

Abstract
Intrinsic properties of neurons that enable them to maintain depolarized, persistently activated states in the absence of sustained input are poorly understood. In short-term memory tasks, individual prefrontal cortical (PFC) neurons are capable of maintaining persistent action potential output during delay periods between informative cues and behavioral responses. Dopamine and drugs of abuse alter PFC function and working memory possibly by modulating intrinsic neuronal properties. Here we use patch-clamp recording of layer 5 PFC pyramidal neurons to identify an action potential burst-evoked intrinsic mGluR5-mediated postsynaptic depolarization that initiates an activated state. Depolarization occurs in the absence of recurrent synaptic activity and is reduced by a postsynaptic dopamine D1/5 receptor pathway. The depolarization is substantially diminished following behavioral sensitization to cocaine; moreover the D1/5 receptor modulation is lost. We propose the burst-evoked intrinsic depolarization to be a novel form of short-term cellular memory that is modulated by dopamine and cocaine experience.
Keywords: metabotropic glutamate receptor, prefrontal cortex, D1 dopamine receptor, bursting, cocaine addiction, persistent activity
Introduction
The prefrontal cortex (PFC) is a highly evolved brain region closely linked to attention and working memory in primates and rodents.1,2 Drugs of abuse like cocaine are known to disrupt attentional and working memory processes, but the mechanism is unknown.3,4 In fact, little is known about the basic neuronal and network properties that sustain attention and working memory in general. In working memory tasks, pyramidal neurons within the PFC are capable of maintaining action potential output during delay periods between an informative cue presentation and the appropriate behavioral response.5 The induction of persistent action potential activity may function to hold information necessary to guide goal-directed behavior in memory until it is no longer necessary (e.g. after a response or reward). Several computational approaches have been put forth to explain delay period activity ranging from highly abstract models to biophysically detailed conductance based models. Most modeling studies of persistent activity have focused on recurrent network models.6 These models require precise tuning and weighting of synaptic inputs to maintain stable persistent activity and are limited by minimal biophysical validity and pronounced susceptibility to interference.6,7 The robustness and resistance to mistuning of these models can be substantially improved mathematically by prolonging the synaptic decay time constants or incorporating cellular bistability that allows neurons to fluctuate between hyperpolarized resting and depolarized activated states.6,7 Strong bursts of synaptic stimulation and concomitant activation of NMDA channels, voltage-gated Ca2+ channels and Ca2+-activated nonselective cation channels could provide the biological mechanism necessary to maintain activated states under conditions of diminishing excitatory drive during the delay period.7–9
We used an experimental approach to identify an intrinsic mechanism capable of converting subthreshold inputs into persistent suprathreshold output for several seconds in deep layer PFC pyramidal neurons. To date, research on the role of glutamate in working memory has mainly focused on the fast, excitatory ionotropic AMPA and NMDA glutamate receptors, however, recently the group 1 G-protein-coupled metabotropic glutamate receptors (mGluR) and their slow excitatory transmission have gained attention due to their role in controlling prefrontal cortical activity, working memory processes, psychostimulant reward and hippocampal and cerebellar synaptic plasticity.10–15
From a circuit perspective, several inputs converge onto layer 5 pyramidal neurons of the PFC, including local PFC inputs, hippocampal/subicular, medial dorsal thalamic glutamatergic inputs, and DA input from the ventral tegmental area (VTA)16,17 Extracellular DA levels increase in the PFC during working memory tasks in primates and rats.18,19 Furthermore, an optimal level of D1/5R activation is important for proper performance on PFC-dependent working memory tasks.20 D1/5R receptors modulate PFC neuronal activity both in vivo20–21 and in vitro,22–26 where they have been reported to exert inhibitory and excitatory effects. For example, D1/5 receptors interact with NMDA receptors, to enhance cellular excitability,22 and mGluRs to induce LTD.13 The mGluR5s have been recently implicated in PFC-dependent executive functions because inhibition of mGluR5 impairs working memory.10 It is clear that a proper balance of glutamate and DA tone is important for working memory and executive cognitive control, all of which can be disrupted by exposure to psychostimulant drugs of abuse, like amphetamine and cocaine.3,4,10,18–21,27
The goals of this study were to: 1) Identify a novel Ca2+ -dependent postsynaptic mGluR-mediated delayed afterdepolarization (dADP) mechanism capable of providing sustained activity in deep layer PFC pyramidal neurons; 2) Test if this dADP could be modulated by postsynaptic dopamine receptor D1 or D2 family signaling cascades, and lastly; 3) To determine if a behavioral sensitizing regimen of cocaine would lead to long-lasting disruption of this mechanism.
Results and Discussion
Metabotropic glutamate receptor activation induces a burst-triggered delayed afterdepolarization
We report that a single burst of gamma frequency (∼20–50 Hz) synaptic stimulation induces a mGluR-mediated calcium-dependent dADP capable of converting subthreshold input and low frequency activity to prolonged action potential output lasting tens of seconds in layer 5 pyramidal neurons of the rat PFC (Figure 1b and d; Figure 2a; Figure 7a;Supplemental Figure 2). Because most (>90%) fast excitatory transmission was blocked, we hypothesized that the delayed cellular response was due to activation of mGluRs. To test the role of postsynaptic mGluRs more directly, action potentials were triggered with brief current injections through the patch pipette that induced a dADP in the presence of mGluR agonists ACPD (100 µM) or DHPG (50–100 µM) either locally applied with a puffer pipette (150 µm from the base of the apical dendrite) or in the bath as noted (Figure 1c). At a broad range of frequencies (2–100 Hz), bursts of 2 or more action potentials were more effective than single spikes at producing the dADP (Supplemental Figure 1a). We found that the dADP amplitude and integral was independent of the frequency, yet was dependent on the number of spikes, which saturated the dADP after four spikes (n = 6; Supplemental Figure 1b). Using a physiologically relevant simulated synaptic stimulus that triggered 5 action potentials delivered at 50 Hz, the ACPD-induced dADP had average amplitude of 3.7 ± 0.6 mV, peaked at 1.2 s, and decayed with a time constant of 2.5 ± 0.03 s (n = 19). Synaptic activation of 5 and 30 stimuli (50 Hz) delivered between 100 and 200 µm from the soma along the apical dendrite produced a similar prominent dADP in the presence of fast synaptic blockers in ∼63% of pyramidal neurons (7 of 11 neurons). 30 stimuli produced a slightly larger response compared to 5 pulses for dADP peak (4.6 ± 0.8 mV (n=5; 5 stimuli) vs. 7.9 ± 0.9 mV (n=7; 30 stimuli; *p<0.03) and integral and instantaneous frequency (Supplemental Figure 2b, c).
Figure 1.

Patch-clamp recording from layer 5 pyramidal PFC neurons showing a metabotropic glutamate receptor activated dADP and persistent activity. (a) (Left) Neurolucida reconstruction of a representative biocytin (0.1%) filled PFC layer 5 pyramidal neuron. (Right) Schematic of the slice showing the deep layer 5/6 regions in the prelimbic and infralimbic regions (the shaded area) of the PFC where the recordings were made. (b) Voltage response of a pyramidal neuron in layer 5 of PFC following synaptic stimulation with a stimulating electrode positioned along the apical dendrite in cortical layer 3. (Left) A single burst of synaptic stimulation (5 × 50 Hz) induced a dADP in the presence of GABAA and B (SR 95531, 2 µM) and NMDA blockade (AP-5, 50 µM). (c) In the presence of an mGluR agonist ACPD (100 µM) a single burst of 5 brief (2 ms) suprathreshold current step pulses (Left) elicited the dADP with similar kinetics as the synaptically activated dADP in the presence of GABA and ionotropic glutamate blockers (see Methods). The scale bar in (b) applies to (c) as well. (d) Action potential activity from a bursting (inset, 10 ms scale bar) layer 5 pyramidal neuron in response to a 45 second suprathreshold current step. The color map below the trace shows the instantaneous firing rate that averaged ∼2.2 Hz. A single burst of spike triggering synaptic stimulation induced a pronounced increase in the instantaneous firing rate that was sustained for the duration of the current step injection. The color map shows the instantaneous firing rate change from 1–20 Hz.
Figure 2.

Patch-clamp recording from layer 5 pyramidal PFC neurons showing response to an ascending and descending input stimulus before and after mGluR activation. (a) Schematic showing proximal apical dendritic synaptic stimulation and recording Neurolucida reconstruction of a biocytin filled layer 5 pyramidal neuron. (Right Top) A representative trace showing the response to a 5 s ascending and 40 s descending ramp with a burst stimulus (5 × 20 Hz) current command. Note that under control conditions the instantaneous firing rate follows the current command input. (Right Bottom) The enhanced response to a synaptic burst stimulus (5 × 20 Hz) delivered at the peak of the ramp to the apical dendrite. The open circle represents the cell shown in (b and c). Color map shows the instantaneous frequency (0–15 Hz) before and after synaptic stimulation at the peak of the ramp. (b, c) Scatter plot shows the increased duration of action potential firing (n = 7; ***p < 0.001) during the descending ramp and the increased instantaneous firing rate (n = 7; ***p < 0.001) following a burst of 5 synaptic stimuli compared to 5 brief current injection delivered at the peak of the ramp. Open circle shows the representative neuron from (a). (d) Schematic showing a pipette puffing either ACSF or DHPG (100 µM) on the soma, proximal apical and basilar dendrites and the whole-cell patch clamp pipette. A double-barreled pipette located 50 µm from the soma filled with ACSF in one barrel and DHPG in another was used. Gray trace shows a representative action potential response to the ICommandramp stimulus (40 seconds ascending and 40 seconds descending) and a brief 1 second pressure application of ACSF at the peak of the ramp (black arrow). The black trace shows the action potential response of the same cell with a 1 second application of DHPG (100 µM; black arrow) at the peak of the ICommand. Recovery trace puffing ACSF was taken 30 seconds after DHPG. (e and f) Scatter plot shows the increased duration of action potential firing during the descending ramp (n = 7; ***p < 0.005) and the increased instantaneous firing rate (n = 7; **p < 0.02) following a 1 second duration puff of DHPG compared to Control ACSF puff. (g) A raster plot shows the ramp induced spiking in response to a pressure application of Control ACSF (Gray trace) or DHPG (Black trace) delivered at the peak of the ramp (Black Arrow). An instantaneous frequency histogram (1 s bins) for the traces shown in the raster plot is depicted with time zero set to the onset of the first spike. Note the peak frequency following DHPG in this recording occurs 8 seconds after the peak of the ramp current stimulus. Open circle shows the representative recording in (e) and (f).
