitution of p21 completely reversed the apoptotic susceptibility in those podocytes pre-incubated with rosuvastatin (P < 0.005). By contrast, rosuvastatin did not alter the percentage of apoptosis in p21 −/− podocytes transduced with vector, GFP, alone (data not shown).
Figure 6C summarizes data from the three podocyte lines that show convincingly that rosuvastatin protects podocytes from ADR-induced apoptosis through a p21 prosurvival pathway. As depicted, wildtype, p21 −/− and p21 −/− with p21 reconstituted podocytes were similarly susceptible to ADR. However, rosuvastatin protected against ADR-induced apoptosis only in the presence of p21. There were similar percentages of apoptosis among p21 −/− podocytes exposed to ADR with or without pre-exposure to rosuvastatin. These data confirm that rosuvastatin's prosurvival role in podocyte apoptosis is p21-dependent.
Discussion
Podocytes, terminally differentiated resident cells of the glomerulus, are vital for maintaining the structural integrity of the glomerular filtration barrier. It is well established that podocyte damage and subsequent decrement in podocyte number are critical precursors to the clinical manifestation of proteinuria and histological development of glomerulosclerosis. Emerging clinical and experimental data show that apoptosis, or programmed cell death, is an important cause of podocyte depopulation. Accordingly, therapies targeting prosurvival pathways may stabilize podocyte number and delay progression of chronic glomerular disease.
In the last two decades, researchers have directed much attention to HMG-CoA reductase inhibitors, or statins, as potential therapies for treating kidney disease. Studies by Hommel and Nielsen et al. [23,24] first identified a potential role for statins in modulating the progression of renal disease. More recently, retrospective and prospective studies, as well as, meta-analyses of small clinical trials suggest that statins may be renoprotective by reducing proteinuria and slowing the decline in renal function in early stages of chronic kidney disease [8,9,11–14]. The benefits appear to be independent of lipid-lowering, raising the possibility of localized tissue activity in the kidney [10,15]. While statins have been shown to have anti-proliferative effects and to suppress pro-inflammatory pathways in mesangioproliferative disorders [25,26], their effects in podocyte disease remain largely unexplored.
Accordingly, in the current study, we test the hypothesis that statins are prosurvival in podocytes and thus may safeguard against the subsequent development of glomerulosclerosis by preserving overall podocyte number. Conditionally immortalized mouse podocytes were exposed to puromycin aminonucleoside (PA), or alternatively, ADR as a second independent apoptotic trigger. The first major finding of this study is that pre-incubation with rosuvastatin significantly reduced podocyte apoptosis induced by either agent, suggesting that statins may have a generalized prosurvival effect on podocytes. These findings are in stark contrast to other non-renal cell types where statins have been shown to be pro-apoptotic [27–30]. Given the evidence from our lab and others supporting a role for the cell cycle protein p21 in modulating prosurvival pathways, we next queried whether rosuvastatin may be acting in a p21-dependent fashion. Following exposure to rosuvastatin, p21 protein levels in podocytes were significantly increased, consistent with the effects of statins on p21 in mesangial cells [25]. Using a conditionally immortalized mouse podocyte line lacking p21, the second major finding in this study was that rosuvastatin's protective effect was completely negated in the absence of p21. Furthermore, when p21 was stably re-expressed in p21 −/− podocytes, the prosurvival effects of rosuvastatin were restored. Taken together, these findings suggest that rosuvastatin safeguards against podocyte apoptosis in a p21-dependent fashion.
A growing body of evidence supports the notion that statins act directly on podocytes. Studies by Nakamura and colleagues of patients with chronic glomerulonephritis demonstrated that cerivastatin therapy ameliorated proteinuria and reduced podocyte loss in the urine [31]. Camussi's group provided clues to the anti-proteinuric effects of statins by showing that statins act through the AKT pathway to prevent down-regulation of nephrin and preserve the integrity of a permselective filtration barrier following exposure to oxidized LDL [32]. Furthermore, they reported that statin therapy protected against podocyte apoptosis consistent with our findings in the current study. More recent studies show that statins maintain the integrity of the slit diaphragm and prevents foot process effacement both in vivo [33–35] and in vitro [33,36].
