

Abstract
Lethality in hybrids between Drosophila melanogaster and its sibling species Drosophila simulans is caused in part by the interaction of the genes Hybrid male rescue (Hmr) and Lethal hybrid rescue (Lhr). Hmr and Lhr have diverged under positive selection in the hybridizing species. Here we test whether positive selection of Hmr is confined only to D. melanogaster and D. simulans. We find that Hmr has continued to diverge under recurrent positive selection between the sibling species D. simulans and Drosophila mauritiana and along the lineage leading to the melanogaster subgroup species pair Drosophila yakuba and Drosophila santomea. Hmr encodes a member of the Myb/SANT-like domain in ADF1 (MADF) family of transcriptional regulators. We show that although MADF domains from other Drosophila proteins have predicted ionic properties consistent with DNA binding, the MADF domains encoded by different Hmr orthologs have divergent properties consistent with binding to either the DNA or the protein components of chromatin. Our results suggest that Hmr may be functionally diverged in multiple species.
Keywords: adaptive evolution, hybrid incompatibility, speciation, MADF domain, positive selection
Introduction
Progress has been made in understanding the genetics of speciation by reducing the complexities of speciation to investigation of the genetic basis of reproductive isolation (Coyne 1992). A population undergoing divergent evolution can ultimately result in the creation of reproductively isolated populations or new species. The evolution of reproductive isolation requires the establishment of barriers to gene flow, often multiple barriers acting together at various stages in the life cycle of the organism. Hybrid incompatibility (HI), the sterility and inviability of interspecific offspring, is a postzygotic barrier to gene flow. As it is relatively easy to measure the viability and fertility of hybrid progeny, HI has been more amenable to genetic dissection than other reproductive isolating mechanisms. The study of HI loci is also of great interest because it addresses how developmental pathways may diverge between taxa, a process that characterizes both genome evolution and speciation.
Five HI genes have been described: Xmrk-2 from the fish Xiphophorus and the Drosophila genes OdsH, Hmr, Nup96, and Lhr (Wittbrodt et al. 1989; Ting et al. 1998; Barbash et al. 2003; Presgraves et al. 2003; Brideau et al. 2006). Based on sequence homology, two of these genes might be involved with transcriptional regulation or chromatin binding (OdsH and Hmr), Nup96 encodes a component of the nuclear pore complex, Lethal hybrid rescue (Lhr) encodes a heterochromatin-associated factor, and Xmrk-2 encodes a receptor tyrosine kinase. Mitochondrial cytochrome c oxidases show significant reductions in activity in combination with nuclear-encoded cytochrome c proteins that derive from different populations of copepods, making their respective genes strong candidates for causing reduced fitness in copepod hybrids (Rawson and Burton 2002). There is, therefore, no single functional class of genes causing HI. However, four of these HI genes (OdsH, Hmr, Nup96, and Lhr) have diverged rapidly under positive selection (Ting et al. 1998; Presgraves et al. 2003; Barbash et al. 2004; Brideau et al. 2006). The biological significance of HI loci being the target of adaptive evolution is unclear because if HI evolves as a secondary by-product of intraspecific evolution, then the phenotype being selected for is unlikely to be HI. One possible explanation is that if mutations causing HI are rare, then HIs will tend to occur in genes undergoing high rates of substitution. Positive selection would be the engine driving a high substitution rate. Positive selection also implies that the genes may be changing in function in a way that causes developmental breakdown in hybrids. These findings raise several intriguing questions. Is the selection pressure on HI genes limited only to the hybridizing species, or have HI genes experienced recurrent adaptive evolution in other species? Have HI genes changed in their structural properties as well as in primary sequence? Does analysis of HI genes resolve otherwise ambiguous phylogenetic relationships?
Here we address these questions for Hybrid male rescue (Hmr), an HI gene identified in Drosophila melanogaster. Matings between D. melanogaster mothers to fathers from its sibling species Drosophila mauritiana, Drosophila simulans, and Drosophila sechellia produce the same HI phenotype: semiviable but sterile daughters and lethal sons (Sturtevant 1920; Lachaise et al. 1986). Hmr was identified by a loss-of-function mutation in D. melanogaster (Hmr1) that rescues F1 hybrid sons from each of these interspecific crosses (Hutter and Ashburner 1987; Barbash et al. 2003). Population genetic analysis revealed that Hmr has diverged under positive selection in both D. melanogaster and D. simulans (Barbash et al. 2004). In order to obtain a more comprehensive view of Hmr evolution in this study, we have 1) analyzed Myb/SANT-like domain in ADF1 (MADF) domains of Hmr orthologs from 14 species within the Drosophila genus in order to detect possible changes in DNA or chromatin binding, 2) applied maximum likelihood phylogenetic analysis on 7 species within the melanogaster subgroup, and 3) generated and analyzed population sampling data from the three sibling species and from the melanogaster subgroup species pair Drosophila yakuba and Drosophila santomea.
Materials and Methods
Identification and Alignment of Orthologs
Hmr orthologs in D. simulans, D. mauritiana, D. sechellia, and Drosophila erecta were described previously (Barbash et al. 2003, 2004). Additional Hmr orthologs were identified here using D. melanogaster HMR in TBlastN searches of the trace archives or of preliminary assembled contigs from the various Drosophila genome projects (Clark et al. 2007). All putative orthologs were reciprocally blasted back to D. melanogaster, and Hmr was identified as the highest scoring hit. We also looked for conservation of synteny using the flanking genes CG2124 and Rab9D (CG32678). Synteny was conserved in D. yakuba, Drosophila pseudoobscura, and Drosophila willistoni. Only a Rab homolog was found adjacent to Drosophila ananassae and Drosophila virilis Hmr. Neither gene was found adjacent to Hmr in Drosophila mojavensis. Our designation of Hmr orthologs matches those of the published genome assemblies (Clark et al. 2007).
Partial sequence of Drosophila persimilis Hmr was obtained from the National Center for Biotechnology Information (NCBI) trace archives using discontinuous MegaBlast and then completed by sequencing polymerase chain reaction (PCR) products from DNA extracted from a single D. persimilis male from the WSH3 strain that was used for the whole-genome shotgun sequencing project. Drosophila teissieri Hmr was sequenced from template DNA generously provided by Dr John Pool (Cornell University) (GenBank accession number FJ151263). The primers used were the most robust ones from the D. yakuba/D. santomea population study as well as D. teissieri-specific primers.