Figure 7.

Repeated cocaine treatment reduces the mGluR5/burst-mediated conversion of subthreshold input to suprathreshold action potential output and D1/5R modulation. (a) Whole-cell patch clamp recording of a layer 5 pyramidal neuron given a burst of 5 suprathreshold simulated synaptic current injections at 20 Hz followed by a single subthreshold simulated current injection delivered at 85% of threshold at different times from 1 second to 4 seconds post burst (Overlay) in the presence of DHPG (50 µM; Gray Traces) and after SKF81297 (10 µM; Black Traces). DHPG elicited a dADP that converted the fast subthreshold input to suprathreshold spike output in the first 3 seconds post burst. Inset shows the superimposed fast EPSP responses before and after DHPG (Gray trace) and SKF (Black trace) when measured prior to the burst of five action potentials (scale bar is 5 mV and 10 ms). Right panel shows the average spike probability in response to subthreshold simulated postsynaptic input in the presence of DHPG (50 µM; Open squares, Control) and SKF81297 (10 µM; Filled Squares; n = 8, p < 0.01 at 2 and 3 seconds). (b) Short (2 day) withdrawal from repeated cocaine treatment reduces the spike probability (Left Panel; Open squares, Saline/Control, n = 8 and Open circles, Cocaine, n = 11,p < 0.001 at 1 and 2 seconds). The right panel shows the normalized spike probability following DHPG and SKF81297 in the untreated/saline-treated control group (Filled squares, Cont SKF; n = 8) and the cocaine-treated group given DHPG alone (Open circles, COC Cont) or DHPG and SKF81297 (Filled Squares, COC SKF). (c) Late (14 Day) withdrawal from repeated cocaine treatment does not alter the burst triggered dADP (Left Panel; Saline/Control, n = 6 and Cocaine, n = 6; p > 0.05). The right panel shows lack of D1/5R modulation of the normalized spike probability for the cocaine treated group in response to DHPG (Open circles, COC Cont) and after SKF81297 (Filled circles, COC SKF; p > 0.05) compared to non-treated cocaine controls (Filled Squares; Cont SKF).
In vitro simulation of in vivo conditions: The role of subthreshold and suprathreshold activity
In order to understand how the mGluR-induced dADP influences action potential dynamics under simulated in vivo conditions, time varying current inputs were delivered in the presence of brief local or bath mGluR agonist. To test the ability of the dADP to maintain spike output under simulated in vivo conditions in the presence of ongoing action potential activity we programmed an ascending ramp stimulus that simulated the rising excitatory drive associated with attending to an informative stimulus, and a descending ramp to simulate the diminished excitation that occurs following stimulus withdrawal in a behavioral task. Time varying stimuli, like depolarizing ramp current injections, have been used to characterize cortical pyramidal neurons in vitro to mimic the type of input likely to be present in vivo as correlates of preparatory and anticipatory processes in sensorimotor and cognitive behaviors.28 Progressively depolarising and hyperpolarizing inputs are common in cortical parietal and frontal regions, and could participate in muscular recruitment, response preparation, and decision-making processes.28 At the peak of the ascending ramp current command (ICommand) either ACSF alone or 100 µM DHPG was pressure applied using a patch pipette (3 MΩ) for 1 s and the spike output was monitored during the falling phase of the ramp stimulus (Figure 2). The ramp current injection prior to mGluR activation was adjusted to produce peak sustained firing rates between 3 and 12 Hz to correspond to realistic in vivo cortical baseline firing rates.29 Under these conditions mGluR receptors were activated amongst a background of action potential activity as in vivo. Compared to current injection alone brief synaptic stimulation of 5–30 pulses delivered at 20–50 Hz produced a robust increase in the duration (40 ± 7%, p<0.005) and instantaneous frequency (68 ± 12% p<0.005) of action potential firing (Figure 2a,b,c) even under conditions of diminishing excitatory input. Similar results were obtained using local postsynaptic DHPG puff application, which produced a robust increase in the instantaneous frequency (85 ± 25% n = 7, p < 0.01), and duration of action potential firing (64 ± 18% n = 7,p <0.005; Figure 2d,e,f,g). All cells responded to the DHPG puff (7 of 7) and all baseline responses fully recovered within 30 s of DHPG application (Figure 2d; Recovery trace). Therefore, brief postsynaptic activation of mGluRs can prolong and increase the frequency of spike output even in the presence of descending excitatory input (i.e. the firing frequency reaches a delayed maximum firing rate that peaks several seconds after the peak of excitatory input).
Activation of group I mGluRs leads to increases in intracellular Ca2+ in neurons, an effect greatly enhanced by simultaneous action potential generation and/or cellular depolarization.9,11,15,30,31 To determine a necessary role for intracellular Ca2+ we used BAPTA (5 mM) in the recording pipette and monitored the dADP and action potential triggered action potential activity in the presence of DHPG. BAPTA produced a substantial reduction in the dADP (Figure 3a, b; n = 5;p < 0.01), as we have previously reported.9 The functional consequence of the dADP on action potential output was more obvious when we introduced subthreshold membrane potential fluctuations by re-playing a previously recorded near threshold current stimulus command input in order to mimic the background in vivo activity.32 In the subthreshold domain mGluR activation had no significant effect as detected by a low frequency noise current stimulus in the prespike period that produced a 24.7 mV span from −80 mV to −55.3 mV (Figure 3c). In contrast, following a single action potential or doublet burst mGluR activation converted the subthreshold noise input into prolonged spike output which was completely eliminated by intracellular Ca2+ chelation with BAPTA (Figure 3c, d; n = 7, p < 0.01). This demonstrates the Ca2+- dependent nature of the mGluR-induced dADP and its ability to convert brief bursts of activity into sustained output. The sustained activity was maintained even during brief periods (∼100 ms) of hyperpolarization below the resting potential (Figure 3c), indicating that the dADP action potential amplification mechanism is resistant to transient inhibitory interference.
Figure 3.

The dAPD requires intracellular Ca2+ and is capable of amplifying and converting near threshold inputs into sustained output only after an initial action potential. (a) Chelation of intracellular Ca2+ with BAPTA (5 mM) substantially reduces the burst-triggered dADP in the presence of mGluR agonist ACPD (100 µM). The inset shows no differences in the dADP inducing 5 action potential burst after ACPD after initial break-in to whole-cell mode or 15 minutes after intracellular perfusion with BAPTA. Scale bars show 1 mV and 2 s. (b) BAPTA significantly reduces the area under the curve (integral) of the dADP triggered by the burst of action potentials in the presence of bath applied ACPD (n = 5;**p < 0.01). (c) A subthreshold recording from a pyramidal neuron was played back as a current command (ICommand) and scaled to produce an action potential (Black trace). ACPD (100 µM) was applied via pressure injection from 10 seconds prior to sweep until the end (Gray trace). Washout of ACPD 30 seconds after DHPG application indicated rapid recovery to baseline response (data not shown). Scale bars show 20 mV and 1 s. (Right) Subtraction of the ACPD trace (Gray trace) from the baseline (Black trace) shows the amplification of subthreshold activity during the Postspike period. Action potentials, which comprised a very small component of the post spike integral accounted for < 5% of the total area and were subtracted from the total integral calculation as shown. Scale bars show 5 mV and 1 s. (d) Baseline subtraction of the integral of the pre-spike membrane potential (3 seconds) compared to the post-spike membrane potential (3 seconds) after pipette puffing ACPD (100 µM;n = 4) or DHPG (100 µM; n = 3) application (BAPTA alone, n = 6; pooled n = 7; **p < 0.01).
The burst-triggered delayed afterdepolarization is dependent on mGluR5 activation
Group 1 mGluR1/5 agonists (ACPD or DHPG) induce an action potential-triggered nonselective cation current that produces a dADP in the somatosensory cortex,14 subiculum,9 entorhinal cortex,33 and cerebellum,15 In the cerebellum, stimulation of mGluR1 cascades leads to activation of the nonselective cation channel, transient receptor potential canonical 3 (TRPC3) causing slow inward currents in Purkinje neurons.15 The mammalian TRPC family of nonselective cation channels contain seven members (TRPC1–7) that are activated by PLC.34 TRPC4 and 5 PFC are the predominant variants in the PFC and their mRNA and protein expression correlates with the group 1 mGluR-mediated burst-elicited dADP.9 Group 1 mGluRs are comprised of both mGluR1 and mGluR5. The mGluR5s rather than mGluR1s are predominantly expressed on deep layer pyramidal neurons within PFC.35 The mGluR5s are located in the perisynaptic region of the postsynaptic membrane, and are activated by bursts of action potentials.11
Bursting output is significant because it may enhance detection of novel and salient stimuli in goal-directed behaviors and facilitate the induction of synaptic plasticity.36 In fact, Moghaddam and colleagues demonstrated that mGluR5s work together with NMDA receptors in deep layer pyramidal neurons to modulate bursting action potential output.10,11 Interestingly, mGluR5 blockade decreases the spontaneous burst activity of the majority of PFC neurons, but does not alter the nonbursting spontaneous activity of PFC neurons, indicating a preferential role for mGluR5s in regulating bursting spike output.
