

Abstract
The question of when modern birds (Neornithes) first diversified has generated much debate among avian systematists. Fossil evidence generally supports a Tertiary diversification, whereas estimates based on molecular dating favor an earlier diversification in the Cretaceous period. In this study, we used an alternate approach, the inference of historical biogeographic patterns, to test the hypothesis that the initial radiation of the Order Psittaciformes (the parrots and cockatoos) originated on the Gondwana supercontinent during the Cretaceous. We utilized broad taxonomic sampling (representatives of 69 of the 82 extant genera and 8 outgroup taxa) and multilocus molecular character sampling (3,941 bp from mitochondrial DNA (mtDNA) genes cytochrome oxidase I and NADH dehydrogenase 2 and nuclear introns of rhodopsin intron 1, tropomyosin alpha-subunit intron 5, and transforming growth factor ß-2) to generate phylogenetic hypotheses for the Psittaciformes. Analyses of the combined character partitions using maximum parsimony, maximum likelihood, and Bayesian criteria produced well-resolved and topologically similar trees in which the New Zealand taxa Strigops and Nestor (Psittacidae) were sister to all other psittaciforms and the cockatoo clade (Cacatuidae) was sister to a clade containing all remaining parrots (Psittacidae). Within this large clade of Psittacidae, some traditionally recognized tribes and subfamilies were monophyletic (e.g., Arini, Psittacini, and Loriinae), whereas several others were polyphyletic (e.g., Cyclopsittacini, Platycercini, Psittaculini, and Psittacinae). Ancestral area reconstructions using our Bayesian phylogenetic hypothesis and current distributions of genera supported the hypothesis of an Australasian origin for the Psittaciformes. Separate analyses of the timing of parrot diversification constructed with both Bayesian relaxed-clock and penalized likelihood approaches showed better agreement between geologic and diversification events in the chronograms based on a Cretaceous dating of the basal split within parrots than the chronograms based on a Tertiary dating of this split, although these data are more equivocal. Taken together, our results support a Cretaceous origin of Psittaciformes in Gondwana after the separation of Africa and the India/Madagascar block with subsequent diversification through both vicariance and dispersal. These well-resolved molecular phylogenies will be of value for comparative studies of behavior, ecology, and life history in parrots.
Keywords: Cretaceous origin, divergence times, Gondwanan distribution, K/T boundary, molecular phylogeny, parrot, Psittaciformes, Tertiary origin
Introduction
The timing of the diversification of modern birds (Neornithes) is one of the most contentious issues in avian systematics. At the heart of the debate is a mismatch between the fossil record, which includes few modern forms prior to the Cretaceous/Tertiary (K/T) boundary (Feduccia 1999), and estimates based on molecular dating, which consistently place diversification of major avian orders well within the Cretaceous period (Cooper and Penny 1997; Kumar and Hedges 1998; Van Tuinen and Hedges 2001; Slack et al. 2006; Brown et al. 2007; but see Ericson et al. 2006, 2007). One independent line of evidence that has been used to support an earlier diversification date is the inference of biogeographic diversification patterns from modern distributions (Cracraft 2001). A number of avian clades, including Gruiformes, Caprimulgiformes, Apodiformes, and Passeriformes, have taxon distribution patterns consistent with an origin on the Gondwanan continent during the Cretaceous and subsequent vicariant diversification as Gondwana fragmented to form the modern landforms of Africa, India, South America, Antarctica, and Australia (Cracraft 2001). In the case of the Passeriformes, this hypothesis has received further support from molecular phylogenies that show both topologies and timing of major diversification events that are consistent with an origin in Gondwana (Barker et al. 2002; Ericson et al. 2002).
Debates about the origin of the parrots and cockatoos (Order Psittaciformes, hereafter “parrots” or “psittaciforms”) mirror those of the Neornithes in general, with some workers suggesting that they originated in Gondwana during the Cretaceous (Cracraft 1973; Forshaw 1989), whereas others, citing fossil evidence of stem group parrots from Tertiary deposits in Europe, conclude a post-Cretaceous diversification (Dyke and Mayr 1999; Mayr 2002). The general absence of migration among modern psittaciforms and their relatively circumscribed geographic ranges suggests that ancient patterns of diversification within the group have been less obscured by dispersal than in some other orders of birds (Feduccia 2003). Thus, the parrots offer a promising group in which to test alternative hypotheses for the timing of avian diversification. Such an analysis requires a well-supported phylogenetic hypothesis with extensive taxon sampling.
There are other compelling reasons for a broad investigation of phylogenetic relationships among psittaciforms. One reason is that they have one of the highest proportions of endangered species of any avian order, with 95 of 352 extant species listed as critical, endangered, or vulnerable by the International Union for the Conservation of Nature (Snyder et al. 2000). A better understanding of the evolutionary history of the group would facilitate the conservation of evolutionary processes and lineages that represent its evolutionarily significant units (Vane-Wright et al. 1991; Faith 1992; Moritz 2002). A second reason is that parrots are the focus of an increasing number of comparative studies in such areas as vocal communication (Bradbury 2003), brain evolution (Iwaniuk et al. 2005), craniofacial morphology (Tokita et al. 2007), longevity (Brouwer et al. 2000), nesting behavior (Brightsmith 2005), life-history trait evolution (Munshi-South and Wilkinson 2006), invasion biology (Cassey et al. 2004), global patterns of species richness (Davies et al. 2007), and evolution of mitochondrial control region duplications (Eberhard et al. 2001). The availability of a well-supported molecular phylogeny with broad taxonomic sampling would enhance the rigor of such comparative studies.
A third reason for pursuing a molecular phylogeny is that the taxonomy of parrots has been hampered by their relatively homogenous morphology. Early taxonomies varied considerably in the number of families and subfamilies identified within the Order Psittaciformes, with classifications ranging from 7 families, including 1 with 6 subfamilies (Salvadori 1891; Thompson 1899), to a single family with 4 subfamilies (Smith 1975). The influential works by Forshaw (1989, 2006) largely follow Smith (1975) at the tribal level, although in the more recent treatment, the Australasian cockatoos are elevated to family level (see also Homberger 2006). Here we have followed the treatment of Rowley (1997) and Collar (1997) within the “Handbook of the Birds of the World” (del Hoyo et al. 1997), who divide the Order Psittaciformes into 2 families, Cacatuidae and Psittacidae. Rowley (1997) further divides Cacatuidae into 3 subfamilies: Nymphicinae (containing Nymphicus hollandicus), Cacatuinae (containing Callocephalon, Eolophus, and Cacatua), and Calyptorhynchinae (consisting of Probosciger and Calyptorhynchus). The Psittacidae are divided into 2 subfamilies: the Loriinae, containing the 12 genera of the lories and lorikeets, and the Psittacinae, containing 66 genera subdivided among 9 tribes: Psittrichadini, Nestorini, Strigopini, Micropsittini, Cyclopsittacini, Platycercini, Psittaculini, Psittacini, and Arini. The more recent arrangement of Forshaw (2006) differs primarily in reducing the size of Psittacinae by joining tribes Strigopini and Nestorini in a subfamily Strigopinae and elevating the tribes Micropsittini and Psittrichadini to subfamily level, all within the Psittacidae. All the authors of recent classifications have expressed reservations concerning their own arrangements, particularly the unity of the large subfamily Psittacinae, and each have advocated further phylogenetic investigation (Collar 1997; Rowley 1997; Forshaw 2006; Homberger 2006).
A number of recent studies employing molecular methods have produced well-supported phylogenies, but these have generally been restricted to a few genera (Brown and Toft 1999; Ribas and Miyaki 2003; Russello and Amato 2004; Ribas et al. 2005; Astuti et al. 2006; Tokita et al. 2007) or a specific biogeographic region such as the Neotropics (Miyaki et al. 1998; Tavares et al. 2006) and have not provided a broad perspective on the relationships among basal psittaciform lineages. de Kloet RS and de Kloet SR (2005) produced a molecular phylogeny with broader sampling (47 of 82 extant genera) based on a single intron of the Z-linked spindlin gene. Although this analysis supported previous phylogenies and taxonomy in some areas (e.g., monophyly of the Neotropical parrots, tribe Arini), it also suggested that many traditional taxonomic groups (sensu Collar 1997) were polyphyletic (e.g., tribes Platycercini, Psittacini, and Psittaculini). It also found strong support for placement of several New Zealand taxa (kakapo, Strigops habroptilus; kea, Nestor notabilis; and kaka, Nestor meridionalis) as sister to a clade containing the remaining psittaciforms, contrary to most current taxonomies that place the Australasian cockatoos in the family Cacatuidae as sister to family Psittacidae (Smith 1975; Collar 1997; Rowley 1997; Forshaw 2006). Diversification patterns were interpreted as consistent with a Gondwanan origin for psittaciforms (de Kloet RS and de Kloet SR 2005). Broadly, similar patterns were seen a study by Tokita et al. (2007) that reconstructed relationships among representatives of 34 parrot genera using sequence data from the 16S and 12S ribosomal RNA mitochondrial genes. In both studies, the basal relationships among the cockatoos and other parrot lineages were not well resolved, perhaps due to the reliance on single loci for phylogenetic reconstruction. Numerous recent studies have found that sequencing more independently assorting genes and increasing the number of taxa improved the resolution and statistical support for tree topologies (Omland et al. 1999; Braun and Kimball 2002; Pereira et al. 2002, 2007; Bailey et al. 2006; Tavares et al. 2006).
In this paper, we employ a multilocus approach and broad generic sampling to test 2 alternative hypotheses for the timing of the origin and diversification of parrots. Chronograms based on a Cretaceous versus Tertiary origin are evaluated for their fit to the timing of known geological events and the resulting scenarios for psittaciform evolution. We find diversification patterns and timing are most consistent with a Gondwanan origin during the Cretaceous and subsequent diversification by vicariance. We also discuss implications of our phylogeny for current taxonomic classifications and conservation of the parrots.
Materials and Methods
Taxon Sampling
We sampled single representatives of 69 of the 82 extant genera of parrots (Psittaciformes) and representatives of 8 additional avian orders as outgroups for a total of 77 avian taxa. The 8 outgroups were selected from the orders Falconiformes, Passeriformes, Columbiformes, Cuculiformes, Piciformes, Coraciiformes, Strigiformes, and Coliiformes, each of which has been considered an ally of the parrots at one time (Sibley and Ahlquist 1990) or has been identified as a sister group in recent molecular phylogenies (Sorensen et al. 1999; Fain and Houde 2004; Ericson et al. 2006). We used frozen tissue or blood samples from wild birds and vouchered museum specimens as sources of DNA (for sample details, see supplementary table S1, Supplementary Material online). Taxonomic nomenclature at the species level follows Juniper and Parr (1998), whereas higher order classification follows Rowley (1997) and Collar (1997).
