

Abstract
Current evidence suggests that hematopoietic stem/progenitor cell (HSPC) mobilization by granulocyte colony-stimulating factor (G-CSF) is mediated by induction of bone marrow proteases, attenuation of adhesion molecule function, and disruption of CXCL12/CXCR4 signaling in the bone marrow. The relative importance and extent to which these pathways overlap or function independently are uncertain. Despite evidence of protease activation in the bone marrow, HSPC mobilization by G-CSF or the chemokine Groβ was abrogated in CXCR4−/− bone marrow chimeras. In contrast, HSPC mobilization by a VLA-4 antagonist was intact. To determine whether other mobilizing cytokines disrupt CXCR4 signaling, we characterized CXCR4 and CXCL12 expression after HSPC mobilization with Flt3 ligand (Flt3L) and stem cell factor (SCF). Indeed, treatment with Flt3L or SCF resulted in a marked decrease in CXCL12 expression in the bone marrow and a loss of surface expression of CXCR4 on HSPCs. RNA in situ and sorting experiments suggested that the decreased CXCL12 expression is secondary to a loss of osteoblast lineage cells. Collectively, these data suggest that disruption of CXCR4 signaling and attenuation of VLA-4 function are independent mechanisms of mobilization by G-CSF. Loss of CXCL12 expression by osteoblast appears to be a common and key step in cytokine-induced mobilization.
Introduction
Under basal conditions, the great majority of hematopoietic stem and progenitor cells (HSPCs) reside in the bone marrow. In response to injury or after administration of a wide range of pharmacologic agents, HSPCs are mobilized into the circulation. Clinically, HSPC mobilization has been exploited as a means to obtain hematopoietic stem cells for therapeutic transplantation. In fact in current practice, mobilized blood has essentially replaced bone marrow as a source of HSPCs for stem cell transplantation.1,2 The most widely used mobilizing agent in the clinical setting is granulocyte–colony-stimulating factor (G-CSF). Treatment with G-CSF mobilizes sufficient numbers of HSPCs for transplantation in most donors with a minimum of toxicity. Nevertheless, development of more rapid and effective mobilization protocols would be of great clinical interest.2 This could be achieved, in theory, by identifying downstream mechanisms important in HSPC mobilization and targeting those mechanisms directly, either individually or in combination
Although the mechanisms by which G-CSF induces HSPC mobilization are not fully understood, there is evidence that3 general pathways are in involved: activation of proteases, attenuation of adhesion molecule function, and disruption of CXCL12/CXCR4 signaling (reviewed in Levesque et al3 and Papayannopoulou4). G-CSF administration is associated with the activation of several proteases and down-regulation of serpin family protease inhibitors in the bone marrow.5–8 This results in the induction of a proteolytic bone marrow environment that may contribute to HSPC mobilization by degrading extracellular matrix and cleaving key signaling molecules.5,7,9 However, mice genetically deficient in several implicated proteases mobilize normally, suggesting that either protease activation is not a necessary event in HSPC mobilization or that redundant protease pathways compensate during mobilization in knockout mice.10,11 Thus, the role of proteases in HSPC mobilization remains controversial.
Very late antigen-4 (VLA-4/α4β1 integrin) is one of the principal integrins regulating HSPC function.12 Vascular adhesion molecule-1 (VCAM-1, CD106), a major ligand for VLA-4, is constitutively expressed in the bone marrow.13–15 Inhibition of VLA-4 or VCAM-1 function results in rapid HSPC mobilization. Moreover, G-CSF administration results in a loss of VCAM-1 expression in the bone marrow, implicating this pathway in G-CSF–induced mobilization.16
There is accumulating evidence suggesting that CXCL12 (stromal-derived factor-1, SDF-1) through interaction with CXCR4 plays an important role in regulating HSPC trafficking. CXCR4−/− bone marrow chimeras display constitutive HSPC mobilization,17 and treatment with CXCR4 antagonists results in rapid HSPC mobilization.18,19 G-CSF administration is associated with a decrease in CXCL12 protein and mRNA expression in the bone marrow and lower surface expression of CXCR4 on HSPCs.7,20,21 We and others previously reported that G-CSF–induced suppression of osteoblasts may account for the decreased CXCL12 expression in the bone marrow.20,22,23 However, several recent reports question whether osteoblasts express significant amounts of CXCL12, raising the possibility that osteoblast loss during G-CSF treatment represents a coincidental finding that does not play a role in mobilization.24,25
The relative importance of each of these pathways to HSPC mobilization and the extent to which they overlap or function independently are largely unknown. For example, activation of proteases may contribute to cleavage of both CXCL12 and adhesion molecules in the bone marrow.7,16 Similarly, CXCL12 signaling might potentiate adhesion molecule interactions between stromal cells and HSPCs.26 To better understand these relationships, we investigated the function of mobilization pathways in the absence of the CXCL12/CXCR4 signaling. Using CXCR4−/− bone marrow chimeras, we show that in the absence of CXCR4 signaling, G-CSF– and CXCL2 (growth-regulated protein beta [Groβ])– but not anti-VLA4–induced HSPC mobilization is abrogated. Moreover, we provide evidence that loss of CXCL12 expression by osteoblast is a common and key step in cytokine-induced mobilization.
Methods
Mice
CXCR4+/− and congenic C57BL/6 mice carrying the Ly5.1 gene (B6.SJL-Ptprc* Pep3b BoyJ) were obtained from The Jackson Laboratory and pOBCol2.3-GFP was a generous gift from Dr David Rowe (University of Connecticut, Hartford, CT). All mice were inbred onto a C57BL/6 background at least 10 generations. Sex- and age-matched mice between 6 and 16 weeks of age were used in accordance with the guidelines of the Washington University Animal Studies Committee.
Fetal liver transplantation
Fetal liver cells from 12- to 15-dpc CXCR4+/− intercrosses were cryopreserved in RPMI 1640 with 20% fetal calf serum and 20% dimethylsulfoxide (Sigma-Aldrich). Genotyping of genomic DNA prepared from a portion of the embryo was performed to identify CXCR4+/+ and CXCR4−/− embryos, as previously described.27 Recipient mice were conditioned with 900 cGy from a 137Cesium source at a rate of approximately 95 cGy/min 24 hours before transplantation. After thawing, 5 × 106 CXCR4−/− or wild-type fetal liver cells were injected into the lateral tail vein of recipient mouse. Prophylactic antibiotics (Trimethoprim-Sulfamethoxazole; Alpharma) were given during the initial 2 weeks after transplantation.
Mobilization protocols
G-CSF, Flt3 ligand, and SCF.
Recombinant human G-CSF, Flt3 ligand (Flt3L), or stem cell factor (SCF; all from Amgen) were diluted in phosphate-buffered saline (PBS) with 0.1% low endotoxin bovine serum albumin (BSA; Sigma-Aldrich) and administered by daily subcutaneous injection for 7 days at a dose of 250 μg/kg (G-CSF), 10 μg/mouse (Flt3 ligand), or 4 μg/mouse (SCF). Mice were analyzed 3 to 4 hours after the final cytokine dose.