To determine which mGluR was responsible for the dADP we used a pharmacological and gene deletion approach. Group 2 mGluRs were not involved in the dADP induction since bath application of a group 2 antagonist selective concentration of LY 341495 (20 nM) did not significantly change the ACPD-induced dADP amplitude (n = 6, p = 0.27). Group 1 mGluRs appeared to mediate the dADP since higher concentration of the group 1 and 2 mGluR antagonist, LY 341495 (20 µM), significantly decreased the dADP amplitude (from 6.6 ± 0.1 mV to 3.0 ± 0.4 mV, p < 0.002). The group 1 mGluR agonist, DHPG (50 µM) induced the burst-triggered dADP with an average amplitude of 3.4 ± 0.2 mV, peak at 1.5 ± 0.04 s, and decay time constant of 3.4 ± 0.3 s (n = 15).
To dissect which group 1 mGluR was responsible the burst-triggered dADP we applied the selective mGluR1 receptor antagonist, LY 367385 (10 µM) which did not significantly decrease the dADP (n = 6; p = 0.6), while blockade of mGluR5 receptors with the selective mGluR5 antagonist MPEP (10 µM) produced a significant 60% decrease of the DHPG-induced dADP amplitude (Supplemental Figure 2 d, e; n = 6, p < 0.01). This suggested that mGluR5s were responsible for the dADP, but pharmacological blockade of mGluR5s did not completely abolish the DHPG-induced dADP. Therefore, we tested the DHPG-induced dADP in two groups of knockout (KO) mice lacking mGluR1 or 5 (Figure 4). Using voltage- and current-clamp recording we isolated DHPG-induced current (IDHPG) and dADP respectively in mGluR1 KO mice and found no differences compared to wild-type (WT) littermate controls (n =9–11, p > 0.05; Figure 4a, b) indicating a lack of mGluR1 involvement. On the other hand, both the IDHPG and dADP were completely abolished in the mGluR5 KO mice (Figure 4 c–f; n = 9–11;**p < 0.01); therefore, both the IDHPG and burst-elicited dADP are mediated by mGluR5s.
Figure 4.

The burst-induced dADP is mediated by mGluR5 receptors.
(a) Superimposed representative traces of the showing the burst (5 spikes, 50 Hz)-induced dADP following application of DHPG (100 µM) in WT mice (Black trace) and mGluR1 KO mice (Gray trace). Scale bars show 2 mV and 2 s. The inset shows the 5 action potentials during the burst in mGluR1 KO mice. (b) Mice lacking the mGluR1 receptor exhibit normal DHPG-mediated burst-induced dADP amplitude (left panel) and integral (right panel) compared to WT littermate controls (n = 11;p = 0.8). (c) Shows an overlay of representative traces of the burst-triggered dADP in the presence of DHPG from WT mice (Black trace) and its absence in mGluR5 KO mice (Gray trace). (d) Compared to littermate controls, mGluR5 KO mice (Black trace) do not show the DHPG-mediated burst-induced dADP (left, Amplitude; n = 9;**p < 0.01) or (right, Integral; n = 9;**p < 0.01). (e) Top panel shows the average current induced by 30 s local puff application of DHPG (100 µM) 50 µm from the soma of PFC pyramidal neurons held at -70mV in mGluR1 WT mice (Black trace) and KO mice (Gray trace;). Bottom panel shows the absence of a DHPG-induced a slow inward cation current (IDHPG) in mGluR5 KO mice compared to WT controls‥ The scale bars show 10 pA and 10 s. (f) Average IDHPG from mGluR1 WT and KO mice (left panel; n = 9–11; p>0.05) and mGluR5 WT and KO mice (right panel; n = 9–11;**p < 0.01) in response to 30 s pressure application of DHPG.
Functional implications of the mGluR5-mediated delayed afterdepolarization
The mechanism underlying short-term working memory has been elusive because the storage mechanism cannot depend on pre-existing synaptic connections or classic cellular models of synapse selection, like long-term potentiation (LTP) and long-term depression (LTD). The cellular cascades that produce LTP and/or LTD turn on and off too slowly to function as a temporary memory storage buffer that holds novel information on a trial-by-trial basis, much like computer RAM memory. In contrast, short-lived enhancement of intrinsic excitability mediated through mGluR5 delayed depolarization is an attractive mechanism as a working memory buffer because it is capable of converting strong, “information bearing” input into enduring action potential output that decays over several seconds and then resets when the response has been made and the stored information is no longer needed. In the PFC, both LTP and LTD have been reported to be critically dependent on DA receptor signaling and withdrawal from repeated psychostimulant drugs exposure produces changes in synaptic and intrinsic excitability of deep layer 5 pyramidal neurons (Supplemental Table).13,24,25
We propose that robust synaptic and perisynaptic glutamatergic activation of the layer 5 pyramidal neurons in the PFC leads to Ca2+ entry through NMDA and voltage-gated channels, and initiation of a slow positive-feedback mGluR5-coupled TRPC channel-like depolarization underlying persistent activity.8,9,37 The robustness and tuning of recurrent network computational models of persistent activity can be greatly improved by lengthening the synaptic time constant beyond ∼100 ms, but thus far have lacked a plausible biophysical mechanism .6,7 The mGluR5-mediated dADP with a time constant of ∼3 s provides such a mechanism that enables synaptically triggered output to be maintained for several seconds under conditions of diminishing excitatory drive or with only background synaptic activity.
Dopamine D1/PKA modulation of the mGluR5-mediated delayed afterdepolarization
Because of the important role that DA exerts on working memory and PFC function, we examined the effects of DA on the mGluR5-induced burst-triggered dADP. In the presence of DA (10µM), the DHPG-induced dADP amplitude was significantly decreased (Figure 5; n = 8, p < 0.05). Other properties of the dADP, such as the time to peak or the kinetics of decay remained unchanged. DA normally exerts its cellular effects by activation of D1R and/or D2R classes. Pyramidal neurons in the PFC express both types of receptors, although the D1-type is expressed 20-fold higher amounts than the D2-type.38 In the rat, layer 5 receives the strongest DA input, as shown by electron microscopy of DA terminals.17
Figure 5.

Dopamine reduces the mGluR5/burst-induced dADP through a D1/5 receptor protein kinase A pathway. (a) Dopamine (DA; n=8;**p<0.01) reduces the DHPG-mediated burst-triggered dADP (Cont; n = 30) and the synaptically-triggered dADP (White Bar; n = 6; **p < 0.01). The D2 agonist, quinpirole (10 µM) fails to modulate the dADP (n = 8) while the D1R agonist, SKF81297 (10 µM; Gray bar) decreases the dADP (n = 28; **p < 0.002). Bath applied Forskolin (10 µM) reduced the DHPG-induced dADP (n = 8;**p<0.01) and Forskolin (10 µM) applied to the internal electrode solution reduced synaptically-triggered dADP (n=6;**p<0.01). The PKA inhibitor, H89 (10 µM) and the D1/5R antagonist, SCH 23390 (5 µM), blocked the D1/5R mediated inhibition. H89 alone (10 µM) had no effect (n = 6). The inset demonstrates the PKA inhibitor, H89, blocks the ability of SKF81297 to elevate PKA activity using an immunoblot for the PKA phosphorylation site of DARPP-32 (Thr 34) after incubating slices for 10 minutes in DHPG (50 µM;Cont) and SKF81297 (10 µM) or 1hour in H-89 (10 µM) prior to DHPG+SKF81297 (n = 4; *p < 0.05). (Bottom Panel) Superimposed representative traces showing the modulation of the DHPG (50 µM)-induced dADP by the D1/5R agonist SKF81297 (10 µM; Gray trace) compared to DHPG alone (Black trace). Scale bars of 2 mV and 2 s. Inset shows the 5 action potentials triggered by a 50 Hz train of brief current stimulation before and after DHPG or DHPG+ SKF81297. (b) Dopamine (DA; 10 µM) inhibits the synaptically-evoked (30 × 50 Hz) dADP (n = 5; **p < 0.01; right gray bar) more than the fast EPSP (n = 5;*p < 0.05; black bar) integral either before or after compensation for the reduced fast EPSP (Norm dADP). The middle gray bar shows the restored fast EPSP (Norm EPSP) in response to DA obtained by increasing the stimulation intensity. (c). Representative traces show the Control EPSP (Dark Gray) , the EPSP after DA (Black trace) or following compensation of the fast EPSP response by increasing the stimulus intensity back to the control level after DA (Norm DA; Light Gray trace). Scale bars show 2 mV and 5 ms. (d) Representative traces show the synaptically-triggered dADP control (Dark Gray Traces) and after DA (Black Traces) and after increasing the stimulus strength to restore the fast EPSP back to control levels (Light Gray Traces). Scale bars show 2 mV and 2 s
To determine if bath applied DA would reduce the synaptically triggered dADP we tested the effects of DA on the cells responding to a synaptically elicited dADP. As with bath applied DHPG, the synaptically triggered dADP was reduced ∼50% by DA (10 µM). Because DA is capable of reducing presynaptic glutamate release we monitored the fast EPSPs and paired pulse ratio and found that at this low concentration (10 µM) of DA there was a significant albeit small 11% reduction in the fast EPSP (Figure 5b,c,d) and a 20% reduction in the paired pulse ratio (PPR;n = 5, p < 0.05). A similar D1/5R-mediated reduction in PPR was reported by Goldman-Rakic and colleagues recording PFC pyramidal neuron pairs under similar experimental conditions.42 To compensate for the slightly reduced fast EPSP in the presence of DA we increased the stimulation intensity to effectively normalize the EPSP response before delivering the dADP triggering burst of synaptic stimulation (Figure 5b,c,d). After adjusting the synaptic stimulation intensity in the presence of DA we still observed a substantial reduction in the dADP. The finding that synaptically elicited dADP is reduced by about 50% while the fast EPSP is reduced by only 11% indicates that the dADP is a more sensitive target for DA modulation compared to the fast AMPA-mediated EPSP (Figure 5b,c,d).
