

Abstract
ErbB2 (or Her2/Neu) overexpression in breast cancer signifies poorer prognosis, yet it has provided an avenue for targeted therapy as demonstrated by the success of humanized monoclonal antibody Trastuzumab (Herceptin™). Resistance to Trastuzumab and eventual failure in most cases, however, necessitate alternate ErbB2-targeted therapies. HSP90 inhibitors such as 17-allylaminodemethoxygeldanamycin (17-AAG), potently downregulate the cell surface ErbB2. While the precise mechanisms of Trastuzumab or 17-AAG action remain unclear, ubiquitinylation-dependent proteasomal or lysosomal degradation of ErbB2 appears to play a substantial role. As Trastuzumab and 17-AAG induce the recruitment of distinct E3 ubiquitin ligases, Cbl and CHIP respectively, to ErbB2, we hypothesized that 17-AAG and Trastuzumab combination could induce a higher level of ubiquitinylation and downregulation of ErbB2 as compared to single drug treatments. We present biochemical and cell biological evidence that combined 17-AAG and Trastuzumab treatment of ErbB2-overexpressing breast cancer cell lines leads to enhanced ubiquitinylation, downregulation from the cell surface and lysosomal degradation of ErbB2. Importantly, combined 17-AAG and Trastuzumab treatment induced synergistic growth arrest and cell death specifically in ErbB2-overexpressing but not in ErbB2-low breast cancer cells. Our results suggest the 17-AAG and Trastuzumab combination as a mechanism-based combinatorial targeted therapy for ErbB2-overexpressing breast cancer patients.
Keywords: ErbB2, 17-AAG, Trastuzumab, synergy, ubiquitin ligase
Introduction
Overexpression of ErbB2 (Her2/Neu), a member of the Epidermal Growth Factor Receptor (EGFR) family, is involved in the pathogenesis of nearly 20-30% of invasive breast cancers and is associated with poor prognosis. ErbB2 therefore represents a therapeutic target and a humanized anti-ErbB2 monoclonal antibody, Trastuzumab (Herceptin™, Genentech, San Francisco, CA), is now an essential part of treatment of ErbB2-overexpressing breast cancers. Trastuzumab is currently administered with other chemotherapeutics, like microtubule stabilizing agents (Docetaxel, Paclitaxel), DNA binding drugs (Doxorubicin, Epirubicin, Cisplatin) or alkylating agents (Cyclophosphamide). However, clinical data indicate a less than satisfactory response rate in patients1 as well as a somewhat increased risk of cardiac toxicity when Trastuzumab is used with doxorubicin2, 3. Importantly, most patients that do respond initially eventually develop Trastuzumab resistance1, 4. The molecular factors that impart Trastuzumab resistance remain unknown, in part because the mechanism of Trastuzumab action itself is unclear. Current models suggest that one or more of the following mechanisms mediates Trastuzumab action: (a) induction of antibody-dependent cellular cytotoxicity (ADCC)5-10, (b) a block in ErbB2-signaling via the PI3K-Akt pathway and activation of PTEN11, 12, and (c) enhancement of the endocytic downregulation of ErbB2 via the lysosomal/proteasomal pathways1, 13. Recent reports have suggested that ADCC is the predominant mechanism of action5-10. While Trastuzumab action may be in part due to its ability to activate immune responses, it does not explain why some patients are intrinsically resistant while others eventually develop resistance. Recent studies have also identified the tumor suppressor PTEN as a factor imparting Trastuzumab resistance12, 14, 15. Furthermore, the ability of various chemotherapeutics (taxane-, anthracycline- or platinum-based) to synergize with Trastuzumab cannot be explained by ADCC. These observations suggest additional mechanism of Trastuzumab and its ability to disrupt ErbB2 signaling networks.
Earlier work by Klapper et al.16 (and this study) strongly suggest a role for the endocytic pathway in ErbB2 downregulation, a mechanism similar to EGF-induced downregulation of EGFR (ErbB1)17-19. In this pathway, receptor ubiquitinylation plays a crucial role in regulating the traffic of endocytosed receptor to the lysosomes and subsequent degradation17-19. EGF induces rapid ubiquitinylation and lysosomal trafficking of EGFR. However ErbB2 ubiquitinylation and lysosomal trafficking is far less efficient than that of EGFR20-22. A potential reason for this inefficiency is that mature ErbB2, unlike EGFR, exists in association with the HSP90 molecular chaperone complex23-26, which may reduce the efficiency of ubiquitinylation through steric hindrance or reduced interaction with E3 ubiquitin ligases. Indeed, inhibition of HSP90 function using ansamycin antibiotics geldanamycin or 17-allylaminodemethoxygeldanamycin (17-AAG) induces a chaperone complex rearrangement leading to the dissociation of HSP90 and the association of HSP70 which is followed by rapid ubiquitin-dependent degradation of ErbB226, 27. Phase I clinical trials of 17-AAG have recently been completed28-32 and this agent has entered phase II clinical trials as a potential anti-cancer agent (www.clinicaltrials.gov).
Klapper et al16 have previously demonstrated a role for Cbl E3 ligase in ErbB2 ubiquitinylation and degradation and the tumor-inhibitory activity of a monoclonal anti-ErbB2 antibody16. By inference, Cbl is thought to mediate Trastuzumab-induced ErbB2 ubiquitinylation, and Trastuzumab treatment of ErbB2-overexpressing breast cancer cells induces a Cbl-binding site on ErbB226 but whether Cbl indeed plays a role in Trastuzumab-induced ErbB2 ubiquitinylation and downregulation is unknown. 17-AAG-induced ErbB2 ubiquitinylation, on the other hand, has been linked to chaperone assisted recruitment of an E3 ligase CHIP (C-terminus of Hsc70 Interacting Protein)26, 27. 17-AAG-treatment induces a rearrangement of the chaperone complex with dissociation of ErbB2-bound HSP90 and recruitment of Hsc70-CHIP complex26, 27. As 17-AAG-induced ErbB2 ubiquitinylation and degradation does not require a Cbl-binding site on ErbB226, it is clear that treatment with 17-AAG or Trastuzumab results in the recruitment of distinct E3s to ErbB2. We therefore hypothesized that a combination of 17-AAG and Trastuzumab could recruit both Cbl and CHIP E3 ubiquitin-ligases to ErbB2, resulting in its enhanced ubiquitinylation and degradation. Accordingly we proposed that a combination of Trastuzumab and 17-AAG could have additive/synergistic effects by virtue of the enhanced ErbB2 ubiquitinylation and lysosomal degradation mediated by the combined action of Cbl and CHIP E3-ligases (see model presented in Fig. 7). We present findings that combination of 17-AAG and Trastuzumab allows effective ErbB2 ubiquitinylation and lysosomal degradation at substantially lower concentrations than either drug alone; importantly, the combination of these drugs is also exhibited pharmacological synergy in inducing proliferative arrest and apoptosis selectively in ErbB2-overexpressing breast cancer cell lines. Our findings suggest that drug-induced combinatorial recruitment of E3s is a potential mechanism to enhance the efficacy of ErbB2-targeted therapeutics and provide rationale for further development of HSP90 inhibitor and Trastuzumab combination therapy against ErbB2-overexpressing breast cancers.
Fig. 7. Schematic representation of the hypothesis of this study.

The thickness of arrows represents relative dominance of recycling versus lysosomal targeting, with stippled arrows being the least dominant route. A. Normal, predominantly recycling (solid arrow) route of ErbB2 traffic activated via heregulin (HRG), which binds to ErbB3/4 and activates ErbB2 through hetero-dimerization. B. Anti-ErbB2 antibody-induced ErbB2 down-regulation via Cbl recruitment and increased lysosomal targeting (and reduced recycling). C. Our model of how 17-AAG binding to HSP90 recruits CHIP, promoting ErbB2 down-regulation by lysosomal targeting. D. Our hypothesis that combining Cbl (in response to anti-ErbB2) and CHIP (in response to 17-AAG) ubiquitinylation pathways will lead to synergistic down-regulation of ErbB2 by switching to predominantly lysosomal fate (solid arrow) instead of recycling (stippled arrow).
