

Abstract
The genomic organizations and functions of many miRNA genes have been described in recent years, but the origin and evolution of miRNAs in the exons of protein-coding genes are not well understood. The overlap of miR-650 genes with the protein-coding region of immunoglobulin lambda variable (IGVL) region genes has given a unique opportunity to witness a birth of miRNA gene. Both sequence comparisons and structure predictions indicate that the miR-650 genes are present in multiple copies and overlap in the same transcription orientation with the leader exon of primate IGVL genes of a specific phylogenetic clan (clan II). By reconstructing the phylogeny of the clan II IGVL genes, the stages in which the mutations accumulated in the leader exon and gave rise to a stable hairpin structure of miR-650 could be documented. The copy number variation of miR-650 genes among different species is the result of the duplication or deletion of the IGVL genes. To my knowledge, this is the first report of a genomic association between miRNA and the protein-coding genes of a multigene family. Analysis of the upstream region of the leader exon suggests that the IGVL and the mir-650 genes use the same promoter region for their transcription. However, in contrast to the general expectation about the expression of miRNAs that overlap with other genes in the same transcriptional orientation, this analysis provides evidence that the miR-650 gene is apparently transcribed independently of the IGVL gene with which it overlaps because they are expressed in different cell types.
Keywords: micro-RNA evolution, immunoglobulin lambda variable region genes, micro-RNA host genes, micro-RNA transcription, overlapping genes, multigene family
Introduction
Micro-RNAs (miRNAs) are single-stranded, endogenously expressed small RNA molecules about 22 nucleotide (nt) long, which do not encode proteins but regulate gene expression (Lagos-Quintana et al. 2001; Lau et al. 2001; Lee and Ambros 2001). They are transcribed in long precursors known as primary miRNAs (pri-miRNAs), containing characteristic stem-loop structures that are processed in the nucleus by a complex (called microprocessor) of RNase III enzyme Drosha and RNA-binding protein DGCR8 (Denli et al. 2004; Han et al. 2004). After their processing, the stem loop–shaped intermediates, pre–micro-RNAs, are exported to the cytoplasm where functionally matured miRNAs of about 22-nt length are excised by the RNase III enzyme Dicer (Hutvagner et al. 2001; Bartel and Chen 2004; Cullen 2004). The miRNAs regulate gene expression at the posttranscriptional level by blocking the translation or by degenerating the target mRNAs (Bartel 2004). Several hundred miRNA genes are currently known, and most of them are differentially expressed in different tissues (Lagos-Quintana et al. 2002; Aravin et al. 2003; Liu et al. 2004; Landgraf et al. 2007). Some miRNA genes show a high degree of conservation, and some are relatively poorly conserved among distantly related species (Lim et al. 2003; Bentwich et al. 2005; Houbaviy et al. 2005; Zhang et al. 2008). However, the functional significance of the conservation versus divergence of miRNAs has not been well established. It has been suggested that in comparison with highly conserved miRNAs, poorly conserved miRNAs are a potentially more important source of functional novelties during evolution (Zhang et al. 2007).
Initially, it was thought that miRNA genes were located in the intergenic regions, but recent studies have shown that most of the mammalian miRNA genes are present in the defined transcription units (TUs) (Lau et al. 2001; Rodriguez et al. 2004). On the basis of their location in the annotated TUs, a majority of the miRNA genes can be categorized into three groups: 1) intronic miRNA genes in protein-coding TUs, 2) intronic miRNA genes in noncoding TUs, and 3) exonic miRNA genes in noncoding TUs (Rodriguez et al. 2004; Kim and Nam 2006). In a few cases, miRNA genes are located in either an exon or an intron depending on the splicing pattern (Rodriguez et al. 2004; Kim and Nam 2006). Micro-RNAs can also appear in clusters on a single polycistronic transcript (Tanzer and Stadler 2004; Glazov et al. 2008). The miRNA genes located in the TUs with the same transcription orientation are thought to be cotranscribed with the host genes, whereas the miRNA genes located in intergenic regions or within TUs in antisense orientation have their own promoters (Lagos-Quintana et al. 2002; Aravin et al. 2003). Recent studies indicate that miRNAs regulate gene expression in many different biological processes such as cellular differentiation (Kawasaki and Taira 2003), embryonic development (Wienholds et al. 2005), immune regulation (Hoefig and Heissmeyer 2008; Lindsay 2008), heterochromatin formation (Verdel and Moazed 2005), tumorigenesis (Lu et al. 2005), etc. Because of their heterogeneity in genomic organization as well as in functional involvements, different miRNA genes might be subjected to different modes of evolution. However, the origin and evolution of different miRNA genes are not well studied. In particular, the evolutionary origins of miRNA genes in the exon of protein-coding TUs are not well understood. Previously, it was reported that new miRNAs are sometimes generated from non-miRNA sequences that accumulate nucleotide substitutions to become miRNA genes (Svoboda and Di Cara 2006; Lu et al. 2008). Some others have arisen by duplication of the existing miRNA genes (Tanzer and Stadler 2004) or have been derived from transposable elements (Piriyapongsa et al. 2007). A survey of the genomic context of human miRNA genes in miRBase (Release 11.0) (Griffiths-Jones et al. 2008) shows that the hsa-miR-650 gene is located in the immunoglobulin lambda light chain variable (IGVL) genes. The hsa-miR-650 gene was originally identified from the expressed miRNAs of human colorectal cells (Cummins et al. 2006). Here I have analyzed 10 completely sequenced mammalian genomes to determine the genomic organization, origin, and evolution of miR-650 genes. This comprehensive study of miR-650 genes along with the host IGVL genes elucidates not only the origin and evolution of the miR-650 gene family but also facilitates the understanding of the functional aspects of their novel structural association.