How are the prosurvival effects of statins on podocytes regulated through the p21 pathway? There is a complex role for p21 in regulating podocyte biology in both states of health and disease. While best known as a cyclin-dependent kinase (cdk) inhibitor, emerging data show that p21 plays a wide variety of physiological roles and importantly, is a critical player in cell survival. Interestingly, p21 seems to vacillate between prosurvival and proapoptotic phenotypes. This bipolarity with respect to cell viability is rooted in a number of only recently recognized factors including relative protein levels, subcellular localization and protein–protein interactions [37–40]. It is fairly well established that p21 plays a vital prosurvival role when localized to the cytosol. Cytoplasmic p21 has been shown to bind to and prevent activation of procaspase 3, thus interfering with Fas-mediated apoptosis [41]. Also, in the cytosol, p21 has been shown to inhibit the pro-apoptotic kinase ASK1 [42]. Taken together, p21 is a protein with multiple, oft-diverging functions. Based on available data and our findings in this study, we can conjecture that rosuvastatin facilitates a prosurvival role for p21 in injured podocytes. Among the plausible mechanisms, rosuvastatin may influence the localization, phosphorylation or even stability of this pleiotropic protein.
In summary, we have reported that rosuvastatin is prosurvival in cultured podocytes by safeguarding against apoptosis in a p21-dependent fashion. These findings provide insights into the possible mechanisms by which statin therapy may preserve overall podocyte number, thereby slowing glomerulosclerosis characteristic of many forms of chronic kidney disease.
Acknowledgments
The Authors wish to thank AstraZeneca for providing rosuvastatin compound for use in these studies and Dr Brian Kestenbaum for assistance with statistical analyses. This work was supported by National Institute of Health grants to SJS (DK60525, DK56799, DK51096), and by funding from the American Diabetes Association.
Conflict of interest statement. S.J.S. is an established investigator of the American Heart Association. R.V.D. is a recipient of a NIDDK K08 award and Investigator-Initiated Sponsord Award from AstraZeneca. The authors are solely responsible for formulating the underlying hypotheses, experimental design, and data interpretation. The results presented in this manuscript represent our original research and have not been published previously in whole or part, except in abstract format.
References
- 1.Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol. 2002;13:3005–3015. doi: 10.1097/01.asn.0000039661.06947.fd. [DOI] [PubMed] [Google Scholar]
- 2.Kriz W, Elger M, Nagata M, et al. The role of podocytes in the development of glomerular sclerosis. Kidney Int Suppl. 1994;45:S64–S72. [PubMed] [Google Scholar]
- 3.Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int. 1998;54:687–697. doi: 10.1046/j.1523-1755.1998.00044.x. [DOI] [PubMed] [Google Scholar]
- 4.Kriz W, Hosser H, Hahnel B, et al. From segmental glomerulosclerosis to total nephron degeneration and interstitial fibrosis: a histopathological study in rat models and human glomerulopathies. Nephrol Dial Transplant. 1998;13:2781–2798. doi: 10.1093/ndt/13.11.2781. [DOI] [PubMed] [Google Scholar]
- 5.Pagtalunan ME, Miller PL, Jumping-Eagle S, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 7.Susztak K, Raff A, Schiffer M, et al. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 8.Athyros VG, Mikhailidis DP, Papageorgiou AA, et al. The effect of statins versus untreated dyslipidaemia on renal function in patients with coronary heart disease. A subgroup analysis of the Greek atorvastatin and coronary heart disease evaluation (GREACE) study. J Clin Pathol. 2004;57:728–734. doi: 10.1136/jcp.2003.012989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bianchi S, Bigazzi R, Caiazza A, et al. A controlled, prospective study of the effects of atorvastatin on proteinuria and progression of kidney disease. Am J Kidney Dis. 2003;41:565–570. doi: 10.1053/ajkd.2003.50140. [DOI] [PubMed] [Google Scholar]