Materials and Methods
Cell Culture
The following ErbB2-overexpressing breast cancer cell lines were used in this study: SKBr-3, BT-474 and 21MT-1 (derived from tumors overexpressing ErbB2) and MCF-7-ErbB2 (MCF-7 cells retrovirally transduced with ErbB2 using pMSCV-puro-ErbB233). The ErbB2-low cell lines included the breast cancer cell lines MCF-7 and MDA-MB-231 and immortal non-tumorigenic cell line MCF-10A. BT-474, SKBr-3, MCF-7 and MDA-MB-231 cell lines were maintained in α-MEM medium supplemented with 5% fetal bovine serum (FBS), 1 % L-glutamine, 1% Penicillin-Streptomycin, 20 mM HEPES, Non-essential amino acids and sodium-pyruvate (complete α-MEM; all components from Invitrogen Carlsbad, CA, U.S.A). 21MT-1 cells were maintained in complete α-MEM, further supplemented with 12.5 ng/ml epidermal growth factor (EGF) and 1 μg/ml hydrocortisone (Sigma). MCF-10A cells were grown in DFCI-1 medium34.
Antibodies and other reagents
The primary antibodies (Ab) used for this study and the conditions of their use are described in Supplementary Table 1. HRP-conjugated secondary antibodies or Protein-A as well as fluorescently-labeled secondary antibodies were from Invitrogen Corporation (Carlsbad, CA, U.S.A). Trastuzumab (Herceptin™) was obtained through Evanston Northwestern Healthcare pharmacy while 17-AAG was provided by the Drug Synthesis and Chemistry Branch, Division of Cancer Treatment and Diagnosis of the NCI, or was purchased from Biomol International (Plymouth, PA, U.S.A.). Bafilomycin A was from EMD Biosciences (San Diego, CA, U.S.A). Annexin V staining kit was from Invitrogen (Carlsbad, CA, U.S.A.).
Flow cytometry and confocal immunofluorescence microscopy (CIM)
Flow cytometry analysis of cell-surface ErbB2 was performed on live cells (without fixation and permeabilization). Following drug-treatments, cells were trypsinized, washed in ice-cold fluorescence-activated cell sorter (FACS) buffer (2 % FBS, 2 % BSA in PBS containing 0.02 % sodium azide) and serially stained with primary and secondary antibodies on ice in the same buffer. Stained cells were analyzed using a FACScalibur (San Jose, CA, U.S.A.). For CIM, cells grown on coverslips were treated with the drugs, fixed in 4% paraformaldehyde (PFA), and permeabilized using CIM buffer (10 % FBS, 0.2 % BSA and 0.05 % Saponin in PBS). Cells were stained with primary Ab and Alexafluor™-conjugated secondary Ab diluted in CIM, and mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA) containing DAPI (4′, 6-diamidino-2-phenylindole) to visualize the nuclei. Images were acquired using a Nikon C1 confocal microscope under 100× magnification. Merged pictures were generated using Nikon C1 software.
Immunoprecipitation (IP) and Western-blotting (WB)
IPs and IBs were carried out as described previously 35.
Cytotoxicity Assays and analysis of 17-AAG/Trastuzumab interactions
2000-3000 cells per well were plated in 96-well plates and 8 replicate wells used for each condition. Drug treatments included the following: (1) a serial 2-fold dilution of 17-AAG (200 to 0.19 nM); (2) a serial 2-fold dilution of Trastuzumab (80 to 0.078 μg/ml); and (3) a serial 2-fold dilution of 17-AAG (200 to 0.19 nM) with selected concentrations of Trastuzumab (0.1, 1 or 10 μg/ml). Controls included untreated cells as well as cells treated with DMSO. The cells were treated for 7 days with change of medium (containing the drugs) every other day. At the end of the treatment, cell viability was assessed using an MTT assay kit (Invitrogen, Carlsbad, CA, U.S.A.). Absorbance values with cells at day 0 (before drug treatment) were subtracted from values at day 7, and the corrected values used to calculate the effect as a fraction of untreated controls. All absorbance values were in the linear range of the calibration curve. The data were used to calculate the fraction affected (Fa). Multiple-drug effect analysis was done using the computer software program Calcusyn, from Biosoft (Cambridge, UK) to calculate the median dose (Dm) and Combination Index (CI). C.I. value < 1 indicates drug synergy, = 1 indicates additive effects, whereas a value >1 indicates antagonistic interaction36. Each experiment was repeated at least three times with replicates. To ensure the quality of data, linear correlation coefficient (r) of the median-effect plot was required to be greater than 0.85.
Apoptosis and Cell cycle analysis
Annexin V (Invitrogen, Carlsbad, CA, U.S.A.) staining of drug-treated cells was performed according to the manufacturer's protocol to evaluate drug-induced cell death. Cell cycle arrest was assessed by FACS analysis of PI-stained cells using standard protocols.
Results
Combined treatment of ErbB2-overexpressing breast cancer cell lines with 17-AAG and Trastuzumab induces a more pronounced downregulation of surface ErbB2
While a previous study did not observe any significant difference in the rate of ErbB2 degradation upon combined geldanamycin plus Trastuzumab as compared to 17-AAG alone22, we reasoned that this result was likely due to different kinetics of ErbB2 degradation induced by 17-AAG and Trastuzumab as well as the dose of geldanamycin used. Whereas relatively high concentrations of 17-AAG (0.1-1 μM) induce a nearly complete loss of ErbB2 on ErbB2-overexpressing breast cancer cells within 6-8 h of treatment, Trastuzumab-induced ErbB2 degradation is slow, requiring 48-72 hours (Fig 1A and 1B). Similar to Austin et al22, we observed little effect of combining Trastuzumab (10 μg/ml) with high concentrations of 17-AAG (100 nM) as 17-AAG effect dominated under these conditions (Fig. 1C). We reasoned that any positive effects of the combination were more likely to be seen at lower concentrations of 17-AAG and upon longer treatment times. Indeed, treatment of ErbB2-overexpressing breast cancer cell lines SKBr-3 and 21MT-1 with a low concentration (10 nM) of 17-AAG in combination with Trastuzumab (10 μg/ml) led to a substantially more efficient ErbB2 degradation within 48 h compared to either agent alone (Fig. 1D; compare lanes 6 and 10 with the remaining lanes). The effectiveness of the combined treatment was confirmed by flow cytometry (on live cells). 17-AAG (10 nM) plus Trastuzumab (10 μg/ml) was significantly more effective in causing downregulation of ErbB2 from the cell surface in both 21MT-1 (as shown in Fig. 2A) and SKBr-3 cells (data not shown) as compared to single drug treatments. Finally, confocal immunofluorescence microscopy also verified that 17-AAG and Trastuzumab combination led to significantly lower cell-surface levels of ErbB2 (Fig. 2B; follow changes in localization and intensity of green fluorescence). ErbB2 was seen to accumulate in punctate intracellular structures after longer treatment periods, consistent with ErbB2 endocytosis22, 37, 38. Similar results were obtained in BT-474 cells; these cells are more sensitive to Trastuzumab as well as the combination as ErbB2-downregulation was seen in less than 24 hrs (see Fig. 3D).
Fig. 1. Combination of 17-AAG at low concentrations and Trastuzumab induces more effective ErbB2-degradation.