Materials and Methods
Retrieval of Homologous hsa-miR-650 Sequences
Publicly accessible human hsa-miR-650 sequence (accession number MI0003665) was obtained from miRBase (Release 11.0) (Griffiths-Jones et al. 2008). To retrieve the homologs of the hsa-miR-650 gene (96 nt in length) in mammalian species, I performed two-round BlastN search with the cutoff E-value of 10−30 against the genome sequences of human (Homo sapiens) (assembly: NCBI Build 36.2, September 2006), chimpanzee (Pan troglodytes) (assembly: CHIMP 2.1, March 2006), orangutan (Pongo pygmaeus abelii) (assembly: PPYG2, September 2007), macaque (Macaca mulatta) (assembly: MMUL 1.0, February 2006), mouse (Mus musculus) (NCBI m36, December 2005), rat (Rattus norvegicus) (RGSC 3.4, December 2004), cow (Bos taurus) (Btau_3.1, August 2006), horse (Equus caballus) (EquCab 2, September 2007), opossum (Monodelphis domestica) (monDom5, October 2006), and platypus (Ornithorhynchus anatinus) (Ornithorhynchus_anatinus-5.0, December 2005) from Ensembl Genome Browser. The first-round Blast best-hit sequences of each genome were used as queries in the second round of BlastN search to find additional candidates for homologs of hsa-miR-650 genes, if any.
Secondary Structure Prediction
Secondary structures of miRNAs were predicted by RNAfold program (available at http://rna.tbi.univie.ac.at) (Gruber et al. 2008). The program predicts the stem loop of the input sequences based on minimum free energy (MFE) structures using the dynamic programing algorithm (Zuker and Stiegler 1981) and base-pair probabilities using the partition function algorithm (McCaskill 1990). The reliability of the secondary structures was verified by comparing the mountain plot presentations of the MFE structure and the centroid structure (available as graphical output from the RNAfold server). The centroid structure is the structure with the minimal base-pair distance to all structures in the thermodynamic ensemble (Ding et al. 2005). Thus, the centroid structure can be considered as the single structure that best represents the central tendency of a set of structures. The higher the extent of overlap of the mountain plots of the MFE structure and the centroid structure, the more reliable the structural prediction (Gruber et al. 2008).
Sequence Alignments
All the retrieved homologous sequences were aligned with the query sequence (MI0003665) using the ClustalW program (Thompson et al. 1994) and the alignments were inspected manually to maximize similarity. The alignments of the predicted secondary structures were carried out based on the linear alignments of nucleotide sequences.
Identification of IGVL Genes
I used IGVL sequences that were identified in a previous study (Das, Nikolaidis, et al. 2008) on seven mammalian species (i.e., human, mouse, rat, cow, horse, opossum, and platypus). To retrieve chimpanzee, orangutan, and macaque IGVL genes, I performed two rounds of TBlastN search against the genome sequences with the cutoff E-value of 10−15. In the first round, the amino acid sequences of three known IGVL sequences (accession numbers X53936, X57811, and M99606) of human were used as queries. Because these queries are similar to one another, they hit the same genomic regions. I extracted only nonoverlapping sequences given by the best hit (with the lowest E-value). On the basis of sequence similarity, some immunoglobulin light chain variable sequences of another isotype (kappa) were found as significant hits in the Blast search with the cutoff E-value of 10−15. I identified the kappa variable genes (IGVK) using IGVK-specific molecular markers (Das, Nikolaidis, et al. 2008) and excluded them from the analysis. The retrieved IGVL sequences that aligned with the query sequence without any frameshift mutations and/or premature stop codons in the leader sequence and the V-exon, encoded the two conserved Cys residues in framework regions (FRs) 1 and 3, respectively, and had a proper recombination signal sequence (RSS) were regarded as potentially functional IGVL genes. Other sequences (including truncated ones) were regarded as IGVL pseudogenes. The first-round Blast best-hit sequences of a specific organism were used as queries in the second round of TBlastN search to find additional IGVL sequences in the chimpanzee, orangutan, and macaque.
Phylogenetic Analysis
The phylogenetic trees were constructed by the Neighbor-Joining (NJ) (Saitou and Nei 1987) method based on the pairwise deletion option using the MEGA4.0 program (Tamura et al. 2007). The p-distance method (Nei and Kumar 2000) was used to calculate evolutionary distances. The reliability of the tree was assessed by bootstrap resampling with a minimum of 1,000 replications.
Hydropathy Profiles of Signal Peptides
Hydropathicity of IGVL signal peptides was calculated using Kyte and Doolittle scale (Kyte and Doolittle 1982).
Repetitive Sequence Analysis
The analysis of repetitive elements present in the 10-kb flanking regions of the 5′ and 3′ ends of IGVL genes was carried out using CENSOR software tool (Kohany et al. 2006).
Results
Overlap of miR-650 Genes and IGVL Genes
The study of genomic organization revealed that the hsa-miR-650 gene (accession number MI0003665) overlaps with the leader exon of IGVL gene (fig. 1A). The IGVL genes consist of two exons (leader and V-exons), which are separated by an intron (∼100 nt in length). The leader exon is ∼89 nt long and encodes 15 amino acid (aa) residues of the signal peptide (19 aa in length). Both the miR-650 and IGVL genes are almost completely overlapping with each other except 2 and 5 nt at the upstream and downstream regions of the leader exon, respectively, and both genes are found in the same transcription orientation (fig. 1A and supplementary fig. 1, Supplementary Material online). The Untranslated Region (UTR) of the IGVL leader exon contains the mature miRNA sequence, whereas its complementary sequence is located in protein-coding sequences (CDSs). The promoter region of the IGVL gene consists of the conserved octamer sequence (ATTTGCAT) and the TATA box located within 100 nt upstream of the leader exon (Vasicek and Leder 1990). Analysis of the DNA sequences 300 nt upstream of the leader exon using SIGNAL SCAN program (Prestridge 1991) revealed the presence of the IGVL-specific octamer transcriptional element but no micro-RNA–specific transcriptional element (data not shown). It can, therefore, be assumed that the IGVL and the miR-650 genes use the same promoter region for their transcription. This is consistent with the previous proposition that miRNAs located in TUs with the same transcriptional orientation usually use the same promoter (Bartel 2004; Eis et al. 2005; Kim and Kim 2007).