Character Sampling
Total genomic DNA was isolated using DNEasy extraction kits following manufacturer's protocols (Qiagen, Valencia, CA). We amplified 5 different gene regions using the polymerase chain reaction (PCR). Two of these regions were mitochondrial protein-coding genes: a portion of the cytochrome oxidase I gene (COI) and the entire protein-coding region of NADH dehydrogenase 2 gene (ND2) with portions of the adjoining tRNAs. Three noncoding nuclear introns were amplified with primers sited in the adjoining exons: tropomyosin alpha-subunit intron 5 (TROP), rhodopsin intron 1 (RDPSN), and transforming growth factor ß-2 (TGFB2) intron 5. Primer sequences and references are listed in table 1. Products were PCR amplified in 20 μl volumes with final concentrations of 1× PCR buffer, 0.8 mM of deoxynucleoside triphosphates, 2.0–3.0 mM of MgCl2, 1 μM of each primer, 2 μl of dilute template, 0.8 U of polymerase, and ultrapure water to volume. Magnesium concentrations and polymerase type (AmpliTaq or AmpliTaq Gold, Applied Biosystem, Foster City, CA) were optimized for each locus (conditions available upon request). PCR was performed on a PTC-200 Thermocycler (MJ Research, Waltham, MA) with a “touchdown” profile of 95 °C for 5 min (AmpliTaq) or 8 min (AmpliTaq Gold) followed by 30 cycles of 95 °C for 25 s, 60 °C for 30 s (dropping by 0.5 °C each cycle), and 72 °C for 90 s, then an additional 5 cycles of 95 °C for 25 s, 45 °C for 30 s, and 72 °C for 90 s followed by 10 min of extension at 72 °C. Products were examined by gel electrophoresis to confirm that only a single product of the expected size was produced per reaction.
Table 1.
Primers Used to Amplify and Sequence Gene Regions in This Study
| Primer Name | Gene Region | Sequence 5′–3′ | Source |
| LCOIp | COI | TTCTGATTYTTYGGMCACCCAG | a |
| HCOIp | COI | GTRTAGGCRTCTGGRTAGTC | a |
| L5216 | ND2 | GGCCCATACCCCGRAAATG | b |
| H5766 | ND2 | RGAKGAGAARGCYAGGATTYTTKCG | b |
| L5758 | ND2 | GGNGGNTGAATRGGNYTNAAYCARAC | b |
| H6313 | ND2 | ACTCTTRTTTAAGGCTTTGAAGGC | b |
| TROP-F | Tropomyosin | AATGGCTGCAGAGGATTA | c |
| TROP-R | Tropomyosin | TCCTCTTCAAGCTCAGCACA | c |
| TGFB2-F | TGFB2 | GAAGCGTGCTCTAGATGCTG | d |
| TGFB2-R | TGFB2 | AGGCAGCAATTATCCTGCAC | d |
| RDPSN-F | RDPSN | TGCTACATCGAGGGCTTCTT | e |
| RDPSN-R | RDPSN | CGAGTGACCAGAGAGCGATT | e |
We sequenced both strands of the resulting product using the Big Dye Terminator version 3.1 cycle sequencing kit and a 3100 Genetic Analyzer running Sequencing Analysis 3.7 (Applied Biosystems). We did not obtain sequences for all taxa from all gene regions. Data were coded as missing for the following gene region/taxon combinations: COI for the outgroup Colius colius; TROP for the parrot taxa Forpus passerinus and Psephotus varius; TGFB2 for the parrots Cyanopsitta spixii, Hapalopsittaca amazonina, and Nandayus nenday and the outgroup Picus canus; and RDPSN for the parrots Barnardius zonarius, Coracopsis vasa, C. spixii, Eunymphicus uvaeensis, Guaruba guarouba, H. amazonina, Leptosittaca branickii, N. nenday, Orthopsittaca manilata, Poicephalus robustus, P. varius, and Psittacus erithacus and the outgroups Columbina passerina, Falco peregrinus, P. canus, and Tockus flavirostris. Sequences were combined and edited to produce a single consensus sequence for each taxon and gene region using Sequencher version 4.5 (Gene Codes, Ann Arbor, MI). Consensus sequences were deposited in GenBank under the accession numbers EU327596–EU327672 (ND2), EU621593–EU621668 (COI), EU660234–EU660306 (TGFB2), EU665562–EU665636 (TROP), and EU665501–EU665561 (RDPSN).
Phylogenetic Analysis
Sequences for each gene region were aligned separately in ClustalW (Chenna et al. 2003) using the default settings for gap opening and extension penalties followed by limited manual correction of gap placement. Gap opening and extension penalties of 5 and 2.5, respectively, were used for the RDPSN gene region to improve the alignment of clearly homologous regions at the terminal ends of sequences. The resulting alignments were combined in PAUP* version 4.0b10 (Swofford 1999) to create a data set of 3,915 aligned nucleotide positions for 77 taxa. Gaps in this sequence alignment created by insertions and deletions (indels) were treated as missing data in some of the subsequent phylogenetic analyses; this data set is referred to as the “uncoded-gaps” data set.
We tested the combinability of partitions using the incongruence length difference (ILD) test (Farris et al. 1994). Although there has been considerable debate regarding the utility of this test (Cunningham 1997; Yoder et al. 2001; Barker and Lutzoni 2002; Dowton and Austin 2002), recent work has suggested that it remains a valid first approach to examining the degree of conflict between different data partitions (Hipp et al. 2004). We used PAUP* to conduct pairwise ILD tests between each data partition, treating the 2 mitochondrial DNA (mtDNA) segments as a single partition. In all ILD tests, we excluded uninformative characters and taxa that were missing from either data set. Heuristic searches were performed with a single random addition and tree bisection-reconnection (TBR) branch swapping over 500 replicates with a maximum of 1,000 trees saved each replicate. The fraction of ILD null replicates greater than the initial partition values was used to assess significance at the alpha = 0.05 level after a table-wide Bonferroni correction for multiple tests (Sokal and Rohlf 1995). In addition, we performed separate phylogenetic analyses on each data partition to assess conflict among them. Analyses were conducted in PAUP* using the maximum parsimony (MP) criterion. We used heuristic searches with 10 random taxon-addition replicates and TBR branch swapping. All characters were equally weighted, and gaps were treated as missing data.
Combined phylogenetic analyses of the uncoded-gaps data set were conducted in PAUP* using both MP and maximum likelihood (ML) criteria. For MP, we used heuristic searches with 100 random taxon-addition replicates and TBR branch swapping. All characters were equally weighted and gaps were treated as missing data. To assess nodal support we 1) performed bootstrap analyses with 1,000 replicates, each consisting of heuristic searches with 10 random taxon-addition replicates and TBR branch swapping, and b) calculated Bremer decay index values using TreeRot version 2c (Sorenson 1999). To further assess the thoroughness of our parsimony searches, we conducted a parsimony ratchet analysis (Nixon 1999) implemented in TNT version 1.1 (Goloboff et al. 2000) accessed through Winclada version 1.62 (Nixon 2002). We performed 20 ratchets, each consisting of 200 iterations with default parameters of a sectorial search, 4% of characters upweighted and downweighted, 1,000 trees held at each iteration, and 5 rounds of tree fusion. For ML, we used the general time-reversible model plus invariant sites and a gamma distribution (GTR + I + G) model of nucleotide substitution selected by the Akaike information criteria implemented in Modeltest version 3.7 (Posada and Crandall 1998). The proportion of invariable sites (0.19) and gamma shape parameters (0.52) were determined empirically (table 2). A single heuristic search was conducted using the MP topology as the starting tree and TBR branch swapping in PAUP*. Nodal support was assessed with 100 bootstrap replicates, each consisting of a single ML run implemented in GARLI version 0.951 (Zwickl 2006) using the same ML model, random starting trees, and default parameters for the genetic search algorithm.
Table 2.
Data Characteristics for the 5 Gene Regions and the Combined Data Set
| COIa | ND2a | TROP | TGFB2 | RDPSN | Combined | |
| Aligned base pairs | 570 | 1,098 | 552 | 814 | 881 | 3,915 |
| Number variable (%) | 238 (42) | 781 (71) | 218 (38) | 454 (56) | 630 (72) | 2,321 (59) |
| Number parsimony informative (%) | 220 (38) | 678 (62) | 95 (17) | 246 (30) | 351 (40) | 1,590 (41) |
| Number coded indel characters | 0 | 17 | 38 | 72 | 118 | 245 |
| % Ab | 0.37 (28) | 0.36 (33) | 0.22 (21) | 0.24 (24) | 0.20 (21) | 28 (27) |
| % Cb | 0.40 (29) | 0.45 (36) | 0.20 (21) | 0.20 (22) | 0.26 (24) | 30 (28) |
| % Gb | 0.07 (17) | 0.04 (8) | 0.18 (18) | 0.23 (23) | 0.30 (30) | 17 (18) |
| % Tb | 0.16 (25) | 0.15 (22) | 0.40 (39) | 0.32 (31) | 0.24 (26) | 25 (28) |
| rACb | 0.44 | 0.07 | 2.02 | 1.00 | 1.61 | 1.11 |
| rAGb | 14.85 | 4.40 | 12.55 | 5.27 | 5.33 | 4.76 |
| rATb | 1.16 | 0.14 | 0.66 | 0.68 | 0.96 | 0.65 |
| rCGb | 0 | 0.16 | 1.68 | 1.25 | 1.43 | 0.49 |
| rCTb | 19.89 | 1.52 | 4.91 | 4.33 | 4.41 | 6.94 |
| Proportion invariant | 0.57 | 0.24 | 0 | 0 | 0 | 0.19 |
| Gamma | 0.73 | 0.64 | 0.55 | 1.70 | 2.04 | 0.52 |
| No. of treesc | 2 (2) | >200,000 (32,498) | >200,000 (>200,000) | 352 (98) | 4 (21) | |
| Number of resolved nodes in consensusc | 74 (74) | 30 (33) | 47 (43) | 50 (50) | 71 (65) | |
| Tree lengthc | 9,029 (8,996) | 472 (413) | 1,137 (1,051) | 1,608 (1,468) | 12,324 (12,001) | |
| Consistency Indexc | 0.204 (0.203) | 0.655 (0.656) | 0.651 (0.636) | 0.675 (0.659) | 0.323 (0.311) | |
| Retention Indexc | 0.364 (0.363) | 0.758 (0.768) | 0.724 (0.726) | 0.739 (0.739) | 0.459 (0.456) | |
NOTE.—RI, retention index CI, Consistency index.
The 2 mtDNA partitions were treated as a single partition in parsimony searches.
Base frequencies and substitution rates from the best fit model found by Mr. Modeltest version 2.2 (Modeltest version 3.7 for combined data set) using the Akaike information criteria. Observed base frequencies are given in parentheses.
Results from parsimony searches for the coded-gaps data set (results from uncoded-gaps data set in parentheses).
Alignment gaps at the sequence level caused by indels are particularly common in nuclear introns and may contain substantial phylogenetic information (Prytchitko and Moore 2000; Kimball et al. 2001; Fain and Houde 2004). To assess the level of phylogenetic signal provided by indels, we coded gaps as presence–absence characters using the “simple indel coding method” (Simmons and Ochoterena 2000) as implemented in the software GapCoder (Young and Healy 2003). The 5 regions were concatenated before coding and gap characters appended to sequence alignments in PAUP* to create a data set of 4,160 characters for 77 taxa; this data set is referred to as the “coded-gaps” data set. We tested for combinability of partitions as above with the scored gap characters for each gene region included in that data partition, employing both ILD tests and comparisons of MP trees for each separate partition.
