

Abstract
Abnormal centrosome and centriole numbers are frequently detected in tumor cells where they can contribute to mitotic aberrations that cause chromosome missegregation and aneuploidy. The molecular mechanisms of centriole overduplication in malignant cells, however, are poorly characterized. Here, we show that the core SCF component CUL1 localizes to maternal centrioles and that CUL1 is critical for suppressing centriole overduplication through multiplication, a recently discovered mechanism whereby multiple daughter centrioles form concurrently at single maternal centrioles. We found that this activity of CUL1 involves the degradation of Polo-like kinase 4 (PLK4) at maternal centrioles. PLK4 is required for centriole duplication and strongly stimulates centriole multiplication when aberrantly expressed. We found that CUL1 is critical for the degradation of active PLK4 following deregulation of cyclin E/CDK2 activity, as is frequently observed in human cancer cells, as well as for baseline PLK4 protein stability. Collectively, our results suggest that CUL1 may function as a tumor suppressor by regulating PLK4 protein levels and thereby restraining excessive daughter centriole formation at maternal centrioles.
Introduction
Centrosomes function as major microtubule organizing centers in most mammalian cells (1). The single centrosome of a non-dividing cell consists of a pair of centrioles, short microtubule cylinders, surrounded by pericentriolar material. In order to organize a bipolar mitotic spindle, the centrosome duplicates precisely once prior to mitosis (2). This process is frequently disrupted in tumor cells, where centrosome overduplication has been implicated in mitotic defects, chromosome missegregation and aneuploidy (3–6).
Supernumerary centrosome numbers may arise as a consequence of mitotic defects that lead to the formation of tetraploid and/or binucleated cells that will contain four centrosomes if they re-enter the cell division cycle. Whereas tetraploidy has been implicated as a gateway to aneuploidy, it is a remarkably stable cellular state, suggesting that additional mutations are necessary that cause chromosome missegregation and aneuploidy (7). Importantly, however, some oncogenic events, for example the HPV-16 E7 oncoprotein (8), directly interfere with the process of centriole synthesis resulting in the formation of multiple daughter centrioles from a single maternal centriole during a single round of cell division as was originally postulated by Boveri (9).
The duplication of the centrosome is initiated by the disengagement of the two centrioles (10). This is followed by activation of Polo-like kinase 4 (PLK4) and the subsequent recruitment of hSAS-6 and other centriolar components to maternal centrioles, which are critical steps in procentriole assembly (11–13).
During a normal centriole duplication cycle, a single maternal centriole generates one and only one daughter centriole. However, recent studies have revealed that single maternal centrioles have the ability to mediate the concurrent formation of multiple daughter centrioles at the same time (centriole multiplication). The structures that arise during this process, a single maternal centriole surrounded by a ring of concurrently synthesized daughters, have been referred to as centriole flowers (8, 12). Besides overexpression of PLK4 or hSAS-6, (12–15), inhibition of the proteasome (8), overexpression of the HPV-16 E7 oncoprotein (8) and inactivation of CDK1 in Drosophila wing disc cells (16) have been reported to stimulate the formation of multiple daughters at single maternal centrioles. In addition, the centriolar pathway of basal body production in oviduct cells has also been shown to involve the concurrent formation of multiple daughter centrioles (17, 18).
Given the intrinsic potential of maternal centrioles to give birth to multiple daughters, the question arises what the molecular mechanisms that normally restrain procentriole formation to only one per cell division cycle may be.
The centrosome has been shown to harbor components of the ubiquitin-proteasome machinery including subunits of SKP1-Cullin-F-Box (SCF) E3 ubiquitin ligases and components of the 26S proteasome (19–22). Although SCF complexes have been implicated in centriole duplication control as well as centriole separation (19, 23), their precise functions are not understood in detail.
Here, we show that the SCF core component, cullin 1 (CUL1), localizes to maternal centrioles and that these centrioles serve as assembly platforms for oncogene-induced centriole overduplication. Moreover, SCF ubiquitin ligase activity was found to be critically involved in suppressing centriole multiplication in human tumor cells by regulating PLK4 protein levels. We provide evidence that SCF ubiquitin ligase activity is critical for the degradation of active PLK4 following overexpression of cyclin E/CDK2, as well as for baseline PLK4 protein stability. Our results suggest that SCF ubiquitin ligase activity provides an important mechanism for restraining excessive daughter centriole formation at single maternal centrioles and hence centrosome-mediated cell division errors and chromosomal instability. Since recurrent deletions of the CUL1 locus on chromosome 7q36.1 have been detected in human malignancies, our findings provide insights into possible tumor suppressor activities of CUL1.