FIG. 1.—

Genomic organization of the miR-650 gene in relation to the IGVL gene. (A) Top: Overlap of the hsa-miR-650 gene (MI0003665) and the leader exon of IGVL gene. The position of the hsa-miR-650 gene and the positions of the octamer, TATA box, leader exon, V-exon, RSS of the IGVL gene are shown. Below: Enlarged view of the association between the hsa-miR-650 gene and the leader exon of IGVL. The CDS is indicated by a horizontal solid line, and the UTR is indicated by a broken line. The arrowhead indicates the transcription direction. The positions of the MR, seed sequence, TL, and CR are shown on the reference hsa-miR-650 sequence. The region that overlaps with the CDS of the leader exon of the IGVL is underlined in the nucleotide sequence of the hsa-miR-650. The positional information of the genomic organization of IGVL gene and miR-650 is based on the human miR-650-bearing IGVL gene number 2 shown in figure 1B for which hsa-miR-650 has the exact match. The other miR-650-bearing IGVL genes have basically the same genomic organization. (B) Positions of miR-650-bearing IGVL genes in the human immunoglobulin lambda locus (not to scale). Short vertical lines show IGVL genes, whereas long vertical lines indicate either immunoglobulin lambda constant genes (solid line) or joining genes (dotted line). The numbers below indicate the positions of miR-650-bearing IGVL genes in the lambda locus numbered from the proximity to the immunoglobulin lambda constant genes.
Genomewide Search for miR-650 Genes
To find hsa-miR-650 homologs in the draft genome sequences of 10 mammalian species, the BlastN search was carried out using 96-nt-long hsa-miR-650 sequence (accession number MI0003665) as a primary query and the best-hit sequences of the first-round search in each species as queries of the second-round search. With the exception of four primate species (human, chimpanzee, orangutan, and macaque), the homology search did not detect any sequence within a cutoff E-value of 10−30 in other mammalian genomes (BlastN search with a lower cutoff E-values up to 10−5 found no other hits except 12 horse sequences and 4 cow sequences that overlap with the horse and cow IGVL genes included in the analyses shown in the later section). In the genomes of the four primate species, I found multiple copies of hsa-miR-650 homologs and their numbers varying from species to species (table 1). There were 9, 10, 11, and 11 miR-650 genes present in the human, chimpanzee, orangutan, and macaque genomes, respectively. The annotations of all retrieved hsa-miR-650 homologs in primates indicate that IGVL genes with miR-650 genes are localized in clusters (see fig. 1B and supplementary table 1, Supplementary Material online). The human, chimpanzee, and orangutan miR-650 genes are located on chromosome 22, and the macaque miR-650 genes are on chromosome 10. However, because of the incompleteness of the genome assemblies, four miR-650 genes in chimpanzee and five genes in macaque could not be assigned to a specific region.
Table 1.
Number of Clan II IGVL and miR-650 Genes
| Clan II IGVL | miR-650 | |||
| Species | F | NF | F | NF |
| Human | 5 | 4 | 4 | 5 |
| Chimpanzee | 4 | 4(2) | 5 | 5 |
| Orangutan | 6 | 4(1) | 4 | 7 |
| Macaque | 8 | 0(3) | 4 | 7 |
| Mouse | 0 | 0 | 0 | 0 |
| Rat | 0 | 0 | 0 | 0 |
| Horse | 5 | 7 | 0 | 0 |
| Cow | 2 | 2 | 0 | 0 |
| Opossum | 0 | 0 | 0 | 0 |
| Platypus | 0 | 0 | 0 | 0 |
NOTE.—F, functional gene; NF, nonfunctional gene (pseudogene).
The numbers in the parentheses refer to partial genes.
The genomic context of all miR-650 genes with respect to the leader exon of IGVL gene is exactly the same (see fig. 1A). The mature region (MR) of the query sequence (hsa-miR-650) is 21 nt long. This MR acts as a posttranscriptional regulatory element, which contains 7-nt-long (GGAGGCA) seed sequence important for recognition of target genes by complementally binding to the 3′ UTRs of mRNA. To determine whether the hsa-miR-650 homologs in four primate species are potential candidates for functional miRNA genes, I used two criteria: 1) ≥15 nt base pairing (>70% arm base pairing) in the mature and complementary regions (CRs) of the predicted hairpin structure (Ambros et al. 2003; Stark et al. 2007). 2) Position of the terminal loop (TL), which does not include the MR (Ambros et al. 2003). If these two criteria were fulfilled, the miR-650 genes were regarded as functional. The sequences of functional miR-650 genes were very similar differing by only a few nucleotides, and all potentially functional miR-650 genes had perfect conservation of the seed sequences (fig. 2). The numbers of potentially functional miR-650 genes in the four primate species are given in table 1.
FIG. 2.—

Alignments of functional miR-650 sequences and their secondary structures. The hsa-miR-650 (accession no. MI0003665) is used as the reference sequence. The MR, TL, and CR are indicated by arrows. (A) Sequences of potential functional miR-650 genes in human, chimpanzee, orangutan, and macaque. Dots represent the same nucleotide as that of the first sequence. The seed sequences are shown in the box. (B) Predicted secondary structure of miR-650 sequences. “.” and “( )” denote unpaired and paired nucleotides in miR-650 hairpins.