BIO5192.
The VLA-4 antagonist BIO5192, a generous gift from Genzyme was reconstituted to a final concentration of 0.2 mg/mL in sterile 10:36:54 ethanol-propylene glycol-water and administered as a single intravenous injection at a dose of 1 mg/kg. Mice were analyzed 3 hours after injection.
Groβ.
Recombinant human Groβ, a generous gift from Genzyme, was diluted in sterile PBS with 0.1% BSA and administered by subcutaneous injection at a dose of 2.5 mg/kg. Mice were analyzed at indicated times after injection.
AMD3100.
AMD3100 (Sigma-Aldrich) was reconstituted in sterile PBS with 0.1% BSA and administered by subcutaneous injection at a dose of 5 mg/kg.
Peripheral blood and bone marrow analysis
Blood was obtained by retro-orbital venous plexus sampling in polypropylene tubes containing EDTA. Bone marrow cells were isolated by flushing femurs and tibias with 3 to 4 mL ice-cold PBS. Bone marrow extracellular fluid was obtained by flushing each femur with 400 to 1000 μL PBS, and the supernatant was harvested after centrifugation at 400g for 3 minutes.
Metalloproteinase and NE assay
Metalloproteinase activity in bone marrow extracellular fluid was measured with the EnzChek Gelatinase kit using the DQ Gelatin fluorescein conjugate substrate, according to the manufacturer's instructions (Invitrogen). Neutrophil elastase (NE) activity was measured by incubating bone marrow plasma (50 μL) with the NE-specific substrate N-methoxysuccinyl Ala-Ala-Pro-Val P-nitroanilide (Sigma-Aldrich), as previously described.28
CXCL12 ELISA
Quantification of CXCL12 protein in bone marrow extracellular fluid was performed using commercially available enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems) according to the manufacturer's instructions.
CFU-C assay
Blood, bone marrow, and spleen cells were harvested from mice using standard techniques and the number of nucleated cells in these tissues was quantified using a Hemavet (Drew Scientific) automated cell counter. We plated 7 to 35 μL blood, 105 nucleated spleen cells, or 2.0 × 104 nucleated bone marrow cells in 2.5 mL methylcellulose media supplemented with a cocktail of recombinant cytokines (MethoCult 3434; StemCell Technologies). Cultures were plated in duplicate and placed in a humidified chamber with 6% carbon dioxide (CO2) at 37°C. Colonies containing at least 50 cells were counted on day 7 of culture. The total number of bone marrow colony-forming cells (CFU-Cs) was calculated assuming that one femur represents 6% of the animal's total bone marrow.29 Similarly, total peripheral blood CFU-C was calculated assuming a total blood volume of 1.8 mL.29
Real-time quantitative RT-PCR
Femurs were each flushed with 1 mL Trizol reagent (Invitrogen) and RNA was isolated according to the manufacturer's instructions. Quantitative real-time reverse transcriptase-polymerase chain reaction (RT-PCR) was performed using the TaqMan One-step RT-PCR Master Mix Reagents Kit (Applied Biosystems) on a GeneAmp 7300 Sequence Detection System (Applied Biosystems). The reaction mix consisted of 5 μL RNA, 12.5 μL RT-PCR reaction mix, 200 nM forward primer, 200 nM reverse primer, 280 nM internal probe, and 0.625 μL Multiscribe reverse transcriptase (Applied Biosystems) in a total reaction volume of 25 μL. Reactions were repeated in the absence of reverse transcriptase to confirm that DNA contamination was not present. RNA content was normalized to murine β-actin. PCR conditions were 48°C for 30 minutes and 95°C for10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for1 minute. Primers used are as follows: CXCL12 forward primer, 5′-GAGCCAACGTCAAGCATCTG-3′; CXCL12 reverse primer, 5′-CGGGTCAATGCACACTTGTC-3′; CXCL12 dT-FAM/TAMRA probe, 5′-TCCAAACTGTGCCCTTCAGATTGTTGC-3′; β-actin forward primer, 5′- ACCAACTGGGACGATATGGAGAAGA-3′; β-actin reverse primer, 5′- TACGACCAGAGGCATACAGGGACAA-3′; and β-actin dT-FAM/TAMRA probe, 5′- AGCCATGTACGTAGCCATCCAGGCTG-3′.
CXCR4 cell surface expression
Bone marrow and peripheral blood cells were incubated with allophycocyanin (APC)–conjugated anti–c-Kit and biotinylated anti-CXCR4 antibodies along with the following panel of fluorescein isothiocyanate (FITC)–conjugated lineage-restricted antibodies: Gr-1 (granulocytes), B220(B lymphocytes), CD3e (T lymphocytes), and Ter-119 (erythroid cells); all antibodies were obtained from eBiosciences except anti-CXCR4 (BD Biosciences). After incubation with phycoerythrin (PE)–conjugated streptavidin, cells were analyzed on a FACScan 5-color, 2-laser flow cytometer (BD Biosciences and Cytek Development).
Transwell migration assay
After red blood cell (RBC) lysis, bone marrow blood leukocytes cells were resuspended in RPMI plus 10% FBS and placed in the top chamber of a 24-well transwell plate (Corning). An aliquot of bone marrow (100 000 cells)and peripheral blood (100 μL) was set aside for CFU-C assay to calculate the number of input CFU-Cs. Recombinant SDF-1 (100 ng/mL; R&D Systems) was placed in the bottom well, and the chamber was incubated for 3 hours at 37°C degrees. Cells were then harvested from the bottom well, and the number of migrated CFU-Cs was measured. The number of migrated CFU-Cs was divided by the input CFU-C number to calculate the percentage of migrated CFU-Cs.
CFU-C half-life assay
The bone marrow mononuclear fraction was isolated by density centrifugation using Lympholyte M (Cedarlane). Recipient mice were treated with a single subcutaneous injection of BIO5192 (1 mg/mL) or vehicle alone, and 6 million donor bone marrow mononuclear cells were adoptively transferred via tail vein injection. Recipient peripheral blood was sampled before adoptive transfer and at 2 and 30 minutes after adoptive transfer and the number of circulating CFU-Cs at each time point measured.
Histomorphometry
Osteoblasts in the bone marrow were quantified by histomorphometry, as previously described.20 Briefly, femurs and tibiae were harvested, fixed overnight in 10% neutral formalin, decalcified by incubating in 14% EDTA at 40°C for 2 weeks, and then embedded in paraffin. To ensure that osteoclasts were excluded from the osteoblast count, deparaffinized sections were stained histochemically for tartrate-resistant acid phosphatase (TRAP) and counterstained with hematoxylin. Osteoblasts were counted in a blinded fashion in 4 to 6 200× fields per section. In some cases, 2 sections 75 micrometers apart were taken from the same sample and osteoblast number was averaged. The number of osteoblasts per millimeter bone perimeter (N.Ob/mm) and percentage osteoblast surface (Ob.S/BS) were calculated using the OsteoMeasure Histomorphometry System (OsteoMetrics, Atlanta, GA).
