

Abstract
Opioids, although fundamental to the treatment of pain, are limited in efficacy by side effects including tolerance and hyperalgesia. Using an in vitro culture system, we report that morphine increased microglial migration via a novel interaction between μ-opioid and P2X4 receptors, which is dependent upon PI3K/Akt pathway activation. Morphine at 100 nm enhanced migration of primary microglial cells toward adenosine diphosphate by 257, 247, 301, 394, and 345% following 2, 6, 12, 24, and 48 h of stimulation, respectively. This opioid-dependent migration effect was inhibited by naloxone and confirmed to be μ-opioid receptor-dependent through the use of selective agonists and antagonists. PPADS [pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid)], a P2X1–3,5–7 antagonist, had no effect on microglial migration; however, TNP-ATP [2′,3′-O-(2,4,6-trinitrophenyl)-ATP], a P2X1–7 antagonist, inhibited morphine-induced migration, suggesting a P2X4 receptor-mediated effect. The PI3K inhibitors wortmannin and LY294002 decreased morphine-induced microglial migration. Iba1 protein, a microglial marker, and P2X4 receptor expression were significantly increased after 6, 12, 24, and 48 h of morphine stimulation. Together, these results provide evidence for two phases of morphine effects on microglia. The initial phase takes place in minutes, involves PI3K/Akt pathway activation and leads to acutely enhanced migration. The longer-term phase occurs on the order of hours and involves increased expression of Iba1 and P2X4 receptor protein, which imparts a promigratory phenotype and is correlated with even greater migration. These data provide the first necessary step in supporting microglial migration as an attractive target for the prevention or attenuation of morphine-induced side effects including tolerance and hyperalgesia.
Keywords: μ-opioid receptor, Iba1, PI3K, Akt, tolerance, hyperalgesia
Introduction
Opioids are a standard for treatment of chronic pain; however, their efficacy is limited by side effects including tolerance, hyperalgesia, sedation, mental clouding, hormonal imbalance and immune modulation. While many of these side effects can be mitigated pharmacologically, tolerance and hyperalgesia remain as major limitations to opioid use.
Glia, once thought to be merely supporting cells of the CNS, are now recognized to play a central role in the formation and maintenance of morphine tolerance (Song and Zhao, 2001). We have previously shown that chronic morphine treatment increased spinal microglial reactivity as measured by expression of the microglial marker CD11b (Raghavendra et al., 2002; Tawfik et al., 2005). We and others have reported that the glial modulating agents minocycline and propentofylline inhibit spinal microglial reactivity and attenuate the formation of morphine tolerance in vivo (Raghavendra et al., 2004; Cui et al., 2008). We have also shown that ADP is a robust chemoattractant in transwell migration experiments and that minocycline and propentofylline attenuate microglial migration toward ADP in vitro (Nutile-McMenemy et al., 2007; Horvath et al., 2008). This suggests a potential role for microglial migration in the development of morphine tolerance. We hypothesize that morphine enhances microglial reactivity, inducing the release of proinflammatory cytokines and chemokines and direct signaling between microglia and nociceptive neurons. Furthermore, morphine-induced migration of reactive microglia produce locally elevated levels of these diffusible factors, inducing neuronal sensitization which manifests as tolerance and/or hyperalgesia.
Microglia express μ-, δ-, and κ-opioid receptors (Calvo et al., 2000; Turchan-Cholewo et al., 2008), as well as ionotropic (P2X4 and P2X7) and metabotropic (P2Y2, P2Y6, and P2Y12) purinergic receptors (Inoue, 2006). Several of these purinergic receptors have been implicated in microglial migration (Honda et al., 2001; Ohsawa et al., 2007) and the gating of chronic pain behaviors (Tsuda et al., 2003; Zhang et al., 2008). Exogenous ATP/ADP has been shown to activate microglia (Nakajima and Kohsaka, 2001) and induced chemotaxis via P2Y12 and P2X4 receptors and the PI3K/Akt pathway (Honda et al., 2001; Ohsawa et al., 2007; Irino et al., 2008). P2X4 receptors and microglial migration may be integral to the development and maintenance of neuropathic pain. It is recognized that neuropathic pain and opioid tolerance share common mechanisms (Mayer et al., 1999; Mika, 2008; Romero-Sandoval et al., 2008b).
The present study was designed to determine the modulatory affect of morphine on microglial migration in vitro as a first step toward exploring the greater role of immune competent cells, i.e., microglia, in opioid actions. We hypothesize that morphine enhances the migration of primary cultured microglia via acute PI3K/Akt pathway activation and chronic increases in Iba1 and P2X4 receptor expression. To this end, we assessed the affect of morphine on microglial migration toward ADP, determined the response of P2X4 and opioid receptor antagonism on morphine-induced microglial migration and measured the expression of the microglial marker, Iba1, and P2X4 receptors after morphine stimulation.
Materials and Methods
Cell culture.
All procedures used in these studies were approved by the Dartmouth College Institutional Animal Care and Use Committee. Highly purified microglial cultures were prepared as described previously (Nutile-McMenemy et al., 2007; Horvath et al., 2008). Briefly, cortices were harvested from postnatal day 2 (P2)–P3 Harlan Sprague Dawley pups, minced and incubated with Trypsin/EDTA (Mediatech). The supernatant was then replaced with DMEM (Mediatech) supplemented with 10% fetal bovine serum (Hyclone), 1.1% GlutaMax (Invitrogen), and 1% penicillin/streptomycin (100 U/ml penicillin, 100 μg/ml streptomycin, Mediatech) containing 2000 units DNase (Sigma). The tissue was mechanically disrupted by trituration, the cell suspensions were centrifuged and the cells resuspended in the media described above without DNase. A small aliquot of cells were stained for trypan blue exclusion and cells were plated at 10 million cells per 75 cm2 flask. Cultures were maintained at 37°C with 5% CO2, and media was changed every 3–4 d. After 10 d in vitro (DIV 10), microglial cells were harvested by gently shaking the flasks by hand for 1 min. The resulting cells were found to be >98% microglia by staining with OX-42 antibody (generous gift from Dr. William Hickey, Dartmouth Medical School, Hanover, NH), a marker for the microglial CD11b receptor. Cells were either used immediately for migration experiments or plated for Western blot analysis.
Migration.