Association between IGVL and miR-650 Genes in a Specific Phylogenetic Clan
As shown in figure 1B, the miR-650 genes are associated with some IGVL genes only. To determine whether the miR-650 genes are associated with evolutionary closely related IGVL genes or whether they are randomly associated with any group of IGVL genes, I analyzed the phylogenetic relationship of the amino acid sequences encoded in all IGVL genes (except truncated pseudogenes) in each primate species separately. The human IGVL sequences have been found to fall into six major clans (clans I–VI), clearly supported by high (>80%) bootstrap values (fig. 3A). All the IGVL genes with the miR-650 genes in humans are clustered together and belong to clan II (fig. 3A). Similarly in chimpanzee, orangutan, and macaque all miR-650-bearing IGVL genes clustered together in a single clan on phylogenetic trees with high bootstrap support (data not shown). When the phylogenetic tree was constructed for human IGVL sequences and the miR-650-bearing IGVL sequences of each of the nonhuman primates separately, a specific cluster was found for all miR-650-bearing IGVL sequences. One example is shown in figure 3B, which represents the phylogenetic tree for all the human IGVL sequences and the macaque miR-650-bearing IGVL sequences. These observations suggest that all miR-650-bearing IGVL genes (i.e., clan II genes) are evolutionary closely related to one another and that they have a common origin.
FIG. 3.—
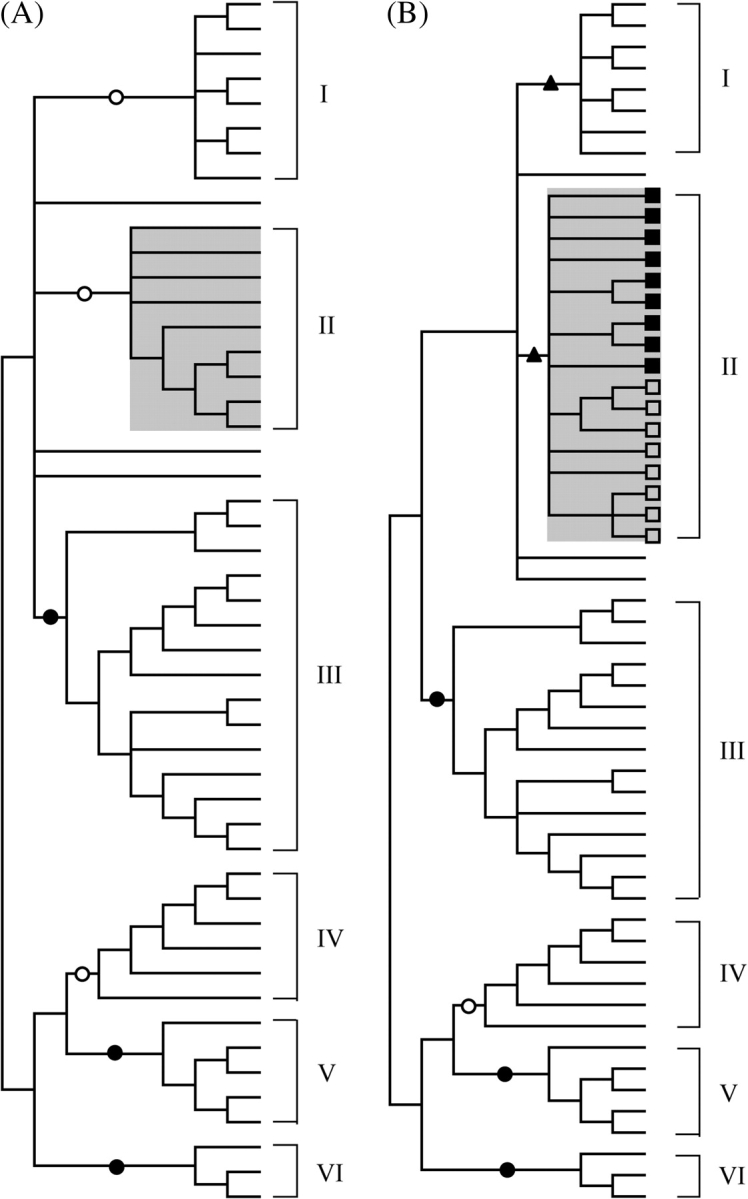
NJ trees of IGVL sequences. The trees are condensed at the 50% bootstrap value level. Filled triangles, open circles, and filled circles indicate that the interior branches are supported by ≥70%, ≥80%, and ≥90% bootstrap value, respectively. The trees are constructed by using the pairwise deletion option and the p-distance method. (A) The phylogenetic tree of all IGVL sequences from human. There are six major clans (clans I–VI) of IGVL genes identified from this tree. The miR-650-bearing IGVL genes are clustered in clan II. (B) The phylogenetic tree of the human and macaque IGVL sequences including the miR-650-bearing IGVL sequences (clan II) of macaque. The filled rectangles and the open rectangles indicate the clan II IGVL genes of human and macaque, respectively.
Clan II IGVL Homologs in Nonprimate Mammals
To study whether clan II IGVL homologs are present also in nonprimate species, the phylogenetic trees were constructed for human sequences combining the sequences from a given nonprimate species (i.e., human–mouse, human–cow, human–horse, etc.). By these phylogenetic analyses, no clear-cut homologs of clan II IGVL could be found in nonprimate species (see supplementary fig. 2, Supplementary Material online). This result is in agreement with the previous findings (Das, Nikolaidis, et al. 2008) that the evolutionary relationships of the IGVL genes, which are short and evolve relatively fast (Ota et al. 2000), are difficult to resolve between distantly related species by phylogenetic tree building methods. Therefore, I used an alternative approach to find clan II IGVL homologs in nonprimate species, an approach based on the definition of molecular cladistic markers (Das, Nikolaidis, et al. 2008). The clan II–specific markers were found both in the signal peptides and in the encoded amino acid sequences of V-exons (fig. 4 and supplementary fig. 3, Supplementary Material online). The encoded signal peptides (19 aa long) of clan II IGVL genes have fairly conserved motifs at positions 5–6 (LL) and positions 9–13 (T(S)LLTQ). In addition, the clan II signal peptides have a characteristic hydropathy profile, which distinguishes them from the signal peptides encoded in other IGVL genes (supplementary fig. 4, Supplementary Material online). Furthermore, two specific motifs, QS(T)V(I)TI and SK(T)SGNTAS(T)LTI(V)SGLQA, are present in frameworks 1 and 3 (FR1 and FR3) regions encoded in the clan II V-exon, respectively (fig. 4 and supplementary fig. 3, Supplementary Material online). All of these markers have shown ≥90% conservation in the clan II sequences.