The gene structures of Hmr orthologs were predicted by GeneWise software (Birney et al. 2004) guided by D. melanogaster Hmr and manually checked for exon–intron conservation. We were unable to identify a homologous exon 1 in D. pseudoobscura, D. ananassae, D. virilis, or D. mojavensis. We therefore annotated Hmr in these species as having the longest conceptual open reading frame initiating in the large exon that is orthologous to exon 2 of D. melanogaster. Some of our Hmr annotations differ from Clark et al. (2007), but these differences do not affect the MADF domains analyzed in table 1.
Table 1.
Predicted Charge and Isoelectric Points (pI) of MADF Domains within Hmr Orthologs from 14 Drosophila Species
| MADF1 |
MADF2 |
MADF3 |
MADF4 |
|||||
| Charge | pI | Charge | pI | Charge | pI | Charge | pI | |
| Drosophilamelanogaster | 16.52 | 11.18 | 2.66 | 8.33 | −2.47 | 5.95 | 1.06 | 8.16 |
| Drosophilasimulans | 11.85 | 10.55 | 2.52 | 8.51 | −0.61 | 6.6 | 2.59 | 9.21 |
| Drosophilamauritiana | 12.02 | 10.32 | 2.72 | 8.78 | −0.61 | 6.6 | 2.56 | 8.82 |
| Drosophilasechellia | 11.18 | 10.37 | 2.69 | 8.51 | −0.61 | 6.6 | 2.56 | 8.83 |
| Drosophilayakuba | 13.19 | 11.55 | 3.55 | 9.24 | −2.5 | 5.95 | 1.76 | 8.52 |
| Drosophilasantomea | 13.19 | 11.64 | 3.55 | 9.24 | −1.51 | 6.32 | 1.76 | 8.52 |
| Drosophilateissieri | 12.02 | 11.54 | 4.89 | 9.74 | −3.34 | 5.85 | 2.39 | 9.16 |
| Drosophilaerecta | 12.36 | 10.35 | 4.52 | 9.39 | −2.44 | 5.96 | −0.57 | 6.62 |
| Drosophilaananassae | 9.86 | 10.31 | 2.55 | 8.72 | −2.67 | 5.58 | 0.76 | 7.54 |
| Drosophilapseudoobscura | 12.36 | 10.35 | 2.25 | 9.13 | 1.72 | 8.19 | 2.39 | 8.8 |
| Drosophilapersimilis | 9.36 | 10.03 | 1.42 | 8.48 | 1.55 | 8.17 | 0.43 | 7.42 |
| Drosophilawillistoni | 8.88 | 10.31 | 2.25 | 9.1 | 2.12 | 8.17 | 1.43 | 8.48 |
| Drosophilamojavensis | 16.69 | 11.41 | −1.24 | 6.49 | −0.17 | 6.5 | −0.4 | 6.77 |
| Drosophilavirilis | 16.52 | 11.56 | 4.74 | 9.62 | −1.87 | 5.69 | 3.09 | 9.18 |
NOTE.—MADF domains with predicted negative charges and acidic isoelectric points are in bold.
Population Samples
Hmr was isolated from 12 lines of D. simulans including 5 lines used in a previous study (Barbash et al. 2004) and an additional 7 lines collected in Zimbabwe, Africa. Twelve lines of D. mauritiana were obtained from Dr Shun-Chern Tsaur (Academia Sinica, Taipei, Taiwan; “W” lines) or from the Tucson Drosophila Stock Center (other lines). Five lines of D. sechellia were obtained from the Tucson Drosophila Stock Center. Primers for PCR amplification and sequencing are described in Barbash et al. (2004). GenBank accession numbers for these sequences are D. simulans FJ151256–FJ151262, D. mauritiana FJ151229–FJ151240, and D. sechellia FJ151252–FJ151255. Hmr was also isolated from 11 D. yakuba and 11 D. santomea strains. Flies for the different strains of D. yakuba and D. santomea were obtained via Dr David Begun (University of California, Davis) from collections of Dr Peter Andolfatto (University of California, San Diego) and Dr Manyuan Long (University of Chicago). Drosophila yakuba lines were collected in Cameroon and are described in Bachtrog et al. (2006); the D. santomea samples were from several populations. PCR primers were designed from the genome sequence of D. yakuba, to cover the entire Hmr gene in five overlapping amplicons of approximately 1.2 kb each. Multiple attempts using several primer sets to PCR amplify the 3′-most amplicon failed from many of the samples. This necessitated that we restrict our population genetic analysis to the Hmr region ending 1,067 bp upstream of the stop codon in the reference D. yakuba Hmr coding sequence. GenBank accession numbers for these sequences are D. yakuba FJ151264–FJ151274 and D. santomea FJ151241–FJ151251. A low-complexity region that contained polymorphic indels in both species was excluded from our analyses, it corresponds to amino acid positions 802–885 and 787–855 for D. yakuba and D. santomea reference sequences, respectively. Due to the difficulty in sequencing the 3′-most amplicon from single-fly preps, a complete Hmr gene sequence from D. santomea was generated by synthesizing a composite allele. The 3′-most block was amplified and sequenced from DNA extracted from 25 D. santomea flies (GenBank accession number FJ151275). This was joined to one D. santomea Hmr allele chosen at random.
DNA from a single male fly for each strain was used as a template in the PCRs in order to obtain a single allele of the X-linked Hmr. DNA was prepared by the method of sodium dodecyl sulfate lysis followed by phenol–chloroform extraction. The PCR products were purified either with Qiagen PCR cleanup columns or gel purified using the QIAEX II Gel Extraction Kit (Qiagen, Valencia, CA). The PCR products were then sequenced directly using Big Dye version 1.1, 3.1 (Applied Biosystems, Foster City, CA) reagents on an ABI capillary sequencer.
Sequences were aligned using MegAlign from the Lasergene v.6 package (DNASTAR Inc., Madison, WI) and the alignments corrected by eye.
Analyses of Divergence
ClustalW was used for multiple sequence alignment of Hmr within and between species (Thompson et al. 1994). All the coding sequence alignments were obtained by first aligning their protein products. Using RepeatMasker (Smit et al. 1996–2004), two low-complexity regions located in the second and fourth exons were found in melanogaster subgroup species and were removed from some analyses to ensure accurate alignment. The total length of removed material ranged from 22 amino acids in D. melanogaster to 122 amino acids in D. yakuba. Two low-complexity regions were also found in the first exon of D. persimilis (81 amino acids) and D. pseudoobscura (121 amino acids). Alignments without these low-complexity regions were used for the construction of the eight-species phylogeny (supplementary fig. S3, Supplementary Material online) and the PAML analyses (fig. 3). The complete Hmr sequences from D. melanogaster, D. simulans, D. mauritiana, and D. sechellia were used for McDonald–Kreitman (MK) tests (table 3). For D. yakuba and D. santomea, alignments without the low-complexity regions were used for MK tests (table 3).