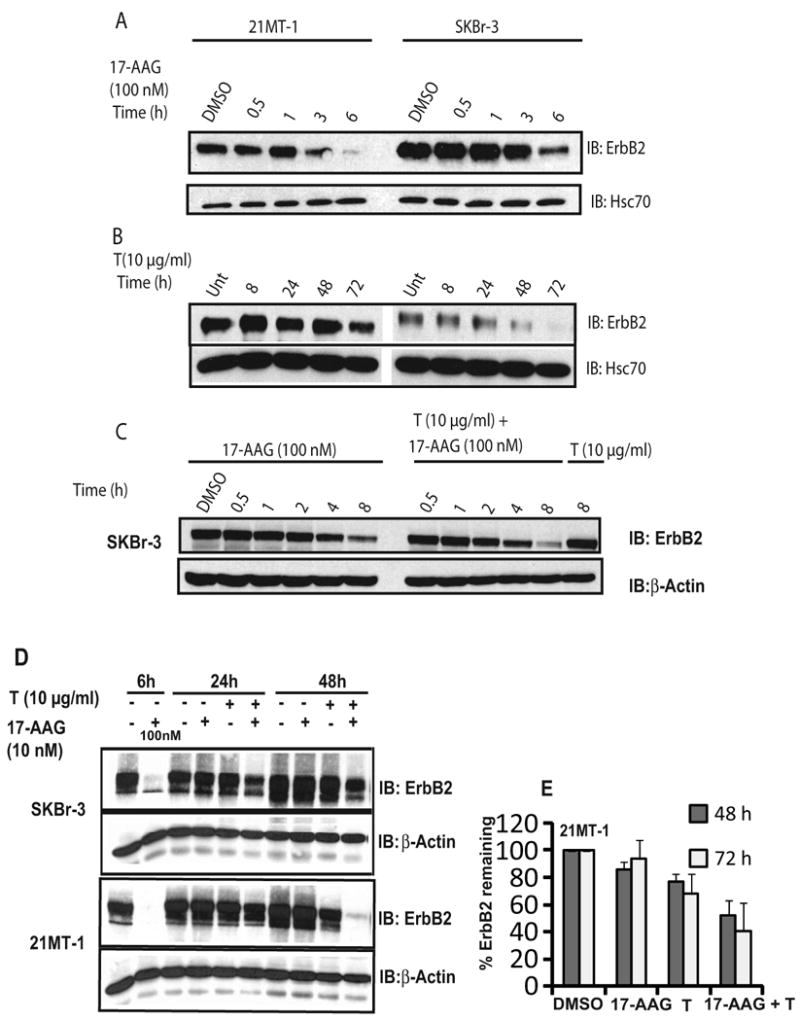
A, B. Differential kinetics of ErbB2-downregulation induced by 17-AAG and Trastuzumab; SKBr-3 and 21MT-1 cells were treated with 100 nM 17-AAG (A) or 10 μg/ml Trastuzumab (B) for the indicated time periods, followed by cell lysis. 50 μg aliquots of the cell lysate were analyzed by SDS-PAGE/WB. Hsc70 served as a loading control. C. Co-operative interactions between Trastuzumab and 17-AAG are not appreciable at high concentrations of 17-AAG; SKBr-3 cells were treated with 17-AAG (100 nM) alone, 17-AAG (100 nM) + T (10 μg/ml), or T (10 μg/ml) for the indicated time periods. Cells were lysed and 25 μg aliquots analyzed by SDS-PAGE/WB and probed for ErbB2 and β-actin (loading control). D. Combination of 17-AAG and Trastuzumab is more effective in degradation of ErbB2; SKBr-3 or 21MT-1 cells were treated with 17-AAG alone (10 nM) or Trastuzumab (T) alone (10 μg/ml) or a combination (10 nM; 10 μg/ml) for 24 or 48h. The cells were also treated with 100 nM 17-AAG for 6h as a comparison of relative potency. Following treatment, the cells were lysed in Triton X-100 containing lysis buffer. 50 μg aliquots of lysate were directly analyzed by SDS-PAGE/Western blotting and probed for ErbB2. An immunoblot for β-actin served as a loading control. E. Quantitation of effect of 17-AAG plus Trastuzumab on ErbB2-degradation in 21MT1 cells analyzed by SDS-PAGE/WB followed by densitometry (± SEM; n = 6)
Fig. 2. 17-AAG plus Trastuzumab combination reduces cell-surface levels of ErbB2.
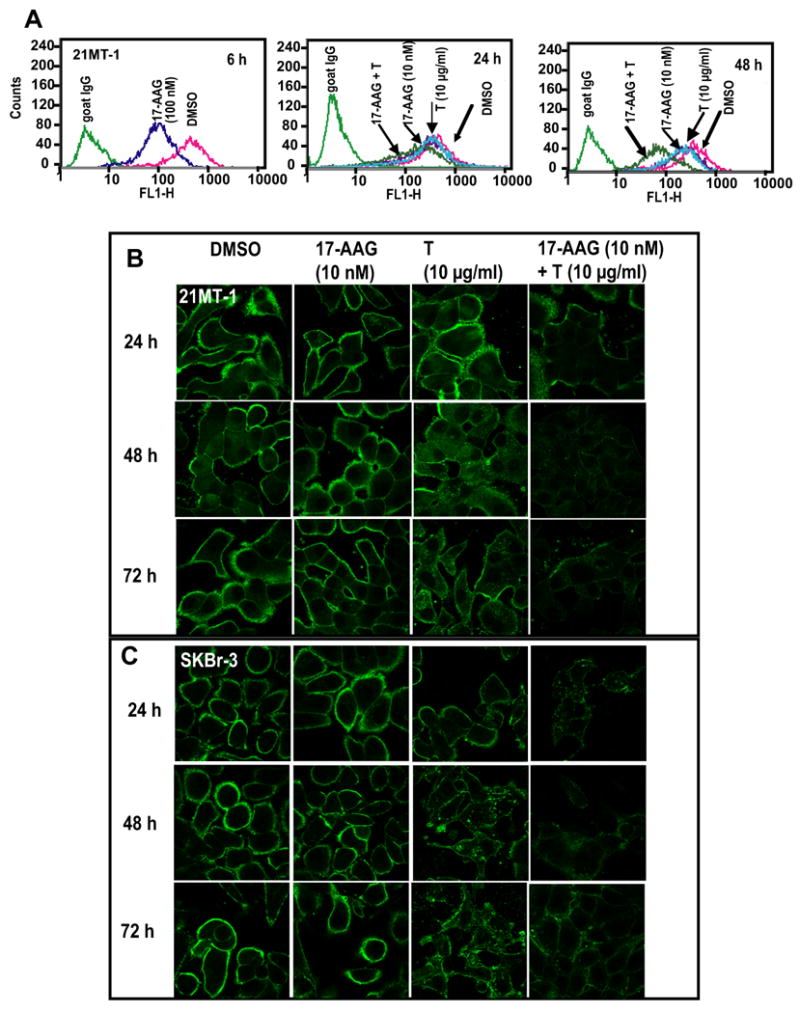
A. 21MT-1 cells grown in 6-well plates were treated with DMSO (crimson), 10 nM 17-AAG (purple), 10 μg/ml T (Cyan) or 17-AAG + T (dark green) for the indicated time periods. A 100 nM 17-AAG-treatment (dark blue) was also included as a control for comparison. Following treatment, the cells were trypsinized, washed in FACS buffer and the live cells stained for ErbB2 as described in the methods section. Cell surface levels were assessed using flow cytometry. B. 21MT-1 or SKBr-3 cells grown on glass coverslips, were treated with the indicated concentrations of either 17-AAG alone, Trastuzumab alone or the combination for the indicated time periods. Following treatment, the coverslips were fixed in 4% paraformaldehyde (PFA) and immunostained for ErbB2 (Green) as detailed in the methods section. Shown here are the Confocal Immunofluorescence microscopy images.
Fig. 3. Combinatorial treatment with 17-AAG and Trastuzumab induces enhanced ubiquitinylation and lysosomal degradation of ErbB2.