FIG. 4.—

Alignment of clan II functional IGVL sequences. The cladistic molecular markers that are the characteristics of clan II sequences are highlighted. The conserved motifs are defined by ignoring amino acids that appeared in only one sequence. Dots represent the same amino acid residue as in the first sequence. The numbering of the amino acid positions in the V-segment is based on human immunoglobulin light chain variable sequences (IGVK and IGVL) according to Das, Nikolaidis, et al. (2008). The arrow indicates the absence of Ser/Thr of position 7 (a gap relative to IGVK sequences), which is used as a molecular marker for immunoglobulin light chain isotypes differentiation.
Based on the similarities in molecular markers present in the signal peptides and in the encoded amino acid sequences of V-exons, 4 and 12 homologs of clan II IGVL genes are identified in cow (two functional and two pseudogenes) and horse (five functional and seven pseudogenes), respectively (table 1). A list of clan II IGVL genes is given in supplementary table 2M(Supplementary Material online). Any homolog of clan II genes is not found in rodents (mouse and rat), prototheria (platypus), and metatheria (opossum) (table 1). These results suggest that the clan II IGVL genes probably evolved in eutherian mammals and were lost in the rodent lineage.
Comparison of Clan II Leader Exons between Primates and Nonprimates
To determine whether the leader sequences of the clan II IGVL genes in nonprimate species are potential candidates for the functional miR-650 genes, I compared the differences in the sequences of clan II leader exons between primate and nonprimate species. I have found that the CDSs of clan II leader exons in primates differ from those of nonprimate species by 2-nt positions, whereas the UTRs have differences in nucleotides at 10 different positions relative to nonprimate species (fig. 5). The computed net p-distance (i.e., the average p-distance between primates and nonprimates subtracting the differences within primates and nonprimates) in UTRs of clan II leader exons between primates and nonprimates is 0.146 (SE ± 0.045), whereas it is 0.008 (SE ± 0.008) in CDSs. To see whether the clan II IGVL genes in cow and horse produce the characteristic hsa-miR-650-like folded structure, I analyzed their predicted secondary structures (fig. 6). I have found that none of the predicted secondary structures of the hsa-miR-650 homologous regions in cow and horse clan II IGVL genes produce the characteristic stem loop of hsa-miR-650 gene.
FIG. 5.—

Alignment of leader exons of clan II functional IGVL genes. The sequences include 2 and 5 nt upstream and downstream of the leader exons, respectively (similar to miR-650 genes in primates). Dots represent the same nucleotide as in the first sequence. For primates, one representative of clan II leader exons from each species is shown. The differences in the specific sites as compared with functional miR-650 genes in primate species are indicated by “*” (for one nonprimate species) and “**” (for both nonprimate species), respectively. If the majority of the nonprimate sequences have a nucleotide different from that of the primate sequences, the site is considered to be different between primate and nonprimate species. The “+” indicates the nonconserved sites (<80% conservation) among functional miR-650 genes in primates (see alignment of functional miR-650 sequences in fig. 2).
FIG. 6.—

Predicted secondary structures of (A) hsa-miR-650 sequence, (B) homologous sequence in horse, and (C) homologous sequence in cow. The nucleotides in the MR and CR of hsa-miR-650 sequence and their homologous region in horse and cow sequences are marked in yellow and red colors, respectively. The nucleotide at the 5′ end is marked in blue color.
Orthologous Relationship between miR-650-Bearing IGVL Genes
To understand the short-term evolution of miR-650-bearing IGVL genes, I determined the orthologous relationship between clan II IGVL genes. Because of the shortness and fast evolution of the immunoglobulin variable genes (Ota et al. 2000), orthologous relationships are difficult to determine between sequences from distantly related species on the basis of phylogenetic analysis alone (Das, Nikolaidis, et al. 2008; Das, Nozawa, et al. 2008). Even between closely related species, phylogenetic analysis gave ambiguous results for some sequences as indicated by relatively low bootstrap values (Das, Nozawa, et al. 2008). As the divergence times between human–chimpanzee (∼6 Ma) and human–orangutan (∼13 Ma) are relatively short (Sibley and Ahlquist 1987), I examined the orthologous relationships between the miR-650-bearing IGVL sequences (clan II) of human, chimpanzee, and orangutan. As in a previous study of orthologous relationships between immunoglobulin heavy chain variable genes in human and chimpanzee (Das, Nozawa, et al. 2008), the orthologous relationships of clan II IGVL genes were determined by phylogenetic analysis using >80% bootstrap support as a criterion of orthology and by the similarity of repetitive elements that flank them. The clan II IGVL genes in macaque were not used as outgroup because both the phylogenetic analysis and the analysis of flanking repetitive elements failed to detect orthologous relationship for more than 50% of the sequences. One limitation of the analysis is the incompleteness of the chimpanzee genomic sequences. The contigs of IGVL locus are fragmented and the genomic positions of some regions are not determined in chimpanzee, though all the miR-650-bearing IGVL genes are in the continuous region in human and orangutan.
The results of the analysis revealed that although the number of miR-650-bearing IGVL genes in human, chimpanzee, and orangutan is apparently similar (9, 10, and 11 in human, chimpanzee, and orangutan, respectively) multiple events of duplications or deletions of clan II IGVL genes occurred in these three species (fig. 7). Three and one possible duplication events occurred in the orangutan (for IGVL gene numbers 2, 3, 4, 7, and 8) and chimpanzee (for IGVL gene numbers 1, 10) lineages, respectively. One ortholog of human (gene number 6) and chimpanzee (gene number 5) clan II IGVL genes is lost in the orangutan lineage. The analysis shows that some IGVL genes became nonfunctional after speciation. For example, in the human (gene number 8) and orangutan (gene number 8), one nonfunctional clan II IGVL is found whose ortholog in chimpanzee are functional. Like IGVL genes, several miR-650 genes became nonfunctional after speciation. The duplications or deletions of miR-650 genes exactly follow the duplications or deletions of the host IGVL genes. However, the miR-650 genes can become nonfunctional independently of the IGVL genes.