The optimal experimental procedures for microglial migration in Costar Transwell plates (6.5 mm diameter insert, 8.0 μm pore size, polycarbonate membrane, Corning) have been previously reported by our lab (Nutile-McMenemy et al., 2007; Horvath et al., 2008). Briefly, DIV 10 primary microglia were harvested as described above, counted, and resuspended in serum-free media at 100 × 103 cells per 100 μl, and placed in siliconized low-adhesion microcentrifuge tubes for 2–3 h treatments or in 12 well plates for 6–48 h treatments. Cells were treated with antagonists [d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP), pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid) (PPADS), 2′,3′-O-(2,4,6-trinitrophenyl)-ATP (TNP-ATP), wortmannin, or LY294002, Sigma] for 1 h, then stimulated with agonists [morphine, [d-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin (DAMGO), [d-Pen2,5]-enkephalin (DPDPE), or U-69593, Sigma)] for 2, 6, 12, 24, or 48 h. Cells were counted post-treatment with trypan blue to insure survival (>99% viability), then added (100 × 103 cells in 100 μl) to the top chamber of a transwell plate with fibronectin-coated membranes and 500 μl of 10 μm ADP in the bottom well. After 2 h of migration, any cells remaining on top of the membrane were removed. The membranes were then rinsed with PBS, the migrated cells were fixed with 2% formaldehyde in PBS, permeabilized with 0.01% Triton X-100 (Sigma) in PBS, stained with crystal violet (Sigma), and rinsed twice with dH2O. The membranes were then dried, inverted, and mounted on microscope slides for analysis. Images of 10 random fields (20× objective) for each membrane were captured via a Q-Fired cooled CCD camera attached to an Olympus microscope and counted by hand with aid of SigmaScan Pro imaging analysis software. Counts for all 10 fields were averaged to give a mean cell count for each membrane. All experiments were completed at least three times with n = 3 per trial. Results are expressed as mean cell migration relative to vehicle control ± SEM.
Western blot analysis.
For all Western blot experiments, DIV 10 primary microglial cells were harvested as described above and then resuspended in complete media. Cells were plated at 250 × 103 cell per well in poly-d-lysine coated 12 well plates. For Iba1 and P2X4 receptor expression experiments, cells were incubated with 0, 1, 10, or 100 nm morphine for 0, 5, 15, 30, or 60 min or 2, 6, 12, 24, or 48 h. For pAkt time course expression experiments, cells were incubated with 100 nm morphine for 0, 5, 15, 30, 60, or 120 min. For PI3K inhibition experiments, cells were incubated with media, 100 nm wortmannin, or 50 μm LY294002 for 15 min then with media or 100 nm morphine for 15 min. After treatments, cells were washed with PBS and dissociated in 40 μl of 1× Laemmli buffer (Bio-Rad) containing 2-mercaptoethanol. Approximately 30–40 μg of protein (40 μl) and standard protein marker were subjected to SDS-PAGE (10% or 18% gels, Bio-Rad), transferred to polyvinylidene difluoride (Bio-Rad) membranes, and blocked with 5% milk in TBS-Tween 20 (0.05%, Sigma). The 10% membranes were incubated with rabbit anti-phospho-Akt primary antibody (pAkt Ser 473, 1:500, Cell Signaling) and 18% membranes with rabbit anti-Iba1 primary antibody (1:1000, Wako) overnight at 4°C. Membranes were then incubated with goat anti-rabbit HRP-conjugated secondary antibody (1:3000, Pierce) for 1 h at 22°C and visualized with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce) for 5 min and imaged using the Syngene G-Box (Synoptics). Membranes were then stripped, blocked, and 10% membranes reprobed with rabbit anti-Akt primary antibody (1:500, Cell Signaling). The 18% membranes were reprobed with rabbit anti-P2X4 receptor primary antibody (1:250, Alomone Labs) then stripped and probed with mouse monoclonal anti-β-actin (1:1000, Abcam). Band intensities were assessed using the analysis software package provided with the Syngene G-Box. For Iba1 and P2X4 receptor expression, data were quantified as relative intensity of band of interest divided by the intensity of β-actin. All treatments were completed at least three times and data were expressed graphically as relative intensity normalized to vehicle control ± SEM.
Statistical analysis.
All experiments were completed at least three times and data are expressed as mean ± SEM. Statistical analyses were completed using GraphPad Prism 4 (GraphPad Software). Unpaired t tests were used to determine significance between groups for all experiments. Significance was determined at a level of p < 0.05.
Results
Morphine enhances microglial migration toward ADP
Two hour morphine treatment robustly increased microglial migration toward 10 μm ADP (Fig. 1a). Morphine dose-dependently increased microglial migration with a peak of 257.3 ± 8.65% (p < 0.001) after treatment with 100 nm morphine compared with 102.0 ± 4.62% in the control group (Fig. 1b). An EC50 value of 9.46 × 10−10 m [4.11 × 10−10 to 1.99 × 10−9 m, 95% confidence interval (C.I.)] was determined for morphine-induced microglial migration. Naloxone, a nonselective opioid receptor antagonist, dose-dependently decreased morphine-induced microglial migration with an IC50 value of 1.45 × 10−10 m (9.64 × 10−11 to 2.19 × 10−10 m, 95% C.I.). Preincubation with 100 nm naloxone for 1 h decreased migration from 257.3 ± 8.65% in 100 nm morphine alone to 92.3 ± 2.91% (p < 0.001) (Fig. 1c).
Figure 1.

Morphine dose-dependently enhanced microglial migration which is inhibited by naloxone. Primary microglia were treated for 2 h with 0, 1, 10, or 100 nm morphine and then allowed to migrate toward 10 μm ADP. a, Images of stained microglia which migrated to the bottom of the transwell membrane. b, Quantification of microglial migration after morphine treatment, represented as mean ± SEM. Microglia were treated for 1 h with 0 or 100 pm or 1, 10, or 100 nm naloxone, then 0 or 100 nm morphine was added. Cells were allowed to migrate toward 10 μm ADP for 2 h; then the migrated cells were counted. c, Quantification of microglial migration after naloxone and morphine treatment, represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with media. ##p < 0.01 compared with 1 nm morphine. Med, Media; mor, morphine.
DAMGO, a μ-receptor-selective agonist, dose-dependently increased microglial migration with an EC50 value of 6.33 × 10−10 m (3.38 × 10−10 to 1.19 × 10−9 m, 95% C.I.). Treatment with 100 nm DAMGO increased microglial migration from 100.0 ± 8.54% in control to 245.0 ± 1.00% (p < 0.001) (Fig. 2a). DPDPE and U-69593, δ- and κ-receptor-selective agonists, respectively, had no effect on microglial migration (Fig. 2b,c). Finally, pretreatment of microglia with 1 or 100 nm CTAP, a μ-opioid receptor selective antagonist, before treatment with 100 nm morphine dose-dependently reduced migration from 255.3 ± 6.01% to 115.0 ± 1.50% (p < 0.001) and 92.0 ± 2.65% (p < 0.001), respectively (Fig. 2d).