FIG. 3.—

A maximum likelihood phylogenetic tree of Hmr built by the free-ratio model in PAML. The likelihood of this model was significantly better than the one-ratio model (2Δl = 54.224, degrees of freedom = 11, P < 10−7). The tree length is defined as the number of nucleotide substitutions per codon. The number shown above each lineage is the estimated DN/DS value of that lineage.
Table 3.
MK Tests of Neutrality
| Divergence |
Polymorphism |
|||||
| Pairwise Comparison | Species Pair or Lineage | Synonymous | Nonsynonymous | Synonymous | Nonsynonymous | Fisher's exact test P (two tailed) |
| 1 | Drosophilasimulans versus Drosophilamauritiana | 3 | 28 | 90 | 105 | 6.138 × 10−05*** |
| D. simulans lineage | 2 | 16 | 48 | 61 | 8.537 × 10−03** | |
| D. mauritiana lineage | 1 | 8 | 45 | 44 | 0.03366* | |
| 2 | D. simulans versus Drosophilasechellia | 16 | 43 | 53 | 68 | 0.03450* |
| 3 | D. mauritiana versus D. sechellia | 16 | 42 | 49 | 49 | 0.007209** |
| 4 | Drosophilayakuba versus Drosophilasantomea | 22 | 22 | 92 | 41 | 0.02889* |
| D. yakuba lineage | 10 | 7 | 72 | 32 | 0.4113 | |
| D. santomea lineage | 10 | 13 | 21 | 8 | 0.04823* | |
| 5 | D. yakubaversus Drosophila teissieri | 93 | 145 | 72 | 32 | 3.301 × 10−07*** |
| D. yakuba lineage | 40 | 70 | 71 | 32 | 2.597 × 10−06*** | |
| 6 | D. santomea versus D.teissieri | 93 | 146 | 21 | 8 | 1.113 × 10−03** |
| D. santomea lineage | 43 | 73 | 22 | 8 | 4.382 × 10−04*** | |
NOTE.—Six pairwise comparisons were tested, using total polymorphism in comparisons 1–4. Within some pairwise comparisons, both lineages (comparisons 1 and 4) or one lineage (comparisons 5 and 6) were then tested by polarizing each lineage using an outgroup sequence. For D. simulans and D. mauritiana (comparison 1), Drosophila melanogaster Hmr was used as the outgroup sequence; for D. yakuba and D. santomea (comparison 4), D. teissieri Hmr was used as the outgroup sequence; and for comparisons 5 and 6, Drosophila erecta Hmr was used as the outgroup sequence.
*P < 0.05; **P < 0.01; ***P < 10−3.
Neutrality tests were carried out in DnaSP v.4.5 (Rozas et al. 2003). Tests of whether synonymous sites are evolving toward preferred or unpreferred codons were made using the method of DuMont et al. (2004) with the “Biased” mutations options. Significance was tested using Fisher's exact test (two tailed).
Phylogenetic Analysis
Phylogenetic trees were built by MEGA 3.1 using parsimony and Neighbor-Joining methods (Kumar et al. 2004). PAML was used for the maximum likelihood method of phylogenetic analysis (Yang 1997). The lineage-specific models in PAML allow for the variation of DN/DS ratios among different lineages. The M0 (one-ratio) model was compared with a two-ratio model as well as a free-ratio model along each lineage. P values were calculated in R 2.2.0 using the likelihood ratio test of each comparison (Yang and Nielsen 2002). Figures of phylogenetic trees were prepared by retracing the primary images in Adobe Illustrator.
Structural Analyses
Secondary structure predictions were made using Jpred (Cuff and Barton 2000). The charge and isoelectric points (pI) were predicted using the Editseq program that is part of the Lasergene v.6 package (DNASTAR, Inc.).
Results
Domains and Structure of Hmr
Hmr from D. melanogaster encodes a predicted protein of 1,413 amino acids, in which two MADF domains were identified previously (Barbash et al. 2003). The MADF domain was discovered in the Drosophila protein ADF1 based on its sequence similarity to the DNA-binding domain of MYB and is required for the DNA-binding activity of ADF1 (England et al. 1992; Cutler et al. 1998). The predicted MADF secondary structure is also similar to the SANT domain, which is found in a large number of DNA- and chromatin-associated proteins (Aasland et al. 1996). We identified orthologs of Hmr from 14 Drosophila species using previously published work (Barbash et al. 2004), our sequencing here, and preliminary assemblies of whole-genome shotgun data (Clark et al. 2007). While examining these Hmr orthologs, we identified two additional candidate MADF domains (fig. 1A). These four HMR MADF domains are generally highly conserved among all Drosophila species (supplementary fig. S1, Supplementary Material online).
FIG. 1.—

Domain structure and MADF domains of Hmr. (A) The positions of four MADF domains and the putative BEAF, Su(var)3-7 and Stonewall (BESS) domain (Brideau et al. 2006) encoded by Drosophila melanogaster Hmr are shown. (B) Alignment and comparison of MADF, MYB, and SANT domains. The four MADF domains from D. melanogaster HMR and the MADF domain from D. melanogaster (Dm) ADF1 were aligned with the second and third MYB domains from Mus musculus (Mm) c-MYB and the SANT domain from D. melanogaster ISWI. This alignment was generated by extracting the alignment of the MADF domains from supplementary figure S1 (Supplementary Material online) and then manually adjusted to the published alignment of MYB and SANT domains (Bhaskar and Courey 2002; Boyer et al. 2004). Asterisks underneath the alignment indicate the conserved tryptophan residues, and the shaded boxes indicate predicted α-helical regions. The ionic properties of each domain are indicated within parentheses after the domain name as (charge, isoelectric point), respectively. HMR MADF1 and ADF1 MADF have ionic properties similar to the DNA-binding MYB domain, whereas HMR MADF3 is distinct in being more similar to the chromatin-associated SANT domain. For comparison, DIP3, another MADF domain–containing transcription factor that has been experimentally shown to bind DNA (Bhaskar and Courey 2002), has a MADF domain with a charge of +8.25 and an isoelectric point of 10.52, which is again similar to the MYB domain. The NCBI Entrez GeneIDs are Mm c-Myb, 17863; Dm Iswi, 36390; Dm Adf1, 47082; and Dm Dip3, 53579.