- 10.Campese VM, Nadim MK, Epstein M. Are 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors renoprotective? J Am Soc Nephrol. 2005;16(Suppl 1):S11. doi: 10.1681/asn.2004110958. [DOI] [PubMed] [Google Scholar]
- 11.Douglas K, O’Malley PG, Jackson JL. Meta-analysis: the effect of statins on albuminuria. Ann Intern Med. 2006;145:117–24. doi: 10.7326/0003-4819-145-2-200607180-00009. [DOI] [PubMed] [Google Scholar]
- 12.Fried LF, Orchard TJ, Kasiske BL. Effect of lipid reduction on the progression of renal disease: a meta-analysis. Kidney Int. 2001;59:260–269. doi: 10.1046/j.1523-1755.2001.00487.x. [DOI] [PubMed] [Google Scholar]
- 13.Sandhu S, Wiebe N, Fried LF, et al. Statins for improving renal outcomes: a meta-analysis. J Am Soc Nephrol. 2006;17:2006–2016. doi: 10.1681/ASN.2006010012. [DOI] [PubMed] [Google Scholar]
- 14.Tonelli M, Moye L, Sacks FM, et al. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol. 2003;14:1605–1613. doi: 10.1097/01.asn.0000068461.45784.2f. [DOI] [PubMed] [Google Scholar]
- 15.Oda H, Keane WF. Recent advances in statins and the kidney. Kidney Int Suppl. 1999;71:S2–S5. doi: 10.1046/j.1523-1755.1999.07101.x. [DOI] [PubMed] [Google Scholar]
- 16.Jat PS, Noble MD, Ataliotis P, et al. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci USA. 1991;88:5096–5100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mundel P, Reiser J, Kriz W. Induction of differentiation in cultured rat and human podocytes. J Am Soc Nephrol. 1997;8:697–705. doi: 10.1681/ASN.V85697. [DOI] [PubMed] [Google Scholar]
- 18.Mundel P, Reiser J, Zuniga Mejia Borja A, et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–258. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 19.Shankland SJ, Pippin JW, Reiser J, et al. Podocytes in culture: past, present, and future. Kidney Int. 2007;72:26–36. doi: 10.1038/sj.ki.5002291. [DOI] [PubMed] [Google Scholar]
- 20.Wada T, Pippin JW, Terada Y, et al. The cyclin-dependent kinase inhibitor p21 is required for TGF-beta1-induced podocyte apoptosis. Kidney Int. 2005;68:1618–1629. doi: 10.1111/j.1523-1755.2005.00574.x. [DOI] [PubMed] [Google Scholar]
- 21.Martin PD, Dane AL, Nwose OM, et al. No effect of age or gender on the pharmacokinetics of rosuvastatin: a new HMG-CoA reductase inhibitor. J Clin Pharmacol. 2002;42:1116–1121. doi: 10.1177/009127002401382722. [DOI] [PubMed] [Google Scholar]
- 22.Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19:117–125. doi: 10.1111/j.1472-8206.2004.00299.x. [DOI] [PubMed] [Google Scholar]
- 23.Hommel E, Andersen P, Gall MA, et al. Plasma lipoproteins and renal function during simvastatin treatment in diabetic nephropathy. Diabetologia. 1992;35:447–451. doi: 10.1007/BF02342442. [DOI] [PubMed] [Google Scholar]
- 24.Nielsen S, Schmitz O, Moller N, et al. Renal function and insulin sensitivity during simvastatin treatment in type 2 (non-insulin-dependent) diabetic patients with microalbuminuria. Diabetologia. 1993;36:1079–1086. doi: 10.1007/BF02374502. [DOI] [PubMed] [Google Scholar]
- 25.Danesh FR, Sadeghi MM, Amro N, et al. 3-Hydroxy-3-methylglutaryl CoA reductase inhibitors prevent high glucose-induced proliferation of mesangial cells via modulation of Rho GTPase/ p21 signaling pathway: implications for diabetic nephropathy. Proc Natl Acad Sci USA. 2002;99:8301–8305. doi: 10.1073/pnas.122228799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SI, Han DC, Lee HB. Lovastatin inhibits transforming growth factor-beta1 expression in diabetic rat glomeruli and cultured rat mesangial cells. J Am Soc Nephrol. 2000;11:80–87. doi: 10.1681/ASN.V11180. [DOI] [PubMed] [Google Scholar]