To determine which receptor might be responsible for DA actions on the dADP, selective agonists of the two different receptor classes were used. Activation of the D2 receptor using the D2 selective agonist, quinpirole (10 µM), exerted no effect on the dADP integral in the presence of DHPG (Figure 5; n = 8; p > 0.4;). On the other hand application of the full D1/5R agonist, SKF81297 (10 µM) mimicked the effect of DA application producing a significant decrease in the dADP integral (Figure 5; n = 28; p < 0.002). The effect of SKF81297 was prevented by prior application of the D1R antagonist, SCH23390 (5 µM), indicating that the effect of SKF81297 is receptor-mediated (Supplemental Figure 3). No differences were found between 1 and 10 µM SKF81297; however, an inverted U-shaped dose response was observed across a full dose response from 0.5–50 uM SKF81297 (Supplemental Figure 3). This is interesting since a D1/5R-mediated inverted U-shaped dose response curve has been noted in PFC working memory tasks with optimal levels of D1/5R stimulation capable of facilitating performance on these tasks.20
We have previously shown that D1/5R stimulation reduces current through voltage-gated Na+ channels in PFC pyramidal neurons.25 Furthermore, behavioral sensitization to cocaine reduces whole-cell Na+ current in the accumbens.42 Despite these reported findings D1/5R modulation of the mGluR-mediated dADP was independent of Na+ channel function (Supplemental Figure 3). To determine if D1/5R activation reduces the dADP by attenuating dendritic backpropagating action potentials initiated by the burst we applied a low TTX concentration (15 nM) that inhibits a substantial (∼60%) fraction of Na+ channels while minimally reducing somatic action potential height (∼5%).44 As we reported previously using high concentrations of TTX (500 nM)9, low TTX (15 nM) failed to alter the DHPG-induced burst triggered dADP or the inhibitory effects of the D1/5R dADP modulation (n = 28, SKF alone; n = 6, TTX+SKF, p > 0.05; Supplemental Figure 3).
The D1/5R couples to Gs proteins, which increases cAMP levels and activates PKA.26 To determine whether a PKA mechanism is involved in mediating the effects of SKF81297, we used forskolin (10 µM), an activator of adenylyl cyclase and PKA. Bath application of forskolin-induced a significant decrease in the DHPG-induced dADP integral (Figure 5). Application of the PKA blocker, H-89 (10 µM), prevented the reduction in the DHPG-induced dADP integral caused by SKF81297 (Figure 5). To confirm that the effects of SKF81297 and H89 were mediated through the PKA pathway we tested the PKA phosphorylation site on p-DARPP-32(Thr-34) which showed a significant increase in phosphorylation after slices were incubated for 10 min in SKF81297 (10 µM; Figure 5 inset). As with the physiology, preincubation of PFC slices in H89 (10 µM) eliminated the effects of SKF81297 on the PKA site of DARPP-32 (Figure 5 inset). It is possible, that the D1/5R can couple to a PKC mechanism.26 However, three findings argue against such a mechanism: 1) the PKC inhibitor BMXI (1–50 µM) failed to reduce the SKF-induced inhibition (n = 5; p > 0.05); 2) The PKA inhibitor completely blocked the effects of SKF81297 (Figure 5); and, 3) Forskolin completely occluded the effects of SKF81297, while it failed to occlude PKC-induced reduction of the dADP (n = 5, p > 0.05;data not shown).
To examine the synaptically triggered dADP and show that the effects of forskolin were postsynaptic we used forskolin (10 µM) inside the patch pipette and monitored the synaptically triggered dADP (Figure 5a; Supplemental Figure 4a). Internal forskolin produced a significant reduction in the dADP that was similar to bath applied DA, SKF81297 and forskolin (Figure 5a). Intracellular Ca2+ response of heterologously expressed mGluR5 to DHPG was inhibited by forskolin (10 µM) by ∼40% (Supplemental Figure 4b). These experiments demonstrate that postsynaptic PKA activation is sufficient to produce an inhibition of mGluR5 functions and hence the reduction in the dADP and rules out a presynaptic PKA contribution.
Plasticity of the delayed afterdepolarization and loss of D1R modulation after repeated cocaine exposure and withdrawal
Repeated noncontingent injections of cocaine (5 × 15 mg/kg; i.p.) have been shown to induce plasticity of intrinsic excitability in the PFC.24 Here we report that repeated cocaine treatment (5 days) and short (2 day) and long (14 day) term withdrawal induces behavioral sensitization to a cocaine challenge injection comparing saline and cocaine treated animals (Figure 6a, n=6/group; p < 0.05). Short-term withdrawal from cocaine produces a substantial reduction in the ability of mGluRs and bursts to elicit the dADP despite a significant increase in measured input resistance observed in the cocaine group (Figure 6 and Figure 7; Supplemental Table 1; n = 21–22/group, p < 0.03). The diminished cocaine-induced dADP was accompanied by a complete loss of the inhibitory effects of D1/5R stimulation (Figure 6c; n = 7–8/group; p < 0.001). After long-term withdrawal the dADP recovered in the cocaine-treated group (Figure 6d; n = 13–15/group; p>0.05); but D1/5R modulation was still absent (Figure 6d; n=11/group; p<0.03). These results are consistent with our previous work in deep layer PFC neurons showing that repeated cocaine and short-term withdrawal leads to increased basal PKA activity and intrinsic changes in ion channel function.24 D1/5R pathway modulation of the dADP was absent at both withdrawal times, yet the cocaine-induced reduction in the dADP recovered fully after longer withdrawal suggesting independent or compensatory mechanisms in the PKA pathway during withdrawal from cocaine.
Figure 6.

Repeated cocaine exposure reduces the mGluR5/Burst-induced dADP and dopamine D1/5 receptor modulation. (a) Behavioral sensitization measured by locomotor ambulation in Cocaine (15 mg/kg × 5 days) or Saline (0.9% 1ml/kg) treated rats. Cocaine challenge (15 mg/kg) injections administered in both groups on withdrawal days 2 and 14 indicated behavioral sensitization in the cocaine treated group (SAL vs COC; n=6/group; *p<0.05). (b) Superimposed representative traces show the DHPG-mediated dADP triggered by brief bursts of 5 action potentials (50 Hz) from Saline (Left Panel) and Cocaine-treated (Right Panel) rats at the early withdrawal (2 day) time point. SKF81297 (10 µM; Black trace) reduced the dAPD in the Saline but not Cocaine treated group at the 2 day withdrawal time point. Insets show the burst stimulus used to evoke the dADP in the presence of DHPG (50 µM). (c) (Left) Average dADP integral response after 2 days withdrawal in Saline (n = 21) or Cocaine (n = 22) treated rats showing the reduced dADP (*p < 0.03). (Right) Average change in dADP integral in response to SKF81297 (10 µM) after 2 days withdrawal from repeated Saline (n = 7) and Cocaine (n=8;**p<0.001) treated rats. (d) (Left) No significant differences were observed in the average dADP integral after 14 days withdrawal in Saline (n=13) and Cocaine (n = 15) treated rats (p > 0.05). (Right) The D1/5R mediated inhibition (SKF81297, 10 µM) of the dADP remained significantly reduced in cocaine-treated (COC/SKF; n = 11;*p < 0.03) compared to Saline (SAL/SKF; n = 11) treated groups at the 14 day withdrawal time point.
The functional implications of the mGluR5/burst-induced dopamine D1/5R PKA-modulated dADP are illustrated in Figure 7, which show the capability of the dADP to convert near threshold simulated synaptic inputs (Figure 7a inset) to suprathreshold output for up to 4 seconds post burst. We have previously used simulated synaptic current injections designed to match the kinetics of the synaptically elicited fast EPSPs in order to probe postsynaptic excitability using a physiologically relevant stimulus without presynaptic interference.32,39 D1/5R inhibition of the dADP using SKF81297 reduces the near threshold conversion of simulated EPSPs to spikes, thus reducing the spike probability and narrowing the window whereby the dADP is capable of boosting subthreshold inputs (Figure 7a;see Appendix). In a normal behaving animal such D1/5R modulation may be important for filtering out stimuli that are not paired closely in time with the stimuli eliciting the initial burst, or this for the reward-related termination of persistent activity.
In cocaine-treated animals given a short withdrawal, the ability of the dADP to convert near threshold input to action potential output (spike probability) was significantly reduced for inputs occurring within 4 seconds after the initial burst (Figure 7b). This cocaine-induced reduction in spike probability recovered after a long-term (Figure 7c left panel) 14-day withdrawal period, however the loss of D1/5R function remained substantially reduced compared to saline controls (Figure 7c right panel). The cocaine-induced increase in input resistance and spike output (Supplemental Table) in response to prolonged current injections indicate increased excitability, while the reduced burst-triggered dADP suggests a reduced in vivo ability to maintain a stable persistent dADP state in the absence of sustained excitatory drive. These cocaine-induced changes may bias the prefrontal cortical neurons to favor only strong sustained input lasting seconds while weakening their ability to shift to a persistent output mode in response to brief burst activation.