A close comparison of the D. melanogaster HMR MADF domains to each other and to the ADF1 MADF domain reveals that the third and fourth putative MADF domains contain insertions and differ at certain conserved residues, which raises the question of whether these four domains have the same function. Interestingly, we find considerable variation in the predicted charge and isoelectric point of each domain (fig. 1B). MADF1 is highly positively charged and thus most closely resembles DNA-binding domains found in MYB or ADF1, whereas MADF3 is negatively charged and thus is more similar to a chromatin-binding domain such as the SANT domain from ISWI. MADF2 and MADF4 have a significantly lower charge compared with the canonical ADF1 MADF domain, although their pI values are still consistent with a putative DNA-binding function. These results imply that each of the four HMR MADF domains may have unique functions with respect to DNA or histone association. These differences also suggest that HMR may bind to both the DNA and the protein components of chromatin.
To determine if these unusual MADF domains are unique to D. melanogaster HMR, we analyzed the MADF domains encoded by Hmr orthologs from 13 other species in the Drosophila genus (table 1). A clear trend was observed, with the first domain within each species’ HMR resembling a canonical MADF domain consistent with DNA-binding function. The remaining three MADF domains, however, have variable ionic properties. Hmr orthologs from taxa within the melanogaster group are in general similar to the D. melanogaster ortholog, with only the third MADF domain having a net negative charge, suggestive of chromatin binding. One exception is the fourth predicted MADF domain from D. erecta that also has a net negative charge. Hmr orthologs from other Sophophora species (D. pseudoobscura, D. persimilis, and D. willistoni) do not encode a negatively charged third MADF domain. The most divergent ortholog is from D. mojavensis with three MADF domains having a net negative charge.
We conducted a similar analysis of MADF domains identified by the SMART database for other D. melanogaster genes, in order to determine how common are MADF domains with a predicted net negative charge. We only found two other genes that encode MADF domains with isoelectric points below seven: CG31627 (pI = 6.75) and CG1603 (two MADF domains with pIs = 6.75 and 9.3). We conclude that most MADF domains are likely to be involved in DNA binding, whereas Hmr encodes unusual MADF domains with potential chromatin-binding properties.
Nearly, all Hmr orthologs also encode simple amino acid repeats, consisting predominantly of serine, alanine, and proline. Such simple sequence repeats are overrepresented among transcription factors (Albà and Guigó 2004; Hancock and Simon 2005). Sequencing of multiple Hmr alleles from the D. yakuba and D. santomea species pair (see below) revealed a unique microsatellite-like polymorphism within the coding region. The kernel of this repeat is present within the melanogaster subgroup, but the expansion is restricted to the lineage leading to D. yakuba, D. santomea, and D. teissieri (supplementary fig. S2, Supplementary Material online). The expansion has resulted in a tandem repeat consisting of nearly perfect alternating “SAT” and “QAA” residues, ranging from 63 amino acids to 87 amino acids in length.
Hmr Phylogenetic Pattern in melanogaster Subgroup
Outside of the melanogaster subgroup, the predicted HMR protein is highly diverged and thus impossible to align fully. Therefore, we aligned Hmr only from 8 species within the melanogaster subgroup (supplementary fig. S2, Supplementary Material online). Phylogenetic trees were built using both the Neighbor-Joining and the maximum parsimony methods. These methods produced similar results, except for the grouping of the D. melanogaster sibling species D. simulans, D. mauritiana, and D. sechellia (supplementary fig. S3, Supplementary Material online). We therefore obtained a population data set from these 3 species to further explore their phylogenetic relationship (table 2).
Table 2.
Summary of Hmr Polymorphism Data
| Drosophilasimulans | Drosophilamauritiana | Drosophilasechellia | Drosophilayakuba | Drosophila santomea | |
| Number of alleles analyzed | 13 | 12 | 5 | 11 | 11 |
| Nucleotide diversity (Pi) | 0.00878 | 0.00606 | 0.00117 | 0.00815 | 0.0028 |
| Total variable sites | 133 | 108 | 12 | 126 | 38 |
| Variable sites within coding sequence | 110 | 90 | 10 | 104 | 30 |
| Total number of singletons | 35 | 55 | 7 | 82 | 21 |
| Number of synonymous singletons | 15 | 30 | 2 | 45 | 10 |
| Number of nonsynonymous singletons | 15 | 17 | 3 | 20 | 6 |
Phylogenetic reconstruction of the population data set using maximum parsimony provided support for D. sechellia branching off prior to the split of D. simulans and D. mauritiana, albeit with relatively low bootstrap support (fig. 2). We therefore further analyzed all the sites that have an unambiguous phylogenetic signal. The definition of unambiguous sites follows Ting et al. (2000): a site is defined as unambiguous only when two of the three species share a derived nucleotide with none of their alleles having the ancestral nucleotide, whereas the third species has the ancestral nucleotide with no alleles having the derived one. We found a total of 10 unambiguous sites for Hmr. Among them, 6 sites support the grouping of D. simulans and D. mauritiana, 3 sites support the grouping of D. simulans and D. sechellia, and only 1 site supports the grouping of D. mauritiana and D. sechellia. This result is consistent with both of our maximum parsimony phylogenetic trees (supplementary fig. S3B [Supplementary Material online] and fig. 2).