Figure 2.

Morphine-induced microglial migration is μ-opioid receptor-dependent. Primary microglia were treated with opioid receptor agonists or antagonist, then allowed to migrate toward 10 μm ADP. Migrated cells were counted and represented as mean ± SEM. a, Quantification of microglial migration after treatment with 0, 1, 10, or 100 nm DAMGO, a selective μ-opioid receptor agonist. b, Quantification of microglial migration after treatment with 0, 1, 10, or 100 nm DPDPE, a selective δ-opioid receptor agonist. c, Quantification of microglial migration after treatment with 0, 1, 10, or 100 nm U-69593, a selective κ-opioid receptor agonist. d, Microglia were pretreated for 1 h with 0, 1, or 100 nm CTAP, a selective μ-opioid receptor antagonist, followed by 2 h of treatment with 0 or 100 nm morphine. **p < 0.01, ***p < 0.001. ##p < 0.01 compared with media control. Med, Media; mor, morphine.
P2X4 inhibition decreases morphine-induced microglial migration
To assess the role of P2X4 receptor antagonism on microglial migration in the presence of morphine stimulation, we used PPADS (P2X1–3,5–7 receptor antagonist) and TNP-ATP (P2X1–7 receptor antagonist). TNP-ATP, and not PPADS, reduced basal ADP-directed and morphine-induced migration (Fig. 3a,b). Pretreatment of microglial cells with 1 or 100 nm TNP-ATP dose-dependently decreased basal ADP-directed migration from 99.7 ± 3.38% in media control to 68.7 ± 2.40% (p < 0.01) and 50.3 ± 1.45% (p < 0.001), respectively (Fig. 3b). Pretreatment with TNP-ATP also decreased morphine-induced microglial migration from 249.7 ± 2.40% in 100 nm morphine alone to 96.67 ± 1.86% (p < 0.001) in the 1 nm TNP-ATP group and 90.3 ± 2.67% (p < 0.001) in the 100 nm TNP-ATP group. Increasing concentrations of TNP-ATP dose-dependently decreased morphine-induced microglial migration with an IC50 value of 6.50 × 10−11 m (3.88 × 10−11 to 1.09 × 10−10 m, 95% C.I.) (Fig. 3c).
Figure 3.
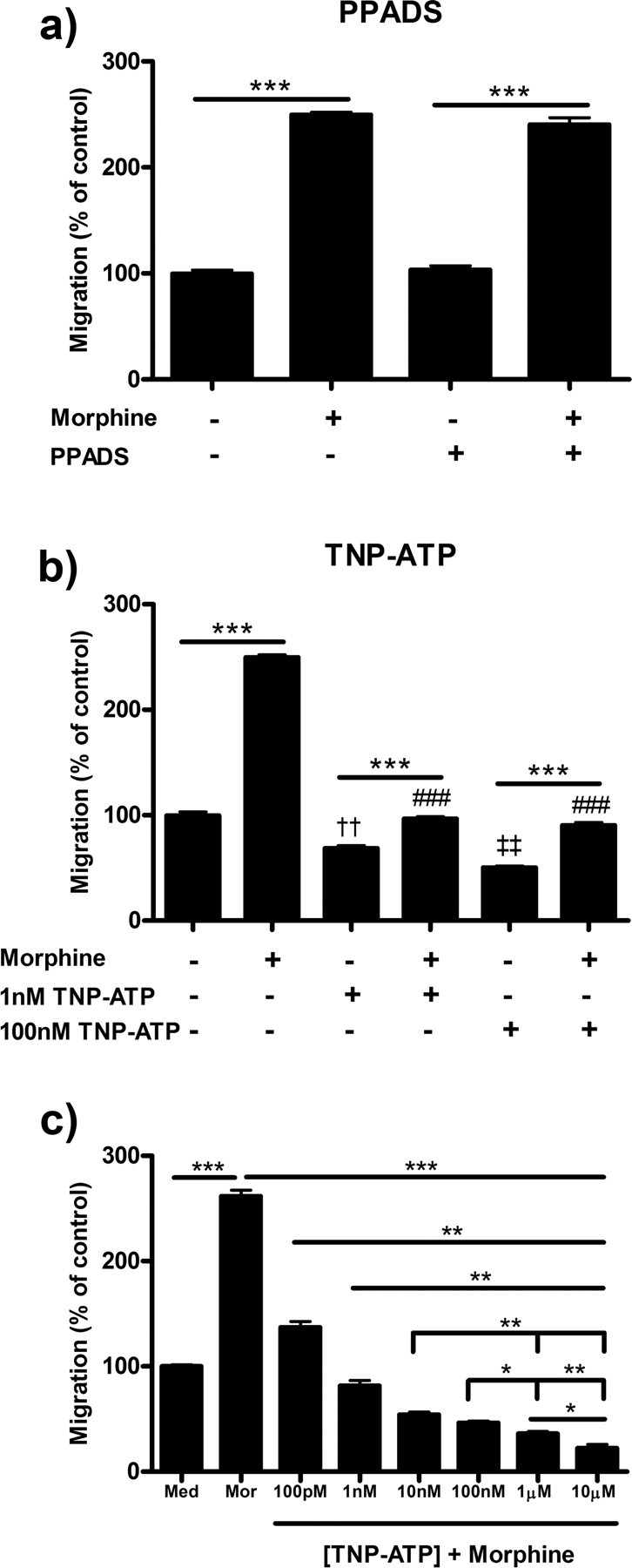
P2X4 inhibition reduced morphine-induced microglial migration. Primary microglia were treated with P2X inhibitors, then allowed to migrate toward 10 μm ADP. Migrated cells were counted and represented as mean ± SEM. a, Microglia were pretreated with 0 or 100 nm PPADS, a P2X1–3,5–7 receptor antagonist, for 1 h, then treated with 0 or 100 nm morphine for 2 h. b, Microglia were pretreated with 0, 1, or 100 nm TNP-ATP, a P2X1–7 receptor antagonist, for 1 h followed by 0 or 100 nm morphine. c, TNP-ATP dose–response inhibition of morphine-induced microglial migration. *p < 0.05, **p < 0.01, ***p < 0.001. ###p < 0.001 compared with 100 nm morphine. ††p < 0.01 compared with media control. ‡‡p < 0.01 compared with 1 nm TNP-ATP. Med, Media; mor, morphine.