Isolation of osteoblast lineage cells by flow cytometry
Bone marrow cells were recovered from the femurs of pOBCol2.3-GFP mice by flushing with PBS. The femurs were then infused with PBS containing 50 mg/mL type II collagenase (Worthington Biochemical) and incubated at 37°C for 15 minutes. The collagenase-treated femurs were flushed again with PBS, cells were pooled, and the process was repeated for a total of 6 digests. Pilot experiments demonstrated that virtually all recoverable GFP-positive cells were found in these 6 digests (data not shown).
To isolate osteoblast lineage and nonosteoblast cells, pooled fractions were stained with PE-conjugated antimouse CD45 and antimouse Ter119 antibodies (eBiosciences). CD45−, Ter119−, GFP+ (osteoblast), and GFP− (nonosteoblast) cells were sorted directly into TRIZOL using a MoFlo high-speed cell sorter (Dako).
In situ hybridization
In situ hybridization using a probe for CXCL12 was performed on deparaffinized sections from mouse long bones using 35S-labeled riboprobes as described previously.30 The sections were counterstained with toluidine blue before analysis. Images were acquired with a Leica DM LB microscope (Leica Microsystems) using a Leica NPlan 10×/.025 objective and the Optronics Magnafire CE digital camera (Optronics). Images were processed using Adobe Photoshop (Adobe Systems).
Statistical analysis
Data are presented as mean plus or minus SEM. Statistical significance was assessed using a 2-sided Student t test.
Results
HSPC mobilization by G-CSF is abrogated in CXCR4−/− bone marrow chimeras
CXCR4−/− bone marrow chimeras were generated by transplanting CXCR4−/− fetal liver cells into lethally irradiated wild-type recipient mice. Six weeks after transplantation, more than 90% of peripheral blood and bone marrow leukocytes and bone marrow c-Kit+ lineage− HSPCs were donor derived (Supplemental Figure 1 [available on the Blood website; see the Supplemental Materials link at the top of the online article] and data not shown). Consistent with previous reports,17 at baseline, the number of HSPCs in the blood and spleen of CXCR4−/− chimeras was significantly increased compared with wild-type bone marrow chimeras (Figure 1A-B). As expected, G-CSF administration to wild-type chimeras resulted in mobilization of HSPCs into the blood (34.2 ± 11.7-fold change from baseline) and spleen (14.3 ± 2.6-fold change). In contrast, no change in the number of HSPCs in the blood (0.78 ± .25-fold change) or spleen (1.1 ± .4-fold change) was observed in CXCR4−/− chimeras. The failure of CXCR4−/− chimeras to mobilize is not simply due to a loss of HSPCs, since the number of CFU-Cs in the bone marrow is comparable with wild-type chimeras (Figure 1C; supplemental Table 1).
Figure 1.

G-CSF treatment does not increase the number of circulating progenitors in CXCR4−/− chimeras. Wild-type (CXCR4+/+) and CXCR4−/− chimeras (n = 7-10 each group) were treated with G-CSF or left untreated (control), and CFU-Cs were measured in (A) peripheral blood, (B) spleen, and (C) bone marrow. Data represent mean ± SEM of pooled data from 2 separate experiments; *P < .01.
The absence of a mobilization response in CXCR4−/− chimeras suggests at least 3 possibilities concerning the role of proteases in G-CSF–induced mobilization: (1) protease activation may be dependent upon CXCR4 signaling; (2) proteases may act by disrupting CXCR4 signaling; or (3) proteases may not be required for G-CSF–induced HSPC mobilization. To test the first possibility, we assayed the activity of neutrophil elastase (NE) and metalloproteinases in the bone marrow after G-CSF administration. Consistent with previous reports,5,16 G-CSF administration resulted in a significant increase in metalloproteinase and NE activity in the bone marrow of wild-type chimeras (Figure 2A-B). Interestingly, a similar increase was observed in CXCR4−/− chimeras. These data show that activation of NE and metalloproteinases is not dependent on CXCR4 signaling and suggests that these proteases do not contribute independently to mobilization.
Figure 2.

Bone marrow metalloproteinases and neutrophil elastase are induced normally in CXCR4−/− chimeras. Bone marrow chimeras (n = 2-4 each group) reconstituted with wild-type (CXCR4+/+) or CXCR4−/− fetal liver cells were treated with G-CSF or left untreated (ctrl) and bone marrow plasma was isolated. Metalloproteinase (A) and neutrophil elastase (B) activity in the bone marrow plasma was estimated by measuring cleavage of labeled substrate and normalizing for protein content. Data represent mean ± SEM; *P < .05; **P < .01.
VLA-4 blockade increases the number of circulating HSPCs in CXCR4−/− bone marrow chimeras
To assess the contribution of VLA-4 signals to HSPC trafficking in the absence of CXCR4, we next treated control and CXCR4−/− chimeras with BIO5192, a small molecule inhibitor of VLA-4.31,32 Treatment of wild-type chimeras with a single injection of BIO5192 resulted in an approximately 2.5-fold increase in circulating CFU-Cs that peaked at 3 hours and returned to baseline by 24 hours (Figure 3A and data not shown). A similar approximately 2.5-fold increase in circulating CFU-Cs was observed in CXCR4−/− chimeras (Figure 3B). However, on an absolute basis, the increase from baseline in circulating CFU-Cs was much greater in CXCR4−/− chimeras (18 520 ± 3800) than wild-type chimeras (196 ± 43).
Figure 3.

VLA-4 antagonism increases number of circulating HSPCs in CXCR4+/+ and CXCR4−/− chimeras. CXCR4+/+ and CXCR4−/− chimeras (n = 6-9 each group) were treated with BIO5192, a specific VLA-4 antagonist, and peripheral blood CFU-Cs were measured in (A) CXCR4+/+ and (B) CXCR4−/− chimeras. (C) Recipient mice (n = 3 each group) were administered VLA-4 inhibitor or vehicle 3 hours before adoptive transfer of 6 × 106 wild-type bone marrow mononuclear cells. The number of circulating CFU-Cs was measured before adoptive transfer (t = 0) and 2 and 30 minutes after adoptive transfer. Data represent mean ± SEM; *P < .01.
Previous studies have implicated VLA-4 in the homing of HSPCs to the bone marrow as well as their retention in the bone marrow,14 raising the possibility that reduced clearance may contribute to the increase in HSPCs in the circulation after BIO5192 administration. To test this possibility, the clearance of wild-type CFU-Cs from the blood after adoptive transfer into nonirradiated wild-type mice treated with BIO5192 or vehicle alone was measured (Figure 3C). A similar increase and subsequent decrease in CFU-Cs in the blood after adoptive transfer were observed in control and BIO5192-treated mice, indicating that altered clearance of CFU-Cs from the circulation does not account for the increase in CFU-Cs after BIO5192 administration. Collectively, these data suggest that disruption of VLA-4 induces HSPC mobilization in a CXCR4-independent fashion.