FIG. 2.—
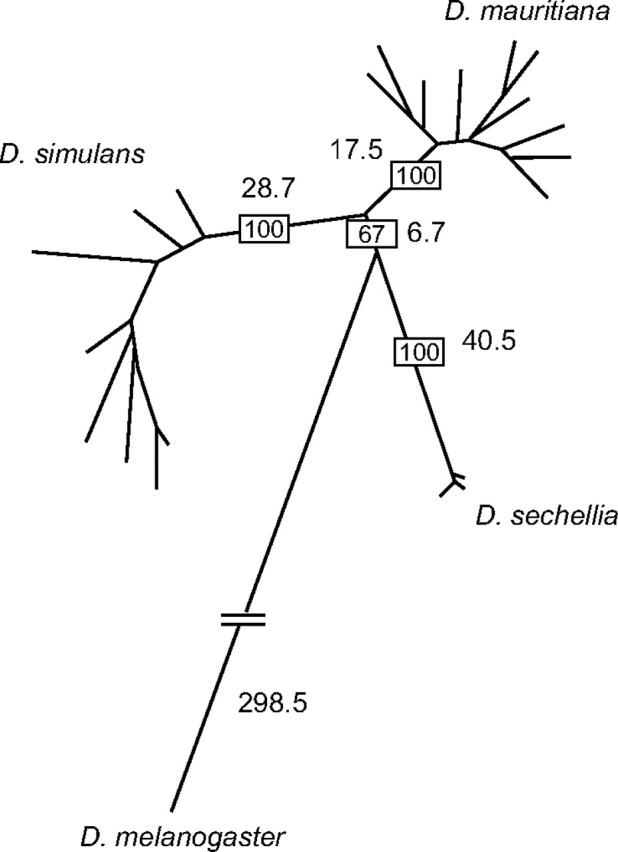
A maximum parsimony phylogenetic tree constructed using Hmr population samples from the three D. melanogaster sibling species. Bootstrap values of major branches are shown in the rectangles (100 replicates). The lengths of major branches, which are proportional to the number of nucleotide changes for the gene, are indicated along the branches. Hmr has clearly diverged and shares no common alleles among the three sibling species.
Tests for Selection: Phylogenetic Analysis among Multiple Drosophila Species
Using a single outgroup sequence, Hmr was previously inferred to have an increased ratio of nonsynonymous to synonymous substitutions (DN/DS ratio) along the branches leading to D. melanogaster and its sibling species (Barbash et al. 2004). In order to obtain a more comprehensive view of Hmr evolution in the melanogaster subgroup, we applied a maximum likelihood analysis on 7 species (fig. 3). Because we obtained conflicting phylogenies for D. simulans and its sister species D. mauritiana and D. sechellia from two different phylogenetic methods (supplementary fig. S3, Supplementary Material online), we excluded D. sechellia in this PAML analysis to avoid possible artifacts caused by using the incorrect evolutionary history. Using a free-ratio model, we confirmed that the estimated DN/DS ratios for Hmr have generally increased along branches leading to D. melanogaster and its sibling species, relative to other lineages in the subgroup. We found DN/DS ratios of approximately one or higher along the lineages leading to D. melanogaster, D. simulans, and D. mauritiana. These values are similar but not identical to those of Barbash et al. (2004), due to the inclusion of different species sequences in the two studies. Note also that the values for DN and DS for D. mauritiana were erroneously switched in figure 3 of Barbash et al. (2004).
DN/DS ratios were also relatively high for other lineages of the subgroup, with the striking exception of the D. yakuba and D. santomea lineages after the split from their common ancestor, where DN/DS was approximately 0.2.
Tests for Selection: Evidence for Positive Selection in D. simulans and D. mauritiana
We analyzed population genetic data sets from different species pairs and groups in order to further explore the very different estimations of divergence in figure 3. We first examined the sibling species of D. melanogaster (table 2). Polymorphism values, including the much lower level for D. sechellia, were generally consistent with observations from other genes (Kliman et al. 2000).
The MK test (McDonald and Kreitman 1991) was carried out for D. simulans and D. mauritiana and rejects the null hypothesis of neutral evolution with a highly significant P value (table 3). This comparison shows a particularly high amount of nonsynonymous substitutions relative to synonymous substitutions. We then polarized substitutions using D. melanogaster as an outgroup and rejected neutrality along both the D. simulans and D. mauritiana lineages. The significance of these tests is most likely caused by an excess of nonsynonymous substitutions. We tested an alternative hypothesis that departures from neutrality may be due to selection on synonymous sites for preferred codons (DuMont et al. 2004). Tests were not significant for either D. mauritiana (P = 0.339) or D. simulans (P = 0.115) using D. melanogaster as an outgroup.
Pairwise comparisons of both D. simulans and D. mauritiana with D. sechellia also reject neutral evolution (table 3). These tests are not independent from the above D. simulans–D. mauritiana comparisons but rather reinforce the inference of positive selection on those two lineages. There is little power to test for nonneutral evolution along the D. sechellia lineage due to the very low amount of polymorphism in this species (table 2), and not surprisingly, MK tests did not reject the null hypothesis for the D. sechellia lineage (data not shown).
In combination with the DN/DS estimates in figure 3, we conclude that Hmr has continued to diverge under positive selection along both the D. mauritiana and the D. simulans lineages after the divergence of their common ancestor from D. melanogaster.
Tests for Selection: Evidence for Positive Selection in the D. santomea Lineage
The MK test for D. yakuba and D. santomea also rejected the null hypothesis of neutral evolution. Like in the aforementioned tests with D. simulans and D. mauritiana, there is a higher relative ratio of nonsynonymous to synonymous variation between species relative to within species. Polarization of these data relative to D. teissieri revealed nonneutral evolution exclusively on the lineage leading to D. santomea. Analysis of the synonymous substitutions suggested that there is not an excess of substitutions leading to preferred codons for either species, using D. teissieri as an outgroup (P = 0.286 for D. yakuba; P = 1.000 for D. santomea).
Tests for Selection: Evidence for Positive Selection in the Lineage Leading to D. yakuba and D. santomea
Although estimated DN/DS ratios were low along the branches leading to both D. yakuba and D. santomea, DN/DS had a high value of 0.6585 in the lineage leading to their common ancestor (fig. 3). We therefore extended our MK test analyses to a pairwise comparison of each species with D. teissieri. In both cases, we rejected neutral evolution. These results were not due to selection on synonymous sites for preferred substitutions (P = 0.845 for D. yakuba using D. erecta as the outgroup; P = 0.839 for D. santomea using D. erecta as the outgroup; similar results were obtained when using D. melanogaster as the outgroup [data not shown]). We also polarized these MK test results and again rejected neutral evolution for both the D. yakuba and the D. santomea lineages. These tests of the D. yakuba and D. santomea lineages are clearly not independent as the majority of the substitutions occurred before the speciation of D. yakuba and D. santomea. However, our results do strongly suggest that Hmr diverged under positive selection in the common ancestor of these two species.
Hmr in Other Species
In order to obtain a view of Hmr evolution outside of the melanogaster subgroup, we assembled and analyzed Hmr orthologs from the species pair D. persimilis and D. pseudoobscura. We estimated DN/DS between these species to be 1.174. This high value is suggestive of possible adaptive evolution between these species but will require further analysis, in part because of the low level of divergence between these species (DS = 0.0345).