PI3K inhibition decreases morphine-induced microglial migration
In this series of experiments, we assessed the role of the PI3K/Akt pathway in morphine-induced microglial migration through the use of two PI3K inhibitors, wortmannin and LY294002. Wortmannin is a bacterially derived semiselective PI3K inhibitor which binds to the ATP-binding domain of the kinase. LY294002 is a synthetic semiselective PI3K inhibitor which binds to catalytic domain of the kinase. We used both inhibitors at moderate concentrations, dissolved in DMSO which was then diluted to a final concentration of than 0.1% DMSO. Pretreatment of microglial cells for 3 h with 0.1% DMSO vehicle did not alter basal or morphine-induced microglial migration (data not shown).
Pretreatment with 100 nm wortmannin decreased basal migration from 100.0 ± 2.52% in media control to 54.0 ± 2.08% (p < 0.001) and morphine-induced migration from 260.0 ± 2.43% to 102.3 ± 4.33% (p < 0.001) (Fig. 4a). In parallel experiments, pretreatment with 50 μm LY294002 decreased basal migration from 100.0 ± 2.52% in media control to 47.7 ± 1.20% (p < 0.001) and morphine-induced migration from 260.0 ± 6.43% to 102.3 ± 5.93% (p < 0.001) (Fig. 4b).
Figure 4.
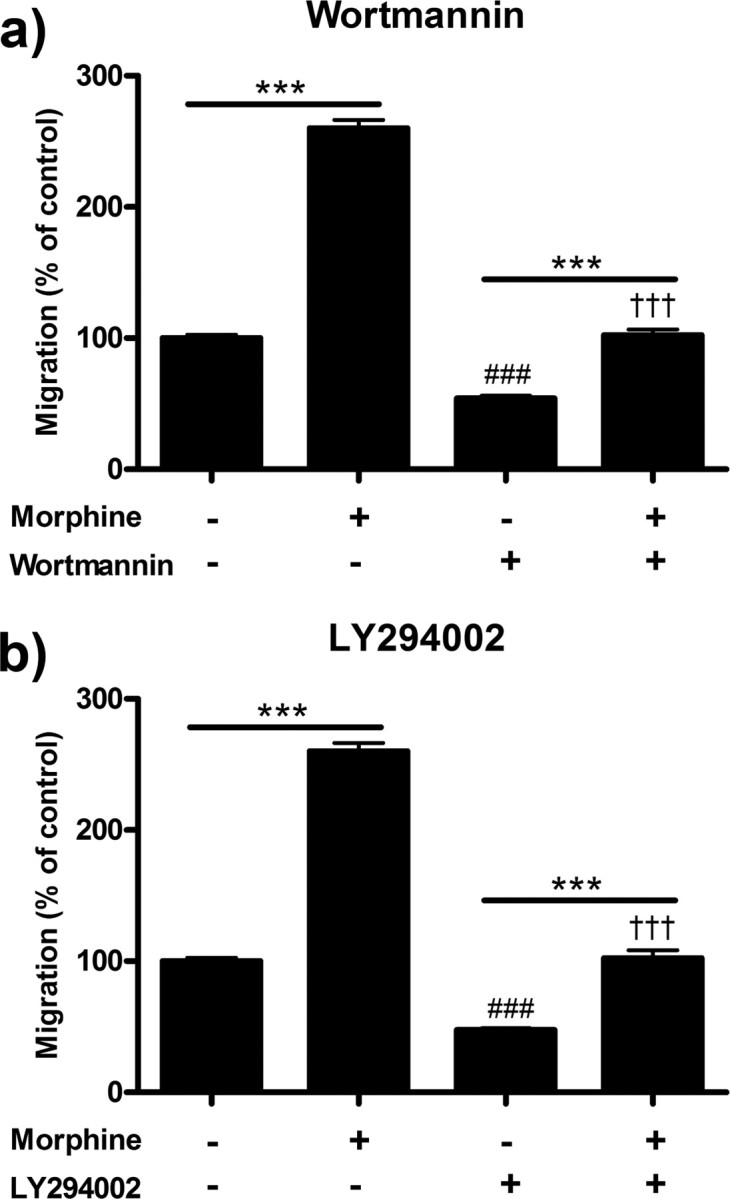
PI3K inhibition reduced morphine-induced microglial migration. Primary microglial cells were treated with two PI3K inhibitors, wortmannin and LY294002, for 1 h then treated for 2 h with morphine and allowed to migrate toward 10 μm ADP. Migrated cells were counted and represented as mean ± SEM. a, Quantification of microglia pretreated with 0 or 100 nm wortmannin followed by 0 or 100 nm morphine. b, Quantification of microglia pretreated with 0 or 50 μm LY294002 followed by 0 or 100 nm morphine. ***p < 0.001. ###p < 0.001 compared with media control. †††p < 0.001 compared with 100 nm morphine.
Morphine increases pAkt in a PI3K and μ-opioid receptor-dependent manner
It has been established that ADP induces Akt phosphorylation (pAkt) acutely and transiently (Irino et al., 2008). The results above indicate that PI3K inhibition reduces morphine-induced microglial migration, indicating a potential role for Akt phosphorylation in morphine-induced migration. Therefore, we completed the following series of experiments to determine if morphine increases the phosphorylation of Akt, via a μ-opioid receptor-dependent mechanism. For these experiments, we exposed microglia to 100 nm morphine for 0, 5, 15, 30, 60, and 120 min, then assessed pAkt and total Akt expression via Western blot analysis. Morphine at 100 nm induced peak Akt phosphorylation at 15 min, which returned to basal levels after 60 min (Fig. 5a). Total Akt levels remained unchanged during the entire 120 min period. To determine if morphine-mediated Akt phosphorylation is PI3K dependent, we again used the PI3K inhibitors, wortmannin and LY294002. Both wortmannin and LY294002 inhibited Akt phosphorylation in the presence and absence of 100 nm morphine (Fig. 5b,c).
Figure 5.