We conducted searches on the combined partitions with MP and assessed nodal support as described above for the uncoded-gaps data set. To assess the effects of uncertainty in the alignment of the RDPSN partition, we used the same MP search parameters in PAUP* to analyze a combined data set of all partitions except for RDPSN and its associated coded gaps and compared the resulting strict consensus tree and bootstrap values with those obtained for all 5 partitions. ML searches were not performed for this data set due to the difficulty in incorporating gap character data into a general model for nucleotide substitutions. Instead, Bayesian analysis was performed using MrBayes version 3.1 (Ronquist and Huelsenbeck 2003). We employed separate models for the sequence data from each of the 5 sequence partitions (treating the 2 mtDNA regions separately) as selected in the program Mr. Modeltest (Nylander 2004) using the Akaike information criteria and the Jukes–Cantor model for a sixth data partition consisting of the gap characters from all the gene regions. We used uninformative priors for substitution rates, and base frequencies and default priors were used for all other parameters. We ran 2 independent runs of Metropolis-coupled Markov chain Monte Carlo analyses, each with 2 replicates of 1 cold chain and 3 heated chains with a default temperature setting of 0.2. Chains were run for 5 million generations and sampled every 100 generations with the first 25% of samples discarded as burn-in. We judged the runs to be stationary when the standard deviation (SD) of split frequencies equaled 0.01 or less and traces of the log-likelihood values showed no distinct trends. We further examined the traces of all estimated parameters using the program Tracer version 1.4 to ensure stationarity had been reached (Rambaut and Drummond 2007a). Fifty percent majority-rule consensus trees were created from the retained trees for each independent run.
Biogeographic Analysis
We used ancestral area reconstruction (Bremer 1992, 1995) and our phylogenetic reconstruction test the hypothesis that the Psittaciformes radiated within Gondwana. The ancestral area reconstruction method treats biogeographic areas as characters for taxa and compares the number of gains and losses of each area on a cladogram using forward or reverse Camin-Sokal parsimony (Bremer 1992). The area with the highest gain/loss ratio is estimated to be the likeliest ancestral area, based on the assumptions that area characters that are reconstructed as plesiomorphic on the cladogram and are more widely represented are more likely to have been part of the ancestral area. We coded each genus represented in our phylogeny as present or absent in each of 5 biogeographic areas: Australasia (Australia, New Zealand, and New Guinea), Asia, Oceania (islands of the Western Pacific excluding Australasia), the New World, and Africa. Gains and losses were calculated on the tree obtained from the Bayesian search using the irreversible parsimony option implemented in MacClade version 4.06 (Maddison DR and Maddison WP 2003).
Timing of Divergences
We estimated divergence times among parrot lineages using 2 different calibration dates representing separate historical events. To evaluate the hypothesis of a Cretaceous origin, we assigned a date of 82 million years ago (MYA) to the basal split between the New Zealand endemics Nestor and Strigops and the clade containing all other psittaciforms. This date corresponds to the minimum age for the current estimate of 82–85 MYA for the splitting of New Zealand from Gondwana (McLoughlin 2001). To evaluate the hypothesis of a Tertiary origin, we applied a minimum date of 50 MYA to the same basal node joining Nestor and Strigops to the remaining extant psittaciforms, corresponding to a hypothesized divergence between modern parrots and fossil forms found in Europe and dated to the Lower Eocene (Mayr 2002). For both calibration points, we estimated divergence times using 2 different approaches.
The first dating approach was a penalized likelihood method implemented in the program r8s version 1.7 (Sanderson 2002) with branch lengths and topology resulting from the tree obtained with Bayesian search criteria. For each calibration point, we performed a separate run using a smoothing factor of 10 obtained through cross-validation, the ancestor–descendant penalty function with an additive scale for rate penalty, and the truncated Newton method of optimization with bound constraints. Ninety-five percent confidence intervals (CIs) were estimated using the r8s bootstrap kit (Eriksson 2007). One hundred bootstrap replicates of the original data set were created using SeqBoot in the PHYLIP package. Branch lengths for each bootstrap replicate were calculated in PAUP*. The resulting 100 trees, each with the same topology but different branch lengths, were run in r8s using the parameters above to calculate a distribution of divergence date estimates. The mean divergence date, the SD of dates, and 95% CI for the mean were calculated for each node across the 100 bootstrap replicates following (Rutschmann et al. 2004).
The second dating approach was a Bayesian relaxed-clock method that allows for the incorporation of uncertainty in calibration dates and of variation in rates of evolution along different branches of the phylogenetic tree (Thorne et al. 1998; Drummond et al. 2006). We performed separate runs of Bayesian relaxed-clock analyses for each of the 2 calibration points using the program BEAST version 1.4.7 (Drummond and Rambaut 2007). We used the uncoded-gaps data set for our 69 parrot taxa only, outgroup rooted with the 2 New Zealand taxa (Nestor and Strigops). We used the GTR + I + G model of nucleotide substitution with estimated base frequencies, gamma shape distribution (with 4 categories), proportion of invariant sites, and a relaxed molecular clock with uncorrelated lognormal distribution of branch lengths. We specified a Yule tree prior and a mean root height of either 50 or 82 MYA, normally distributed with an SD = 2. All other priors were unspecified, and default search operators were retained. The chronograms resulting from the penalized likelihood searches in r8s were used as the starting trees for the 2 searches. Each search consisted of a single chain of 10 million generations sampled every 1,000 generations. We examined the runs for stationarity in Tracer version 1.4 (Rambaut and Drummond 2007a) and used TreeAnnotator version 1.4.7 (Rambaut and Drummond 2007b) to combine the last 7,501 trees from each run into a single 50% majority consensus tree. The resulting chronograms and the associated 95% highest posterior density (HPD) distributions around the estimated node ages were viewed in FigTree version 1.1.1 (Rambaut 2008).
Results
Sequence Characteristics
Sequences for COI were all 570 bp in length, whereas those for ND2 ranged from 1,087 to 1,093 bp due to the presence of small indels in the flanking tRNA regions. Neither mitochondrial segment exhibited stop codons nor frameshift mutations within protein-coding regions, suggesting that nuclear pseudogenes of mitochondrial genes were not mistakenly sequenced. Sequence lengths were more variable for nuclear introns. TROP sequences ranged in size from 510 to 540 bp within the psittaciforms and from 527 to 532 bp in the outgroup taxa. RDPSN sequences ranged from 722 to 795 bp within the psittaciforms and from 805 to 816 bp in the outgroup taxa. TGFB2 sequences generally ranged from 610 to 629 bp within psittaciforms and from 587 to 630 bp in the outgroup taxa although the parrot Charmosyna papou exhibited a large insertion that resulted in a sequence of 766 bp.
After alignment and concatenation, the combined sequences totaled 3,915 bp, with 1,668 bp from mitochondrial DNA (mtDNA) and 2,247 bp from nuclear introns (table 2). As shown in table 2, the 5 aligned regions varied in the percentage of variable sites (38–72%), the percentage of sites that were parsimony informative (17–62%), and the number of indel sites coded as gaps (0–118 gaps). The G + C content was 46% for COI, 44% for ND2, 39% for TROP, 45% for TGFB2, 53% for RDPSN, and 46% for the concatenated sequences (table 2). Chi-square tests of base composition heterogeneity implemented in PAUP*4.0b10 were not significant for any individual segments (COI, χ2 = 92, degrees of freedom [df] = 228, P = 1; ND2, χ2 = 240, df = 228, P = 0.28; TROP, χ2 = 16, df = 228, P = 1; TGFB2, χ2 = 22, df = 228, P = 1; and RDPSN, χ2 = 87, df = 228, P = 1).
Phylogenetic Analysis
Separate MP searches on the combined mtDNA partition and the 3 nuclear intron partitions resulted in strict consensus trees exhibiting varying degrees of resolution and homoplasy (table 2). The 3 nuclear intron partitions showed generally similar levels of homoplasy for both the uncoded-gaps and the coded-gaps data sets, whereas the combined mtDNA partition had higher levels of homoplasy as measured by the consistency and retention indices (table 2). The topology of the strict consensus trees from these MP searches was consistent with this result; the mtDNA tree showed generally good resolution at more terminal nodes, whereas the nuclear introns showed better resolution at the more basal nodes, although the TROP tree was poorly resolved throughout. The TGFB2 and TROP analyses both resulted in >200,000 equally parsimonious trees for the coded-gaps data set.
We evaluated the suitability of combined phylogenetic analyses using both ILD tests and comparisons of topology and nodal support resulting from MP analyses of separate data partitions. For the uncoded-gaps data set, pairwise ILD tests indicated that there were no conflicts among any of the 3 nuclear intron partitions nor between those partitions and the combined mtDNA partition (table 3). Similar results were found with ILD tests among the 4 partitions of the coded-gaps data set (table 3). Examination of the strict consensus trees from MP searches for each separate partition indicated very few points of conflict among partitions in either data set. The most notable area of conflict among the partitions was in the placement of the taxa C. vasa of Madagascar and Psittrichas fulgidus of New Guinea. In the mtDNA MP consensus trees, Coracopsis was grouped in a clade with the Australian taxa Neophema elegans and Neopsephotus bourkii, whereas Psittrichas was grouped with the African taxa P. robustus and P. erithacus. In the TGFB2 consensus tree, these 2 taxa formed a clade together, whereas in the TROP and RDPSN trees, neither species exhibited a close relationship to any other taxa. Given the strong likelihood that this variable placement of Coracopsis and Psittrichas resulted from long-branch attraction and the general lack of conflicts detected among partitions in both data sets, we proceeded with a combined approach in which all partitions were analyzed jointly.
Table 3.
Results of the ILD Tests for Pairwise Comparisons between the Data Partitions
| Partitions | mtDNA | TROP | TGFB2 | RDPSN |
| mtDNA | 1.0 | 0.98 | 0.81 | |
| TROP | 1.0 | 0.14 | 0.74 | |
| TGFB2 | 0.87 | 0.06 | 0.19 | |
| RDPSN | 0.84 | 0.74 | 0.18 |
NOTE.—P values from the uncoded-gaps data set are on the upper half of the matrix and those from the coded-gaps data set are on the lower half in italics.
Analyses of the combined partitions using MP in PAUP* resulted in 4 equally parsimonious trees for the coded-gaps data set and 21 in the uncoded-gaps data set. The parsimony ratchet analysis of the coded-gaps data set found the same 4 most parsimonious trees as the heuristic search in PAUP*; these were located in 6 of the 20 ratchets. A similar result was found for the uncoded-gaps data set, in which the same 21 most parsimonious trees were found in 19 of 20 ratchets. The topologies of strict consensus trees derived from the coded-gap (fig. 1) and uncoded-gap data sets (data not shown) differed only in the degree of resolution at 4 nodes, with the coded-gaps data set showing more resolution at each node. Likewise, the MP tree obtained with the coded-gaps data set with the RDPSN partition excluded differed from the coded-gaps analysis with all 5 partitions at only 9 nodes within the psittaciforms. All these differences consisted of the collapsing of poorly supported nodes in 1 of the 2 trees, suggesting that the uncertainty in the alignment of RDPSN had minimal effect on tree topologies. Based on the similarities between these MP trees, all further comparisons are based on the MP tree resulting from the coded-gaps data set with all 5 partitions included. The 50% majority consensus trees obtained from the 2 independent runs of the Bayesian analyses of the coded-gaps data set were identical in topology to each other (fig. 2) and substantially similar to the MP trees. The ML analyses of the uncoded-gaps data sets (supplementary fig. S1, Supplementary Material online) had similar but not identical topologies to those obtained with the other 2 criteria. Differences among the various trees always corresponded to nodes with lower bootstrap support and Bremer decay indices (in MP analyses), low posterior support values (in Bayesian analyses), and short internode branches (in ML and Bayesian analyses).