Discussion
Recurrent Positive Selection of Hmr
Hmr causes lethality in hybrid progeny of D. melanogaster females mated to males of its sibling species. This lethality reflects a divergence in function of Hmr between these species because lethality is only caused by Hmr+ from D. melanogaster and not by Hmr+ from D. simulans or D. mauritiana (Barbash et al. 2004). These genetic observations might suggest that D. melanogaster Hmr+ has diverged from its ancestral state but sibling species Hmr+ has not. In contrast to this simple evolutionary scenario, Hmr has diverged extensively along both the D. melanogaster and D. simulans lineages and has done so in a manner consistent with positive selection rather than neutral evolution (Barbash et al. 2004).
Here we have found that Hmr has continued to diverge under positive selection between the sibling species D. simulans and D. mauritiana. Both lineages have high DN/DS values (fig. 3), and polymorphism samples show an absence of allele sharing (fig. 2) and a rejection of neutral evolution by MK tests (table 3). Our data also demonstrate positive selection along the lineage leading to the common ancestor of D. yakuba and D. santomea. This branch has a high DN/DS value (fig. 3), and pairwise tests of both species with D. teissieri clearly reject neutral evolution (table 3). We suggest that these cases demonstrate that Hmr has experienced independent episodes of recurrent adaptive evolution in the melanogaster subgroup along at least three evolutionary branches: between D. simulans and D. mauritiana, between D. melanogaster and the common ancestor of its sibling species (the ancestor of the simulans clade), and between D. teissieri and the common ancestor of D. yakuba and D. santomea. Our analysis of the subsequent divergence of D. yakuba and D. santomea also suggests that nonneutral evolution continued in the D. santomea lineage. DN/DS values are low on both branches, but the MK test between these species rejects neutral evolution, and polarization confines this signal to the D. santomea branch (table 3). Might Hmr cause HI between any of these species pairs? Introgression studies in D. mauritiana/D. simulans hybrids (True et al. 1996) and quantitative trait locus analysis in D. yakuba/D. santomea hybrids (Moehring et al. 2006) have found evidence for genes contributing to male sterility in or near the respective regions corresponding to D. melanogaster cytological region 9D, where Hmr is located. Whether Hmr contributes to these phenotypes remains speculative because the mapping resolution was relatively low, and these and other studies (Masly and Presgraves 2007) suggest that there is a high density of X-linked hybrid male sterility factors in Drosophila.
Our results raise the question of how common is recurrent adaptive evolution, for orthologs of other HI genes, and more generally for other classes of genes. The gene Odysseus (OdsH) causes male sterility in D. simulans/D. mauritiana hybrids and has a large excess of nonsynonymous substitutions compared with synonymous substitutions, strongly suggesting that it diverged between these species under positive selection (Ting et al. 1998). In contrast, OdsH orthologs from species of the Drosophila montium subgroup have low DN/DS values, suggesting that it is evolving under purifying selection in these species (Wen et al. 2006). A similar pattern was seen for the Drosophila innate immunity gene Relish, which shows evidence for adaptive evolution in D. simulans but not in several other Drosophila species pairs, based on population genetic sampling (Begun and Whitley 2000; Levine and Begun 2007). In contrast, population genetic analyses indicate that the spermatogenesis gene roughex has undergone at least two independent rounds of recurrent adaptive evolution, between D. melanogaster and D. simulans and between D. yakuba and D. santomea (Llopart and Comeron 2008).
Phylogeny of the simulans Species Complex and Positive Selection
The phylogeny of the simulans complex is not well resolved because for many genes multiple alleles from different species often group with each other instead of resolving only within their species (Kliman et al. 2000). In contrast, phylogenetic analysis of our Hmr population data set fully resolves D. simulans, D. mauritiana, and D. sechellia (fig. 2). Strongly supported phylogenies were previously obtained for OdsH (Ting et al. 2000) and the retroviral envelope-derived gene Iris (Malik and Henikoff 2005). However, the results differed, with OdsH supporting the same pattern as Hmr with D. sechellia branching off first, whereas Iris supports a different phylogeny where D. mauritiana branches off first.
Two explanations may help explain these discrepancies. The first is the hypothesis that reproductive isolation does not occur as a single event but rather that different regions of the genome will become isolated at different times during nascent speciation (Wu 2001). In this view, the genomes of well-defined species will be a mosaic of regions that have different histories of isolation. Although both Hmr and OdsH are X-linked, they are not particularly close to each other (cytological regions 9D and 16D, respectively). Considering that a region less than 2 kb away from OdsH showed a distinct phylogenetic pattern (Ting et al. 2000), Hmr and OdsH cannot share the same phylogenetic pattern due to linkage. There are also no known large inversions between the simulans complex species. Therefore under the mosaic genome hypothesis, this similarity between Hmr and OdsH in their phylogenetic pattern would likely be coincidental.
A second possible explanation for the phylogenetic discrepancies is that these genes may have experienced selection in different subsets of the three simulans complex species (Malik and Henikoff 2005). It is difficult to directly test this possibility for OdsH, Hmr, and Iris because of the use of different methods to detect selection and different data sets. OdsH shows strong evidence for positive selection between D. simulans and D. mauritiana based on a high DN/DS ratio within its homeodomain, but the D. sechellia lineage has not been examined. Iris shows evidence of positive selection between D. melanogaster and the common ancestor of the simulans complex, and codon-based models rejected neutral evolution among 12 Drosophila species, but selection specifically between simulans complex species has not been detected.
Assessment of MADF Domain Functional Properties
The DNA-binding function of the MYB domain has been biochemically well established in a diverse set of transcription factors from a wide variety of eukaryotes (Lipsick 1996; Oh and Reddy 1999). The SANT domain was discovered as a conserved region, closely related to the MYB domain, in the chromatin remodelers and/or transcriptional cofactors SWI3, ADA2, N-CoR, and TFIIIB (Aasland et al. 1996). Where the MYB domain presents a basic surface that contacts the negatively charged DNA phosphate backbone, the SANT domain presents a distinctly acidic surface that is much more likely to contact positively charged histone tails. This difference is also evident in the general ionic properties of each domain, such as c-MYB R2 (+6.38, pI 10.01) compared with ISWI SANT (−4.41, pI 4.49) (Grune et al. 2003) (fig. 1B).