FIG. 7.—

Chromosomal locations of miR-650-bearing IGVL genes (clan II) and their orthologous relationships in human, chimpanzee, and orangutan. The open rectangles indicate miR-650 genes, whereas filled rectangles and filled ovals (for partial genes) indicate IGVL genes. The long and short rectangles represent functional and nonfunctional genes, respectively. Orthologous relationships between IGVL sequences are shown by broken lines. Due to incompleteness of the chimpanzee genomic sequences (indicated by the gaps in the lines), the positions of four IGVL sequences are in the undetermined regions of chromosome 22 (C 22R1 and C 22R2).
Discussion
Organization of miR-650 Genes in Relation to the IGVL Genes
In the present study, I have found multiple copies of miR-650 genes in the primate IGVL locus. They all show the same overlap with the leader exon of IGVL genes. In previous studies, several genomewide screens have been undertaken to determine the genomic locations of miRNA genes relative to other genes. Rodriguez et al. (2004) found that most of the known mammalian miRNAs were located within introns of either protein-coding or noncoding TUs, or in intergenic regions, whereas only a few were located in exons of noncoding RNAs or UTR of protein-coding genes. In a systematic scanning of the entire human genome Bentwich et al. (2005) found 434,239 potential hairpins including 86% of known miRNA genes, but none of them were located in protein-coding regions. It seems, therefore, that the overlap between miR-650 gene and the leader exon of IGVL gene may be a very rare form of association between RNA-based regulatory genes and protein-coding genes. To my knowledge, this is the first report of a genomic association between miRNA genes and the exonic regions of protein-coding genes of a multigene family.
Expression of miR-650 Genes
Most vertebrate miRNAs are expressed in a developmentally regulated or tissue-specific manner (Lagos-Quintana et al. 2002; Cullen 2004). Usually, if the miRNA genes are located in the intergenic regions, they have their own promoter, but if they are located in the TUs in the same transcriptional orientation, they generally cotranscribe with host genes using the same promoter (Bartel 2004; Eis et al. 2005; Kim and Kim 2007). The miRNA genes located in the intronic regions are either processed out from intron lariats or from the primary transcripts before splicing (Rodriguez et al. 2004; Kim and Kim 2007). In the case of miRNA genes found within the UTRs, the processing of miRNAs may sometime inhibit the expression of the linked protein-coding genes by removing the mRNA poly(A) tail (Cullen 2004). In the case of the association between IGVL and miR-650 genes, the situation may be different. Like other antibody-coding genes, the IGVL genes require IGVL–IGJL recombination to produce functional proteins and this event occurs during an early stage of B-cell differentiation (Klein and Hořejší 1997). The transcription of IGVL genes takes place only after the occurrence of recombination between IGVL and IGJL genes. However, because the miR-650 gene is known to be expressed in human colorectal cells (Cummins et al. 2006) in which the IGVL gene remains in a germline configuration (i.e., IGVL does not recombine with IGJL), the transcription of miR-650 gene may not require such recombination. Several studies in which miR-650 expression was not detected in B-cells (Liu et al. 2004; Landgraf et al. 2007; Lawrie et al. 2008) support this conclusion. Hence, contrary to a previous proposition for miRNA genes located in the TU (Bartel 2004; Eis et al. 2005; Kim and Kim 2007), cotranscription may not be necessary for the expression of miR-650 and IGVL genes because they express themselves in colorectal and B-cells, respectively. However, the IGVL and the miR-650 genes may nevertheless use the same promoter region for their transcription as no miRNA-specific transcriptional element was found in the 300 nt upstream of the leader exon of IGVL genes, and in the overlapping segment the two genes are in the same transcriptional orientation. It is proposed therefore that depending on the cell type, the promoter region of the miR-650-bearing IGVL gene may function in two separate processes: in miRNA biogenesis (recombination-independent transcription) and in protein synthesis (recombination-dependent transcription). If so, it would be a novel RNA-processing control in an overlapping structural association between miRNA and protein-coding genes.
Origin and Evolution of the Association between miR-650 and IGVL Genes
The miR-650-bearing IGVL genes belong to clan II of phylogenetically closely related genes (see fig. 3). The analysis using molecular markers suggests that the clan II IGVL genes are not restricted to primates but are also found in nonprimate species, such as cow and horse. However, the clan II IGVL genes have not been found in the mouse and rat. No clan II homologs have also been found in platypus and opossum. It is, therefore, possible that clan II IGVL genes may have originated in eutherian mammals and afterward they were lost in certain eutherian lineage(s) (i.e., rodents). Although clan II IGVL genes are present in nonprimate species, both sequence comparison and structure predictions indicate that the leader exon of clan II IGVL genes may act as functional miR-650 genes only in primates (see figs. 5 and 6). The absence of clan II IGVL genes in rodents also supports the notion that expression of miR-650 genes might be restricted to primates only. No experimentally determined target genes of miR-650 have been reported thus far. A list of computationally predicted target genes is, however, available in the miRBase (Griffiths-Jones et al. 2008). Interestingly, >95% of the predicted targets of human miR-650 genes have orthologs in the mouse (not shown). Although miRNA target-gene prediction methods have several limitations, this finding, together with previous reports that one miRNA can target several mRNAs and one mRNA can be targeted by multiple miRNAs (Houbaviy et al. 2005; Lim et al. 2005), suggests that possibly in nonprimate species some other miRNAs may regulate the common targets that in primates are regulated by miR-650.