There is a growing body of research indicating diminished PFC functioning after chronic psychostimulant use in humans and in animal models.3,4,24,25,27,39,43,45,47 For example, cocaine abusers manifest behavioral abnormalities similar to those observed in patients with orbitofrontal damage, such as choosing immediate high-risk rewards, despite the knowledge of the future negative consequences of the decision.45 Human brain imaging studies of cocaine dependent individuals show increased activity (fMRI) of the PFC following cocaine alone and in response to cocaine-paired cues.46,47 In the rodent PFC, glutamatergic transmission is critical for cocaine-primed reinstatement of drug-seeking behavior and is mediated by a glutamatergic projection from the PFC to the NAc .48 mGluR5-mediated transmission is necessary for the normal psychostimulant response to cocaine (Supplemental Figure 5).12 Cocaine self-administration produces deficits in impulsivity and attention that is correlated with changes in mGluR5 mRNA in the frontal cortex.3 Furthermore, repeated d-amphetamine exposure has been shown to produce a progressive enhancement of PFC inhibition of neurons that respond to task-related events and is correlated with decline in working memory performance.27 We found that the mGluR5 mediated dADP mechanism is impaired during the abstinence period after repeated cocaine exposure and this would be predicted to diminish PFC intrinsic persistent activity.
Overall, our findings implicate the mGluR5 dADP as a novel intrinsic cellular mechanism capable of maintaining persistent activity. As in working memory tasks, the dADP is reduced by mGluR5 antagonists, modulated by D1R/PKA activation and diminished following repeated cocaine exposure/withdrawal.
Materials and Methods
Animals
Male rats (Charles River, Kingston, RI) were group housed, in a temperature and humidity controlled vivarium, under a 12 hr light/dark cycle. Food and water were available ad libitum. The mGluR1 and 5 mice were obtained from Dr. Kim Huber (UT Southwestern Medical Center, Dallas, TX) and were 5–6 weeks old. Animals arrived at four-weeks of age and were allowed to acclimate for 3–5 days before being used for experiments. All animal procedures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the IACUC.
Slice preparation
Animals were perfused, under halothane anesthesia, with cold and oxygenated artificial cerebrospinal fluid (aCSF) containing (in mM) NaCl 124, KCl 2.5, NaHCO3 26, MgCl2 2, CaCl2 2 and glucose 10; pH = 7.4; 310 mOsm/l. Following decapitation, the brain was removed, immersed in ice-cold aCSF. The part containing the PFC was glued onto the specimen holder, and submerged in the buffer tray of a Leica VT1000S vibratome (Leica Microsystems, Nussloch, Germany), containing cold and oxygenated aCSF. Brain slices were cut in 300 µm thick sections and transferred to a custom-made, nylon-support chamber, filled with oxygenated aCSF, where they were kept at 34°C for 30 min, and then transferred to room temperature until recording 1–4 hours later.
Electrophysiological recordings
Brain slices were place in a custom temperature-controlled (32–35°C) recording chamber and placed under a Olympus (Tokyo, Japan) BX-51WI upright light microscope placed on a Vibraplane 9100/9200 Series Vibration Isolation Table (Kinetic Systems, Inc, Roslindale, MA). The slice was perfused by gravity-fed, oxygenated aCSF at a flow rate of 2–3 ml/min. Whole-cell patch clamp recordings were obtained from layer 5 PFC pyramidal neurons, visually-identified on a Sony monitor using differential interference contrast (DIC) optics and a Dage MTI camera (Michigan City, IN). Following the formation of a tight seal (>1GΩ), small, whole-cell configuration was obtained. A patch-clamp amplifier (BVC 700A, current-clamp; 3900A, voltage-clamp, Dagan Corporation, Minneapolis, MN) was used for current and voltage-clamp recording. Voltage signals were digitized with an Instrutech (Port Washington, NY) ITC-18 analog to digital converter. The data was stored in a Pentium computer using DataPro IGOR Pro software (Wavemetrics, Inc. Lake Oswego, OR). Recordings were accepted for study if the neuron’s resting membrane potential was more negative than −60mV, had overshooting spikes and a series resistance less than 30 MΩ. Slices were perfused with ACSF containing 2.5 mM Kynurenic acid (Sigma), 2 µM SR 95531(Tocris), 1µM Atropine to block ionotropic (NMDA and AMPA) and GABA-A receptors. At the end of each experiment the slice was then placed in 4% paraformaldehyde solution, and kept at 4°C until histological processing.
Electrophysiological protocols
Current-Clamp recording
All current and voltage-clamp recordings were obtained with glass electrodes (2–3 MΩ) in whole-cell configuration. Recording pipettes (2–4MΩ) were pulled from Corning 7056 (Corning, NY) borosilicate glass capillaries with a horizontal pipette puller (Flaming/Brown P-97, Sutter Instruments, CA) and filled with (in mM): K+-Gluconate 115, HEPES 10, KCl 20, MgCl2 2, Na2ATP 3, Na2GTP 0.3, biocytin 0.1%, pH = 7.3, 280mOsm. Cells in current clamp (IClamp) were recorded with a holding potential of −65 mV unless otherwise noted. Synaptic responses were recorded in response to stimulation of the layer 3 axons using a 50 Hz bipolar stimulus (5 or 30 pulses). To monitor the input resistance, a series of 600 ms step pulses from −200pA to +300pA were delivered and the steady-state voltage deflection was calculated. To trigger the dADP, a train of five suprathreshold 5 ms step pulses delivered 50 Hz was used. The current injection trains were applied every 30 seconds. Unless otherwise noted, all experiments were performed using the above train protocol. When traces were obtained for dADP measurements, the membrane potential of the cell was held at −67 mV. All recordings were made in the presence of kynurenic acid (2 mM) to block iontotropic glutamate (∼90% block) and SR 95531 (2 µM) to block GABAA receptors and atropine to block muscarinic acetylcholine receptors.
The simulated synaptic current injections (sEPSCs) were injected into the soma via the recording patch pipette followed 400 ms later by a 5 ms square step pulse scaled to half the sEPSC amplitude to verify accurate bridge balance and capacitance compensation. We set the sEPSCs kinetics with a rise time of 0.2 ms and a decay time of 6 ms, yielding sEPSPs that closely resembled actual EPSPs.32,39
Voltage-clamp recordings
A Cs-gluconate based internal was used to record IDHPG (in mM:115 Cs-gluconate, 10 Na2-phosphocreatine, 10 HEPES, 2 EGTA, 2 MgATP, and 0.3 Na2GTP, 20 mM CsCl). Membrane potentials reported were not corrected for an −8 mV liquid junction potential present when using gluconate-based internal solutions. The recorded current was filtered at 3 kHz and sampled at 50 kHz. Data analysis was performed with IGOR pro software. Currents were acquired using a P/6 to subtract leak and capacitive currents. Analysis was performed using averages of 2–10 responses. Values are reported as mean ± SEM. DHPG (100 µM) was puffed on for 1 second duration and the corresponding inward current (IDHPG) was measured at a holding potential of −65 mV.
Drugs used
(1S,3R)ACPD, DHPG, LY341495, LY367385, MPEP, SR95531 were obtained from Tocris. DHPG, kynurenic acid, DA, SKF81297, Forskolin, quinpirole and atropine were obtained from Sigma. Drugs were dissolved in H2O or ACSF.
Analysis
The amplitude of the dADP was calculated by measuring the difference between the average membrane potential 50 ms prior to the stimulus onset and the membrane potential peak after the spike train. The decay phase of the dADP was fitted by a single exponential function (y = a*e−bx), and the time constant of decay was 1/b. Throughout the experiment, the input resistance of the cell was monitored, using a 600 ms step pulse protocol with current injections from −100 pA to +100 pA. The points of the potential difference between the Vm, and the response potential were plotted against the current used for the points between −100 pA and +100 pA. The points were fitted to a line (y = ax+b), and input resistance was the slope of that line.
Histology
After recording the slice was removed from recoding chamber and placed in 4% paraformaldehyde and washed with phosphate buffered saline (PBS), incubated in a solution containing 10% methanol, 3% hydrogen peroxide dissolved in PBS for 30min, washed again and incubated in the ABC reagent solution for at least 2 hrs at RT. Following another wash, the slice was incubated in 0.1% gluteraldehyde for 4min, washed and incubated in the DAB solution and then cover-slipped with Mowiol. Slices were observed under an Olympus BX-51WI microscope to determine the cellular morphology. Following recording, each cell was processed for morphological verification of pyramidal neuronal morphology and placement in layer 5 (Figure 1a).
Statistics
ANOVA followed by paired or unpaired t-tests were used where appropriate. A result was considered significant when the p value was less than 0.05 using a two-tailed analysis. Bonferroni correction was used to hold p constant at 0.05 when performing multiple comparisons among treatment means.
Locomotor Activity
Mice were habituated to the locomotor apparatus for 1 hr prior to testing on each day. Mice were tested for control locomotor activity for 2 days of saline (SAL) injections prior to an ascending dosing regimen of cocaine challenge injections (10, 20 and 40 mg/kg; 2 ml/kg volume). Between cocaine injection days mice were tested with SAL to avoid conditioning to the effects of cocaine.