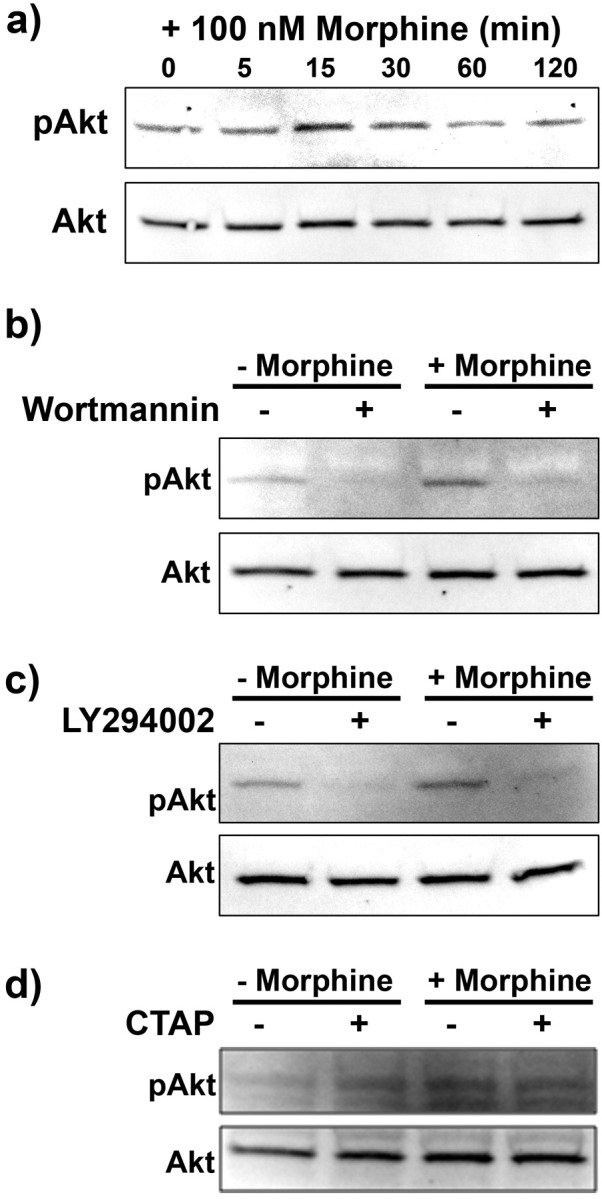
Morphine enhanced Akt phosphorylation in a PI3K and μ-opioid receptor-dependent manner. Primary microglial cells were treated with 100 nm morphine for 0, 5, 15, 30, 60, and 120 min, subjected to Western blot analysis and probed with anti-pAkt then Akt antibody (a). b, Image of Western blot membrane of microglia pretreated for 15 min with 0 or 100 nm wortmannin, then treated for 15 min with 0 or 100 nm morphine. c, Image of Western blot membrane of microglia pretreated for 15 min with 0 or 50 μm LY294002, then treated for 15 min with 0 or 100 nm morphine. d, Image of Western blot membrane of microglia pretreated for 15 min with 0 or 100 nm of the μ-opioid receptor antagonist CTAP, then treated for 15 min with 0 or 100 nm morphine.
To confirm that morphine-mediated Akt phosphorylation is μ-opioid receptor-dependent, we used the μ-opioid receptor-selective antagonist, CTAP. CTAP reduced acute morphine-induced Akt phosphorylation (Fig. 5d).
Morphine enhances microglial Iba1 expression
Enhanced expression of the microglial marker Iba1, is associated with microglial reactivity (Romero-Sandoval et al., 2008a) and has been shown to play a role in microglial migration (Ohsawa et al., 2000, 2004). In this series of experiments, we sought to determine if morphine directly induces Iba1 expression in microglia. Treatment with morphine for 5, 15, 30, 60, or 120 min had no effect on Iba1 expression (data not shown). However, Iba1 expression was significantly increased after 6 to 48 h of morphine stimulation (Fig. 6). Iba1 immunoreactivity was increased to a maximum of 1.81 ± 0.20 (p < 0.001), 2.84 ± 0.75 (p < 0.05), 2.29 ± 0.29 (p < 0.01), and 3.11 ± 0.40 (0 < 0.01) relative to media control after 6, 12, 24, and 48 h of morphine treatment, respectively (Fig. 6a–e).
Figure 6.

Morphine enhances Iba1 expression in microglial cells. Primary microglia were treated with 0, 1, 10, or 100 nm morphine for 6 (a), 12 (b), 24 (c), or 48 (d, e) h, lysed, and subjected to Western blot analysis. a–d, Graphs indicate relative Iba1 expression normalized to β-actin loading control ± SEM. e, Image of membrane after 48 h treatment probed with anti-Iba1 and anti-β-actin antibodies. Graphs show data from at least four individual Western blots, n = 2 per blot. *p < 0.05, **p < 0.01, ***p < 0.001.
Morphine enhances P2X4 receptor expression
In this series of experiments, we examined the time course of P2X4 receptor protein expression which is one possible explanation for the morphine effects observed on microglial migration. Treatment with morphine for 5, 15, 30, 60, or 120 min had no effect on P2X4 receptor expression (data not shown). However, morphine increased P2X4 receptor expression at later time points (Fig. 7). P2X4 receptor expression reached a maximum of 1.96 ± 0.30 (p < 0.05), 2.52 ± 0.60 (p < 0.05), 2.70 ± 0.55 (p < 0.05), 2.61 ± 0.30 (p < 0.01) relative to media control after 6, 12, 24, and 48 h of morphine treatment, respectively (Fig. 7a–e).
Figure 7.

Morphine enhances P2X4 receptor expression in microglial cells. Primary microglia were treated with 0, 1, 10, or 100 nm morphine for 6 (a), 12 (b), 24 (c), or 48 (d, e) h, lysed, and subjected to Western blot analysis. a–d, Graphs indicate relative P2X4 receptor expression normalized to β-actin loading control ± SEM. e, Image of membrane after 48 h treatment probed with anti-P2X4 receptor and anti-β-actin antibodies. Graphs show data from at least four individual Western blots, n = 2 per blot. *p < 0.05, **p < 0.01, ***p < 0.001.
Morphine-induced Iba1 and P2X4 receptor expression is μ-opioid receptor-dependent
To confirm that the morphine-induced changes in Iba1 and P2X4 protein expression were μ-opioid receptor-dependent, we completed a series of experiments using the μ-opioid receptor-selective antagonist, CTAP. Primary microglia were pretreated with CTAP, then exposed to morphine for 24 h. Morphine-induced Iba1 expression was reduced from 2.09 ± 0.46 to 1.04 ± 0.12 with CTAP pretreatment (p < 0.05) (Fig. 8a,b). CTAP pretreatment similarly reduced morphine-induced P2X4 receptor expression from 1.81 ± 0.07 to 1.21 ± 0.14 (p < 0.01) (Fig. 8c,d).