By reconstructing the phylogeny of the clan II IGVL genes, the stages in which the point mutations accumulated in the leader exon to give rise to a stable hairpin structure as the precursor of miR-650 could be documented. As shown in figure 1A, the UTR regions of the clan II leader exons contain the mature miRNA sequence in primates, whereas its complementary sequence is located in protein-coding regions. To understand the mode of origin of miR-650, I compared the sequences of clan II leader exons between primate and nonprimate species. The sequence comparison shows that the protein-coding regions are mostly conserved between primate and nonprimate species except at 2-nt positions (see fig. 5), but as compared with nonprimate species, the UTRs of primates clan II IGVL genes are different at several nucleotide positions. The predicted secondary structures indicate that the sequences of clan II IGVL genes in cow and horse can fold but that none of the folds has the characteristic hairpin structure of the primate miR-650 genes (see fig. 6). It may be, therefore, possible that accumulation of nucleotide changes in the leader exons of clan II IGVL genes, particularly in the UTRs have led to the birth of functionally stable miRNA hairpins in primates. Once the leader exons of clan II IGVL genes became stable hairpins, the target of the miR-650 may have been chosen at random, based on complementarity between the potential seed sequence and the 3′ UTR of mRNAs. This supports the random selection hypothesis that assumes accumulation of nucleotide changes and subsequent target acquisition of miRNA (Svoboda and Di Cara 2006). Figure 8 shows the hypothetical evolutionary scheme of miR-650 along with clan II IGVL genes, based on the evolutionary tree of the species of mammals. The duplications or deletions of miR-650 genes in the different primates followed the duplications or deletions of the host gene (IGVL gene) (see fig. 7). The functionality of the miR-650 gene is, however, not dependent on the functionality of the overlapping IGVL gene, except in the situation where IGVL becomes nonfunctional because of a mutation in the promoter region.
FIG. 8.—
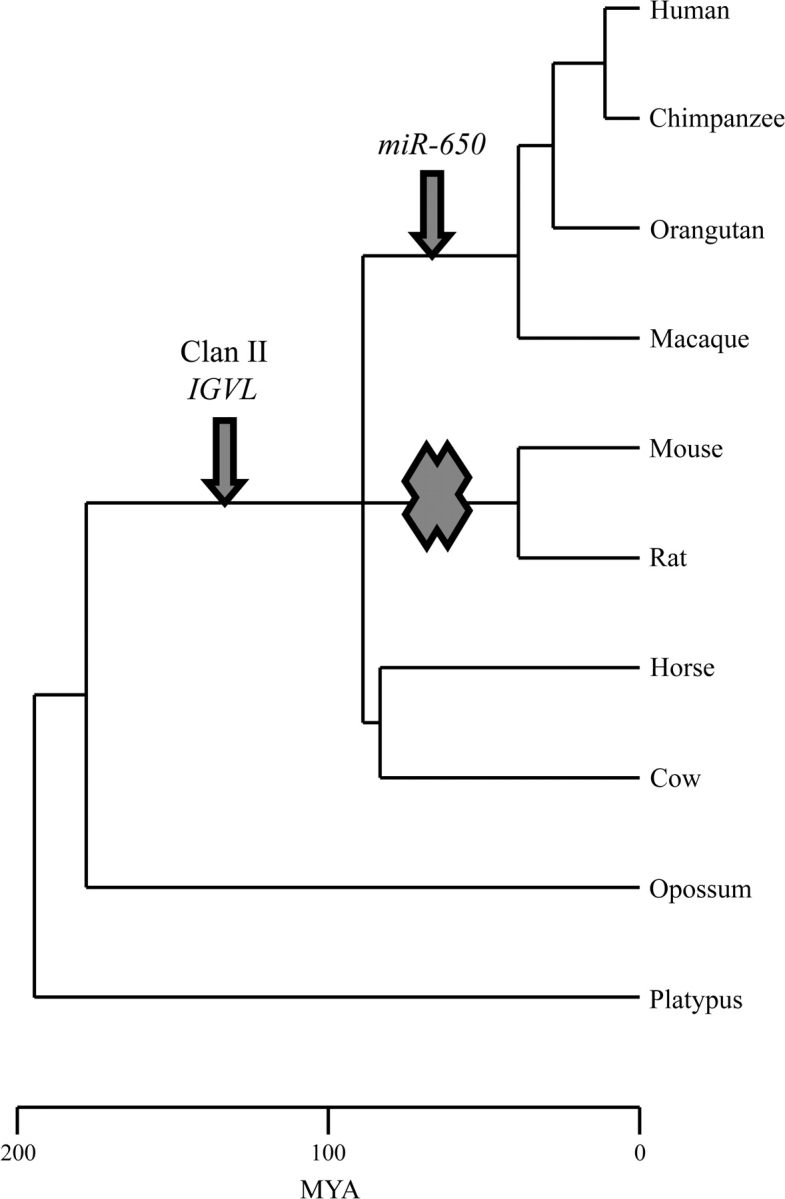
The hypothetical evolutionary scheme of miR-650 genes with relation to clan II IGVL genes. The arrows indicate the possible time of appearance of the clan II IGVL and miR-650 genes. The cross mark indicates the complete loss of clan II IGVL genes in the rodent lineage.
Supplementary Material
Supplementary tables 1 and 2 and supplementary figures 1–4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
I thank Jan Klein, Sayaka Miura, Masatoshi Nei, Naoko Takezaki, Masayuki Hirano, Nikolas Nikolaidis, Kazuhiko Kawasaki, Masafumi Nozawa, Dimitra Chalkia, Zhenguo Lin, and Hiroki Goto for their help during this study and manuscript preparation. This work was supported by the National Institutes of Health (grant GM020293-35 to M. Nei).