For rats, after at least 5 days acclimation period, animals were assigned randomly into 2 groups that received an intraperitoneal injection of either saline (1 ml/kg) or cocaine HCl (15 mg/kg) for 5 days. After a 2 or 14 day withdrawal period rats were then tested with a challenge injection (15 mg/kg) to probe for locomotor sensitization. For the five treatment days and challenge days locomotor activity was recorded during their light cycle in a darkened circular test chambers with a 12 cm wide runway, equipped with four pairs of photocells located at 90-degree intervals around the 1.95 m perimeter.
Supplementary Material
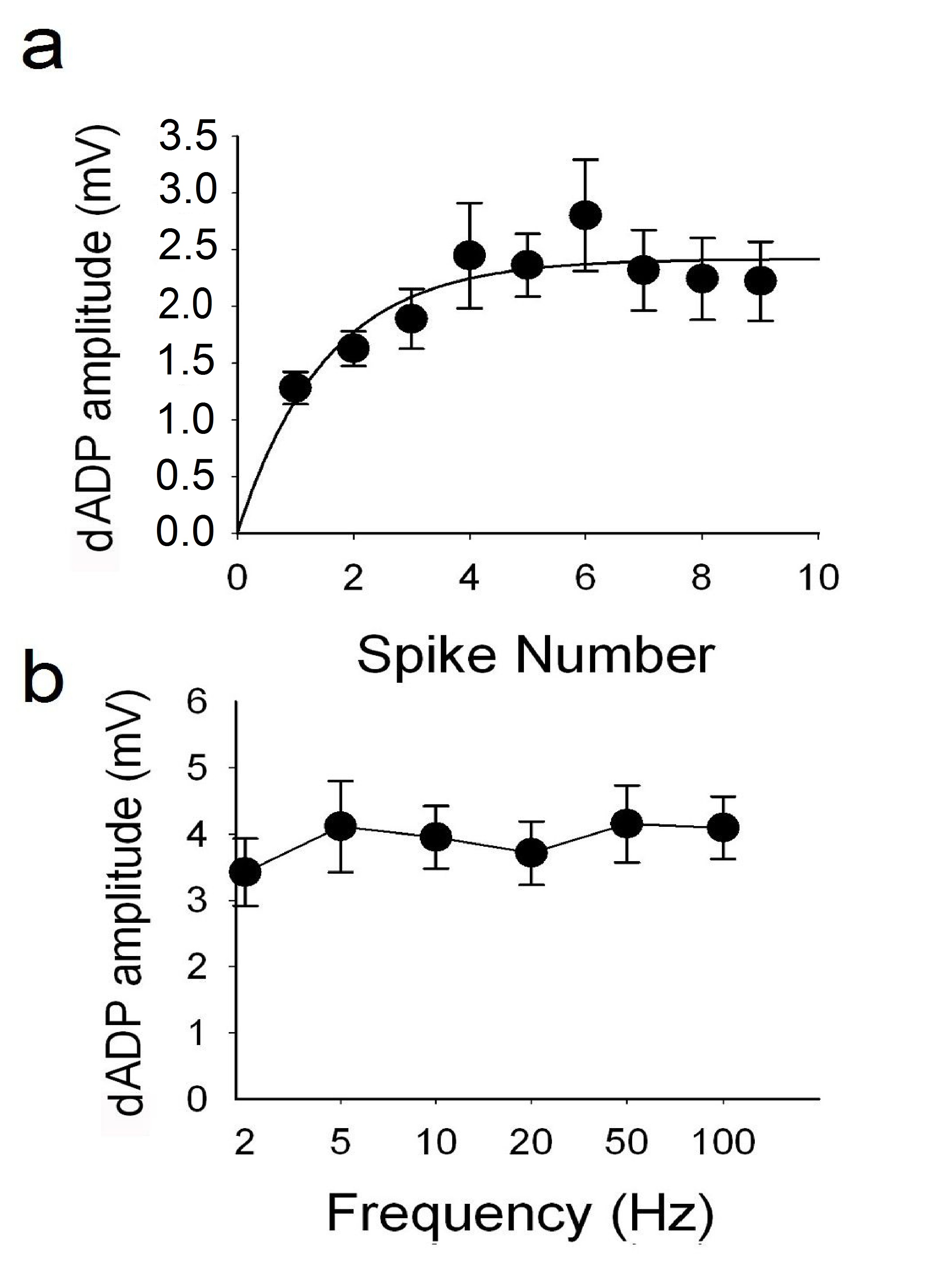
The DHPG (50 µM)-induced dADP amplitude reaches an asymptote after a burst of 4 action potentials (A) and is frequency independent at frequencies greater than 2 Hz (b; n = 6).
{kind=link}
Synaptic stimulation of the proximal apical dendrite of deep layer 5/6 pyramidal neurons produces a dADP and a persistent increase in the instantaneous frequency. (a) 30 synaptic pulses delivered at 50 Hz produces a substantial dADP in the presence of 2.5 mM kynurenic acid, which blocks ∼90% of AMPA and NMDA glutamate receptors. (Inset) Shows a schematic of the stimulation site in the apical dendrite. For each cell the minimum stimulation intensity required to produce a single action potential was chosen for subsequent 5 and 30 pulse experiments (b,d). (a) Middle traces show a current injection (50-100 pA) that was delivered for 600ms to induce stable action potential output (Black trace and Black open circles) for subsequent testing before and after 30 pulse synaptic stimulation (* Black filled circles). (b) 30 synaptic pulses (n=7) delivered at 20 Hz produces a large dADP slightly larger dADP compared to 5 (n = 5) synaptic pulses (*p < 0.03). (c) 5 synaptic pulses (50 Hz) produces an increase in the peak instantaneous frequency compared to control current injection delivered at the same 50 Hz frequency as synaptic stimulation (n = 8;**p < 0.002) and 30 pulses produce a similar increase in the peak instantaneous frequency (n = 13;***p < 0.0001) current injections. There were no differences comparing 5 and 30 pulses (p > 0.2) (d) Representative traces showing the selective mGluR5 antagonist significantly reduces the dAPD triggered by synaptic stimulation of the apical dendritic tree in deep layer 5/6 pyramidal neurons. The inset shows the same synaptic stimulation of action potentials before and after MPEP. (e) Shows the average dADP block by MPEP (30 µM;n = 5;**p < 0.01).
{kind=link}
The dopamine D1 receptor agonist SKF81297 produces an inverted U-shaped dose response relationship to DHPG (50 µM)-induced dADP (n =4 for 0.5 µM, n = 5 for 1µM, n=28 for 10 µM and n=5 for 50 µM;Black Circles). The D1R antagonist, SCH 23390 (5 µM, n = 4; p > 0.05, Open Diamond) blocks the inhibitory effects of SKF81297 at the maximally effective dose (10 µM). Low TTX (15 nM) does not alter the dADP and does not diminish the inhibitory effect of SKF 10µM (TTX+SKF) on the dADP (n = 6). **p < 0.01,*p < 0.05
{kind=link}
(a) Forskolin reduces the mGluR5-mediated postsynaptic responses. Representative traces showing the post synaptic reduction of the synaptically triggered dADP by internal Forskolin. Black traces show the dAPD after initial break-in to whole-cell configuration and Red traces show 20 minutes after Forskolin perfusion in the internal pipette solution. Insets show no changes in the synaptic stimulation and action potentials before and after Forskolin (average data shown in Figure 5). (b) To test whether mGluR5 function can be modulated by PKA, HEK293 cells stably expressing mGluR5 were seeded in wells of a 96-well plate for 20 hrs before being washed and loaded with 5 µM Fluo4-AM in an extracellular solution as described previously.84 In order to suppress the constitutive activity of the receptor, the cell culture medium contained mGluR5 antagonist, 2-methyl-6 [phenylethynyl]-pyridine (MPEP, 1 µM). Fluo-4 fluorescence change was read using a fluorescence plate reader, FlexStation (Molecular Devices). Cells were untreated (Control; n = 12) or treated with Forskolin (10µM; n = 24;***p<0.001) for 5 min before DHPG was added. RFU: random fluorescence unit.