Figure 8.

Morphine-induced Iba1 and P2X4 receptor expression is μ-opioid receptor-dependent. Primary microglia were treated with 0 or 100 nm of the μ-opioid receptor-selective antagonist, CTAP, for 1 h, then treated with 0 or 100 nm morphine for 24 h. Cells were lysed and subjected to Western blot analysis. a, Image of Western blot membrane probed with anti-Iba1 and anti-β-actin antibodies. b, Graphical analysis of CTAP inhibition of morphine-induced Iba expression normalized to β-actin loading control ± SEM. c, Image of Western blot membrane probed with anti-P2X4 receptor and anti-β-actin antibodies. d, Graphical analysis of CTAP inhibition of morphine-induced P2X4 receptor expression normalized to β-actin loading control ± SEM. Graphs show data from at least six individual Western blots, n = 1 per blot. *p < 0.05, **p < 0.01, ***p < 0.001.
Morphine enhances microglial migration at 6, 12, 24, and 48 h
Our results show that morphine enhances Akt phosphorylation acutely and transiently and increases Iba1 and P2X4 receptor expression at later time points. Therefore, in these studies, we sought to examine the effect of longer-term opioid exposure on microglial migration. Microglia were treated with 0, 1, 10, or 100 nm morphine for 6, 12, 24, or 48 h and then allowed to migrate toward ADP for 2 h. At each time point, increasing concentrations of morphine dose-dependently enhanced microglial migration (Fig. 9). After 2, 6, 12, 24, and 48 h of treatment with 100 nm morphine, microglial migration was increased to a maximum of 257.3 ± 8.65%, 247.2 ± 5.67%, 300.6 ± 3.84%, 394.2 ± 14.43%, and 345.1 ± 17.30%, respectively. The magnitudes of morphine-enhanced microglial migration were no different in the 2 and 6 h treatment groups. Microglial migration at 12 h was significantly increased compared with 2 and 6 h (p < 0.001 for 1, 10, and 100 nm morphine groups). Migration at 48 was significantly increased compared with 2, 6, and 12 h (p < 0.001 for 1, 10, and 100 nm morphine groups). Migration was greatest after 24 h, which was significantly increased compared with 2, 6, 12, and 48 h (p < 0.001 for 10 and 100 nm morphine groups).
Figure 9.

Six, twelve, twenty-four, and forty-eight hour morphine treatment dose-dependently enhanced microglial migration. Primary microglial were treated for 2, 6, 12, 24, or 48 h with 0, 1, 10, or 100 nm morphine and then allowed to migrate toward 10 μm ADP. Quantification of microglial migration after 2, 6, 12, 24, or 48 h morphine treatment, represented as mean ± SEM.
Discussion
In this study, we show that (1) morphine enhances microglial migration toward ADP. (2) P2X4 receptor antagonism inhibits morphine-induced microglial migration. (3) Morphine-induced microglial migration is μ-opioid receptor and PI3K dependent. (4) Morphine enhances Iba1 and P2X4 receptor protein expression.
The studies presented here are the first to assess the signaling mechanisms through which morphine modulates microglial migration toward the chemoattractant ADP. A study has suggested that morphine in concentrations >1 μm acts as a microglial chemoattractant (Takayama and Ueda, 2005). However, the potential chemoattractant role of morphine in high concentrations is not relevant to in vivo situations where morphine is given exogenously to provide analgesia. Our results indicate that, at nanomolar concentrations, morphine potently enhances microglial migration directed toward ADP. ATP/ADP is present in low concentrations in the CNS and after a pathological event, such as nerve injury, it may be released from dorsal horn neurons (Sawynok et al., 1993) or astrocytes (Salter and Hicks, 1994; Werry et al., 2006), creating a chemoattractant gradient and inducing microglial migration (Kurpius et al., 2007). A recent study also found that basal and evoked ATP release is increased in the L4/L5 dorsal root ganglia after sciatic nerve entrapment (B. Snyder, B. Tran, H. Iwase, Y. Matsuka, T. Ono, S. Mitrirattanakul, and I. Spigleman, unpublished observations). Our data suggest that opioids given to treat these pathological events might exacerbate microglial migration.
There have been conflicting reports on the effect of morphine on macrophage and microglial migration in the literature. Studies have suggested that morphine inhibits macrophage or microglial migration toward chemokines (Chao et al., 1997; Hu et al., 2000; Malik et al., 2002; Hatsukari et al., 2006), while others have shown that morphine itself is a chemoattractant (Singhal et al., 1996; Takayama and Ueda, 2005). There are however some limitations to these studies suggesting an inhibitory effect of morphine on macrophage and microglial migration. Findings reported by Hatsukari et al. (2006) and Malik et al. (2002) are based upon monocytes and macrophages and several of their experiments required long-term opioid dosing and migration toward a milieu of multiple chemoattractants.
The studies completed using microglia similarly have several limitations. Chao et al. (1997) reported an IC50 value of 1 fM morphine for the inhibition of microglial migration toward C5a. This value is well below the accepted Ki value of ∼10 nm for morphine binding to the μ-opioid receptor (Williams et al., 2001). This discrepancy increases the possibility that the effects observed were off target or mediated via nonclassical μ-opioid receptors. The study by Hu et al. (2000) used pharmacologically relevant concentrations of morphine and asserts that morphine inhibits the production of and migration toward RANTES. This suggests that morphine induces a potential “autocrine-like” effect of RANTES on microglial motility where morphine inhibits the production of the chemoattractant and thus decreases migration. These results, when compared with those we present here, suggest that morphine enhances microglial migration toward ATP/ADP, whereas it may reduce migration toward RANTES.