Groβ-induced HSPC mobilization is abrogated in CXCR4−/− chimeras
The rapid kinetics of HSPC mobilization by chemokines (minutes) compared with G-CSF (days) suggests distinct mechanisms of mobilization.3,4,33 Indeed, in contrast to G-CSF, there is no change in CXCL12 expression in the bone marrow after Groβ (CXCL2) administration.34 Instead, Groβ (and other chemokines) are thought to induce HSPC mobilization through the activation of metalloproteinases.34–36 To test these postulates, we characterized HSPC mobilization by Groβ in wild-type or CXCR4−/− chimeras. Consistent with previous studies, treatment with Groβ in wild-type chimeras resulted in a modest (4.4 ± 1.4-fold) but rapid increase in circulating CFU-C and serum metalloproteinase activity (Figures 4A,C).36,37 Surprisingly, despite induction of a similar level of serum metalloproteinase activity, no Groβ-induced HSPC mobilization was observed; in fact, there was a small but significant decrease in circulating CFU-Cs after Groβ administration (Figure 4B). Consistent with these findings, pharmacologically inhibiting CXCR4 with AMD3100 completely blocked mobilization by Groβ in wild-type mice (Figure 4D). These data show that Groβ-induced mobilization is dependent upon CXCR4 signaling.
Figure 4.

Groβ administration decreases the number of circulating HSPCs in CXCR4−/− chimeras. Circulating CFU-Cs were measured in CXCR4+/+ (A, n = 3-6) and CXCR4−/− (B, n = 5-11) chimeras at the indicated time points after administration of Groβ (2.5 mg/kg). (C) CXCR4+/+ (WT) or CXCR4−/− (KO) chimeras (n = 3-4 each group) were treated with Groβ or left untreated (ctrl). Serum was collected 15 minutes after treatment and assayed for metalloproteinase activity. (D) Wild-type mice (n = 4 each group) were given a single injection of AMD3100 (5 mg/kg) and 3 hours later Groβ was administered. Shown is number of circulating CFU-Cs before and 15 minutes after Groβ administration. Data represent mean ± SEM; *P < .05 compared with pretreatment; **P < .01 vs control.
Disruption of CXCL12/CXCR4 signaling is a common feature of cytokine-induced mobilization
Cytokines with distinct target cell populations and biologic activities are able to induce HSPC mobilization (reviewed in Thomas et al38). We hypothesized that disruption of CXCR4 signaling may be a common mechanism of mobilization by hematopoietic cytokines. To test this hypothesis, we characterized CXCL12 and CXCR4 expression after treatment with Flt3 ligand (Flt3L) or stem cell factor (SCF), 2 strong mobilizing cytokines. As expected, treatment of wild-type mice with Flt3L39,40 or SCF41–43 for 7 days resulted in a significant increase in circulating and splenic CFU-Cs (Figure 5A and data not shown). Consistent with previous reports, KL hematopoietic progenitors mobilized by G-CSF into the blood had reduced cell surface expression of CXCR4 compared with BM-resident KL cells (Figure 5B). A similar decrease in CXCR4 expression was observed in KL cells mobilized with Flt3L but not SCF. In addition, both Flt3L- and SCF-mobilized KL cells showed reduced migration to CXCL12, suggesting impaired CXCR4 signaling (Figure 5C). A key step in G-CSF–induced HSPC mobilization is down-regulation of CXCL12 expression in the bone marrow. Indeed, G-CSF treatment resulted in an approximately 5-fold decrease in the level of CXCL12 protein and mRNA in the bone marrow, and a similar decrease in CXCL12 protein and mRNA expression was observed after treatment with Flt3L or SCF (Figure 5D). These data suggest that disruption of CXCR4 signaling is a common feature of G-CSF–, Flt3L-, and SCF-induced HSPC mobilization.
Figure 5.

Disruption of CXCL12/CXCR4 signaling is a common feature in cytokine-induced mobilization. Wild-type mice (n = 6-8 each group) were left untreated (ctrl) or treated with G-CSF, Flt3L, or SCF for 7 days. Shown is the number of CFU-Cs in the blood (A) and bone marrow (B). (C) CXCR4 surface expression of Kit+ lineage− cells in the blood (PB) and bone marrow (BM) was measured by flow cytometry. Shown is the mean fluorescent intensity (MFI). (D) The migration of CFU-Cs isolated from the blood or bone marrow in response to CXCL12 was measured using a transwell assay. Shown is the percentage of input CFU-Cs that migrated in response to CXCL12. (E) CXCL12 mRNA expression in the bone marrow (relative to β-actin) was measured by real-time RT-PCR. (F) CXCL12 protein expression in bone marrow plasma was measured by ELISA. Shown is the expression relative to control bone marrow. Data represent mean ± SEM; *P < .05; **P < .01.
Cytokine administration results in the specific loss of osteoblast CXCL12
We and others have shown that the G-CSF–induced decrease in bone marrow CXCL12 is associated with a decrease in osteoblast number.20,22,23 Since osteoblasts are an important source of CXCL12,20,44 we investigated whether the Flt3L- and SCF-induced decrease in CXCL12 expression was associated with a decrease in osteoblast number. Osteoblasts were enumerated on H&E-stained sections from mice treated with Flt3L, G-CSF, or SCF and compared with untreated mice. A similar decrease in endosteal and trabecular osteoblasts was noted in mice treated with each cytokine, indicating that loss of osteoblasts is a common feature of cytokine-induced mobilization (Figure 6).
Figure 6.