FIG. 1.—
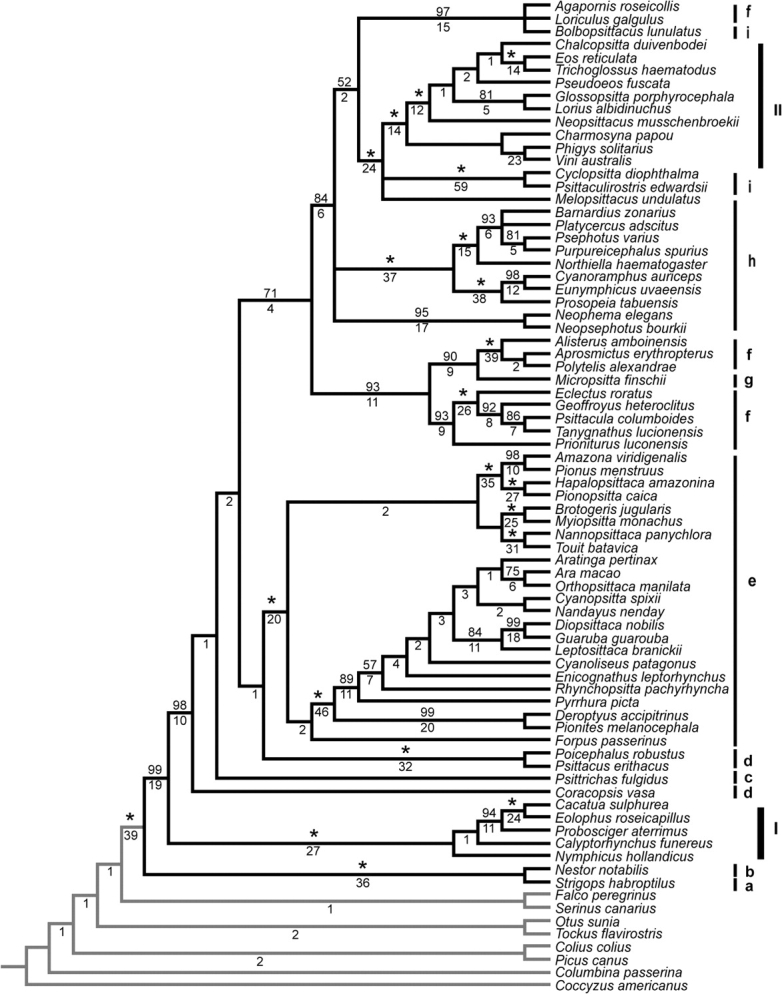
Cladogram of relationships among representatives of 69 parrot genera and 8 outgroup taxa based on a strict consensus of 4 trees obtained from combined parsimony (MP) analysis of the 4 sequence partitions (COI + ND2, TROP, TGFB2, and RDPSN) with coded gaps. MP bootstrap support values for each node are shown above the branches with values of 100% indicated by asterisks; Bremer decay index values are shown below. The classifications of Rowley (1997) and Collar (1997) are indicated by bars on the right: I) family Cacatuidae, II) subfamily Loriinae, and, within the subfamily Psittacinae, the tribes a) Strigopini, b) Nestorini, c) Psittrichadini, d) Psittacini e) Arini, f) Psittaculini, g) Micropsittini, h) Platycercini, and i) Cyclopsittacini.
FIG. 2.—
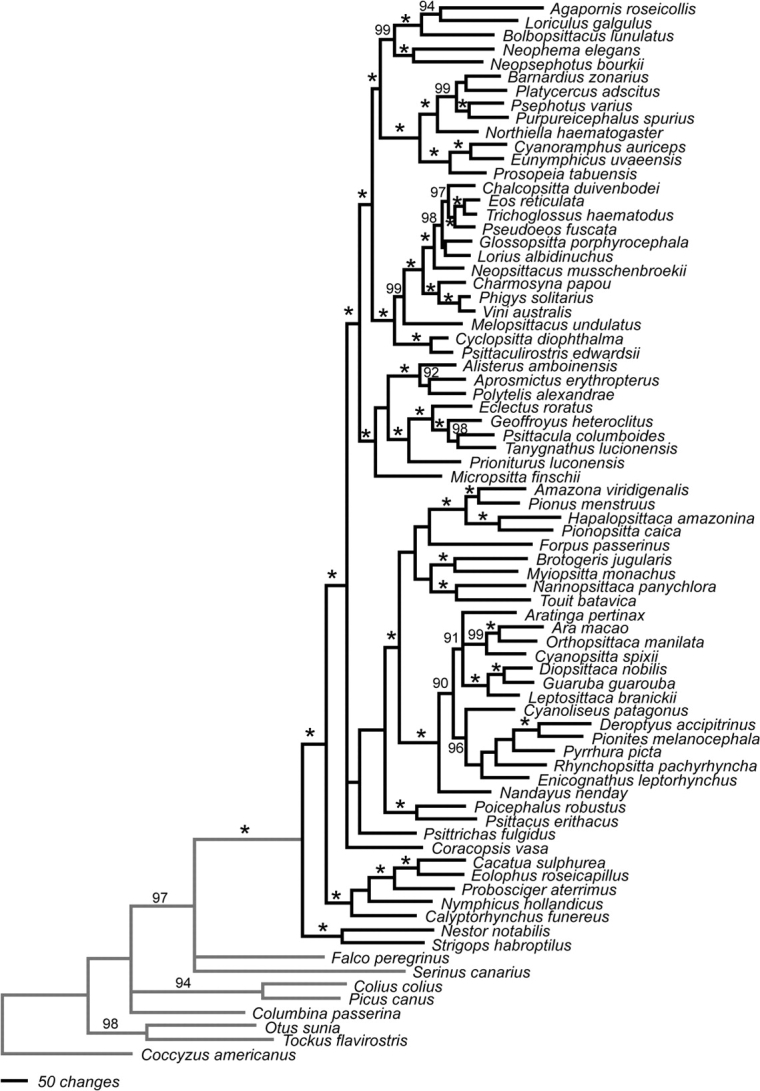
Phylogram of relationships among representatives of 69 parrot genera and 8 outgroup taxa based on Bayesian analysis of the 4 sequence partitions (COI + ND2, TROP, TGFB2, and RDPSN) and a fifth partition consisting of coded gaps from the 4 sequence partitions. Bayesian posterior probabilities ≥0.90 are indicated as percentages above the branches, values of 1.0 are indicated with an asterisk. The scale bar indicated the number of changes (base substitutions or changes in state of indel characters).
The trees obtained from these various analyses provided strong support for a number of notable relationships within the parrots (figs. 1 and 2; supplementary fig. S1, Supplementary Material online). First, the sister relationship between a clade containing the New Zealand taxa N. notabilis (Nestorini within Psittacidae) and S. habroptilus (Strigopini within Psittacidae) and all other psittaciforms was strongly supported. There was also strong support for a sister relationship between the Australasian cockatoos (Cacatuidae) and all remaining psittaciforms (Psittacidae minus Nestorini and Strigopini). The next-most basal nodes were less resolved, with low support levels and short internode distances present in all analyses and topological differences among them. Beyond these nodes (moving toward the terminal nodes), the different analyses again showed general agreement. All analyses recovered 5 well-supported clades within the Psittacidae: (1) P. fulgidus, the vulturine parrot endemic to New Guinea (Psittrichadini within subfamily Psittacinae); (2) C. vasa, a Madagascar endemic (Psittacini within the Psittacinae); (3) the African genera Poicephalus and Psittacus (both Psittacini within Psittacinae); (4) a large clade consisting of the remaining Old World parrots (subfamily Loriinae and tribes Micropsittini, Cyclopsittacini, Platycercini, Psittaculini, and Psittacini within Psittacinae); and (5) a clade consisting of all the Neotropical parrots (Arini within Psittacinae). In the strict consensus tree obtained in the coded-gap MP analyses, C. vasa was sister to the other 4 clades, and P. fulgidus was sister to the remaining 3 clades; both these nodes had less than 50% support in the coded-gaps MP bootstrap analysis (fig. 1). In the ML and Bayesian analyses, C. vasa was found in a polytomy with the large Old World clade (clade [d] above) and a clade containing P. fulgidus, the African clade of Poicephalus and Psittacus and the Neotropical parrots (fig. 2; supplementary fig. S1, Supplementary Material online). The Bayesian, ML, and MP trees all recovered the African genera Poicephalus and Psittacus as sister to the Neotropical parrots.
Five distinct subclades within the Neotropical radiation (Tribe Arini) were strongly supported with 100% bootstrap support (MP and ML) and posterior probability (Bayes) values (figs. 1 and 2; supplementary fig. S1, Supplementary Material online). These are (a) Amazona, Pionus, Hapalopsittaca, and Pionopsitta; (b) Ara, Orthopsittaca, Aratinga, Cyanopsitta, Nandayus, Diopsittaca, Guarouba, Leptosittaca, Cyanoliseus, Enicognathus, Rhynchopsitta, Pyrrhura, Deroptyus, and Pionites; (c) Nannopsittaca and Touit; (d) Brotogeris and Myiopsitta; and (e) F. passerinus. Branching relationships among these subclades were not well supported in any analysis and varied among them (figs. 1 and 2; supplementary fig. S1, Supplementary Material online).
The large Old World clade composed of members of the subfamilies Psittacinae and Loriinae can be subdivided into 5 subclades supported by nodes with >95% bootstrap support in MP and ML analyses and >95% posterior probabilities in Bayesian analyses (figs. 1 and 2; supplementary fig. S1, Supplementary Material online). The first of these 6 subclades consists of representatives of the African Agapornis and the Australasian Loriculus, both placed in the tribe Psittaculini, and the Philippine endemic, Bolbopsittacus lunulatus of tribe Cyclopsittacini. The second subclade consists of the Australian Neophema and Neopsephotus, both members of the tribe Platycercini. The third subclade includes the Australian Barnardius, Platycercus, Psephotus, Purpureicephalus, Northiella, and Prosopeia and the western Pacific genera Cyanoramphus and Eunymphicus all traditionally classified in the tribe Platycercini.