MADF domains from two Drosophila proteins, ADF1 and DIP3, have been shown to bind directly to DNA (Cutler et al. 1998; Bhaskar and Courey 2002), and both are positively charged (fig. 1B). MADF domains from most other Drosophila proteins are also positively charged, as are three out of four of the predicted MADF domains from HMR in most species (table 1). HMR MADF3, however, is negatively charged in many species, including D. melanogaster, suggesting that HMR may have both DNA- and chromatin-binding properties.
Molecular Signatures of an HI Gene
Analyses of a handful of experimental model organisms have led to the identification of many conserved genes that have critical structural and regulatory roles in species throughout the eukaryotic kingdom. Will the identification of speciation genes in model organisms such as Drosophila be similarly generalizable? This remains an open question because our understanding of the speciation process is extensive but the collection of known speciation genes is sparse.
Most HI genes identified to date show evidence of positive selection (Ting et al. 1998; Presgraves et al. 2003; Barbash et al. 2004; Brideau et al. 2006). These observations therefore suggest that candidate HI genes may be identifiable from whole-genome comparisons by the criteria of high DN/DS ratios and nonneutral evolution.
The high evolutionary rate of Hmr may also have led to it having unique functional properties. Three out of the four MADF domains of D. melanogaster Hmr have a range of ionic properties different from the canonical MADF domain, and one MADF domain even has ionic properties inconsistent with DNA binding. This apparent functional plasticity is not restricted to D. melanogaster as Hmr orthologs from different species have evolved MADF domains with unique ionic properties (table 1). Most striking is D. mojavensis HMR in which three out of the four MADF domains have a net negative charge, making them more similar to the chromatin-binding SANT domain than to the canonical DNA-binding MADF domain.
Additionally, Hmr from D. yakuba and D. santomea encodes a microsatellite-like tandem repeat within its coding region that has undergone variable-length expansion. Although many molecular evolutionary analyses by necessity ignore variable-length repeats because they introduce gaps into multialignments, they may be of functional importance. Studies have linked length variation of simple amino acid repeats to evolutionary agility, meaning the capacity to generate a phenotypic range. One example found a correlation between morphological changes among dog breeds and repeat-length variation in two developmental regulatory genes, Alx-4 and Runx-2 (Fondon and Garner 2004). Another example is the well-studied clock gene period (per), which has a minisatellite-like coding repeat of alternating threonines (Thr) and glycines (Gly), the length of which shows a significant north–south cline across Europe that appears to have been maintained by natural selection (Costa et al. 1992; Rosato et al. 1997). These examples justify further investigation of whether repeat-length variation in Hmr also contributes to its functional divergence.
The most striking observation from our analysis is that Hmr has undergone recurrent positive selection in multiple Drosophila lineages. We have also found that sequence evolution in Hmr has resulted in large variation in repeat-tract lengths among orthologs and has significantly altered the ionic properties of its MADF domains. Similar investigations of other HI genes will be necessary to address whether features such as recurrent selection and protein sequence plasticity are peculiar to Hmr or instead reflect general features of genes that cause reproductive isolation.
Supplementary Material
Supplementary figures S1–S3 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We thank Dr David Begun and Dr Shun-Chern Tsaur for flies; Ms Vanessa Bauer Dumont for help with preferred codon analyses; Ms Julie Chepovetsky for help with DNA sequencing; Dr Charles Aquadro, Dr David Begun, Dr Nadia Singh, and Dr Hsiao-Pei Yang for useful discussions; and Dr Michael Nachman and the anonymous reviewers for helpful suggestions that improved the manuscript. Supported by National Institutes of Health grant R01GM074737 to D.A.B.
References
- Aasland R, Stewart AF, Gibson T. The SANT domain: a putative DNA-binding domain in the SWI-SNF and ADA complexes, the transcriptional co-repressor N-CoR and TFIIIB. Trends Biochem Sci. 1996;21:87–88. [PubMed] [Google Scholar]
- Albà MM, Guigó R. Comparative analysis of amino acid repeats in rodents and humans. Genome Res. 2004;14:549–554. doi: 10.1101/gr.1925704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D, Thornton K, Clark A, Andolfatto P. Extensive introgression of mitochondrial DNA relative to nuclear genes in the Drosophila yakuba species group. Evol Int J Org Evol. 2006;60:292–302. [PubMed] [Google Scholar]
- Barbash DA, Awadalla P, Tarone AM. Functional divergence caused by ancient positive selection of a Drosophila hybrid incompatibility locus. PLoS Biol. 2004;2:e142. doi: 10.1371/journal.pbio.0020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbash DA, Siino DF, Tarone AM, Roote J. A rapidly evolving MYB-related protein causes species isolation in Drosophila. Proc Natl Acad Sci USA. 2003;100:5302–5307. doi: 10.1073/pnas.0836927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Whitley P. Adaptive evolution of relish, a Drosophila NF-kappaB/IkappaB protein. Genetics. 2000;154:1231–1238. doi: 10.1093/genetics/154.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar V, Courey AJ. The MADF-BESS domain factor Dip3 potentiates synergistic activation by Dorsal and Twist. Gene. 2002;299:173–184. doi: 10.1016/s0378-1119(02)01058-2. [DOI] [PubMed] [Google Scholar]
- Birney E, Clamp M, Durbin R. GeneWise and genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Latek RR, Peterson CL. The SANT domain: a unique histone-tail-binding module? Nat Rev Mol Cell Biol. 2004;5:1–6. doi: 10.1038/nrm1314. [DOI] [PubMed] [Google Scholar]
- Brideau NJ, Flores HA, Wang J, Maheshwari S, Wang X, Barbash DA. Two Dobzhansky–Muller genes interact to cause hybrid lethality in Drosophila. Science. 2006;314:1292–1295. doi: 10.1126/science.1133953. [DOI] [PubMed] [Google Scholar]
- Clark AG, Eisen MB, Smith DR, et al. (415 co-authors) Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Costa R, Peixoto AA, Barbujani G, Kyriacou CP. A latitudinal cline in a Drosophila clock gene. Proc Biol Sci. 1992;250:43–49. doi: 10.1098/rspb.1992.0128. [DOI] [PubMed] [Google Scholar]