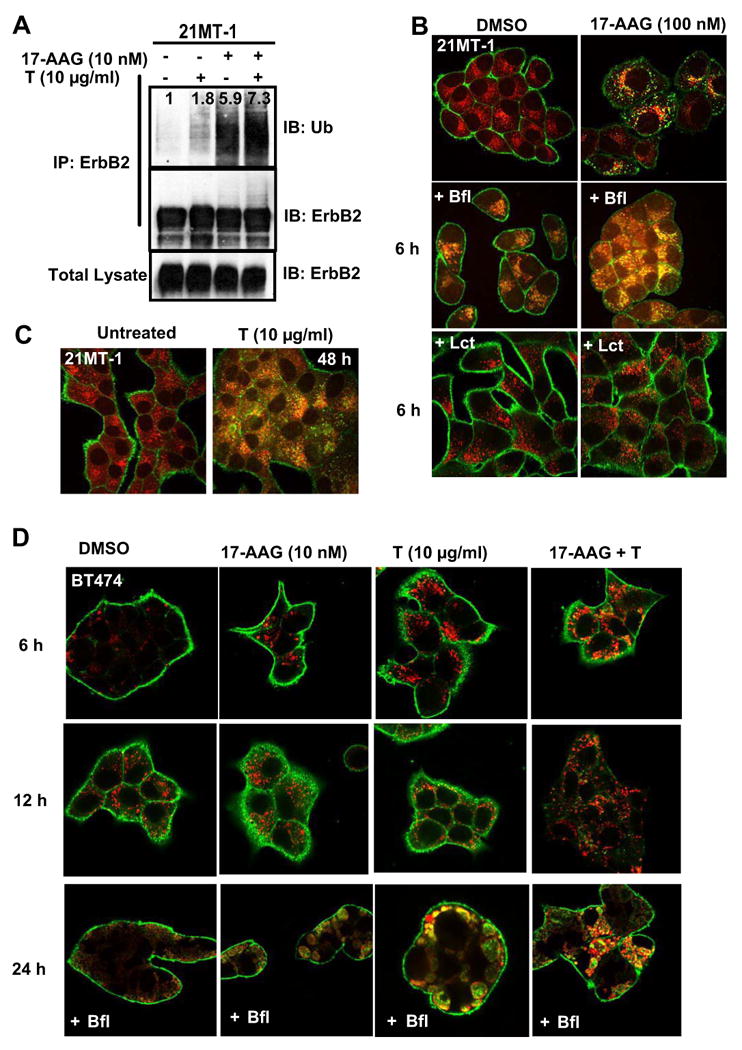
A. 21MT-1 cells were treated with the indicated concentrations of 17-AAG or Trastuzumab or the combination for 6h. Following treatment, the cells lysed and subjected to anti-ErbB2 IP according to previously described method (20). After SDS-PAGE/WB the membrane filter was probed for ubiquitin. The membrane filter was stripped and probed for ErbB2. 50 μg aliquots of total lysate were also similarly analyzed. B. 17-AAG induces ErbB2 accumulation in the lysosome; 21MT-1 cells grown on coverslips were left untreated (top row) or pretreated with 100 nM of the lysosomal inhibitor bafilomycin A1 (middle row) or 10 μM proteasomal inhibitor, lactacystin (bottom row) for 1 h. The cells were then either untreated (left column) or treated with 100 nM 17-AAG (right column). Following treatment, the coverslips were fixed in 4% paraformaldehyde (PFA) and immunostained for ErbB2 (green) and LAMP-1 (red) as detailed in the methods section. Shown here are the Confocal Immunofluorescence microscopy images. C. Trastuzumab-treatment also induces lysosomal localization of ErbB2; 21MT-1 cells, grown on coverslips, were untreated or treated with 10 μg/ml Trastuzumab for 48 h, before fixation and immunostained for ErbB2 (green) and LAMP-1 (red). D. BT-474 cells, grown on coverslips were treated with the indicated concentrations of single drugs or combination for the indicated time periods, with or without Bafilomycin (10 nM). The coverslips, after treatment, were processed as described in the legend for Fig. 3A. Shown here is ErbB2 staining in green and LAMP-1 in red; colocalized regions are seen in yellow.
Combination of 17-AAG and Trastuzumab induces enhanced ubiquitinylation and lysosomal degradation of ErbB2
In order to further establish whether the mechanistic basis for enhanced efficiency of 17-AAG plus Trastuzumab combination towards ErbB2-degradation lies in its ability to enhance ErbB2-ubiquitinylation, we compared the extent of ubiquitinylation induced by 17-AAG, Trastuzumab, and their combination at same concentrations as those used for ErbB2 degradation studies described in Fig. 2D. 21MT-1 cells were pre-treated with proteasome inhibitor lactacystin (10 μM) to minimize any degradation-induced loss of ubiquitin signal. The cells were then treated with the indicated drug concentrations (see Fig. 3A and legend) for 6h followed by immunoprecipitation (IP) of ErbB2 and Western blotting (WB) for ubiquitin. As seen in Fig. 3A (lane 4), ErbB2 ubiquitinylation was appreciably higher in lanes representing combined treatment with 17-AAG plus Trastuzumab.
Ubiquitinylation of proteins is linked to their degradation via the proteasome or the lysosome. While soluble proteins are typically degraded in the proteasome, membrane-proteins, such as RTKs, primarily undergo lysosomal degradation following endocytosis. Receptor ubiquitinylation has become established as a critical signal for sorting of endocytosed receptors from early endosome through multi-vesicular bodies to the lysosome17-19. The mechanism of 17-AAG-induced ErbB2 degradation (lysosomal versus proteasomal) remains controversial. While proteasomal degradation has been implicated, based on the ability of proteasomal inhibitors to block ErbB2-degradation39, recent studies by Lerdrup et al.37 have suggested that proteasomal activity is required for endocytic internalization of ErbB2 and that proteasomal inhibitors may instead block ErbB2 degradation by inhibiting receptor endocytosis. Similar to studies by Lerdrup et al., we observed that following 17-AAG treatment ErbB2 colocalizes with a late endosomal/lysosomal marker, LAMP-1 (stained in red) (Fig, 3B; increase in yellow staining). Inhibition of lysosomal function using bafilomycin A1 further increased the accumulation of ErbB2 in LAMP-1 positive compartments. In contrast, pretreatment with a proteasome inhibitor lactacystin prevented loss of ErbB2 from the cell surface. Notably, Trastuzumab treatment also induced the ErbB2 endocytosis and colocalization with LAMP-1, confirming that receptor degradation via the lysosomal pathway is indeed one of the mechanisms of Trastuzumab action (Fig. 3C).
Since the two drugs (17-AAG and Trastuzumab) individually mediated ErbB2 degradation in the lysosome, we next sought to establish if the combination also leads to ErbB2 degradation in the lysosome. We chose BT-474 cell line for these analyses as its higher sensitivity to drugs being tested made it more amenable for ErbB2-LAMP-1 colocalization in presence of bafilomycin A1. To reduce bafilomycin toxicity during extended periods of observation, we reduced the concentration of bafilomycin A1 to 10 nM (from 100 nM used for shorter times in Fig. 3B). As shown in Fig 3D (top and middle panel), the drug combination induced marked ErbB2 degradation within 24h. Colocalization studies demonstrated that 17-AAG increased the localization of ErbB2 (green) in late endosome/lysosome (LAMP-1-positive, red), resulting in enhancement of regions of yellow/orange staining. This was further confirmed by comparing ErbB2-LAMP-1 colocalization in presence of bafilomycin-A1, (Fig. 3D; bottom panel).
17-AAG plus Trastuzumab combination synergistically induces selective killing of ErbB2-overexpressing breast cancer cells
Having established that a combination of low concentration of 17-AAG with Trastuzumab induced more effective ErbB2 ubiquitinylation and lysosomal-degradation, we wished to assess the biological effects of their combination on proliferation of ErbB2-overexpressing breast cancer cell lines. These included naturally ErbB2-overexpressing breast cancer lines SKBr-3, BT-474 and 21MT-1, as well as an engineered line MCF7-ErbB2 derived by stable retroviral transduction of ErbB2 into ErbB2-low MCF7 cell line. These were compared with three ErbB2-low cell lines MCF-7, MDA-MB-231 (breast cancer cell lines) and MCF-10A (non-tumorigenic immortal mammary epithelial line). The extent of cell proliferation following a 7 day-treatment was assessed using MTT-assay. Dose response curves were generated for cells treated with Trastuzumab alone (see Supplementary data Fig. 1), 17-AAG alone or various combinations of Trastuzumab and 17-AAG (as described in Materials and Methods section). The results are presented in Fig. 4 (top row; ErbB2-overexpressing lines BT-474, SKBr-3 and 21MT1) and Fig. 5A (showing the effect on ErbB2-low cell lines, MCF-7, MDA-MB-231 and MCF-10A; MCF-7-ErbB2 is included as a control).