The fourth subclade within the large Old World clade is composed of the Australasian Cyclopsitta and Psittaculirostris (both tribe Cyclopsittacini), the Australian Melopsittacus (tribe Platycercini), and a large group consisting of 10 genera from Australasia and Oceania traditionally placed in the subfamily Loriinae: Chalcopsitta, Eos, Trichoglossus, Pseudeos, Glossopsitta, Lorius, Neopsittacus, Charmosyna, Phigys, and Vini. Bayesian, ML, and MP analyses all supported the monophyly of the Loriinae and placed Cyclopsitta and Psittaculirostris in a group distinct from Melopsittacus, but the branching order between these 3 groups differed between the 3 analyses. The fifth subclade includes the genus Micropsitta (tribe Micropsittini) and representatives of 8 genera traditionally placed in the tribe Psittaculini: Alisterus, Aprosmictus, Polytelis, Eclectus, Geoffroyus, Psittacula, Tanygnathus, and Prioniturus. Both ML and MP analyses nested Micropsitta well within the representatives of the Psittaculini (fig. 1; supplementary fig. S1, Supplementary Material online), whereas the Bayesian analysis placed this genus sister to the other genera (fig. 2). Thus, 3 of these 5 subclades in this Old World assemblage contain members of more than one tribe or subfamily and both the subfamily Psittacinae and 3 of the tribes within it (Cyclopsittacini, Platycercini, and Psittaculini) are polyphyletic (fig. 1).
There was strong support at the base of the tree for the monophyly of the psittaciforms relative to the 8 outgroups. Branching relationships outside the psittaciforms were less strongly supported, differed between the MP, ML, and Bayesian analyses, and showed no clear indication of a close sister relationship between the parrots and any of the outgroup taxa examined in our study (figs. 1 and 2; supplementary fig. S1, Supplementary Material online).
Biogeographic Analysis
When represented as a character on the Bayesian phylogenetic reconstruction, the biogeographic region of Australasia was both widely represented in our phylogeny and tended to be plesiomorphic (data not shown). In total, 32 of the 69 genera represented in our phylogeny are restricted in distribution to Australasia, and an additional 5 genera have some representatives found there. The genera Nestor and Strigops, which are sister to the clade containing all other psittaciforms, are restricted to New Zealand, and the lineage represented by the 5 genera of Australasian cockatoos is sister to all remaining psittaciforms.
These patterns were reflected in the results from ancestral area analysis and provide strong support for Australasia as the geographic origin of the psittaciforms. For the 5 alternative biogeographic regions of origin, the ratio of gains to losses was highest for Australasia. After rescaling by this value, the ancestral area quotient for all other regions was ≤0.11 (table 4).
Table 4.
Ancestral Area Reconstruction (Bremer 1992, 1995) of the Psittaciformes Using 5 Alternative Biogeographic Regions
| Biogeographic Region | Gains | Losses | G/La | Ancestral Area Quotientb |
| Australasia | 17 | 7 | 2.43 | 1.00 |
| Asia | 5 | 18 | 0.28 | 0.11 |
| Oceania | 2 | 10 | 0.20 | 0.08 |
| New World | 1 | 6 | 0.17 | 0.07 |
| Africa | 4 | 15 | 0.27 | 0.11 |
Ratio of inferred gains to inferred losses.
Determined by dividing each G/L ratio by the highest G/L ratio in the analysis.
Timing of Divergences
Divergence times were estimated using both a penalized likelihood approach with CIs assessed through bootstrap replicates (supplementary fig. S2, Supplementary Material online ; table 5) and a Bayesian relaxed-clock approach that allowed incorporation of uncertainty associated with our 2 contrasting calibration dates (fig. 3 and table 5). With the first calibration date of 82 MYA, the split between the cockatoos and the remaining psittaciforms was estimated with the Bayesian relaxed-clock method to have occurred at 74.13 MYA (63.94–82.93 MYA) (mean [95% HPD]), prior to the K/T boundary at 65 MYA. Subsequently, there was a diversification of parrot lineages between 60 and 65 MYA corresponding to the initial separation of Australia and New Guinea from East Antarctica that led to the separation of the lineages leading to the Neotropical tribe Arini, its African sister clade of Poicephalus and Psittacus, Psittrichas of New Guinea, Coracopsis of Madagascar, and the large clade consisting of the remaining Old World lineages (fig. 3). Diversification of this large Old World clade into the major subclades identified earlier was complete by 30 MYA, by which point Australia, Antarctica, and South America were completely separated. Divergence date estimates from the penalized likelihood approach were generally in agreement with those from the Bayesian relaxed-clock approach.
Table 5.
Means and 95% HPDs for Divergence Dates from the Bayesian Relaxed-Clock Approach, and Means, SD, and 95% CIs for Divergence Dates from 100 Bootstrap Replicates of the Penalized Likelihood Analysis for Alternative Calibration Dates of 82 and 50 MYA for the Basal Psittaciform Node
| Calibration of 82 MYA |
Calibration of 50 MYA |
|||||||||
| Bayesian Relaxed-Clock |
Penalized Likelihood |
Bayesian Relaxed-Clock |
Penalized Likelihood |
|||||||
| Nodea | Mean (MYA) | 95% HPDb (MYA) | Mean (MYA) | SD (MY) | 95% CIc (MYA) | Mean (MYA) | 95% HPDb (MYA) | Mean (MYA) | SD (MY) | 95% CIc (MYA) |
| 1 | 81.91 | 78.1–85.9 | 82 | 49.84 | 45.9–53.7 | 50 | ||||
| 2 | 74.13 | 63.9–82.9 | 70.05 | 2.04 | 69.6–70.4 | 45.04 | 38.0–51.4 | 42.74 | 1.23 | 42.4–42.9 |
| 3 | 53.11 | 38.1–67.2 | 59.14 | 3.07 | 58.5–59.7 | 32.78 | 23.3–42.4 | 36.05 | 1.85 | 35.6–36.4 |
| 4 | 51.24 | 39.1–62.4 | 53.98 | 2.96 | 53.3–54.5 | 30.82 | 23.6–38.7 | 32.93 | 1.82 | 32.5–33.2 |
| 5 | 64.01 | 54.8–72.9 | 59.40 | 2.04 | 58.9–59.8 | 38.83 | 32.5–44.6 | 36.25 | 1.24 | 35.9–36.5 |
| 6 | 39.19 | 27.2–51.1 | 39.30 | 2.71 | 38.7–39.8 | 23.57 | 16.2–31.0 | 23.99 | 1.65 | 23.6–24.3 |
| 7 | 55.28 | 46.9–63.2 | 49.53 | 2.09 | 49.1–49.9 | 33.67 | 28.1–39.1 | 30.23 | 1.26 | 29.9–30.4 |
| 8 | 60.67 | 51.6–69.6 | 58.57 | 2.15 | 58.1–58.9 | 37.03 | 30.9–43.0 | 35.75 | 1.27 | 35.5–36.0 |
| 9 | 51.83 | 42.2–61.9 | 53.43 | 2.32 | 52.9–53.8 | 31.66 | 25.2–37.8 | 32.63 | 1.37 | 32.3–32.8 |
| 10 | 52.84 | 44.1–61.0 | 51.76 | 2.37 | 51.2–52.2 | 32.41 | 26.6–38.0 | 31.60 | 1.43 | 31.3–31.8 |
| 11 | 44.77 | 36.1–52.8 | 44.71 | 2.62 | 44.1–45.2 | 27.54 | 21.8–33.1 | 27.28 | 1.60 | 26.9–27.5 |
| 12 | 40.97 | 31.3–49.6 | 42.44 | 2.60 | 41.9–42.9 | 25.42 | 19.3–31.6 | 25.92 | 1.56 | 25.6–26.2 |
| 13 | 55.97 | 47.4–64.6 | 54.21 | 2.21 | 53.7–54.6 | 34.25 | 28.3–40.1 | 33.09 | 1.32 | 32.8–33.3 |
| 14 | 37.15 | 29.5–44.8 | 40.24 | 2.33 | 39.7–40.6 | 22.38 | 17.4–27.6 | 24.56 | 1.40 | 24.2–24.8 |
| 15 | 38.78 | 31.1–45.9 | 34.75 | 2.12 | 34.3–35.1 | 23.43 | 18.5–28.2 | 21.23 | 1.28 | 20.9–21.4 |
See figure 3 and supplementary figure S2 (Supplementary Material online) for placement of nodes.
95% HPD.
95% CI on the mean node age.
FIG. 3.—

Chronograms showing divergence times among parrot genera based a Bayesian relaxed-clock approach, with 2 alternative dates for the basal divergence between the New Zealand endemics Nestor and Strigops and the remaining psittaciforms: (a) node dated to 50 MYA, based on the hypothesized divergence between modern psittaciforms and fossils dated to the Lower Eocene; and (b) node dated to 82 MYA, based on the split between New Zealand and Gondwana. Uncertainty in the timing of these alternative divergences is incorporated with both calibration estimates representing means of normal distributions with SD = 2; error bars on the nodes illustrate the 95% HPD of node ages. Shaded vertical bars indicate timing of major geological events that may have contributed to diversification through vicariance: separation of New Zealand from Gondwana, 80–85 MYA; the K/T boundary, 65 MYA; the initial separation of Australia from East Antarctica, 58–61 MYA; the final separation of Australia from East Antarctica, 36–41 MYA; the final separation of South America from West Antarctica, 28–32 MYA; and Australia and New Guinea approach southeast Asia, 20–24 MYA.
In contrast, when the basal parrot node was calibrated to 50 MYA, the split between the cockatoos and the remaining psittaciforms was estimated to have occurred 45.04 MYA (38.04–51.40 MYA) (mean [95% HPD]), and the split between Psittrichas, Coracopsis, the Poicephalus/Psittacus clade, Tribe Arini, and the large Old World clade occurred 38–42 MYA, just prior to the time Australia is thought to have split from Antarctica (fig. 3a; table 5). Subsequent diversification of the Neotropical parrot lineages occurred starting 33 MYA, a period roughly coincident with the separation of South America from West Antarctica (table 5).
Discussion
We employed multiple genetic loci, extensive taxon sampling, and several different analytical approaches to reconstruct the evolutionary history of the parrots. The resulting phylogenies had generally strong nodal support and similar topologies. Biogeographic analyses based on these phylogenies support a hypothesis of an origin and initial diversification of psittaciforms in Gondwana during the Cretaceous but subsequent to the separation of the Africa and India/Madagascar block. Below we summarize these results, compare them to other recently published phylogenies and discuss their implications for parrot diversification, taxonomy, conservation, and comparative studies.
Geographic and Temporal Origin of Parrots
The parrots have long been thought to originate in Gondwana based on their present distribution across the southern continents that formerly composed this supercontinent (Cracraft 1973, 2001). The results of our ancestral area analyses support this hypothesis with Australasia as the likeliest region of origin. This result stems from the high number of genera endemic to the region, several of which are rooted at or near the base of the psittaciform tree. The basal split between the New Zealand taxa Nestor and Strigops further suggests that this origin occurred although New Zealand was still connected to Australasia as part of the Gondwana supercontinent during the Cretaceous period. Analyses using similar approaches suggest that both Passeriformes (Barker et al. 2002) and Columbiformes (Pereira et al. 2007) also originated in Gondwana during the Cretaceous.