- Coyne JA. Genetics and speciation. Nature. 1992;355:511–515. doi: 10.1038/355511a0. [DOI] [PubMed] [Google Scholar]
- Cuff JA, Barton GJ. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins. 2000;40:502–511. doi: 10.1002/1097-0134(20000815)40:3<502::aid-prot170>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Cutler G, Perry KM, Tjian R. Adf-1 is a nonmodular transcription factor that contains a TAF-binding Myb-like motif. Mol Cell Biol. 1998;18:2252–2261. doi: 10.1128/mcb.18.4.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuMont VB, Fay JC, Calabrese PP, Aquadro CF. DNA variability and divergence at the notch locus in Drosophila melanogaster and D. simulans: a case of accelerated synonymous site divergence. Genetics. 2004;167:171–185. doi: 10.1534/genetics.167.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England BP, Admon A, Tjian R. Cloning of Drosophila transcription factor Adf-1 reveals homology to Myb oncoproteins. Proc Natl Acad Sci USA. 1992;89:683–687. doi: 10.1073/pnas.89.2.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fondon JW, 3rd, Garner HR. Molecular origins of rapid and continuous morphological evolution. Proc Natl Acad Sci USA. 2004;101:18058–18063. doi: 10.1073/pnas.0408118101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grune T, Brzeski J, Eberharter A, Clapier CR, Corona DF, Becker PB, Muller CW. Crystal structure and functional analysis of a nucleosome recognition module of the remodeling factor ISWI. Mol Cell. 2003;12:449–460. doi: 10.1016/s1097-2765(03)00273-9. [DOI] [PubMed] [Google Scholar]
- Hancock JM, Simon M. Simple sequence repeats in proteins and their significance for network evolution. Gene. 2005;345:113–118. doi: 10.1016/j.gene.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Hutter P, Ashburner M. Genetic rescue of inviable hybrids between Drosophila melanogaster and its sibling species. Nature. 1987;327:331–333. doi: 10.1038/327331a0. [DOI] [PubMed] [Google Scholar]
- Kliman RM, Andolfatto P, Coyne JA, Depaulis F, Kreitman M, Berry AJ, McCarter J, Wakeley J, Hey J. The population genetics of the origin and divergence of the Drosophila simulans complex species. Genetics. 2000;156:1913–1931. doi: 10.1093/genetics/156.4.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Tamura K, Nei M. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Lachaise D, David JR, Lemeunier F, Tsacas L, Ashburner M. The reproductive relationships of Drosophila sechellia with Drosophila mauritiana, Drosophila simulans, and Drosophila melanogaster from the Afrotropical region. Evolution. 1986;40:262–271. doi: 10.1111/j.1558-5646.1986.tb00468.x. [DOI] [PubMed] [Google Scholar]
- Levine MT, Begun DJ. Comparative population genetics of the immunity gene, Relish: is adaptive evolution idiosyncratic? PLoS ONE. 2007;2:e442. doi: 10.1371/journal.pone.0000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsick JS. One billion years of Myb. Oncogene. 1996;13:223–235. [PubMed] [Google Scholar]
- Llopart A, Comeron JM. Recurrent events of positive selection in independent Drosophila lineages at the spermatogenesis gene roughex. Genetics. 2008;179:1009–1020. doi: 10.1534/genetics.107.086231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS, Henikoff S. Positive selection of Iris, a retroviral envelope-derived host gene in Drosophila melanogaster. PLoS Genet. 2005;1:429–443. doi: 10.1371/journal.pgen.0010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masly JP, Presgraves DC. High-resolution genome-wide dissection of the two rules of speciation in Drosophila. PLoS Biol. 2007;5:e243. doi: 10.1371/journal.pbio.0050243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Moehring AJ, Llopart A, Elwyn S, Coyne JA, Mackay TF. The genetic basis of postzygotic reproductive isolation between Drosophila santomea and D. yakuba due to hybrid male sterility. Genetics. 2006;173:225–233. doi: 10.1534/genetics.105.052985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh IH, Reddy EP. The myb gene family in cell growth, differentiation and apoptosis. Oncogene. 1999;18:3017–3033. doi: 10.1038/sj.onc.1202839. [DOI] [PubMed] [Google Scholar]
- Presgraves DC, Balagopalan L, Abmayr SM, Orr HA. Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature. 2003;423:715–719. doi: 10.1038/nature01679. [DOI] [PubMed] [Google Scholar]
- Rawson PD, Burton RS. Functional coadaptation between cytochrome c and cytochrome c oxidase within allopatric populations of a marine copepod. Proc Natl Acad Sci USA. 2002;99:12955–12958. doi: 10.1073/pnas.202335899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato E, Peixoto AA, Costa R, Kyriacou CP. Linkage disequilibrium, mutational analysis and natural selection in the repetitive region of the clock gene, period, in Drosophila melanogaster. Genet Res. 1997;69:89–99. doi: 10.1017/s001667239700267x. [DOI] [PubMed] [Google Scholar]
- Rozas J, Sánchez-DelBarrio JC, Messegyer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. RepeatMasker Open-3.0. 1996–2004. Available from: http://www.repeatmasker.org. [Google Scholar]
- Sturtevant AH. Genetic studies of Drosophila simulans. I. Introduction. Hybrids with Drosophila melanogaster. Genetics. 1920;5:488–500. doi: 10.1093/genetics/5.5.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting C-T, Tsaur S-C, Wu C-I. The phylogeny of closely related species as revealed by the genealogy of a speciation gene, Odysseus. Proc Natl Acad Sci USA. 2000;97:5313–5316. doi: 10.1073/pnas.090541597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting C-T, Tsaur S-C, Wu M-L, Wu C-I. A rapidly evolving homeobox at the site of a hybrid sterility gene. Science. 1998;282:1501–1504. doi: 10.1126/science.282.5393.1501. [DOI] [PubMed] [Google Scholar]
- True JR, Weir BS, Laurie CC. A genome-wide survey of hybrid incompatibility factors by the introgression of marked segments of Drosophila mauritiana chromosomes into Drosophila simulans. Genetics. 1996;142:819–837. doi: 10.1093/genetics/142.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen S-Y, Shimada K, Kawai K, Toda MJ. Strong purifying selection on the Odysseus gene in two clades of sibling species of the Drosophila montium species subgroup. J Mol Evol. 2006;62:659–662. doi: 10.1007/s00239-005-0101-8. [DOI] [PubMed] [Google Scholar]
- Wittbrodt J, Adam D, Malitschek B, Maueler W, Raulf F, Telling A, Robertson SM, Schartl M. Novel putative receptor tyrosine kinase encoded by the melanoma-inducing Tu locus in Xiphophorus. Nature. 1989;341:415–421. doi: 10.1038/341415a0. [DOI] [PubMed] [Google Scholar]
- Wu C-I. The genic view of the process of speciation. J Evol Biol. 2001;14:851–865. [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.