Fig. 4. Synergistic interactions of 17-AAG and Trastuzumab combination in cytotoxicity against ErbB2-overexpressing breast cancer cells.

Top row: Comparison of effect of single drug treatments or combination (at select concentrations of 17-AAG from the dose response curve) on BT-474, SKBr-3 and 21MT-1, showing efficacy of combinatorial treatment over single drugs. The Trastuzumab (T) concentrations were fixed at 0.1 μg/ml (for BT-474 cells) or 10 μg/ml for SKBr-3 and 21MT-1. Middle row: Median-Effect plots for 17-AAG and Trastuzumab combination. The arrows at the x-intercept indicate the median dose of 17-AAG. Note the shift towards the left for the combination as compared to 17-AAG alone. Bottom row: Combination index (CI) values for various fractional effects at varying concentrations of 17-AAG. CI < 1 indicates synergy; CI ∼ 1 indicates additive effects; CI > 1 indicates antagonism.
Fig. 5. 17-AAG and Trastuzumab combination synergistically induces proliferative arrest selectively in ErbB2-overexpressing breast cancer cells.
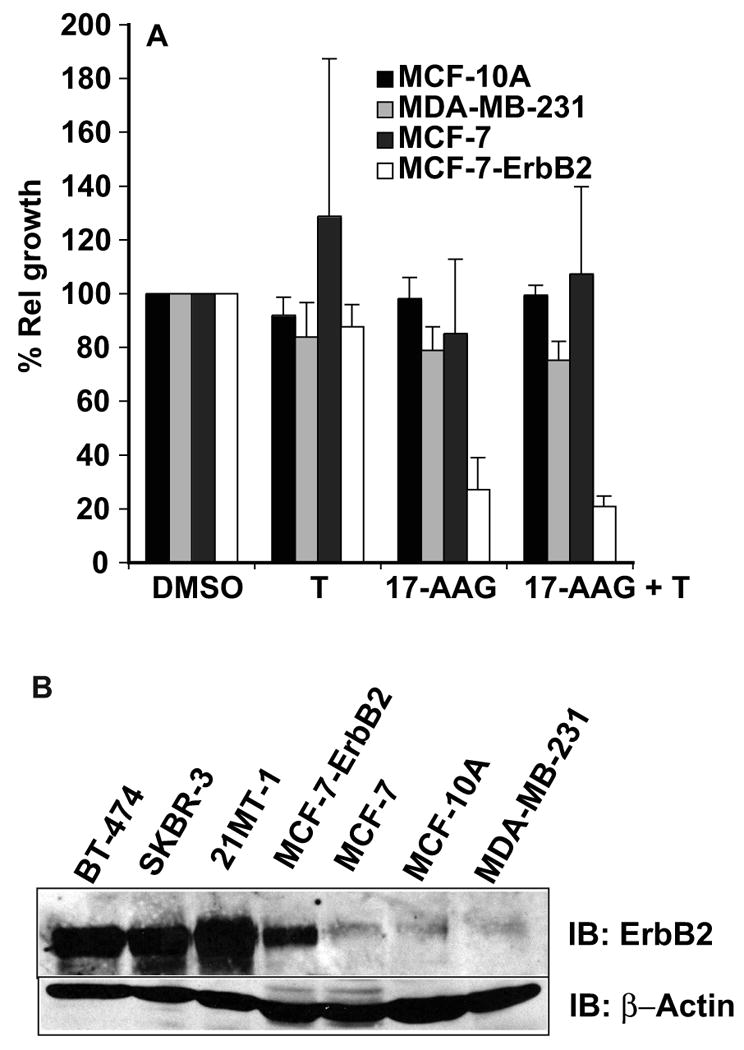
A. ErbB2-low cells including MCF-10A, MDA-MB-231 and MCF-7 cells were treated with 17-AAG alone (20 nM) or Trastuzumab (10 μg/ml) alone or combination at the indicated concentrations and the relative growth assessed by MTT assay. For comparison, MCF-7-ErbB2 (retrovirally transduced) was also used. B. Comparison of ErbB2 levels in the cell lines used for studies described above; 100 μg aliquots of cell lysates from BT-474, SKBr-3, 21MT-1, MCF-ErbB2, MCF-7, MCF-10A, and MDA-MB-231 were analyzed by SDS-PAGE/WB.
As shown in Fig. 4 (top row), treatment with 17-AAG plus Trastuzumab more effectively inhibited the proliferation of three distinct ErbB2 over-expressing cell lines (SKBr-3, BT-474, 21MT-1). Median Dose (Dm) values (the concentration of the drug, 17-AAG, required to inhibit proliferation by 50%; shown in Fig. 4 middle row) were calculated from the median-effect analysis36. Analysis of drug interaction by computing the Dm values showed that 17-AAG and Trastuzumab combination was clearly more effective than single drug treatments (Fig. 4 middle row) with about 160, 20 or 11-fold reduction in the Dm values of 17-AAG for BT-474, SKBr-3 and 21MT-1, respectively, in presence of Trastuzumab as compared to 17-AAG alone. BT-474 cells were more sensitive to Trastuzumab alone treatment and therefore synergistic interactions of Trastuzumab with 17-AAG could be appreciated only at the low concentrations Trastuzumab (<0.1 μg/ml). Further analysis of the combination index (C.I.) using Chou-Talalay analysis36, as shown in Fig. 4 (bottom row), confirmed that the drug interactions using each of the three cell lines were synergistic (C.I. < 1). In comparison to ErbB2-overespressing lines, the proliferation of ErbB2-low cell lines was unaffected by either single drugs or the combination (see Fig. 5A). Retroviral overexpression of ErbB2 into the MCF-7 cell line imparted the drug sensitivity (Fig. 5A). The levels of ErbB2-expression in all the cell lines used under the conditions are as shown in Fig. 5B.
We also analyzed colony forming ability (Crystal violet staining), cell cycle distribution (PI-staining) and proportion of cells undergoing apoptosis (using annexin V assay) to complement our MTT assay-based analyses of the effectiveness of combined treatment with 17-AAG plus Trastuzumab. Crystal violet staining clearly showed markedly reduced number of colonies in wells treated with the combination as compared to single drugs (See Fig. 6A; compare columns 2 and 3 containing treatments at 17-AAG at 5 and 10 nM alone, Trastuzumab alone or combination; column 1 shows control treatments). Cell cycle analysis of SKBr-3 (Fig. 6B) and BT-474 (data not shown) cell lines treated with 17-AAG or Trastuzumab alone or 17-AAG plus Trastuzumab confirmed that the combination induces comparatively more G1 cell cycle arrest. Furthermore, 17-AAG plus Trastuzumab resulted in a significantly higher percentage of annexin V-positive cells (See Fig. 6C). SDS-PAGE analysis of lysates of BT-474 cells treated with single drugs or combination clearly showed that the combination was more efficient at attenuating pro-survival signaling outputs downstream of ErbB2, including phospho-AKT, phospho-mTOR, phospho-p70S6K, cyclin D1 and survivin (See Fig. 6D). Downregulation of cyclin D1 is consistent with published reports on molecular events leading to 17-AAG and Trastuzumab-induced G1 arrest. Thus, targeting ErbB2 and HSP90 by a combination of 17-AAG and Trastuzumab results in enhanced ubiquitinylation and lysosomal degradation of ErbB2 and effectively disengages the critical proliferation and survival signaling networks in ErbB2-overexpressing breast cancer cells.
Fig. 6. 17-AAG plus Trastuzumab is more efficient in inducing cell cycle arrest and apoptosis.