The timing of the origin and diversification of parrots is more difficult to determine given the patchy nature of the parrot fossil record (Collar 1997; Dyke and Mayr 1999; Mayr 2002). We have used our phylogenetic reconstruction and 2 different divergence dating approaches to evaluate 2 contrasting scenarios for the timing of parrot diversification. The first scenario follows other recent studies of passerines and psittaciforms (Ericson et al. 2002; de Kloet RS and de Kloet SR 2005; Tavares et al. 2006) that apply a date of 82 MYA to the basal node between a New Zealand group sister to the remainder of the order, a date that corresponds to the split of New Zealand from Gondwana (fig. 3a). This date is consistent with the hypothesis of an origin and initial diversification of modern avian orders during the late Cretaceous (Cooper and Penny 1997; Kumar and Hedges 1998; Van Tuinen and Hedges 2001; Slack et al. 2006). The resulting chronograms (fig. 3b; supplementary fig. S2b, Supplementary Material online) are both broadly consistent with a hypothesis of vicariant diversification of parrots as the various modern continents and New Zealand split from Gondwana. Under this scenario, most of the initial diversification in psittaciforms, including the origin of the Neotropical tribe Arini, is dated to 58–65 MYA with either estimation approach, a period coincident with the opening of the Coral Sea between New Guinea and Australia and the initial opening of a seaway between Australia and East Antarctica (McLoughlin 2001). Uplift of the Transantarctic Mountains and associated accumulation of ice in Antarctica around 50 MYA also may have contributed to vicariance between the South American and Australian lineages (Wilford and Brown 1994), although the deep marine Drake Passage is thought to have opened between South America and West Antarctica only 32–28 MYA (McLoughlin 2001). Cycles of cooling and warming in Australia and nearby East between 40 and 55 MYA could have spurred further diversification of lineages within those continents prior to their final separation by a deep oceanic channel around 35.5 MYA (Wilford and Brown 1994; McLoughlin 2001). The approach of Australia and New Guinea to their present positions relative to Southeast Asia (20–25 MYA) would have provided a bridge for dispersal to mainland Asia and across to Africa and might have contributed to further cladogenesis.
The second scenario employs a minimum date of 50 MYA for the split between Nestor and the remaining psittaciforms based on fossil records of stem parrots in the Eocene of Europe (Mayr 2002). This date is consistent with the hypothesis of an explosive radiation of modern avian orders during the Tertiary (Feduccia 2003). The resulting chronograms (fig. 3a; supplementary fig. S2a, Supplementary Material online) place most of the diversification of psittaciforms around 40 MYA, after the separation of Australia from West Antarctica and South America. This scenario requires a colonization of New Zealand, Madagascar, and South America by oversea dispersal from Australia, a scenario we consider to be less plausible, although it is consistent with recent work suggesting that New Zealand was submerged during the Oligocene and early Miocene (Landis et al. 2008). It should be noted, however, that both timing scenarios require substantial overwater dispersal to explain the spread of the Loriinae to the distant islands of the Pacific, although the distribution of some of these species may have been altered by human transport (Forshaw 1989).
Psittaciform Taxonomy
Our sampling includes representatives of all families and subfamilies of parrot identified by Rowley (1997) and Collar (1997) and all but 1 of the 9 tribes within Psittacinae. The resulting phylogenies show only partial support for either of the most recent classifications of parrots (Collar 1997; Rowley 1997; Forshaw 2006). We find that the family Psittacidae sensu Collar (1997) is paraphyletic, with the New Zealand kea (N. notabilis) and kakapo (S. habroptilus) sister to a group that includes all the remaining psittaciforms. Likewise, the subfamily Psittacinae sensu Collar (1997) is paraphyletic due to the nested position of the other subfamily, Loriinae within it.
In contrast, we find support for the monophyly of the subfamily Loriinae, albeit with a strongly supported sister relationship to Melopsittacus, Cyclopsitta, and Psittaculirostris, 3 taxa typically classified within the Psittacinae. We also find consistent and strong support for the monophyly of the family Cacatuidae (sensu Rowley 1997), but less support for its division into 3 subfamilies, with the 2 genera of Calyptorhynchinae paraphyletic with respect to Cacatuinae.
A similar pattern is seen at lower taxonomic levels within the Psittacinae (sensu Collar 1997). Within this subfamily, 4 tribes (Psittrichadini, Nestorini, Micropsittini, and Strigopini) contain only a single genus each. Three of the remaining tribes (Cyclopsittacini, Platycercini, and Psittaculini) are represented by multiple taxa in our phylogeny and each appear to be polyphyletic assemblages. In contrast, the 2 representatives of the Afrotropical tribe Psittacini are recovered in a single well-supported clade, as are the 20 genera representing the Neotropical tribe Arini.
Our results generally agree with 4 other recent molecular phylogenies of parrots with more limited taxon and gene sampling (de Kloet RS and de Kloet SR 2005; Astuti et al. 2006; Tavares et al. 2006; Tokita et al. 2007). The phylogeny of de Kloet RS and de Kloet SR (2005) based on intron 3 of the sex-linked spindlin genes and the phylogeny of Tokita et al. (2007) based on mtDNA 12S and 16S ribosomal genes both placed the New Zealand taxa Nestor and Strigops in a basal position sister to all other psittaciforms as found in our analyses. Strigops was also sister to all other parrots in the tree of Tavares et al. (2006) based on a number of mtDNA genes and the nuclear exon RAG-1, with the cockatoo genus Cacatua the next to diverge, as found in our phylogenies and all other recent analyses (Boon 2000; Sainsbury 2004; de Kloet RS and de Kloet SR 2005; Astuti et al. 2006; Chan 2006; Tokita et al. 2007). The tree based on the Z spindlin intron (de Kloet RS and de Kloet SR 2005) differed in placing the cockatoos within a clade composed of members of the Psittacinae, but this arrangement had low nodal support. Our results and those from mtDNA 12S and 16S (Tokita et al. 2007), the 2 spindlin introns (de Kloet RS and de Kloet SR 2005), and a recent study of variation in hypotarsal morphology (Mayr 2008) show further concordance in grouping Melopsittacus, Psittaculirostris, and members of the subfamily Loriinae together. The latter 2 studies, however, differed from ours in their placement of Agapornis as sister to this clade. Recent studies consistently confirm the monophyly of the tribe Arini and the paraphyly of the tribes Platycercini and Psittaculini. Taken in concert, these results suggest that some revision of current parrot taxonomy is warranted, particularly in regard to the composition of family Psittacidae, subfamily Psittacinae, and the tribes Cyclopsittacini, Platycercini, and Psittaculini.
Although the Order Psittaciformes has long been recognized as a distinctive and relatively homogenous group, there has been considerable uncertainty concerning its relationship to other avian orders. Various sister groups have been nominated on the basis of one or a few shared features (Forshaw 1989); these include falcons, hawks, or owls (sharing curved bills), woodpeckers and allies, cuckoos and turacos, and trogons (zygodactyl feet), passerines (vocal learning), and pigeons (fleshy ceres, frugivory, and plumage patterns). Data from egg white proteins suggested a sister relationship between Psittaciformes and Columbiformes (Sibley and Ahlquist 1972). A recent avian phylogeny with comprehensive taxon sampling based on intron 7 of the beta fibrinogen gene found that all parrots shared a large (300 bp) insertion that could not be aligned with other taxa and joined a large polytomy that did not include the pigeons but did include most of the other groups discussed above (Fain and Houde 2004). A second large phylogeny of birds based on 5 nuclear gene regions placed the parrots in a polytomy with the passerine families Tyrannidae and Laniidae and the families Falconidae and Cariamidae (Ericson et al. 2006). Other studies with more limited taxon sampling have suggested sister relationships between psittaciforms and owls based on whole or partial mitochondrial genomes (Sorensen et al. 2003; Gibb et al. 2006) and between psittaciforms and passerines based on intron 9 of the nuclear phosphoenolpyruvate carboxykinase gene (Sorensen et al. 2003). In all cases, the hypothesized sister relationship enjoyed low nodal support measures and, in the case of parsimony analyses, may have resulted from long-branch attraction. Although our study was not designed to identify the sister group to parrots, we did include as outgroups representatives of most of the taxa that have previously been suggested as potential sister groups to the parrots, namely a falcon, a passerine, an owl, a hornbill, a mousebird, a woodpecker, a dove, and a cuckoo. Our various phylogenies produced no consistent placement of outgroups as sister to the parrots, reinforcing the idea that they have no close sister relationship with other modern birds. Studies employing wider taxon sampling and many additional loci may better resolve this issue.
Conservation and Comparative Studies
Of all the larger orders of birds, Psittaciformes contains the most threatened species, with 27% of species currently classified as vulnerable or endangered (Snyder et al. 2000). With such a large proportion of threatened species, it is important to set priorities for conservation that optimize the use of limited resources. One criterion with which conservation priorities may be assigned is “phylogenetic diversity,” in which the overall goal is to protect a subset of species that represents the maximum amount of evolutionary history as measured by branch lengths across a phylogeny (Faith 1992). Priority protection for such taxa has been advocated because it protects not only the current patterns of diversity but also the processes that gave rise to them (Crozier 1997; Moritz 2002). Although we have not attempted a formal analysis based the phylogenetic diversity criterion here, our results do provide an initial basis on which to identify species or genera that represent lineages with particularly long periods of independent evolutionary history. The first of these taxa that deserve special consideration are the New Zealand genera Nestor and Strigops. Our results show that these genera to be sister to all other parrots and may have diverged from the common parrot ancestor as long as 82 MYA. Strigops has been the focus of intensive and ongoing conservation efforts (Snyder et al. 2000) that appear to be fully warranted given the unique evolutionary history of this clade. We sampled many other genera composed of only 1 or 2 species that are also evolutionarily distinct (represented by long branches in our phylogenies) and thus deserve consideration as evolutionarily significant units (e.g., Coracopsis, Bolbopsittacus, Psittrichas, Psittacus, Eclectus, Aprosmictus, Nymphicus, Melopsittacus, Myiopsitta, Cyanoliseus, Nannopsittaca, Nandayus, Leptosittaca, Pionites, Rhynchopsitta, Enicognathus, and Deroptyus). Of these 17 small genera, only Rhynchopsitta and Leptosittaca contain members that are listed as threatened in the current parrot action plan (Snyder et al. 2000), but several others are subject to pressure from hunting for feathers (e.g., Psittrichas), capture for the pet trade (e.g., Psittacus), or have restricted distributions that leave them vulnerable to human-induced habitat change (e.g., Bolbopsittacus, Coracopsis, and Nannopsittaca). Others represent some of the most abundant parrot species both in the wild and captivity (e.g., Eclectus, Nymphicus, Melopsittacus, and Myiopsitta). A more formal analysis based on phylogenetic diversity and other genetically based criteria would likely identify other species and geographic areas that deserve heightened conservation status based on the evolutionary history they represent.
The parrots are the focus of a number of recent and ongoing comparative analyses. Those studies published to date have either utilized current taxonomic classifications (e.g., Davies et al. 2007) or employed a supertree approach to consolidate published phylogenies (e.g., Cassey et al. 2004; Iwaniuk et al. 2005; Munshi-South and Wilkinson 2006). Both approaches may give misleading results: current psittaciform taxonomy requires revision at several levels, and the supertrees constructed to date are poorly resolved at deeper nodes due to the relative paucity of broad-based phylogenetic studies. Our results should help ameliorate these problems and improve future comparative studies of this fascinating group.