Our results suggest a novel interaction between μ-opioid receptor and P2X4 receptor systems in microglia, which is likely mediated via the PI3K/Akt pathway. However, P2X4 antagonism blocks morphine-induced microglial migration, indicating that ADP is the main driver of migration in our system. The role of purinergic receptors in ATP and ADP directed microglial migration has recently received much attention. Honda et al. (2001) were the first to describe the effects of extracellular ATP and ADP on microglial membrane ruffling and chemotaxis. Ohsawa et al. (2007) refined these findings and through the use of purinergic antagonists determined that ATP-induced microglial migration involved both P2X4 and P2Y12 receptors. They found that PI3K inhibition reduced migration and ATP-induced Akt phosphorylation, implicating the PI3K/Akt pathway in purinergic receptor mediated migration. Irino et al. (2008) then provided further evidence that Akt is involved in ADP-induced microglial migration. These studies provided the foundation for our present work assessing the role of μ-opioid and P2X4 receptor activation in microglial migration. Our data indicate that activation of μ-opioid receptors by morphine induces Akt phosphorylation in the same time frame as has been previously shown for ADP (Irino et al., 2008). We also showed that morphine-induced Akt phosphorylation is PI3K dependent, similar to the results shown for ADP (Irino et al., 2008). These findings suggest that μ-opioid and P2X4 receptors share a common intracellular pathway regulating migration in microglia.
We have previously shown that chronic in vivo morphine administration produces marked microglial reactivity as shown by increased CD11b immunoreactivity (Raghavendra et al., 2002, 2003, 2004; Tawfik et al., 2005). It remained unknown, however, whether this effect on microglia was a direct effect of morphine or a result of morphine stimulation of neurons or astrocytes. Here, we show that morphine directly enhances Iba1 and P2X4 protein expression in microglia. In these studies, we chose to assess the expression of Iba1 instead of CD11b because of the difficulty in probing CD11b via Western blot analysis. Another advantage of measuring Iba1 protein is that it binds to the actin cytoskeleton and has been suggested to play a role in migration (Ohsawa et al., 2000, 2004).
Microglial activation is no longer viewed as an “all or nothing event” (Hanisch and Kettenmann, 2007). Microglia were once thought to be quiescent or resting in their nonactivated state. However, it is now recognized that in their nonactivated state microglia actively engage in surveillance of their environment in the CNS (Hickey, 2001). Microglial reactivity includes enhanced expression of surface or cytosolic proteins, morphological changes, proliferation, and migration. Lipopolysaccharide (LPS) has been considered a robust and broad inducer of microglial activation. However, we have recently shown that while LPS stimulation produces profound proinflammatory cytokine production, it does not significantly increase the expression of Iba1, a marker of microglial reactivity (Horvath et al., 2008). Here, we provide evidence for morphine-induced phosphorylation of intracellular proteins, enhanced cytosolic and surface protein expression, and migration toward a chemoattractant in microglia. We suggest that the term “activation” is ill suited to describe the multivariable response of microglia to distinct stimuli and propose that more specific terms related to antigen expression or altered functional state be used to describe the changes observed.
The present study provides evidence for two phases of morphine effects on microglial cells. The initial phase occurs on the order of minutes, inducing activation of the PI3K pathway and phosphorylation of Akt, which increases motility acutely. The simplest explanation for the increase in migration observed acutely would be enhanced P2X4 protein expression, which would increase migration toward the chemoattractant. However, no changes in protein expression were observed until 6 h of morphine stimulation, suggesting that the acute effect of morphine on microglial migration is not dependent on enhanced P2X4 receptor expression. Rather, our results suggest that the acute effect of morphine on ADP-directed microglial migration is mediated through activation of an intracellular PI3K motility pathway. The long-term phase occurs on the order of hours, increasing the expression of Iba1 and P2X4 receptor proteins associated with microglial cell migration machinery. Enhanced protein expression was observed after 6 h of morphine stimulation, followed by increased migration at 12–48 h. Iba1 is known to play a significant role in actin reorganization and leading edge membrane ruffling, supporting migration (Ohsawa et al., 2000, 2004). P2X4 receptor expression is also known to be enhanced after insult to the CNS, increasing pain responses (Tsuda et al., 2003; Zhang et al., 2008). Morphine-induced expression of both Iba1 protein and P2X4 receptors impart microglia with a long-lasting promigratory phenotype which is associated with greater enhancement of microglial migration by morphine at 24 and 48 h, compared with 2 h.
Microglia express μ-, δ-, and κ-opioid receptors (Calvo et al., 2000; Turchan-Cholewo et al., 2008). Studies have shown that while both neurons and glia have similar μ-opioid receptor-binding affinities for morphine, neurons contain nearly 5× more μ-opioid receptors than glia (Maderspach and Solomonia, 1988). We propose that morphine, when given acutely and in low doses, binds to abundant neuronal μ-opioid receptors, producing analgesia. When given chronically and in high doses, morphine binds to sparser microglial μ-opioid receptors, activating microglia and inducing protein expression and migration. Morphine-induced migration of reactive microglia may then lead to elevated local concentrations of proinflammatory cytokines and chemokines, inducing neuronal sensitization. This counteracts the analgesic effect mediated by neurons and is manifest by morphine tolerance or hyperalgesia.
The data presented here identify a novel interaction between opioid and purinergic receptor pathways mediating microglial migration. This study is an integral first step toward assessing the role of microglial migration in morphine tolerance, hyperalgesia, and other side effects. The functional properties of microglia may prove to be valuable targets for the development of therapeutics aimed at attenuating opioid-induced side effects or targets for novel analgesics with enhanced efficacy and better side effect profiles compared with current therapies.
Footnotes
This work was supported by National Institute on Drug Abuse Grants 1F30DA024933-01 (R.J.H.) and DA11276 (J.A.D.), National Institutes of Health Grant T32 AI07363 (W. R. Green, Director), and Immunobiology of Myeloid and Lymphoid Cells (R.J.H.), all of whom we thank.