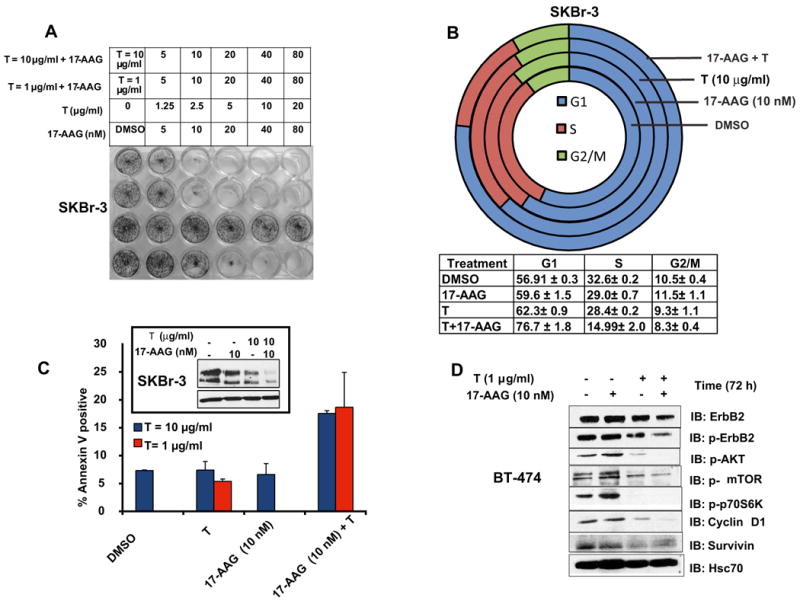
A. SKBr-3 cells grown for 7-days in 24-well plates (in duplicates) were treated with the indicated concentrations of single drugs or combinations. At the end of the drug-treatment, the cells were fixed and stained with 0.25% Crystal Violet dye in 20 % methanol in water. Shown here is a representative plate. B. Combination of 17-AAG and Trastuzumab induce G1-arrest in ErbB2-overexpressing cells; SKBr-3 cells grown in P100 plates were treated (in triplicates) as indicated for 2 days, were subjected to cell cycle analysis. Shown here are representative cell cycle analysis profiles for each condition. The % of cells in each phase (± S.D.) is also shown. The combination induces a significantly more effective G1-arrest as compared to 17-AAG alone (p < 0.01) or Trastuzmab (p < 0.05). C. Annexin V assay of drug treated SKBr-3 cells; SKBr-3 cells, grown in P100 plates were treated (in triplicates) with DMSO or 17-AAG (10 nM), Trastuzumab (1 or 10 μg/ml) or 17-AAG (10 nM) plus Trastuzumab (1 or 10 μg/ml) for 5 days. Following drug-treatment, cells were trypsinized and stained for Annexin V according to the manufacturer's protocol to assess, dead/dying cells. Part of the sample was used for SDS-PAGE analysis of the ErbB2-levels at the end of the experiment. The % of Annexin V positive cells ± S.D is shown here. The inset shows the ErbB2-levels at the end of the treatment. D. Effect of single or combinatorial treatment of ErbB2-overexpressing breast cancer cells on signaling downstream of ErbB2; BT-474 cells were treated with the indicated concentrations of single drugs or the combination for 3 days. 100 μg aliquots of protein from cell lysates were analyzed by SDS-PAGE/WB for the pro-survival signals, p-AKT, p-mTOR and p-P70S6K, cyclin D1 and survivin.
Discussion
Current targeted therapy trials of ErbB2-overexpressing breast cancer patients involves combination of Trastuzumab with non-targeted chemotherapy drugs that interfere with DNA synthesis, such as doxorubicin, cisplatin, cyclophosphamide, 5-fluorouracil and gemcitabane, or that interfere with microtubule function, such as docetaxel (http://www.ClinicalTrials.gov). These combinations are based on the rationale that chemotherapeutic drugs are already clinically approved as well as empirical observations using in vitro cell culture and pre-clinical mouse models rather than a clear mechanistic link between the targets of these drugs and that of Trastuzumab. On the other hand, we have used a rational combination of Trastuzumab and the HSP90 inhibitor 17-AAG that would be expected to bring about the concerted action of distinct E3 ubiquitin ligases, Cbl and CHIP respectively, to enhance the ubiquitinylation of ErbB2 and its subsequent degradation via the lysosomal pathway. The ability of these drugs to act together at a more proximal step (concurrent recruitment of two distinct E3 ligases to ErbB2 by both drugs) within the same pathway rather than in disparate pathways (such as ErbB2-receptor downregulation by Trastuzumab and DNA-damage or inhibition of DNA synthesis by chemotherapeutic agents) was anticipated to produce potentially additive or synergistic effects. The studies reported here, support our hypothesis that combination of 17-AAG and Trastuzumab presumably by bringing about the concerted action of two independent E3 ligases, Cbl and CHIP, can indeed lead to enhanced ErbB2-ubiquitinylation (model presented in Fig. 7).
Biochemical and cell biological analyses clearly demonstrate that enhanced ubiquitinylation of ErbB2 upon treatment of ErbB2-overexpressing breast cancer cells with 17-AAG and Trastuzumab combination leads to lysosomal degradation of ErbB2. Importantly, more pronounced depletion of ErbB2 upon combined treatment with 17-AAG and Trastuzumab results in better attenuation of proliferation- and survival-associated signaling pathways. These biochemical effects are associated with more pronounced proliferative arrest and cell death with a combination of 17-AAG and Trastuzumab as compared to individual drugs, consistent with their dependence on ErbB2-driven survival signaling. While the effects of the two drugs at the proximal step of ErbB2-ubiquitinylation and degradation appear to be additive at most, the final biological outcome (antiproliferative effects, which are several steps downstream of ErbB2-degradation) of combined vs. the individual drug treatments demonstrated a synergistic interaction between 17-AAG and Trastuzumab. This interaction was seen with ErbB2-overexpressing but not in ErbB2-low breast cancer cell lines, indicating that these drugs work together in an ErbB2-targeted manner.
While our biochemical, cell biological and functional analyses together provide strong support for the idea that increased depletion of ErbB2 upon combined 17-AAG and Trastuzumab treatment of ErbB2-overexpressing cells contributes to synergistic biological interaction of these drugs, it is likely that effects of 17-AAG at nodal points in ErbB2 signaling cascade also contribute. For example, while depletion of p-AKT (Fig. 6d) could be a result of more pronounced loss of active ErbB2, AKT itself is a HSP90 client protein40 and therefore 17-AAG is likely to also target it directly. Additional proteins involved in ErbB2 signaling, such as c-RAF-1, c-Src and HIF-1α, are also HSP90 clients and therefore potential direct targets of 17-AAG. In this context, targeting of c-Src may play a particularly important role since Trastuzumab has been shown to inhibit c-Src and thereby reduce c-Src-dependent inhibitory phosphorylation of PTEN; indeed, PTEN levels and activity correlate with Trastuzumab sensitivity1, 12, 41. Combined inhibitory effects of Trastuzumab and 17-AAG on c-Src could therefore contribute to enhanced downregulation of the PI3-kinase axis and reduce survival signaling via AKT. Notably, c-Src activity has also been suggested to promote the degradation of c-Cbl via its phosphorylation, ubiquitinylation and proteasomal degradation42. By implication, enhanced inhibition of c-Src could facilitate ErbB2 downregulation upon combined 17-AAG and Trastuzumab treatment by preventing the loss of Cbl.
Our observation that relatively low concentrations of 17-AAG can synergize with Trastuzumab to enhance the ErbB2 ubiquitinylation and its lysosomal degradation are consistent with recent studies suggesting that 17-AAG-induced ErbB2 degradation takes place in the lysosome37, 38. In this regard, the previous observations that proteasome inhibitors prevent ErbB2 degradation in response to 17-AAG appear to reflect the role of the proteasome in facilitating a cleavage event that makes ErbB2 capable of lysosomal traffic as opposed to the predominant recycling behavior of intact ErbB238. Further studies to understand the molecular determinants of ErbB2 resistance to lysosomal trafficking as well as the purported role of CHIP (for 17-AAG) and Cbl (for Trastuzumab) ubiquitin ligases is likely to identify additional targets to refine ErbB2-targeted therapy. Future analyses using Cbl and CHIP knockdown as well as knockout cells should help determine the relative contributions of Cbl and CHIP towards ErbB2 ubiquitinylation by 17-AAG plus Trastuzumab combination, as well as the requirement of ubiquitin-dependent ErbB2 degradation towards biological synergism seen with combined drug treatment. In this context, the role of selective association of HSP90 with ErbB2 (as opposed to EGFR, for example) to inhibit lysosomal trafficking of ErbB2 also requires further study.