Supplementary Material
Supplementary table S1 and figures S1 and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We thank the following institutions and people for the generous loan of tissue specimens: American Museum of Natural History (P. Sweet), Academy of Natural Sciences of Philadelphia (L. Joseph), Smithsonian Museum of Natural History, Loro Parque Fundación, and the Vertebrate Museum of New Mexico State University (P. Houde). We particularly thank David Waugh of the Loro Parque Fundación and Mathew Chan of Victoria University of Wellington for generous assistance in obtaining data from critical specimens. Noel Carr assisted with testing of nuclear intron primers with support from a Smithsonian Summer Internship. Donovan Bailey provided valuable assistance with phylogenetic methods, particularly the parsimony ratchet analysis; we thank both he and Peter Houde for their thoughtful comments on the manuscript. Research support was provided by a Robinson Postdoctoral Fellowship from Friends of the National Zoo (T.F.W.), the Abbott Fund of the Smithsonian Institution (T.F.W., G.R.G. and R.C.F.), and National Institutes of Health grant S06 GM008136 (T.F.W.). Tissue collection by G.R.G. on the Rio Xingu was supported by the Academia Brasileira de Ciencias through a grant from Electronorte administered by P. E. Vanzolini and by the Smithsonian IESP Neotropical Lowland Research Program. This study was approved by the Institutional Animal Care and Use Committee of New Mexico State University.
References
- Astuti D, Azuma N, Suzuki H, Higashi S. Phylogenetic relationships within parrots (Psittacidae) inferred from mitochondrial cytochrome-b gene sequences. Zool Sci. 2006;23:191–198. doi: 10.2108/zsj.23.191. [DOI] [PubMed] [Google Scholar]
- Bailey CD, Koch MA, Mayer M, Mummenhoff K, O'Kane SI, Jr, Warwick SL, Windham MD, Al-Shehbaz IA. Toward a global phylogeny of the. Brassicaceae. Mol Biol Evol. 2006;23:2142–2160. doi: 10.1093/molbev/msl087. [DOI] [PubMed] [Google Scholar]
- Barker FK, Barrowclough GF, Groth JG. A phylogenetic hypothesis for passerine birds: taxonomic and biogeographic implications of an analysis of nuclear DNA sequence data. Proc R Soc Lond B Biol Sci. 2002;269:295–308. doi: 10.1098/rspb.2001.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker FK, Lutzoni FM. The utility of the incongruence length difference test. Syst Biol. 2002;51:625–637. doi: 10.1080/10635150290102302. [DOI] [PubMed] [Google Scholar]
- Boon WM. Molecular systematics and conservation of the Cyanoramphus parakeet complex and the evolution of parrots [dissertation] Wellington (New Zealand): Victoria University of Wellington; 2000. [Google Scholar]
- Bradbury JW. Vocal communication in wild parrots. In: DeWaal FBM, Tyack PL, editors. Animal social complexity: intelligence, culture and individualized societies. Cambridge (MA): Harvard University Press; 2003. pp. 293–316. [Google Scholar]
- Braun EL, Kimball RT. Examining basal avian divergences with mitochondrial sequences: model complexity, taxon sampling, and sequence length. Syst Biol. 2002;51:614–625. doi: 10.1080/10635150290102294. [DOI] [PubMed] [Google Scholar]
- Bremer K. Ancestral areas: a cladistic reinterpretation of the center of origin concept. Syst Biol. 1992;41:436–445. [Google Scholar]
- Bremer K. Ancestral areas: optimization and probability. Syst Biol. 1995;44:255–259. [Google Scholar]
- Brightsmith DJ. Competition, predation and nest niche shifts among tropical cavity nesters: phylogeny and natural history evolution of parrots (Psittaciformes) and trogons (Trogoniformes) J Avian Biol. 2005;36:64–73. [Google Scholar]
- Brouwer K, Jones ML, King CE, Schifter H. Longevity records for Psittaciformes in captivity. Int Zoo Yearb. 2000;37:299–316. [Google Scholar]
- Brown DM, Toft CA. Molecular systematics and biogeography of the cockatoos (Psittaciformes: Cacatuidae) Auk. 1999;116:141–157. [Google Scholar]
- Brown JW, Payne RB, Mindell DP. Nuclear DNA does not reconcile ‘rocks’ and ‘clocks’ in Neoaves: a comment on Ericson et al. Biol Lett. 2007 doi: 10.1098/rsbl.2006.0611. doi:10 1098/rsbl 2006 0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt DW, Paton IR. Molecular cloning and primary structure of the chicken transforming growth factor-beta2 gene. DNA Cell Biol. 1991;10:723–734. doi: 10.1089/dna.1991.10.723. [DOI] [PubMed] [Google Scholar]
- Cassey P, Blackburn TM, Russell GJ, Jones KE, Lockwood JL. Influences on the transport and establishment of exotic bird species: an analysis of the parrots (Psittaciformes) of the world. Glob Chang Biol. 2004;10:417–426. [Google Scholar]
- Chan C-H. Conservation genetics and hybridisation of the Forbes' parakeet (Cyanoramphus forbesi) in the Chatham Islands [dissertation] Wellington (New Zealand): Victoria University of Wellington; 2006. [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collar NJ. Family Psittacidae (parrots) In: del Hoyo J, Elliot A, Sargatal J, editors. Handbook of the birds of the world. Barcelona (Spain): Lynx Editions; 1997. pp. 280–477. [Google Scholar]
- Cooper A, Penny D. Mass survival of birds across the Cetaceous-Tertiary boundary: molecular evidence. Science. 1997;275:1109–1113. doi: 10.1126/science.275.5303.1109. [DOI] [PubMed] [Google Scholar]
- Cracraft J. Continental drift, paleoclimatology and evolution and biogeography of birds. J Zool. 1973;169:455–545. [Google Scholar]
- Cracraft J. Avian evolution, Gondwana biogeography and the Cretaceous-Tertiary mass extinction event. Proc R Soc Lond B Biol Sci. 2001;268:459–469. doi: 10.1098/rspb.2000.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier RH. Preserving the information content of species: genetic diversity, phylogeny and conservation worth. Annu Rev Ecol Syst. 1997;28:243–268. [Google Scholar]
- Cunningham CW. Can three incongruence tests predict when data should be combined? Mol Biol Evol. 1997;14:733–740. doi: 10.1093/oxfordjournals.molbev.a025813. [DOI] [PubMed] [Google Scholar]
- Davies RG, Orme CDL, Webster AJ, Jones KE, Blackburn TM, Gaston KJ. Environmental predictors of global parrot (Aves: Psittaciformes) species richness and phylogenetic diversity. Global Ecol Biogeogr. 2007;16:220–233. [Google Scholar]
- de Kloet RS, de Kloet SR. The evolution of the spindlin gene in birds: sequence analysis of an intron of the spindlin W and Z gene reveals four major divisions of the Psittaciformes. Mol Phylogenet Evol. 2005;36:706–721. doi: 10.1016/j.ympev.2005.03.013. [DOI] [PubMed] [Google Scholar]
- del Hoyo J, Elliot A, Sargatal J. Handbook of the Birds of the World. Vol. 4. Sandgrouse to Cuckoos. Barcelona (Spain): Lynx Editions; 1997. [Google Scholar]
- Dowton M, Austin AD. Increased congruence does not necessarily indicate increased phylogenetic accuracy—the behavior of the incongruence length difference test in mixed-model analyses. Syst Biol. 2002;51:19–31. doi: 10.1080/106351502753475853. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLOS Biol. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary software analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyke JG, Mayr G. Did parrots exist in the Cretaceous period? Nature. 1999;399:317–318. [Google Scholar]
- Eberhard JR, Wright TF, Bermingham E. Duplication and concerted evolution of the mitochondrial control region in the parrot genus Amazona. Mol Biol Evol. 2001;18:1330–1342. doi: 10.1093/oxfordjournals.molbev.a003917. [DOI] [PubMed] [Google Scholar]
- Ericson PGP, Anderson CL, Britton T, Elzanowski A, Johansson US, Källersjö M, Ohlson JI, Parsons TJ, Zuccon D, Mayr G. Diversification of Neoaves: integration of molecular sequence data and fossils. Biol Lett. 2006;2:543–547. doi: 10.1098/rsbl.2006.0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson PGP, Anderson CL, Mayr G. Hangin' on to our ‘rocks’ and ‘clocks’. Biol Lett. 2007 doi:10 1098/rsbl 2007 0103. [Google Scholar]
- Ericson PGP, Christides L, Cooper A, Irestadt M, Jackson J, Johansson US, Norman JA. A Gondwanan origin of passerine birds supported by DNA sequences of the endemic New Zealand wrens. Proc R Soc Lond B Biol Sci. 2002;269:235–241. doi: 10.1098/rspb.2001.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson T. r8s bootstrap kit. Published by the author: 2007. [Google Scholar]
- Fain MG, Houde P. Parallel radiations in the primary clades of birds. Evolution. 2004;58:2558–2573. doi: 10.1111/j.0014-3820.2004.tb00884.x. [DOI] [PubMed] [Google Scholar]
- Faith D. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61:1–10. [Google Scholar]
- Farris JS, Kallersjo M, Kluge AG, Bult C. Testing significance of incongruence. Cladistics. 1994;10:315–319. [Google Scholar]
- Feduccia A. The origin and evolution of birds. New Haven (CT): Yale University Press; 1999. [Google Scholar]
- Feduccia A. ‘Big bang’ for tertiary birds? Trends Ecol Evol. 2003;18:172–176. [Google Scholar]
- Forshaw JM. Parrots of the World. Willoughby (Australia): Landsdowne Editions; 1989. [Google Scholar]
- Forshaw JM. Parrots of the world: an identification guide. Princeton (NJ): Princeton University Press; 2006. [Google Scholar]
- Gibb GC, Kardailsky O, Kimball RT, Braun EL, Penny D. Mitochondrial genomes and avian phylogeny: complex characters and resolvability without explosive radiations. Mol Biol Evol. 2006;24:269–280. doi: 10.1093/molbev/msl158. [DOI] [PubMed] [Google Scholar]
- Goloboff P, Farris S, Nixon K. TNT (tree analysis using new technology) Published by the author: 2000. [Google Scholar]
- Hipp AL, Hall JC, Sytsma KJ. Congruence versus phylogenetic accuracy: revisiting the incongruence length difference test. Syst Biol. 2004;53:81–89. doi: 10.1080/10635150490264752. [DOI] [PubMed] [Google Scholar]
- Homberger DG. Classification and the status of wild populations of parrots. In: Luescher AU, editor. Manual of parrot behavior. Ames (IA): Blackwell Publishing; 2006. pp. 3–11. [Google Scholar]