- 27.Kaneko R, Tsuji N, Asanuma K, et al. Survivin down-regulation plays a crucial role in 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor-induced apoptosis in cancer. J Biol Chem. 2007;282:19273–19281. doi: 10.1074/jbc.M610350200. [DOI] [PubMed] [Google Scholar]
- 28.Marz P, Otten U, Miserez AR. Statins induce differentiation and cell death in neurons and astroglia. Glia. 2007;55:1–12. doi: 10.1002/glia.20422. [DOI] [PubMed] [Google Scholar]
- 29.Tsujimoto A, Takemura G, Mikami A, et al. A therapeutic dose of the lipophilic statin pitavastatin enhances oxidant-induced apoptosis in human vascular smooth muscle cells. J Cardiovasc Pharmacol. 2006;48:160–165. doi: 10.1097/01.fjc.0000245989.89771.1b. [DOI] [PubMed] [Google Scholar]
- 30.Liang SL, Liu H, Zhou A. Lovastatin-induced apoptosis in macrophages through the Rac1/Cdc42/JNK pathway. J Immunol. 2006;177:651–656. doi: 10.4049/jimmunol.177.1.651. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura T, Ushiyama C, Suzuki S, et al. Urinary excretion of podocytes in patients with diabetic nephropathy. Nephrol Dial Transplant. 2000;15:1379–1383. doi: 10.1093/ndt/15.9.1379. [DOI] [PubMed] [Google Scholar]
- 32.Bussolati B, Deregibus MC, Fonsato V, et al. Statins prevent oxidized LDL-induced injury of glomerular podocytes by activating the phosphatidylinositol 3-kinase/AKT-signaling pathway. J Am Soc Nephrol. 2005;16:1936–1947. doi: 10.1681/ASN.2004080629. [DOI] [PubMed] [Google Scholar]
- 33.Shibata S, Nagase M, Fujita T. Fluvastatin ameliorates podocyte injury in proteinuric rats via modulation of excessive Rho signaling. J Am Soc Nephrol. 2006;17:754–764. doi: 10.1681/ASN.2005050571. [DOI] [PubMed] [Google Scholar]
- 34.Whaley-Connell A, Habibi J, Nistala R, et al. Attenuation of NADPH oxidase activation and glomerular filtration barrier remodeling with statin treatment. Hypertension. 2008;51:474–480. doi: 10.1161/HYPERTENSIONAHA.107.102467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whaley-Connell A, DeMarco VG, Lastra G, et al. Insulin resistance, oxidative stress, and podocyte injury: role of rosuvastatin modulation of filtration barrier injury. Am J Nephrol. 2008;28:67–75. doi: 10.1159/000109394. [DOI] [PubMed] [Google Scholar]
- 36.Gu L, Ni Z, Qian J, et al. Pravastatin inhibits carboxymethyllysine-induced monocyte chemoattractant protein 1 expression in podocytes via prevention of signalling events. Nephron Exp Nephrol. 2007;106:e1–e10. doi: 10.1159/000100498. [DOI] [PubMed] [Google Scholar]
- 37.Agell N, Jaumot M, Rodriguez-Vilarrupla A, et al. The diverging roles of calmodulin and PKC in the regulation of p21 intracellular localization. Cell Cycle. 2006;5:3–6. doi: 10.4161/cc.5.1.2270. [DOI] [PubMed] [Google Scholar]
- 38.Child ES, Mann DJ. The intricacies of p21 phosphorylation: protein/protein interactions, subcellular localization and stability. Cell Cycle. 2006;5:1313–1319. doi: 10.4161/cc.5.12.2863. [DOI] [PubMed] [Google Scholar]
- 39.Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;13:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 40.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- 41.Suzuki A, Tsutomi Y, Akahane K, et al. Resistance to Fas-mediated apoptosis: activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene. 1998;17:931–939. doi: 10.1038/sj.onc.1202021. [DOI] [PubMed] [Google Scholar]
- 42.Asada M, Yamada T, Ichijo H, et al. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 1999;18:1223–1234. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]