References
- Ambros V, Bartel B, Bartel DP, et al. A uniform system for microRNA annotation. RNA. 2003;9:277–279. doi: 10.1261/rna.2183803. (13 co-authors) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Lagos-Quintana M, Yalcin A, Zavolan M, Marks D, Snyder B, Gaasterland T, Meyer J, Tuschl T. The small RNA profile during Drosophila melanogaster development. Dev Cell. 2003;5:337–350. doi: 10.1016/s1534-5807(03)00228-4. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- Bentwich I, Avniel A, Karov Y, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–770. doi: 10.1038/ng1590. (13 co-authors) [DOI] [PubMed] [Google Scholar]
- Cullen BR. Transcription and processing of human microRNA precursors. Mol Cell. 2004;16:861–865. doi: 10.1016/j.molcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Cummins JM, He Y, Leary RJ, et al. The colorectal microRNAome. Proc Natl Acad Sci USA. 2006;103:3687–3692. doi: 10.1073/pnas.0511155103. (16 co-authors) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Nikolaidis N, Klein J, Nei M. Evolutionary redefinition of immunoglobulin light chain isotypes in tetrapods using molecular markers. Proc Natl Acad Sci USA. 2008;105:16647–16652. doi: 10.1073/pnas.0808800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Nozawa M, Klein J, Nei M. Evolutionary dynamics of the immunoglobulin heavy chain variable region genes in vertebrates. Immunogenetics. 2008;60:47–55. doi: 10.1007/s00251-007-0270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- Ding Y, Chan CY, Lawrence CE. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA. 2005;11:1157–1166. doi: 10.1261/rna.2500605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazov EA, McWilliam S, Barris WC, Dalrymple BP. Origin, evolution, and biological role of miRNA cluster in DLK-DIO3 genomic region in placental mammals. Mol Biol Evol. 2008;25:939–948. doi: 10.1093/molbev/msn045. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The Vienna RNA website. Nucleic Acids Res. 2008;36:W70–W74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefig KP, Heissmeyer V. MicroRNAs grow up in the immune system. Curr Opin Immunol. 2008;20:281–287. doi: 10.1016/j.coi.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Houbaviy HB, Dennis L, Jaenisch R, Sharp PA. Characterization of a highly variable eutherian microRNA gene. RNA. 2005;11:1245–1257. doi: 10.1261/rna.2890305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Taira K. Hes1 is a target of microRNA-23 during retinoic-acid-induced neuronal differentiation of NT2 cells. Nature. 2003;423:838–842. doi: 10.1038/nature01730. [DOI] [PubMed] [Google Scholar]
- Kim VN, Nam JW. Genomics of microRNA. Trends Genet. 2006;22:165–173. doi: 10.1016/j.tig.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Kim YK, Kim VN. Processing of intronic microRNAs. Embo J. 2007;26:775–783. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J, Hořejší V. Immunology. Oxford: Blackwell Science Ltd; 1997. [Google Scholar]
- Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. (51 co-authors) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lawrie CH, Saunders NJ, Soneji S, et al. MicroRNA expression in lymphocyte development and malignancy. Leukemia. 2008;22:1440–1446. doi: 10.1038/sj.leu.2405083. (14 co-authors) [DOI] [PubMed] [Google Scholar]
- Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP. Vertebrate microRNA genes. Science. 2003;299:1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Lindsay MA. microRNAs and the immune response. Trends Immunol. 2008;29:343–351. doi: 10.1016/j.it.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Liu CG, Calin GA, Meloon B, et al. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc Natl Acad Sci USA. 2004;101:9740–9744. doi: 10.1073/pnas.0403293101. (14 co-authors) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. (14 co-authors) [DOI] [PubMed] [Google Scholar]
- Lu J, Shen Y, Wu Q, Kumar S, He B, Shi S, Carthew RW, Wang SM, Wu CI. The birth and death of microRNA genes in Drosophila. Nat Genet. 2008;40:351–355. doi: 10.1038/ng.73. [DOI] [PubMed] [Google Scholar]
- McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- Nei M, Kumar S. Molecular evolution and phylogenetics. Oxford: Oxford University Press; 2000. [Google Scholar]
- Ota T, Sitnikova T, Nei M. Evolution of vertebrate immunoglobulin variable gene segments. Curr Top Microbiol Immunol. 2000;248:221–245. doi: 10.1007/978-3-642-59674-2_10. [DOI] [PubMed] [Google Scholar]
- Piriyapongsa J, Marino-Ramirez L, Jordan IK. Origin and evolution of human microRNAs from transposable elements. Genetics. 2007;176:1323–1337. doi: 10.1534/genetics.107.072553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestridge DS. SIGNAL SCAN: a computer program that scans DNA sequences for eukaryotic transcriptional elements. Comput Appl Biosci. 1991;7:203–206. doi: 10.1093/bioinformatics/7.2.203. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sibley CG, Ahlquist JE. DNA hybridization evidence of hominoid phylogeny: results from an expanded data set. J Mol Evol. 1987;26:99–121. doi: 10.1007/BF02111285. [DOI] [PubMed] [Google Scholar]
- Stark A, Kheradpour P, Parts L, Brennecke J, Hodges E, Hannon GJ, Kellis M. Systematic discovery and characterization of fly microRNAs using 12 Drosophila genomes. Genome Res. 2007;17:1865–1879. doi: 10.1101/gr.6593807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda P, Di Cara A. Hairpin RNA: a secondary structure of primary importance. Cell Mol Life Sci. 2006;63:901–908. doi: 10.1007/s00018-005-5558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Tanzer A, Stadler PF. Molecular evolution of a microRNA cluster. J Mol Biol. 2004;339:327–335. doi: 10.1016/j.jmb.2004.03.065. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasicek TJ, Leder P. Structure and expression of the human immunoglobulin lambda genes. J Exp Med. 1990;172:609–620. doi: 10.1084/jem.172.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdel A, Moazed D. RNAi-directed assembly of heterochromatin in fission yeast. FEBS Lett. 2005;579:5872–5878. doi: 10.1016/j.febslet.2005.08.083. [DOI] [PubMed] [Google Scholar]
- Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH. MicroRNA expression in zebrafish embryonic development. Science. 2005;309:310–311. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- Zhang R, Peng Y, Wang W, Su B. Rapid evolution of an X-linked microRNA cluster in primates. Genome Res. 2007;17:612–617. doi: 10.1101/gr.6146507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Wang YQ, Su B. Molecular evolution of a primate-specific microRNA family. Mol Biol Evol. 2008;25:1493–1502. doi: 10.1093/molbev/msn094. [DOI] [PubMed] [Google Scholar]
- Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.