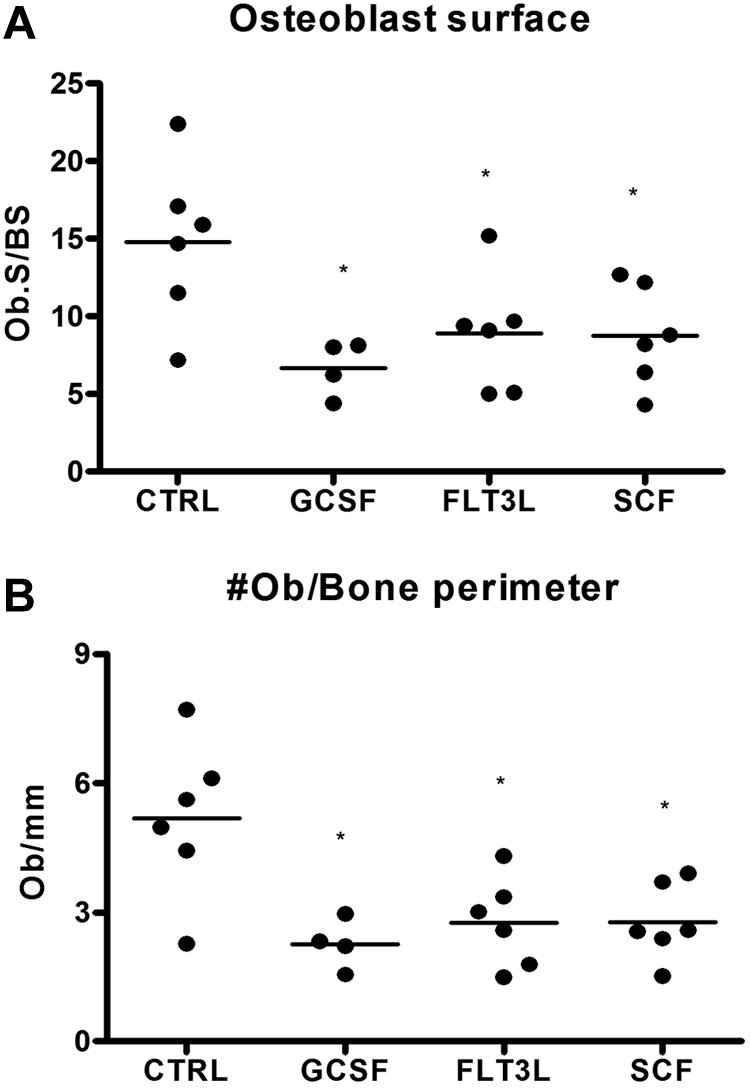
Loss of bone marrow osteoblasts is associated with cytokine-induced mobilization. Mice (n = 4-6 each group) were treated with G-CSF, Flt3L, or SCF for 7 days. Bone marrow osteoblasts were enumerated in H&E-stained paraffin sections using standard histomorphometric technique. Shown is the percentage of osteoblast-covered bone surface (A) and number of osteoblasts per bone perimeter (B). Data represent mean ± SEM; *P < .05.
Controversy exists as to the contribution of osteoblast-expressed CXCL12 to total bone marrow CXCL12.24,25 Besides osteoblasts, other stromal cell types express CXCL12, including endothelial cells and CXCL12-expressing adventitial reticular (CAR) cells scattered throughout the bone marrow space.24,25,44 To further define the cellular source of CXCL12 in the bone marrow, we performed RNA in situ hybridization for CXCL12 (Figure 7A). At baseline, CXCL12 mRNA was detected on endosteal and trabecular bone surfaces as well as in the intramedullary regions of the bone marrow. After G-CSF treatment, abundant scattered cells in the bone marrow continue to express CXCL12 (Figure 7A arrowheads), although CXCL12 expression disappears from bone surfaces (Figure 7A arrows), suggesting that specifically osteoblast CXCL12 is targeted during G-CSF treatment. To confirm this finding, we used transgenic mice expressing GFP under control of a 2.3-kb fragment of the collagen I promoter. These mice express GFP in osteoblast lineage cells, including mature osteoblasts, bone lining cells, and osteocytes.45 The bone marrow stromal cell population (defined as CD45− Ter119−) can be divided into GFP+ osteoblasts and GFP− stromal cells (Figure 7B).23 Osteoblast enrichment in the GFP+ fraction was confirmed by quantitative RT-PCR showing that expression of osteoprotogerin (OPG) and osteocalcin (OC), 2 markers of mature osteoblasts, were increased more than 2500-fold compared with the GFP− stromal cell fraction (Supplemental Figure 1). Using this procedure, the osteoblast and stromal cell fractions were isolated from mice after G-CSF, Flt3L, or SCF treatment, and expression of CXCL12 mRNA in each fraction was measured. Fewer GFP-positive cells were observed in cytokine-treated mice versus controls, consistent with the loss of osteoblasts observed histologically (data not shown). Within the remaining GFP-positive population, CXCL12 mRNA in mice treated with G-CSF, Flt3L, or SCF was markedly reduced (Figure 7C). In contrast, no decrease in CXCL12 mRNA expression was observed in GFP− stromal cells (data not shown). Together, these results suggest that loss of osteoblast-produced CXCL12 may represent a common pathway in cytokine-induced mobilization.
Figure 7.

Cytokine-induced mobilization results in the specific loss of osteoblast CXCL12 expression. (A) Representative photomicrograph of CXCL12 RNA in situ from G-CSF–treated (left) and untreated (right) mouse long bones. Specific CXCL12 signal (red) was detected along endosteal surfaces (arrows) and within the bone marrow (arrowheads; n = 3-5, each group). (B) Transgenic (pOBCol2.3-GFP) mice expressing GFP in osteoblast lineage cells were treated with cytokines(n = 4-5 each group), and stromal cells were isolated and fractionated by flow cytometry into nonosteoblast and osteoblast (GFP+) fractions. Shown are representative dot plots showing the sorting strategy; data are gated on CD45− Ter119− stromal cells. (C) CXCL12 mRNA expression (relative to β-actin) in the GFP+ (osteoblast) cell fraction is shown. Data represent mean ± SEM; *P < .05.
Discussion
HSPC mobilization is a complex biologic process that likely is mediated by several mechanisms. For G-CSF, the most intensively studied mobilizing agent, there is evidence that at least 3 general mechanisms of mobilization are induced: activation of proteases in the bone marrow microenvironment with subsequent cleavage of key regulatory molecules such as VCAM-1 and c-Kit; attenuation of adhesion molecule interactions, especially between VLA-4 and its ligand VCAM-1; and disruption of CXCL12/CXCR4 signaling. In theory, G-CSF may activate these different pathways in parallel, with each pathway contributing independently to mobilization. Alternatively, these mechanisms may functionally interact and/or converge on a common mobilization pathway. In this study, we used a genetic approach to characterize G-CSF–induced HSPC mobilization in the absence of CXCR4 signals. The data show that G-CSF–induced HSPC mobilization is abrogated in the CXCR4−/− bone marrow chimeras. This failure to mobilize does not result from the loss of the mobilizable HSPC pool, since CFU-Cs are efficiently mobilized by BIO5192 in CXCR4−/− chimeras. These findings suggest a model in which all signals induced by G-CSF that contribute to mobilization converge on the CXCL12/CXCR4 pathway. In this model, protease activation either plays a minor role in G-CSF–induced HSPC mobilization or works through disrupting CXCR4 signals. Consistent with the latter possibility, proteases activated by G-CSF have been shown to cleave CXCL12 and CXCR4.7,21
Interestingly, there was a trend toward a reduced number of HSPCs in the bone marrow after G-CSF treatment in both CXCR4+/+ and CXCR4−/− chimeras. This decrease is not likely the result of HSPC mobilization from the bone marrow because (1) the magnitude of the decrease in bone marrow HSPCs is greater than the number of HSPCs mobilized and (2) the magnitude of the decrease in bone marrow CFU-Cs was similar in CXCR4+/+ and CXCR4−/− chimeras, despite the latter's failure to mobilize (Figure 1A-C; supplemental Table 1). Instead, though additional study is required, we favor a model in which changes in the bone marrow microenvironment (eg, loss of osteoblasts) result in a loss of HSPCs through decreased proliferation and/or survival.