In conclusion we demonstrate that 17-AAG plus Trastuzumab combination can more potently downregulate ErbB2 compared to individual agents, and can induce synergistic anti-proliferative and apoptotic affects selectively in ErbB2-overexpressing breast cancer cells. Further preclinical and clinical studies should help determine if the mechanism-based synergistic interaction between Trastuzumab and 17-AAG could be used to treat ErbB2-overexpressing breast cancer patients, in particular those that have developed resistance to currently used combination of chemotherapeutic agents and Trastuzumab. In fact, Modi et al. 28 have recently reported both pre-clinical (using BT474 xenografts in mice) as well as a phase I dose escalation study using a combination of 17-AAG (Tanespimycin or KOS-953 from Kosan Biosciences, Hayward, CA) with Trastuzumab. The combination not only showed effective reduction of tumor in xenograft models but also was found to be safe in patients with Trastuzumab-refractory Her-2 positive breast cancer28, indicating the feasibility of the combination and extension into phase II studies
Supplementary Material
Supplementary Information:
Fig. S1: Trastuzumab dose-response curves against ErbB2-overexpressing breast cancer cells: The proliferation of ErbB2-overexpressing cells (SKBr-3, BT-474 and 21MT-1) treated with varying doses of Trastuzumab (as described in the methods section) was assessed using MTT-assay. Shown here are the relative growths of the cells as a function of Trastuzumab concentrations.
Supplementary Table 1: List of antibodies and their concentrations used in this study.
Acknowledgments
This work was supported by: the NIH Grants CA 116552, 99900, CA99163, CA 87986 and CA76118 to HB, and CA94143, CA96844 and CA81076 to VB; DOD Breast Cancer Research Grants DAMD17-02-1-0303 (HB), DAMD17-02-1-0508 (VB), W81XWH-05-1-0231(VB) and W81XWH-07-1-0351 (VB); the NCI Center for Cancer Nanotechnology Excellence Grant NCI 1U54 CA119341-01 (HB and VB); Avon Breast Cancer Fund, Northwestern University; and the Jean Ruggles-Romoser Chair of Cancer Research (HB) and the Duckworth Family Chair of Breast Cancer Research (VB). SMR and MN acknowledge support from ENH Research Career Development Award and SMR acknowledges support from the Auxiliary of ENH Breast Cancer Pilot Grant. We thank Dr. Brian Drucker for 4G10 antibody. We thank members of the Band Laboratories for helpful suggestions and discussion.
Abbreviations
- 17-AAG
17-(allylamino)-17-demethoxygeldanamycin
- ADCC
Antibody Directed Cellular Cytotoxicity
- Cbl
Casitas B-lineage lymphoma
- CHIP
C-terminus of Hsc70 Interacting Protein
- CI
Combination Index
- CIM
Confocal Immunofluorescence Microscopy
- CSF-1R
Colony Stimulating Factor 1Receptor
- DAPI
4′, 6-diamidino-2-phenylindole
- Dm
Median Dose
- ErbB2 or Her2
Epidermal growth factor receptor B2 or Human Epidermal growth factor receptor 2
- EGF
Epidermal Growth Factor
- EGFR
Epidermal Growth Factor Receptor
- FACS
Fluorescence Activated Cell Sorter
- GA
Geldanamycin
- HSP90
Heat Shock Protein 90
- Hsc70
Heat shock cognate 70
- IP
Immunoprecipitation
- IF
Immunofluorescence
- IB
Immunoblotting
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide
- LAMP-1
Lysosome Associated Membrane Protein-1
- PBS
Phosphate Buffered Saline
- PFA
Paraformaldehye
- RTK
Receptor Tyrosine Kinase
- SDS-PAGE
Sodium dodecylsulfate Polyacrylamide Gel Electrophoresis
- WB
Western blotting
References
- 1.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: Understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3:269–280. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- 2.Ng R, Green MD. Managing cardiotoxicity in anthracycline-treated breast cancers. Expert Opin Drug Saf. 2007;6:315–21. doi: 10.1517/14740338.6.3.315. [DOI] [PubMed] [Google Scholar]
- 3.Braga S, dal Lago L, Bernard C, Cardoso F, Piccart M. Use of trastuzumab for the treatment of early stage breast cancer. Expert Rev Anticancer Ther. 2006;6:1153–64. doi: 10.1586/14737140.6.8.1153. [DOI] [PubMed] [Google Scholar]
- 4.Nahta R, Esteva FJ. HER2 therapy: Molecular mechanisms of trastuzumab resistance. Breast Cancer Res. 2006;8:215. doi: 10.1186/bcr1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooley S, Burns LJ, Repka T, Miller JS. Natural killer cell cytotoxicity of breast cancer targets is enhanced by two distinct mechanisms of antibody-dependent cellular cytotoxicity against LFA-3 and HER2/neu. Exp Hematol. 1999;27:1533–1541. doi: 10.1016/s0301-472x(99)00089-2. [DOI] [PubMed] [Google Scholar]
- 6.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 7.Kono K, Takahashi A, Ichihara F, Sugai H, Fujii H, Matsumoto Y. Impaired antibody-dependent cellular cytotoxicity mediated by herceptin in patients with gastric cancer. Cancer Res. 2002;62:5813–5817. [PubMed] [Google Scholar]
- 8.Yamaguchi Y, Hironaka K, Okawaki M, Okita R, Matsuura K, Ohshita A, Toge T. HER2-specific cytotoxic activity of lymphokine-activated killer cells in the presence of trastuzumab. Anticancer Res. 2005;25:827–832. [PubMed] [Google Scholar]
- 9.Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, Migeon C, Cabaret V, Fermeaux V, Bertheau P, Garnier J, Jeannin JF, Coudert B. Trastuzumab-based treatment of HER2-positive breast cancer: An antibody-dependent cellular cytotoxicity mechanism? Br J Cancer. 2006;94:259–267. doi: 10.1038/sj.bjc.6602930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barok M, Isola J, Palyi-Krekk Z, Nagy P, Juhasz I, Vereb G, Kauraniemi P, Kapanen A, Tanner M, Vereb G, Szollosi J. Trastuzumab causes antibody-dependent cellular cytotoxicity-mediated growth inhibition of submacroscopic JIMT-1 breast cancer xenografts despite intrinsic drug resistance. Mol Cancer Ther. 2007;6:2065–2072. doi: 10.1158/1535-7163.MCT-06-0766. [DOI] [PubMed] [Google Scholar]
- 11.Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and akt is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002;62:4132–4141. [PubMed] [Google Scholar]
- 12.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 13.Baselga J, Albanell J. Mechanism of action of anti-HER2 monoclonal antibodies. Ann Oncol. 2001;12 1:S35–41. doi: 10.1093/annonc/12.suppl_1.s35. [DOI] [PubMed] [Google Scholar]
- 14.Lu CH, Wyszomierski SL, Tseng LM, Sun MH, Lan KH, Neal CL, Mills GB, Hortobagyi GN, Esteva FJ, Yu D. Preclinical testing of clinically applicable strategies for overcoming trastuzumab resistance caused by PTEN deficiency. Clin Cancer Res. 2007;13:5883–5888. doi: 10.1158/1078-0432.CCR-06-2837. [DOI] [PubMed] [Google Scholar]
- 15.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 16.Klapper LN, Waterman H, Sela M, Yarden Y. Tumor-inhibitory antibodies to HER-2/ErbB-2 may act by recruiting c-cbl and enhancing ubiquitination of HER-2. Cancer Res. 2000;60:3384–8. [PubMed] [Google Scholar]
- 17.Duan L, Miura Y, Dimri M, Majumder B, Dodge IL, Reddi AL, Ghosh A, Fernandes N, Zhou P, Mullane-Robinson K, Rao N, Donoghue S, Rogers RA, Bowtell D, Naramura M, Gu H, Band V, Band H. Cbl-mediated ubiquitinylation is required for lysosomal sorting of epidermal growth factor receptor but is dispensable for endocytosis. J Biol Chem. 2003;278:28950–60. doi: 10.1074/jbc.M304474200. [DOI] [PubMed] [Google Scholar]