References
- Calvo CF, Cesselin F, Gelman M, Glowinski J. Identification of an opioid peptide secreted by rat embryonic mixed brain cells as a promoter of macrophage migration. Eur J Neurosci. 2000;12:2676–2684. doi: 10.1046/j.1460-9568.2000.00145.x. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Shark KB, Sheng WS, Gekker G, Peterson PK. Activation of mu opioid receptors inhibits microglial cell chemotaxis. J Pharmacol Exp Ther. 1997;281:998–1004. [PubMed] [Google Scholar]
- Cui Y, Liao XX, Liu W, Guo RX, Wu ZZ, Zhao CM, Chen PX, Feng JQ. A novel role of minocycline: attenuating morphine antinociceptive tolerance by inhibition of p38 MAPK in the activated spinal microglia. Brain Behav Immun. 2008;22:114–123. doi: 10.1016/j.bbi.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hatsukari I, Hitosugi N, Dinda A, Singhal PC. Morphine modulates monocyte-macrophage conversion phase. Cell Immunol. 2006;239:41–48. doi: 10.1016/j.cellimm.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Hickey WF. Basic principles of immunological surveillance of the normal central nervous system. Glia. 2001;36:118–124. doi: 10.1002/glia.1101. [DOI] [PubMed] [Google Scholar]
- Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci. 2001;21:1975–1982. doi: 10.1523/JNEUROSCI.21-06-01975.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath RJ, Nutile-McMenemy N, Alkaitis MS, Deleo JA. Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. J Neurochem. 2008;107:557–569. doi: 10.1111/j.1471-4159.2008.05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Chao CC, Hegg CC, Thayer S, Peterson PK. Morphine inhibits human microglial cell production of, and migration towards, RANTES. J Psychopharmacol. 2000;14:238–243. doi: 10.1177/026988110001400307. [DOI] [PubMed] [Google Scholar]
- Inoue K. The function of microglia through purinergic receptors: neuropathic pain and cytokine release. Pharmacol Ther. 2006;109:210–226. doi: 10.1016/j.pharmthera.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Irino Y, Nakamura Y, Inoue K, Kohsaka S, Ohsawa K. Akt activation is involved in P2Y12 receptor-mediated chemotaxis of microglia. J Neurosci Res. 2008;86:1511–1519. doi: 10.1002/jnr.21610. [DOI] [PubMed] [Google Scholar]
- Kurpius D, Nolley EP, Dailey ME. Purines induce directed migration and rapid homing of microglia to injured pyramidal neurons in developing hippocampus. Glia. 2007;55:873–884. doi: 10.1002/glia.20509. [DOI] [PubMed] [Google Scholar]
- Maderspach K, Solomonia R. Glial and neuronal opioid receptors: apparent positive cooperativity observed in intact cultured cells. Brain Res. 1988;441:41–47. doi: 10.1016/0006-8993(88)91381-9. [DOI] [PubMed] [Google Scholar]
- Malik AA, Radhakrishnan N, Reddy K, Smith AD, Singhal PC. Morphine-induced macrophage apoptosis modulates migration of macrophages: use of in vitro model of urinary tract infection. J Endourol. 2002;16:605–610. doi: 10.1089/089277902320913314. [DOI] [PubMed] [Google Scholar]
- Mayer DJ, Mao J, Holt J, Price DD. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci U S A. 1999;96:7731–7736. doi: 10.1073/pnas.96.14.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika J. Modulation of microglia can attenuate neuropathic pain symptoms and enhance morphine effectiveness. Pharmacol Rep. 2008;60:297–307. [PubMed] [Google Scholar]
- Nakajima K, Kohsaka S. Microglia: activation and their significance in the central nervous system. J Biochem. 2001;130:169–175. doi: 10.1093/oxfordjournals.jbchem.a002969. [DOI] [PubMed] [Google Scholar]
- Nutile-McMenemy N, Elfenbein A, Deleo JA. Minocycline decreases in vitro microglial motility, beta1-integrin, and Kv1.3 channel expression. J Neurochem. 2007;103:2035–2046. doi: 10.1111/j.1471-4159.2007.04889.x. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Imai Y, Kanazawa H, Sasaki Y, Kohsaka S. Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J Cell Sci. 2000;113:3073–3084. doi: 10.1242/jcs.113.17.3073. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Imai Y, Sasaki Y, Kohsaka S. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J Neurochem. 2004;88:844–856. doi: 10.1046/j.1471-4159.2003.02213.x. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Irino Y, Nakamura Y, Akazawa C, Inoue K, Kohsaka S. Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia. 2007;55:604–616. doi: 10.1002/glia.20489. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. 2002;22:9980–9989. doi: 10.1523/JNEUROSCI.22-22-09980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, Rutkowski MD, DeLeo JA. Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokines. Pain. 2003;104:655–664. doi: 10.1016/S0304-3959(03)00138-6. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacology. 2004;29:327–334. doi: 10.1038/sj.npp.1300315. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval A, Chai N, Nutile-McMenemy N, Deleo JA. A comparison of spinal Iba1 and GFAP expression in rodent models of acute and chronic pain. Brain Res. 2008a;1219:116–126. doi: 10.1016/j.brainres.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Sandoval EA, Horvath RJ, DeLeo JA. Neuroimmune interactions and pain: focus on glial-modulating targets. Curr Opin Investig Drugs. 2008b;9:726–734. [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Hicks JL. ATP-evoked increases in intracellular calcium in neurons and glia from the dorsal spinal cord. J Neurosci. 1994;14:1563–1575. doi: 10.1523/JNEUROSCI.14-03-01563.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawynok J, Downie JW, Reid AR, Cahill CM, White TD. ATP release from dorsal spinal cord synaptosomes: characterization and neuronal origin. Brain Res. 1993;610:32–38. doi: 10.1016/0006-8993(93)91213-c. [DOI] [PubMed] [Google Scholar]
- Singhal PC, Shan Z, Garg P, Sharma K, Sharma P, Gibbons N. Morphine modulates migration of monocytes. Nephron. 1996;73:526–531. doi: 10.1159/000189135. [DOI] [PubMed] [Google Scholar]
- Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neurosci Res. 2001;39:281–286. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- Takayama N, Ueda H. Morphine-induced chemotaxis and brain-derived neurotrophic factor expression in microglia. J Neurosci. 2005;25:430–435. doi: 10.1523/JNEUROSCI.3170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik VL, LaCroix-Fralish ML, Nutile-McMenemy N, DeLeo JA. Transcriptional and translational regulation of glial activation by morphine in a rodent model of neuropathic pain. J Pharmacol Exp Ther. 2005;313:1239–1247. doi: 10.1124/jpet.104.082420. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Turchan-Cholewo J, Dimayuga FO, Ding Q, Keller JN, Hauser KF, Knapp PE, Bruce-Keller AJ. Cell-specific actions of HIV-Tat and morphine on opioid receptor expression in glia. J Neurosci Res. 2008;86:2100–2110. doi: 10.1002/jnr.21653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werry EL, Liu GJ, Bennett MR. Glutamate-stimulated ATP release from spinal cord astrocytes is potentiated by substance P. J Neurochem. 2006;99:924–936. doi: 10.1111/j.1471-4159.2006.04133.x. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhang ZY, Fauser U, Schluesener HJ. Mechanical allodynia and spinal up-regulation of P2X(4) receptor in experimental autoimmune neuritis rats. Neuroscience. 2008;152:495–501. doi: 10.1016/j.neuroscience.2007.12.042. [DOI] [PubMed] [Google Scholar]