Compared with cytokines, chemokine-induced HSPC mobilization is very rapid, with peak mobilization achieved within 60 minutes of chemokine administration.36,46 This observation strongly suggests that mechanisms of mobilization by chemokines are distinct from that of cytokines. Neutrophils play an indispensable role, since neutrophil depletion prevented interleukin-8 (IL-8)–induced mobilization in mice.47 Moreover, there is evidence that neutrophil proteases contribute to chemokine-induced mobilization. Antibodies against MMP-9 or Serpina1, a serine protease inhibitor, partially blocked HSPC mobilization by Groβ and IL-8.34,48 Surprisingly, our data suggest that chemokine-induced mobilization also is dependent upon CXCR4 signaling. HSPC mobilization by Groβ was blocked in CXCR4−/− chimeras and in wild-type mice treated with AMD3100, although the possibility remains that the relatively weak mobilization seen with Groβ treatment may be masked by the considerably greater mobilization induced by CXCR4 blockade. The mobilization defect does not appear to result from a failure to activate proteases, since serum metalloproteinase activity after Groβ treatment was comparable in CXCR4−/− and control chimeras. The interaction between Groβ/CXCR2 and CXCR4 signaling in HSPC trafficking will require more work to elucidate.
Disruption of adhesive interactions between HSPCs and the bone marrow stroma is also believed to play a role in HSPC mobilization. Specifically, inhibition of VLA-4 or VCAM-1 function, either with neutralizing antibodies or by genetic ablation, induces HSPC mobilization.13,15 Herein, we show that disrupting VLA-4 signaling with BIO5192 results in robust HSPC mobilization in the absence of CXCR4. Remarkably, more than 30 000 CFU-Cs/mL were detected in the blood of CXCR4−/− chimeras after treatment with BIO5192, representing an approximately 3000-fold increase from untreated control mice. These findings suggest that CXCR4 and VLA-4 are the dominant signals regulating HSPC release from the bone marrow. Of note, the ability of BIO5192 to mobilize HSPCs independently of CXCR4 signaling does not rule out the possibility that VLA-4 inhibition occurs downstream of CXCL12/CXCR4 inhibition as well. Indeed, previous studies indicate that CXCR4 signaling leads to cytoskeletal reorganization, which may lead to changes in HSPC motility and adhesion to bone marrow stroma.26,49–51
A striking feature of HSPC mobilization is the diversity of target cell populations and biologic activities of the many different mobilizing cytokines. In the present study, we provide evidence that disruption of CXCL12/CXCR4 signaling may be a common mechanism of cytokine-induced HSPC mobilization. Specifically, treatment with Flt3 ligand and SCF, similar to G-CSF, is associated with a decrease in CXCL12 expression in the bone marrow. The previously observed synergy between G-CSF and either Flt3 ligand and SCF likely reflects more complete inhibition of CXCL12 production.39,40,43
The mechanisms by which G-CSF (and other cytokines) decrease bone marrow CXCL12 expression are controversial. We previously showed that the decrease in CXCL12 protein in the bone marrow after G-CSF treatment strongly correlates with the decrease in CXCL12 mRNA.20 This finding strongly argues against a posttranslational mechanism (eg, proteolytic cleavage). In the bone marrow, CXCL12 is expressed in osteoblasts, endothelial cells, and spindle-shaped CAR cells scattered throughout the bone marrow.20,22,23 Our data suggest that the loss of CXCL12 expression after cytokine treatment is primarily due to the suppression of osteoblast lineage cells. First, loss of osteoblasts is a common finding in G-CSF, Flt3L, and SCF treatment. Second, sorting experiments suggest that CXCL12 expression in osteoblasts but not other stromal cells is decreased after cytokine treatment. This finding is corroborated by RNA in situ hybridization data showing that G-CSF treatment results in loss of CXCL12 expression on endosteal surfaces but not in the intramedullary region of the bone marrow. Together, these data suggest that decreased CXCL12 expression through suppression of osteoblast lineage cells is a common mechanism of cytokine-induced HSPC mobilization.
Consistent with previous reports, CXCR4 expression on mobilized HSPCs is significantly reduced compared with bone marrow–resident HSPCs (as measured by KL cells, Figure 5C).21 It is unclear whether CXCR4 down-regulation represents a necessary step in HSPC mobilization or merely reflects phenotypic differences between mobilized and bone marrow–resident HSPCs.52 Interestingly, CXCR4 expression on bone marrow–resident HSPCs is unchanged or even increased after cytokine treatment.7 Our data suggest that CXCL12 expression is selectively decreased in osteoblasts after cytokine treatment, with relative preservation of CXCL12 expression from endothelial and perivascular cells. These observations raise the intriguing possibility that the shift in CXCL12 expression from endosteal to perivascular sites may result in directed migration of HSPCs to the vascular niche with subsequent emigration into the blood. This hypothesis is currently being tested.
There is considerable clinical interest in developing improved strategies to disrupt the interactions of HSPCs with bone marrow stromal cells. Not only would this potentially lead to better HSPC mobilization regimens for stem cell transplantation, but it may also be relevant to the treatment of hematologic malignancies. Specifically, mobilization of leukemic stem cells from their protective bone marrow microenvironment may render them more sensitive to chemotherapy.53–55 Our data suggest that strategies that more completely inhibit CXCR4 signaling would be highly effective. Consistent with this hypothesis, administration of AMD3100 by continuous intravenous infusion was found to result in a much greater mobilization than administration of a single bolus.56 Our data also suggest that combination therapy with agents that target the CXCR4/CXCL12 and VLA-4/VCAM pathways would act synergistically and be highly effective. Indeed, Ramirez and DiPersio show that combination treatment with AMD3100 and BIO5192 resulted in robust mobilization in mice (Pablo Ramirez and John DiPersio, Washington University, written personal communication).
In summary, our data support a model in which disruption of CXCR4/CXCL12 signaling is a common and dominant mechanism of HSPC mobilization by cytokines and possibly chemokines. Multiple pathways induced during mobilization may contribute to the disruption of CXCR4 signaling, including decreased CXCL12 expression, proteolytic cleavage of CXCR4, and possibly functional inactivation of CXCR4. Suppression of osteoblast lineage cells appears to be a shared feature of cytokine-induced mobilization and likely accounts for the decrease in CXCL12 in the bone marrow. The mechanism by which cytokines suppress osteoblasts is an area of active investigation.
Supplementary Material
Acknowledgments
We thank Amgen for providing G-CSF and Genzyme for providing BIO5192 and Groβ.
This work was supported by National Institutes of Health (Bethesda, MD) RO1 HL60772 (D.C.L.).
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.C.L. designed research and wrote the paper along with M.J.C., who carried out the research; and F. Liu M.J.H., and F. Long performed and or analyzed essential experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daniel C. Link, Division of Oncology, Department of Medicine, 660 South Euclid Ave, Campus Box 8007, St Louis, MO 63110; e-mail: dlink@dom.wustl.edu.