- 18.Grovdal LM, Stang E, Sorkin A, Madshus IH. Direct interaction of cbl with pTyr 1045 of the EGF receptor (EGFR) is required to sort the EGFR to lysosomes for degradation. Exp Cell Res. 2004;300:388–95. doi: 10.1016/j.yexcr.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Marmor MD, Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene. 2004;23:2057–70. doi: 10.1038/sj.onc.1207390. [DOI] [PubMed] [Google Scholar]
- 20.Hommelgaard AM, Lerdrup M, van Deurs B. Association with membrane protrusions makes ErbB2 an internalization-resistant receptor. Mol Biol Cell. 2004;15:1557–67. doi: 10.1091/mbc.E03-08-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baulida J, Kraus MH, Alimandi M, Di Fiore PP, Carpenter G. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J Biol Chem. 1996;271:5251–7. doi: 10.1074/jbc.271.9.5251. [DOI] [PubMed] [Google Scholar]
- 22.Austin CD, De Maziere AM, Pisacane PI, van Dijk SM, Eigenbrot C, Sliwkowski MX, Klumperman J, Scheller RH. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol Biol Cell. 2004;15:5268–5282. doi: 10.1091/mbc.E04-07-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Citri A, Kochupurakkal BS, Yarden Y. The achilles heel of ErbB-2/HER2: Regulation by the Hsp90 chaperone machine and potential for pharmacological intervention. Cell Cycle. 2004;3:51–60. [PubMed] [Google Scholar]
- 24.Xu W, Yuan X, Xiang Z, Mimnaugh E, Marcu M, Neckers L. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat Struct Mol Biol. 2005;12:120–6. doi: 10.1038/nsmb885. [DOI] [PubMed] [Google Scholar]
- 25.Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, Yarden Y, Neckers L. Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem. 2001;276:3702–8. doi: 10.1074/jbc.M006864200. [DOI] [PubMed] [Google Scholar]
- 26.Zhou P, Fernandes N, Dodge IL, Reddi AL, Rao N, Safran H, DiPetrillo TA, Wazer DE, Band V, Band H. ErbB2 degradation mediated by the co-chaperone protein CHIP. J Biol Chem. 2003;278:13829–37. doi: 10.1074/jbc.M209640200. [DOI] [PubMed] [Google Scholar]
- 27.Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci U S A. 2002;99:12847–52. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Bagatell R, Ma W, Wheler J, Rosen N, Norton L, Cropp GF, Johnson RG, Hannah AL, Hudis CA. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: A phase I dose-escalation study. J Clin Oncol. 2007;25:5410–5417. doi: 10.1200/JCO.2007.11.7960. [DOI] [PubMed] [Google Scholar]
- 29.Solit DB, Ivy SP, Kopil C, Sikorski R, Morris MJ, Slovin SF, Kelly WK, DeLaCruz A, Curley T, Heller G, Larson S, Schwartz L, Egorin MJ, Rosen N, Scher HI. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. Clin Cancer Res. 2007;13:1775–1782. doi: 10.1158/1078-0432.CCR-06-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grem JL, Morrison G, Guo XD, Agnew E, Takimoto CH, Thomas R, Szabo E, Grochow L, Grollman F, Hamilton JM, Neckers L, Wilson RH. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J Clin Oncol. 2005;23:1885–1893. doi: 10.1200/JCO.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 31.Weigel BJ, Blaney SM, Reid JM, Safgren SL, Bagatell R, Kersey J, Neglia JP, Ivy SP, Ingle AM, Whitesell L, Gilbertson RJ, Krailo M, Ames M, Adamson PC. A phase I study of 17-allylaminogeldanamycin in relapsed/refractory pediatric patients with solid tumors: A children's oncology group study. Clin Cancer Res. 2007;13:1789–1793. doi: 10.1158/1078-0432.CCR-06-2270. [DOI] [PubMed] [Google Scholar]
- 32.Bagatell R, Gore L, Egorin MJ, Ho R, Heller G, Boucher N, Zuhowski EG, Whitlock JA, Hunger SP, Narendran A, Katzenstein HM, Arceci RJ, Boklan J, Herzog CE, Whitesell L, Ivy SP, Trippett TM. Phase I pharmacokinetic and pharmacodynamic study of 17-N-allylamino-17-demethoxygeldanamycin in pediatric patients with recurrent or refractory solid tumors: A pediatric oncology experimental therapeutics investigators consortium study. Clin Cancer Res. 2007;13:1783–1788. doi: 10.1158/1078-0432.CCR-06-1892. [DOI] [PubMed] [Google Scholar]
- 33.Dimri M, Naramura M, Duan L, Chen J, Ortega-Cava C, Chen G, Goswami R, Fernandes N, Gao Q, Dimri GP, Band V, Band H. Modeling breast cancer-associated c-src and EGFR overexpression in human MECs: C-src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res. 2007;67:4164–72. doi: 10.1158/0008-5472.CAN-06-2580. [DOI] [PubMed] [Google Scholar]
- 34.Band V, Sager R. Distinctive traits of normal and tumor-derived human mammary epithelial cells expressed in a medium that supports long-term growth of both cell types. Proc Natl Acad Sci U S A. 1989;86:1249–1253. doi: 10.1073/pnas.86.4.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonita DP, Miyake S, L LM, Jr, Langdon WY, Band H. Phosphotyrosine binding domain-dependent upregulation of the platelet-derived growth factor receptor alpha signaling cascade by transforming mutants of cbl: Implications for cbl's function and oncogenicity. Mol Cell Biol. 1997;17:4597–610. doi: 10.1128/mcb.17.8.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chou TC, Motzer RJ, Tong Y, Bosl GJ. Computerized quantitation of synergism and antagonism of taxol, topotecan, and cisplatin against human teratocarcinoma cell growth: A rational approach to clinical protocol design. J Natl Cancer Inst. 1994;86:1517–1524. doi: 10.1093/jnci/86.20.1517. [DOI] [PubMed] [Google Scholar]
- 37.Lerdrup M, Bruun S, Grandal MV, Roepstorff K, Kristensen MM, Hommelgaard AM, van Deurs B. Endocytic down-regulation of ErbB2 is stimulated by cleavage of its C-terminus. Mol Biol Cell. 2007;18:3656–66. doi: 10.1091/mbc.E07-01-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lerdrup M, Hommelgaard AM, Grandal M, van Deurs B. Geldanamycin stimulates internalization of ErbB2 in a proteasome-dependent way. J Cell Sci. 2006;119:85–95. doi: 10.1242/jcs.02707. [DOI] [PubMed] [Google Scholar]
- 39.Mimnaugh EG, Chavany C, Neckers L. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J Biol Chem. 1996;271:22796–801. doi: 10.1074/jbc.271.37.22796. [DOI] [PubMed] [Google Scholar]
- 40.Neckers L. Heat shock protein 90 is a rational molecular target in breast cancer. Breast Dis. 2002;15:53–60. doi: 10.3233/bd-2002-15106. [DOI] [PubMed] [Google Scholar]
- 41.Lan KH, Lu CH, Yu D. Mechanisms of trastuzumab resistance and their clinical implications. Ann N Y Acad Sci. 2005;1059:70–75. doi: 10.1196/annals.1339.026. [DOI] [PubMed] [Google Scholar]
- 42.Bao J, Gur G, Yarden Y. Src promotes destruction of c-cbl: Implications for oncogenic synergy between src and growth factor receptors. Proc Natl Acad Sci U S A. 2003;100:2438–43. doi: 10.1073/pnas.0437945100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information:
Fig. S1: Trastuzumab dose-response curves against ErbB2-overexpressing breast cancer cells: The proliferation of ErbB2-overexpressing cells (SKBr-3, BT-474 and 21MT-1) treated with varying doses of Trastuzumab (as described in the methods section) was assessed using MTT-assay. Shown here are the relative growths of the cells as a function of Trastuzumab concentrations.
Supplementary Table 1: List of antibodies and their concentrations used in this study.