{kind=link}
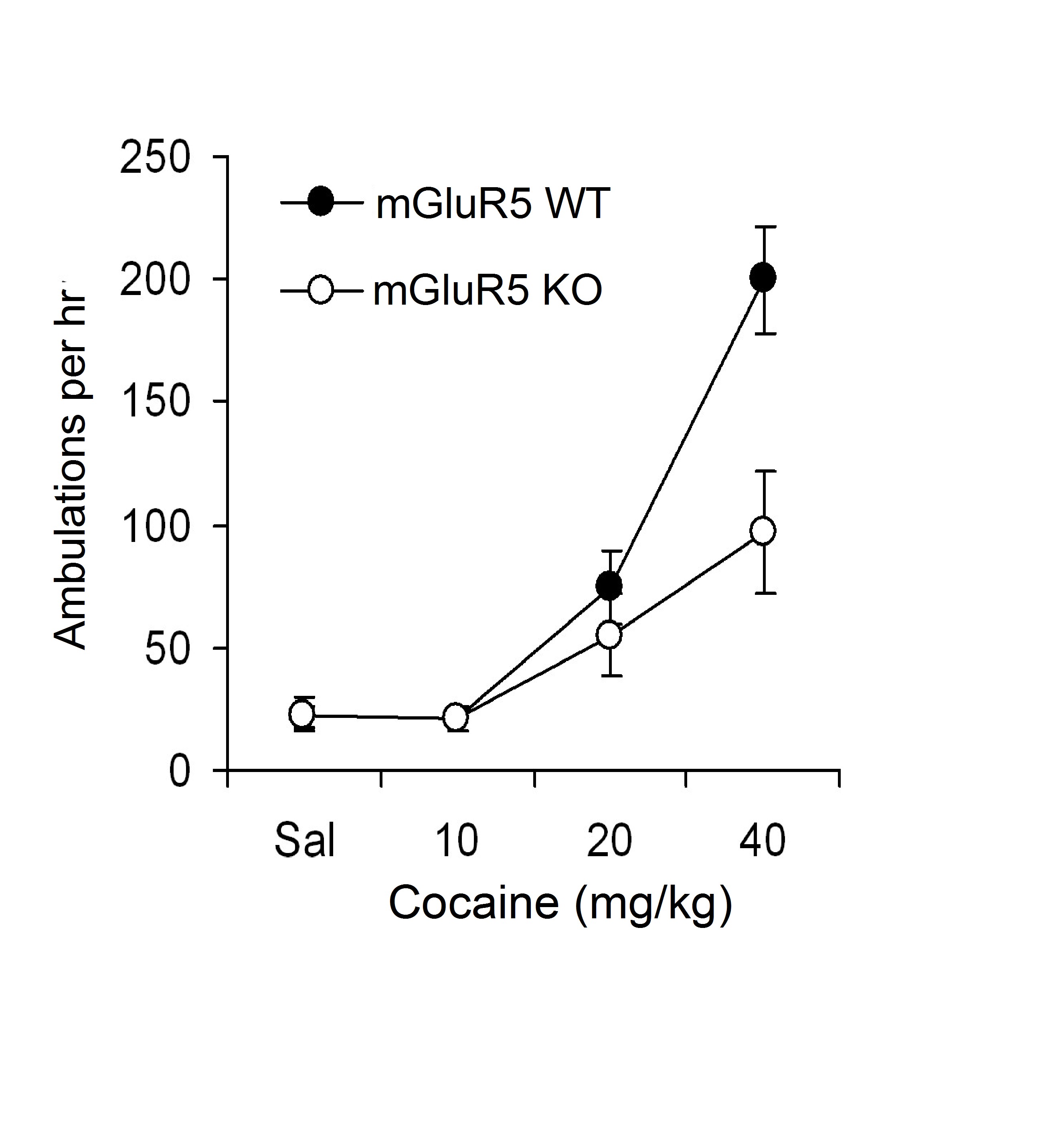
Locomotor response to cocaine in mGluR5 deficient mice (mGluR5 Mut) compared to wild type (mGluR5 WT) controls. Cocaine dose dependently increased locomotor activity producing a significant difference (**p < 0.01) between the Mut (n = 7) and WT (n = 8) mice at the highest dose (40 mg/kg) in a 1 hr test given after 1 hr habituation.
{kind=link}
Acknowledgements
We would like to thank Dr. Kim Huber for providing us with the mGluR1 and 5 wild type and knockout mice. This work was supported by NIDA grant R01-DA24040 (DCC) NIDA K award (K-01DA017750) to DCC, NARSAD Young Investigator award (DCC), a NIDA institutional training grant (T32-DA7290) to MAF, the Onassis Public Benefit Foundation (KS) and a Gulf War Syndrome contract from Dept of Veterans Affairs. This paper is dedicated to the memory of Francis J. White Ph.D., a close friend and mentor.
Appendix
Here we report the ANOVA statistics for the simulated synaptic current injections in the saline and cocaine treated groups given SKF81297 and DHPG presented in Figure 7. Effect of SKF81297 in Saline controls, F(1,40)= 25.99, p < 0.0001; Time (1–4 seconds) effect, F(3,40)= 34.77, p < 0.0001; Drug (SKF81297) by Time (1–4 seconds) interaction, F(3,40) = 0.82, p < 0.42. Bonferroni posttest comparisons indicate significant differences between baseline and Drug (SKF81297) at 2 and 3 seconds (p<0.01).
For saline and cocaine treated groups given short-term, 2 day withdrawal: Group effect SAL vs COC baseline; F(1,68) = 30.04, p < 0.0001; Drug (SKF81297) effect, F(1,68) = 0.01, p = 0.92; Time (1–4 seconds) effect, F(3,68) = 44.55, p < 0.0001; Drug (SKF81297) effect by Time (1–4 seconds) interaction, F(3,68) = 0.94, p = 0.43. Bonferroni posttest comparisons indicate a significant difference between saline and cocaine at 1 and 2 seconds post burst (p < 0.001). No significant differences were detected between baseline and Drug (SKF81297) in the cocaine group at any time point (p > 0.05).
For saline and cocaine treated groups given a long-term, 14 day withdrawal: Group effect SAL vs COC baseline; F(1,40) = 0.79, p = 0.38; Time (1–4 seconds) effect, F(3,40) = 13.60, p < 0.0001; Group by Time interaction, F(3,40) = 0.06, p =0.98. Drug (SKF81297) effect, F(1,40) = 3.13, p = 0.084; Time (1–4 seconds) effect, F(3,40) = 50.81, p < 0.0001); Drug by Time interaction effect, F(3,40) = 0.19, p = 0.90. Bonferroni posttest comparisons indicate no significant differences between saline and cocaine groups or in the baseline and drug (SKF81297) comparison in the cocaine group at any time point (p > 0.05).
Reference List
- 1.Goldman-Rakic PS. Regional and cellular fractionation of working memory. Proc Natl Acad Sci U S A. 1996;93:13473–13480. doi: 10.1073/pnas.93.24.13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Funahashi S, Takeda K. Information processes in the primate prefrontal cortex in relation to working memory processes. Rev Neurosci. 2002;13:313–345. doi: 10.1515/revneuro.2002.13.4.313. [DOI] [PubMed] [Google Scholar]
- 3.Winstanley CA, et al. DeltaFosB induction in orbitofrontal cortex mediates tolerance to cocaine-induced cognitive dysfunction. J Neurosci. 2007;27(39):10497–10507. doi: 10.1523/JNEUROSCI.2566-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.George O, et al. Extended Access to Cocaine Self-Administration Produces Long-Lasting Prefrontal Cortex-Dependent Working Memory Impairments. Neuropsychopharmacology. (2007) Nov 21; doi: 10.1038/sj.npp.1301626. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 6.Seung HS, et al. Stability of the memory of eye position in a recurrent network of conductance-based model neurons. Neuron. 2000;26:259–271. doi: 10.1016/s0896-6273(00)81155-1. [DOI] [PubMed] [Google Scholar]
- 7.Wang XJ. Synaptic reverberation underlying mnemonic persistent activity. Trends Neurosci. 2001;24:455–463. doi: 10.1016/s0166-2236(00)01868-3. [DOI] [PubMed] [Google Scholar]
- 8.Egorov AV, et al. Graded persistent activity in entorhinal cortex neurons. Nature. 2002;420:173–178. doi: 10.1038/nature01171. [DOI] [PubMed] [Google Scholar]
- 9.Fowler MA, et al. Corticolimbic Expression of TRPC4 and TRPC5 Channels in the Rodent Brain. PloS One. 2(6):e573. doi: 10.1371/journal.pone.0000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Homayoun H, et al. Functional Interaction Between NMDA and mGlu5 Receptors: Effects on Working Memory, Instrumental Learning, Motor Behaviors, and Dopamine Release. Neuropsychopharm. 2004;29:1259–1269. doi: 10.1038/sj.npp.1300417. [DOI] [PubMed] [Google Scholar]
- 11.Homayoun H, Moghaddam B. Bursting of prefrontal cortex neurons in awake rats is regulated by metabotropic glutamate 5 (mGlu5) receptors: rate-dependent influence and interaction with NMDA receptors. Cereb Cortex. 2006;16:93–105. doi: 10.1093/cercor/bhi087. [DOI] [PubMed] [Google Scholar]
- 12.Chiamulera C, et al. Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat Neurosci. 2001;4:873–874. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- 13.Otani S, et al. Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J Neurosci. 1999;19:9788–9802. doi: 10.1523/JNEUROSCI.19-22-09788.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greene CC, Schwindt PC, Crill WE. Properties and ionic mechanisms of a metabotropic glutamate receptor-mediated slow afterdepolarization in neocortical neurons. J Neurophysiol. 1994;72:693–704. doi: 10.1152/jn.1994.72.2.693. [DOI] [PubMed] [Google Scholar]
- 15.Chuang, et al. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59(3):392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Groenewegen HJ. Organization of the afferent connections of the mediodorsal thalamic nucleus in the rat, related to the mediodorsal-prefrontal topography. Neuroscience. 1988;24:379–431. doi: 10.1016/0306-4522(88)90339-9. [DOI] [PubMed] [Google Scholar]
- 17.Carr DB, Sesack SR. Dopamine terminals synapse on callosal projection neurons in the rat prefrontal cortex. J Comp Neurol. 2000;425:275–283. doi: 10.1002/1096-9861(20000918)425:2<275::aid-cne9>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe M, Kodama T, Hikosaka K. Increase of extracellular dopamine in primate prefrontal cortex during a working memory task. J Neurophysiol. 1997;78:2795–2798. doi: 10.1152/jn.1997.78.5.2795. [DOI] [PubMed] [Google Scholar]
- 19.Phillips AG, Ahn S, Floresco SB. Magnitude of dopamine release in medial prefrontal cortex predicts accuracy of memory on a delayed response task. J Neurosci. 2004;24:547–553. doi: 10.1523/JNEUROSCI.4653-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vijayraghavan S, et al. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. 2007;10(3):376. doi: 10.1038/nn1846. [DOI] [PubMed] [Google Scholar]
- 21.Williams GV, Goldman-Rakic PS. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 1995;376(6541):572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, O'Donnell P. D(1) dopamine receptors potentiate nmda-mediated excitability increase in layer 5 prefrontal cortical pyramidal neurons. Cereb Cortex. 2001;11:452–462. doi: 10.1093/cercor/11.5.452. [DOI] [PubMed] [Google Scholar]
- 23.Yang CR, $Seamans JK. Dopamine D1 receptor actions in layers 5–6 rat prefrontal cortex neurons in vitro: modulation of dendritic-somatic signal integration. J Neurosci. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong Y, et al. Cocaine-induced Plasticity of Intrinsic Membrane Properties in Prefrontal Cortex Pyramidal Neurons: Adaptations in Potassium Currents. J Neurosci. 2005;26:25(4):936–940. doi: 10.1523/JNEUROSCI.4715-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterson JD, Wolf ME, White FJ. Repeated amphetamine administration decreases D1 dopamine receptor-mediated inhibition of voltage-gated sodium currents in the prefrontal cortex. J Neurosci. 2006;26(12):3164–3168. doi: 10.1523/JNEUROSCI.2375-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Young CE, Yang CR. Dopamine D1/D5 receptor modulates state-dependent switching of soma-dendritic Ca2+ potentials via differential protein kinase A and C activation in rat prefrontal cortical neurons. J Neurosci. 2004;24:8–23. doi: 10.1523/JNEUROSCI.1650-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Homayoun H, Moghaddam B. Progression of cellular adaptations in medial prefrontal and orbitofrontal cortex in response to repeated amphetamine. J Neurosci. 2006;26(31):8025–8039. doi: 10.1523/JNEUROSCI.0842-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanes DP, Schall JD. Neural control of voluntary movement initiation. Science. 1996;274(5286):427–430. doi: 10.1126/science.274.5286.427. [DOI] [PubMed] [Google Scholar]