References
- 1.Gratwohl A, Baldomero H, Horisberger B, Schmid C, Passweg J, Urbano-Ispizua A. Current trends in hematopoietic stem cell transplantation in Europe. Blood. 2002;100:2374–2386. doi: 10.1182/blood-2002-03-0675. [DOI] [PubMed] [Google Scholar]
- 2.Cashen AF, Lazarus HM, Devine SM. Mobilizing stem cells from normal donors: is it possible to improve upon G-CSF? Bone Marrow Transplant. 2007;39:577–588. doi: 10.1038/sj.bmt.1705616. [DOI] [PubMed] [Google Scholar]
- 3.Levesque JP, Winkler IG, Larsen SR, Rasko JE. Mobilization of bone marrow-derived progenitors. Handb Exp Pharmacol. 2007;180:3–36. doi: 10.1007/978-3-540-68976-8_1. [DOI] [PubMed] [Google Scholar]
- 4.Papayannopoulou T. Current mechanistic scenarios in hematopoietic stem/progenitor cell mobilization. Blood. 2004;103:1580–1585. doi: 10.1182/blood-2003-05-1595. [DOI] [PubMed] [Google Scholar]
- 5.Lévesque JP, Hendy J, Winkler IG, Takamatsu Y, Simmons PJ. Granulocyte colony-stimulating factor induces the release in the bone marrow of proteases that cleave c-KIT receptor (CD117) from the surface of hematopoietic progenitor cells. Exp Hematol. 2003;31:109–117. doi: 10.1016/s0301-472x(02)01028-7. [DOI] [PubMed] [Google Scholar]
- 6.Winkler IG, Hendy J, Coughlin P, Horvath A, Levesque JP. Serine protease inhibitors serpina1 and serpina3 are down-regulated in bone marrow during hematopoietic progenitor mobilization. J Exp Med. 2005;201:1077–1088. doi: 10.1084/jem.20042299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 8.Kollet O, Dar A, Shivtiel S, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12:657–664. doi: 10.1038/nm1417. [DOI] [PubMed] [Google Scholar]
- 9.Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol. 2007;82:1375–1381. doi: 10.1189/jlb.0607338. [DOI] [PubMed] [Google Scholar]
- 10.Robinson SN, Pisarev VM, Chavez JM, Singh RK, Talmadge JE. Use of matrix metalloproteinase (MMP)-9 knockout mice demonstrates that MMP-9 activity is not absolutely required for G-CSF or Flt-3 ligand-induced hematopoietic progenitor cell mobilization or engraftment. Stem Cells. 2003;21:417–427. doi: 10.1634/stemcells.21-4-417. [DOI] [PubMed] [Google Scholar]
- 11.Levesque JP, Liu F, Simmons PJ, et al. Characterization of hematopoietic progenitor mobilization in protease-deficient mice. Blood. 2004;104:65–72. doi: 10.1182/blood-2003-05-1589. [DOI] [PubMed] [Google Scholar]
- 12.Papayannopoulou T, Craddock C. Homing and trafficking of hemopoietic progenitor cells. Acta Haematol. 1997;97:97–104. doi: 10.1159/000203665. [DOI] [PubMed] [Google Scholar]
- 13.Papayannopoulou T, Priestley GV, Nakamoto B, Zafiropoulos V, Scott LM, Harlan JM. Synergistic mobilization of hemopoietic progenitor cells using concurrent beta1 and beta2 integrin blockade or beta2-deficient mice. Blood. 2001;97:1282–1288. doi: 10.1182/blood.v97.5.1282. [DOI] [PubMed] [Google Scholar]
- 14.Scott LM, Priestley GV, Papayannopoulou T. Deletion of alpha4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing. Mol Cell Biol. 2003;23:9349–9360. doi: 10.1128/MCB.23.24.9349-9360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craddock CF, Nakamoto B, Andrews RG, Priestley GV, Papayannopoulou T. Antibodies to VLA4 integrin mobilize long-term repopulating cells and augment cytokine-induced mobilization in primates and mice. Blood. 1997;90:4779–4788. [PubMed] [Google Scholar]
- 16.Lévesque JP, Takamatsu Y, Nilsson SK, Haylock DN, Simmons PJ. Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony-stimulating factor. Blood. 2001;98:1289–1297. doi: 10.1182/blood.v98.5.1289. [DOI] [PubMed] [Google Scholar]
- 17.Ma Q, Jones D, Springer TA. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10:463–471. doi: 10.1016/s1074-7613(00)80046-1. [DOI] [PubMed] [Google Scholar]
- 18.Liles WC, Broxmeyer HE, Rodger E, et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood. 2003;102:2728–2730. doi: 10.1182/blood-2003-02-0663. [DOI] [PubMed] [Google Scholar]
- 19.Broxmeyer HE, Orschell CM, Clapp DW, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Semerad CL, Christopher MJ, Liu F, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. 2005;106:3020–3027. doi: 10.1182/blood-2004-01-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lévesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest. 2003;111:187–196. doi: 10.1172/JCI15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katayama Y, Battista M, Kao WM, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–421. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 23.Christopher MJ, Link DC. Granulocyte colony-stimulating factor induces osteoblast apoptosis and inhibits osteoblast differentiation. J Bone Miner Res. 2008;23:1765–1774. doi: 10.1359/JBMR.080612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20:707–718. doi: 10.1016/j.immuni.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 26.Hidalgo A, Sanz-Rodriguez F, Rodriguez-Fernandez JL, et al. Chemokine stromal cell-derived factor-1alpha modulates VLA-4 integrin-dependent adhesion to fibronectin and VCAM-1 on bone marrow hematopoietic progenitor cells. Exp Hematol. 2001;29:345–355. doi: 10.1016/s0301-472x(00)00668-8. [DOI] [PubMed] [Google Scholar]
- 27.Hirbe AC, Rubin J, Uluckan O, et al. Disruption of CXCR4 enhances osteoclastogenesis and tumor growth in bone. Proc Natl Acad Sci U S A. 2007;104:14062–14067. doi: 10.1073/pnas.0705203104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grenda DS, Johnson SE, Mayer JR, et al. Mice expressing a neutrophil elastase mutation derived from patients with severe congenital neutropenia have normal granulopoiesis. Blood. 2002;100:3221–3228. doi: 10.1182/blood-2002-05-1372. [DOI] [PubMed] [Google Scholar]
- 29.Chervenick P, Boggs D, Marsh J, Cartwright G, Wintrobe M. Quantitative studies of blood and bone marrow neutrophils in normal mice. Am J Physiol. 1968;215:353–360. doi: 10.1152/ajplegacy.1968.215.2.353. [DOI] [PubMed] [Google Scholar]
- 30.Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128:5099–5108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- 31.Leone DR, Giza K, Gill A, et al. An assessment of the mechanistic differences between two integrin alpha 4 beta 1 inhibitors, the monoclonal antibody TA-2 and the small molecule BIO5192, in rat experimental autoimmune encephalomyelitis. J Pharmacol Exp Ther. 2003;305:1150–1162. doi: 10.1124/jpet.102.047332. [DOI] [PubMed] [Google Scholar]