- Iwaniuk AN, Dean KM, Nelson JE. Interspecific allometry of the brain and brain regions in parrots (Psittaciformes): comparisons with other birds and primates. Brain Behav Evol. 2005;65:40–59. doi: 10.1159/000081110. [DOI] [PubMed] [Google Scholar]
- Juniper T, Parr M. Parrots: a guide to the parrots of the world. New Haven (CT): Yale University Press; 1998. [Google Scholar]
- Kimball RT, Braun EL, Ligon JD, Lucchini V, Randi E. A molecular phylogeny of the peacock-pheasants (Galliformes: Polyplectron spp.) indicates loss and reduction of ornamental traits and display behaviors. Biol J Linn Soc. 2001;73:187–198. [Google Scholar]
- Kumar S, Hedges SB. A molecular timescale for vertebrate evolution. Nature. 1998;392:917–920. doi: 10.1038/31927. [DOI] [PubMed] [Google Scholar]
- Landis CA, Campbell HJ, Begg JG, Mildenhall DC, Paterson AM, Trewick SA. The Waipounamu erosion surface: questioning the antiquity of the New Zealand land surface and terrestrial fauna and flora. Geol Mag. 2008;145:173–197. [Google Scholar]
- Lemonnier M, Balvay L, Mouly V, Libri D, Fiszman MY. The chicken gene encoding the alpha isoform of tropomyosin of fast twitch muscle fibers: organization expression and identification of the major proteins synthesized. Gene. 1991;107:229–240. doi: 10.1016/0378-1119(91)90323-4. [DOI] [PubMed] [Google Scholar]
- Maddison DR, Maddison WP. MacClade. Sunderland (MA): Sinauer Associates; 2003. [Google Scholar]
- Mayr G. On the osteology and phylogenetic affinities of the Pseudasturidae—lower eocene stem-group representatives of parrots (Aves, Psittaciformes) Zool J Linn Soc. 2002;136:715–729. [Google Scholar]
- Mayr G. The phylogenetic affinities of the parrot taxa Agapornis, Loriculus and Melopsittacus (Aves: Psittaciformes): hypotarsal morphology supports the results of molecular analyses. Emu. 2008;108:23–27. [Google Scholar]
- McLoughlin S. The breakup history of Gondwana and its impact on pre-Cenozoic floristic provincialism. Aust J Bot. 2001;49:271–300. [Google Scholar]
- Miyaki CY, Matioli SR, Burke T, Wajntal A. Parrot evolution and paleogeographical events: mitochondrial DNA evidence. Mol Biol Evol. 1998;15:544–551. [Google Scholar]
- Moritz C. Strategies to protect biological diversity and the processes that sustain it. Syst Biol. 2002;51:238–254. doi: 10.1080/10635150252899752. [DOI] [PubMed] [Google Scholar]
- Munshi-South J, Wilkinson GS. Diet influences life span in parrots (Psittaciformes) Auk. 2006;123:108–118. [Google Scholar]
- Nixon KC. The parsimony ratchet, a new method for rapid parsimony analysis. Cladistics. 1999;15:407–414. doi: 10.1111/j.1096-0031.1999.tb00277.x. [DOI] [PubMed] [Google Scholar]
- Nixon KC. WinClada. Published by the author: 2002. [Google Scholar]
- Nylander JAA. MrModeltest v2.2. Uppsala (Sweden): Evolutionary Biology Centre. Uppsala University; 2004. [Google Scholar]
- Omland K, Lanyon SM, Fritz SJ. A molecular phylogeny of the New World orioles (Icterus): the importance of dense taxon sampling. Mol Phylogenet Evol. 1999;12:224–239. doi: 10.1006/mpev.1999.0611. [DOI] [PubMed] [Google Scholar]
- Palumbi SR. Nucleic acids II: the polymerase chain reaction. In: Hillis DM, Moritz C, Mable BK, editors. Molecular systematics. Sunderland (MA): Sinauer Associates; 1996. pp. 205–221. [Google Scholar]
- Pereira SL, Baker AJ, Wajntal A. Combined nuclear and mitochondrial DNA sequences resolve generic relationships within the Cracidae (Galliformes, Aves) Syst Biol. 2002;51:946–958. doi: 10.1080/10635150290102519. [DOI] [PubMed] [Google Scholar]
- Pereira SL, Johnson KP, Clayton DH, Baker AJ. Mitochondrial and nuclear DNA sequences support a Cretaceous origin of Columbiformes and a dispersal-driven radiation in the Paleogene. Syst Biol. 2007;56:656–672. doi: 10.1080/10635150701549672. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Prytchitko TM, Moore WS. Comparative evolution of the mitochondrial cytochrome b gene and nuclear ß-fibrinogen intron 7 in woodpeckers. Mol Biol Evol. 2000;17:1101–1111. doi: 10.1093/oxfordjournals.molbev.a026391. [DOI] [PubMed] [Google Scholar]
- Rambaut A. FigTree v1.1.1. Published by the author: 2008. [Google Scholar]
- Rambaut A, Drummond AJ. Tracer v1.4. Published by the author: 2007a. [Google Scholar]
- Rambaut A, Drummond AJ. TreeAnnotator v1.4.7. Published by the author: 2007b. [Google Scholar]
- Ribas CC, Gaban-Lima R, Miyaki CY, Cracraft J. Historical biogeography and diversification within the Neotropical parrot genus Pionopsitta (Aves: Psittacidae) J Biogeogr. 2005;32:1409–1427. [Google Scholar]
- Ribas CC, Miyaki CY. Molecular systematics in Aratinga parakeets: species limits and historical biogeography in the “solstitialis” group, and the systematic position of Nandayus nenday. Mol Phylogenet Evol. 2003;30:663–675. doi: 10.1016/S1055-7903(03)00223-9. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Rowley . Family Cacatuidae (Cockatoos) In: del Hoyo J, Elliot A, Sargatal J, editors. Handbook of the birds of the world. Barcelona (Spain): Lynx Editions; 1997. pp. 246–279. [Google Scholar]
- Russello MA, Amato G. A molecular phylogeny of Amazona: implications for Neotropical parrot biogeography, taxonomy and conservation. Mol Phylogenet Evol. 2004;30:421–477. doi: 10.1016/s1055-7903(03)00192-1. [DOI] [PubMed] [Google Scholar]
- Rutschmann F, Eriksson T, Schönenberger J, Conti E. Did Cryteroniaceae really disperse out of India? Molecular dating evidence from rbcL, ndhF, and rpl16 intron sequences. Int J Plant Sci. 2004;165:S69–S83. [Google Scholar]
- Sainsbury JP. Population structure, conservation genetics and evolution of the Kaka (Nestor meridionalis): a microsatellite study [dissertation] Wellington (New Zealand): Victoria University of Wellington; 2004. [Google Scholar]
- Salvadori T. Catalogue of the birds of the British Museum. London: British Museum of Natural History; 1891. [Google Scholar]
- Sanderson MJ. Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach. Mol Biol Evol. 2002;19:101–109. doi: 10.1093/oxfordjournals.molbev.a003974. [DOI] [PubMed] [Google Scholar]
- Sibley CG, Ahlquist JE. A comparative study of the egg white proteins of non-passerine birds. Bull Peabody Mus Nat Hist. 1972;39:1–276. [Google Scholar]
- Sibley CG, Ahlquist JE. Phylogeny and classification of birds: a study in molecular evolution. New Haven (CT): Yale University Press; 1990. [Google Scholar]
- Simmons MP, Ochoterena H. Gaps as characters in sequence-based phylogenetic analysis. Syst Biol. 2000;49:369–381. [PubMed] [Google Scholar]
- Slack KE, Jones CM, Ando T, Harrison GL, Fordyce RE, Arnason U, Penny D. Early penguin fossils, plus mitochondrial genomes, calibrate avian evolution. Mol Biol Evol. 2006;23:1144–1155. doi: 10.1093/molbev/msj124. [DOI] [PubMed] [Google Scholar]
- Smith GT. Systematics of parrots. Ibis. 1975;117:18–68. [Google Scholar]
- Snyder N, McGowan P, Gilardi J, Grajal A. Parrots: status survey and conservation action plan 2000-2004. Cambridge (UK): IUCN; 2000. [Google Scholar]
- Sokal RR, Rohlf FJ. Biometry. New York: W.H. Freeman and Company; 1995. [Google Scholar]
- Sorenson MD. TreeRot v2. Boston (MA): Boston University; 1999. [Google Scholar]
- Sorensen MD, Ast JC, Dimcheff DE, Yuri T, Mindell DP. Primers for PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates. Mol Phylogenet Evol. 1999;12:105–114. doi: 10.1006/mpev.1998.0602. [DOI] [PubMed] [Google Scholar]
- Sorensen MD, Neal E, Garcia-Moreno J, Mindell DP. More taxa, more characters: the hoatzin problem is still unresolved. Mol Biol Evol. 2003;20:1484–1499. doi: 10.1093/molbev/msg157. [DOI] [PubMed] [Google Scholar]
- Swofford DL. PAUP*. Phylogenetic analysis using parsimony (*and other methods) Sunderland (MA): Sinauer Associates; 1999. [Google Scholar]
- Takao M, Yasui A, Tokunaga F. Isolation and sequence determination of the chicken rhodopsin gene. Vision Res. 1988;27:471–480. doi: 10.1016/0042-6989(88)90169-1. [DOI] [PubMed] [Google Scholar]
- Tavares ES, Baker AJ, Pereira SL, Miyaki CY. Phylogenetic relationships and historical biogeography of Neotropical parrots (Psittaciformes: Psittacidae: Arini) inferred from mitochondrial and nuclear DNA sequences. Syst Biol. 2006;55:454–470. doi: 10.1080/10635150600697390. [DOI] [PubMed] [Google Scholar]
- Thompson AW. On characteristic points in the cranial osteology of the parrots. Proc Zool Soc Lond. 1899;1899:9–46. [Google Scholar]
- Thorne JL, Kishino H, Painter IS. Estimating the rate of evolution of the rate of molecular evolution. Mol Biol Evol. 1998;15:1647–1657. doi: 10.1093/oxfordjournals.molbev.a025892. [DOI] [PubMed] [Google Scholar]
- Tokita M, Kiyoshi T, Armstrong K. Evolution of craniofacial novelty in parrots through developmental modularity and heterochrony. Evol Dev. 2007;9:590–601. doi: 10.1111/j.1525-142X.2007.00199.x. [DOI] [PubMed] [Google Scholar]
- Vane-Wright RI, Humphries CJ, Williams PH. What to protect?—systematics and the agony of choice. Biol Conserv. 1991;55:235–254. [Google Scholar]
- Van Tuinen M, Hedges SB. Calibration of avian molecular clocks. Mol Biol Evol. 2001;18:206–213. doi: 10.1093/oxfordjournals.molbev.a003794. [DOI] [PubMed] [Google Scholar]
- Wilford GE, Brown PJ. Maps of late Mesozoic-Cenozoic Gondwana break-up: some paleogeographical implications. In: Hill RS, editor. History of the Australia vegetation: cretaceous to recent. Cambridge: Cambridge University Press; 1994. pp. 5–13. [Google Scholar]
- Yoder AD, Irwin JA, Payseur BA. Failure of the ILD to determine data combinability for slow loris phylogeny. Syst Biol. 2001;50:408–424. [PubMed] [Google Scholar]
- Young ND, Healy J. GapCoder automates the use of indel characters in phylogenetic analysis. BMC Bioinformatics. 2003;4:1–6. doi: 10.1186/1471-2105-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion [dissertation] [Austin (TX)]: University of Texas at Austin; 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.