- 29.Rainer G, Asaad WF, Miller EK. Memory fields of neurons in the primate prefrontal cortex. Proc Natl Acad Sci U S A. 1998;95:15008–15013. doi: 10.1073/pnas.95.25.15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rae MG, et al. Role of Ca2+ stores in metabotropic L-glutamate receptor-mediated supralinear Ca2+ signaling in rat hippocampal neurons. J Neurosci. 2000;20:8628–8636. doi: 10.1523/JNEUROSCI.20-23-08628.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura T, et al. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- 32.Cooper DC, Chung S, Spruston N. Output-mode transitions are controlled by rolonged inactivation of sodium channels in pyramidal neurons of subiculum. PLoS Biol. 2005;3:e175. doi: 10.1371/journal.pbio.0030175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida M, Fransén E, Hasselmo ME. mGluR-dependent persistent firing in entorhinal cortex layer III neurons. Eur J Neurosci. 2008;28(6):1116–1126. doi: 10.1111/j.1460-9568.2008.06409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montell C. The TRP superfamily of cation channels. Sci STKE. 2005;2005(272) doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- 35.Lopez-Bendito G, Shigemoto R, Fairen A, Lujan R. Differential distribution of group I metabotropic glutamate receptors during rat cortical development. Cereb Cortex. 2002;12:625–638. doi: 10.1093/cercor/12.6.625. [DOI] [PubMed] [Google Scholar]
- 36.Cooper DC. The significance of action potential bursting in the brain reward circuit. Neurochem Int. 2002;41:333–340. doi: 10.1016/s0197-0186(02)00068-2. [DOI] [PubMed] [Google Scholar]
- 37.Hagenston AM, Fitzpatrick JS, Yeckel MF. MGluR-mediated calcium waves that invade the soma regulate firing in layer V medial prefrontal cortical pyramidal neurons. Cereb Cortex. 2008;18(2):407–423. doi: 10.1093/cercor/bhm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaspar P, Bloch B, Le Moine C. D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. Eur J Neurosci. 1995;7:1050–1063. doi: 10.1111/j.1460-9568.1995.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 39.Cooper DC, et al. Psychostimulant-induced plasticity of intrinsic neuronal excitability in ventral subiculum. J Neurosci. 2003;23(30):9937–9946. doi: 10.1523/JNEUROSCI.23-30-09937.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura T, et al. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- 41.Lujan R, et al. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- 42.Gao WJ, Krimer LS, Goldman-Rakic PS. Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci U S A. 2001;98(1):295–300. doi: 10.1073/pnas.011524298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang XF, Hu XT, White FJ. Whole-cell plasticity in cocaine withdrawal: Reduced sodium currents in nucleus accumbens neurons. J Neurosci. 1998;18(1):488–498. doi: 10.1523/JNEUROSCI.18-01-00488.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madeja M. Do neurons have a reserve of sodium channels for the generation of action potentials? A study on acutely isolated CA1 neurons from the guinea-pig hippocampus. Eur J Neurosci. 2000;12(1):1–7. doi: 10.1046/j.1460-9568.2000.00871.x. [DOI] [PubMed] [Google Scholar]
- 45.Bechara A, et al. Decision-making deficits, linked to a dysfunctional ventromedial prefrontal cortex, revealed in alcohol and stimulant abusers. Neuropsychologia. 2001;39:376–389. doi: 10.1016/s0028-3932(00)00136-6. [DOI] [PubMed] [Google Scholar]
- 46.Breiter HC, et al. Acute effects of cocaine on human brain activity and emotion. Neuron. 1997;19:591–611. doi: 10.1016/s0896-6273(00)80374-8. [DOI] [PubMed] [Google Scholar]
- 47.Grant S, et al. Activation of memory circuits during cue-elicited cocaine craving. Proc Natl Acad Sci U S A. 1996;93:12040–12045. doi: 10.1073/pnas.93.21.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu HZ, et al. 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2,and TRPV3. J Biol Chem. 2004;279:35741–35748. doi: 10.1074/jbc.M404164200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The DHPG (50 µM)-induced dADP amplitude reaches an asymptote after a burst of 4 action potentials (A) and is frequency independent at frequencies greater than 2 Hz (b; n = 6).
Synaptic stimulation of the proximal apical dendrite of deep layer 5/6 pyramidal neurons produces a dADP and a persistent increase in the instantaneous frequency. (a) 30 synaptic pulses delivered at 50 Hz produces a substantial dADP in the presence of 2.5 mM kynurenic acid, which blocks ∼90% of AMPA and NMDA glutamate receptors. (Inset) Shows a schematic of the stimulation site in the apical dendrite. For each cell the minimum stimulation intensity required to produce a single action potential was chosen for subsequent 5 and 30 pulse experiments (b,d). (a) Middle traces show a current injection (50-100 pA) that was delivered for 600ms to induce stable action potential output (Black trace and Black open circles) for subsequent testing before and after 30 pulse synaptic stimulation (* Black filled circles). (b) 30 synaptic pulses (n=7) delivered at 20 Hz produces a large dADP slightly larger dADP compared to 5 (n = 5) synaptic pulses (*p < 0.03). (c) 5 synaptic pulses (50 Hz) produces an increase in the peak instantaneous frequency compared to control current injection delivered at the same 50 Hz frequency as synaptic stimulation (n = 8;**p < 0.002) and 30 pulses produce a similar increase in the peak instantaneous frequency (n = 13;***p < 0.0001) current injections. There were no differences comparing 5 and 30 pulses (p > 0.2) (d) Representative traces showing the selective mGluR5 antagonist significantly reduces the dAPD triggered by synaptic stimulation of the apical dendritic tree in deep layer 5/6 pyramidal neurons. The inset shows the same synaptic stimulation of action potentials before and after MPEP. (e) Shows the average dADP block by MPEP (30 µM;n = 5;**p < 0.01).
The dopamine D1 receptor agonist SKF81297 produces an inverted U-shaped dose response relationship to DHPG (50 µM)-induced dADP (n =4 for 0.5 µM, n = 5 for 1µM, n=28 for 10 µM and n=5 for 50 µM;Black Circles). The D1R antagonist, SCH 23390 (5 µM, n = 4; p > 0.05, Open Diamond) blocks the inhibitory effects of SKF81297 at the maximally effective dose (10 µM). Low TTX (15 nM) does not alter the dADP and does not diminish the inhibitory effect of SKF 10µM (TTX+SKF) on the dADP (n = 6). **p < 0.01,*p < 0.05
(a) Forskolin reduces the mGluR5-mediated postsynaptic responses. Representative traces showing the post synaptic reduction of the synaptically triggered dADP by internal Forskolin. Black traces show the dAPD after initial break-in to whole-cell configuration and Red traces show 20 minutes after Forskolin perfusion in the internal pipette solution. Insets show no changes in the synaptic stimulation and action potentials before and after Forskolin (average data shown in Figure 5). (b) To test whether mGluR5 function can be modulated by PKA, HEK293 cells stably expressing mGluR5 were seeded in wells of a 96-well plate for 20 hrs before being washed and loaded with 5 µM Fluo4-AM in an extracellular solution as described previously.84 In order to suppress the constitutive activity of the receptor, the cell culture medium contained mGluR5 antagonist, 2-methyl-6 [phenylethynyl]-pyridine (MPEP, 1 µM). Fluo-4 fluorescence change was read using a fluorescence plate reader, FlexStation (Molecular Devices). Cells were untreated (Control; n = 12) or treated with Forskolin (10µM; n = 24;***p<0.001) for 5 min before DHPG was added. RFU: random fluorescence unit.
Locomotor response to cocaine in mGluR5 deficient mice (mGluR5 Mut) compared to wild type (mGluR5 WT) controls. Cocaine dose dependently increased locomotor activity producing a significant difference (**p < 0.01) between the Mut (n = 7) and WT (n = 8) mice at the highest dose (40 mg/kg) in a 1 hr test given after 1 hr habituation.