- 32.Pepinsky RB, Lee WC, Cornebise M, et al. Design, synthesis, and analysis of a polyethelene glycol-modified (PEGylated) small molecule inhibitor of integrin {alpha}4{beta}1 with improved pharmaceutical properties. J Pharmacol Exp Ther. 2005;312:742–750. doi: 10.1124/jpet.104.075648. [DOI] [PubMed] [Google Scholar]
- 33.Pelus LM. Peripheral blood stem cell mobilization: new regimens, new cells, where do we stand. Curr Opin Hematol. 2008;15:285–292. doi: 10.1097/MOH.0b013e328302f43a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pelus LM, Bian H, King AG, Fukuda S. Neutrophil-derived MMP-9 mediates synergistic mobilization of hematopoietic stem and progenitor cells by the combination of G-CSF and the chemokines GRObeta/CXCL2 and GRObetaT/CXCL2delta4. Blood. 2004;103:110–119. doi: 10.1182/blood-2003-04-1115. [DOI] [PubMed] [Google Scholar]
- 35.Pruijt JF, Fibbe WE, Laterveer L, et al. Prevention of interleukin-8-induced mobilization of hematopoietic progenitor cells in rhesus monkeys by inhibitory antibodies against the metalloproteinase gelatinase B (MMP-9). Proc Natl Acad Sci U S A. 1999;96:10863–10868. doi: 10.1073/pnas.96.19.10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pelus LM, Horowitz D, Cooper SC, King AG. Peripheral blood stem cell mobilization: a role for CXC chemokines. Crit Rev Oncol Hematol. 2002;43:257–275. doi: 10.1016/s1040-8428(01)00202-5. [DOI] [PubMed] [Google Scholar]
- 37.King AG, Horowitz D, Dillon SB, et al. Rapid mobilization of murine hematopoietic stem cells with enhanced engraftment properties and evaluation of hematopoietic progenitor cell mobilization in rhesus monkeys by a single injection of SB-251353, a specific truncated form of the human CXC chemokine GRObeta. Blood. 2001;97:1534–1542. doi: 10.1182/blood.v97.6.1534. [DOI] [PubMed] [Google Scholar]
- 38.Thomas J, Liu F, Link DC. Mechanisms of mobilization of hematopoietic progenitors with granulocyte colony-stimulating factor. Curr Opin Hematol. 2002;9:183–189. doi: 10.1097/00062752-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 39.Molineux G, McCrea C, Yan XQ, Kerzic P, McNiece I. Flt-3 ligand synergizes with granulocyte colony-stimulating factor to increase neutrophil numbers and to mobilize peripheral blood stem cells with long-term repopulating potential. Blood. 1997;89:3998–4004. [PubMed] [Google Scholar]
- 40.Brasel K, McKenna HJ, Charrier K, Morrissey PJ, Williams DE, Lyman SD. Flt3 ligand synergizes with granulocyte-macrophage colony-stimulating factor or granulocyte colony-stimulating factor to mobilize hematopoietic progenitor cells into the peripheral blood of mice. Blood. 1997;90:3781–3788. [PubMed] [Google Scholar]
- 41.Bodine DM, Seidel NE, Zsebo KM, Orlic D. In vivo administration of stem cell factor to mice increases the absolute number of pluripotent hematopoietic stem cells. Blood. 1993;82:445–455. [PubMed] [Google Scholar]
- 42.Fleming WH, Alpern EJ, Uchida N, Ikuta K, Weissman IL. Steel factor influences the distribution and activity of murine hematopoietic stem cells in vivo. Proc Natl Acad Sci U S A. 1993;90:3760–3764. doi: 10.1073/pnas.90.8.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molineux G, Migdalska A, Szmitkowski M, Zsebo K, Dexter TM. The effects on hematopoiesis of recombinant stem cell factor (ligand for c-kit) administered in vivo to mice either alone or in combination with granulocyte colony-stimulating factor. Blood. 1991;78:961–966. [PubMed] [Google Scholar]
- 44.Ponomaryov T, Peled A, Petit I, et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J Clin Invest. 2000;106:1331–1339. doi: 10.1172/JCI10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalajzic Z, Liu P, Kalajzic I, et al. Directing the expression of a green fluorescent protein transgene in differentiated osteoblasts: comparison between rat type I collagen and rat osteocalcin promoters. Bone. 2002;31:654–660. doi: 10.1016/s8756-3282(02)00912-2. [DOI] [PubMed] [Google Scholar]
- 46.Pruijt JF, Willemze R, Fibbe WE. Mechanisms underlying hematopoietic stem cell mobilization induced by the CXC chemokine interleukin-8. Curr Opin Hematol. 1999;6:152–158. doi: 10.1097/00062752-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 47.Pruijt JF, Verzaal P, van Os R, et al. Neutrophils are indispensable for hematopoietic stem cell mobilization induced by interleukin-8 in mice. Proc Natl Acad Sci U S A. 2002;99:6228–6233. doi: 10.1073/pnas.092112999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Pel M, van Os R, Velders GA, et al. Serpina1 is a potent inhibitor of IL-8-induced hematopoietic stem cell mobilization. Proc Natl Acad Sci U S A. 2006;103:1469–1474. doi: 10.1073/pnas.0510192103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ganju RK, Brubaker SA, Meyer J, et al. The alpha-chemokine, stromal cell-derived factor-1alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J Biol Chem. 1998;273:23169–23175. doi: 10.1074/jbc.273.36.23169. [DOI] [PubMed] [Google Scholar]
- 50.Wang JF, Park IW, Groopman JE. Stromal cell-derived factor-1alpha stimulates tyrosine phosphorylation of multiple focal adhesion proteins and induces migration of hematopoietic progenitor cells: roles of phosphoinositide-3 kinase and protein kinase C. Blood. 2000;95:2505–2513. [PubMed] [Google Scholar]
- 51.Peled A, Kollet O, Ponomaryov T, et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood. 2000;95:3289–3296. [PubMed] [Google Scholar]
- 52.Lapidot T, Petit I. Current understanding of stem cell mobilization: The roles of chemokines, proteolytic enzymes, adhesion molecules, cytokines, and stromal cells. Exp Hematol. 2002;30:973–981. doi: 10.1016/s0301-472x(02)00883-4. [DOI] [PubMed] [Google Scholar]
- 53.Matsunaga T, Takemoto N, Sato T, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9:1158–1165. doi: 10.1038/nm909. [DOI] [PubMed] [Google Scholar]
- 54.Löwenberg B, van Putten W, Theobald M, et al. Effect of priming with granulocyte colony-stimulating factor on the outcome of chemotherapy for acute myeloid leukemia. N Engl J Med. 2003;349:743–752. doi: 10.1056/NEJMoa025406. [DOI] [PubMed] [Google Scholar]
- 55.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 56.Bonig H, Papayannopoulou T. Insights into the biology of mobilized cells through innovative treatment schedules of the CXCR4 antagonist AMD3100. ASH Annual Meeting Abstracts. 2007;110:2229. doi: 10.1016/j.exphem.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}