Materials and Methods
Cell culture, transfections and inhibitor treatments
U-2 OS/centrin-GFP cells and BJ/TERT/centrin-GFP fibroblasts (centrin-GFP plasmid was kindly provided by Michel Bornens, Institut Curie, France (24); BJ/TERT fibroblasts were kindly provided by Ole Gjoerup, University of Pittsburgh Cancer Institute, Pittsburgh, PA) were maintained as previously reported (8). For transient transfections (48 h), pCMV- or pcDNA3-based plasmids encoding c-MYC (kindly provided by Philip Leder, Harvard Medical School, Boston, MA), HPV-16 E7, E2F-1 (kindly provided by Jacqueline Lees, Massachusetts Institute of Technology, Boston), cyclin E (provided by Robert Weinberg through Addgene, Cambridge, MA, USA (25), CDK2 (provided by Sander van den Heuvel through Addgene (26), Myc-PLK4 or catalytically inactive Myc-PLK4-D154A (kindly provided by Erich A. Nigg (Max Planck Institute of Biochemistry, Martinsried, Germany (14), dominant-negative CUL1 (DN-CUL1; provided by Wade Harper through Addgene (27) or empty vector controls were used and transfected by lipofection (Fugene 6; Roche). Cells were co-transfected with a vector encoding red fluorescent protein targeted to mitochondria (DsRED; BD Biosciences Clontech, Palo Alto, CA) as transfection marker. Cells were treated with 1 μM of the proteasome inhibitor Z-L3VS (Biomol, Plymoth Meeting, PA) or 0.1% DMSO as control. To inhibit CDK activity, cells were treated with 1μM of indirubin-3′-oxime (IO; kindly provided by Laurent Meijer, Station Biologique Roscoff, France) or 0.1% DMSO as control. Cycloheximide (CHX; Calbiochem, San Diego, CA) was used at 30 μg/ml for the indicated time intervals with dH2O as control.
Immunological Methods
Immunoblotting was performed as described previously (8). Primary antibodies used for immunoblotting were directed against CUL1, cyclin E, CDK2 (all Santa Cruz, Santa Cruz, CA), Myc-tag, (Cell Signaling, Danvers, MA), OctA-Probe/Flag® (Santa Cruz) or actin (Sigma, St. Louis, MO).
Immunofluorescence stainings using CUL1 (Neomarkers, Fremont, CA), γ-tubulin (Sigma), CEP170 (Invitrogen, Carlsbad, CA) or PLK4 (mouse monoclonal antibody kindly provided by Erich A. Nigg, Max Planck Institute of Biochemistry, Martinsried, Germany) antibodies were performed as described previously (8). Primary antibodies were detected with FITC-, Rhodamine Red- or AMCA-conjugated secondary antibodies (Jackson Immunoresearch, West Grove, PA) as previously described (8).
Small interfering RNA (siRNA)
Synthetic RNA duplexes to reduce CUL1 expression were obtained commercially (Ambion, Austin, TX) and used according to manufacturer’s protocol.
Statistical Methods
Student’s t test for independent samples was used wherever applicable.
Results
CUL1-positive maternal centrioles are assembly platforms for oncogene-induced centriole overduplication
To explore the role of SCF ubiquitin ligase activity in centriole biogenesis, an immunofluorescence microscopic analysis of the SCF core component CUL1 was performed using U-2 OS cells stably expressing centrin-GFP. CUL1 was found to localize to centrioles (Fig. 1A) whereas weaker CUL1 staining was detected in the nucleus and occasionally in the cytoplasm of cells as previously reported (28). The co-localization pattern between centrin-GFP and CUL1 suggested that CUL1 may be present at older, mature centrioles (Fig. 1A). A co-immunofluorescence microscopic analysis of CUL1 and CEP170, a marker for mature maternal centrioles (29), in U-2 OS/centrin-GFP cells revealed that CUL1 in fact localizes to older, maternal centrioles (Fig. 1B).
Figure 1. CUL1-positive maternal centrioles serve as assembly platforms for oncogene-induced centriole overduplication.

(A) Immunofluorescence microscopic analysis for CUL1 using U-2 OS cells stably expressing centrin-GFP (U-2 OS/centrin-GFP). Arrowheads indicate centrioles shown in inserts. Nuclei stained with DAPI. Scale bar indicates 10 μm.
(B) Co-immunofluorescence microscopic analysis of U-2 OS/centrin-GFP cells for CUL1 and CEP170, a marker for mature maternal centrioles. Arrowheads indicate centrioles with co-localization of CUL1 and CEP170 (see also inserts).
(C) Immunofluorescence microscopic analysis of U-2 OS/centrin-GFP cells for CUL1 following overexpression of E2F-1. Note overduplication of centrioles in the presence of only two CUL1-positive centrioles (bottom panels).
(D) Quantification of the proportion of U-2 OS/centrin-GFP cells with centriole overduplication in the presence of one or two CUL1-positive centrioles after overexpression of c-MYC, HPV-16 E7 or E2F-1. Arrows point to centrioles shown in inserts. Mean and standard error of three independent experiments with at least 100 cells counted per experiment are shown.
Overexpression of the HPV-16 E7 oncoprotein has previously been shown to lead to excessive daughter centriole formation at maternal centrioles (8, 30). U-2 OS/centrin-GFP cells were transiently transfected with HPV-16 E7, or other oncogenes including c-MYC oncogene or the transcription factor E2F-1 followed by immunofluorescence staining for CUL1. Since normal centriole duplication generates a maximum of two mature maternal centrioles and a maximum of two immature daughters, excessive numbers of CUL1-negative centrioles in the presence of one or two CUL1-positive centrioles were counted as abnormal. An increase of cells with more than 4 centrioles in the presence of only one or two CUL1-positive centrioles was detected in cells overexpressing c-MYC (6.7%), HPV-16 E7 (10.7%, p≤0.05) or E2F-1 (13%, p≤0.005) in comparison to empty vector control (3%).
These results indicate that the SCF ubiquitin ligase component CUL1 localizes to maternal centrioles that serve as assembly platforms for excessive daughter centriole formation induced by various oncogenic stimuli.
Inhibition of SCF ubiquitin ligase activity causes centriole multiplication
To determine the role of CUL1 in the control of centriole biogenesis, we used siRNA to deplete cells of CUL1 and a dominant-negative mutant of CUL1 (DN-CUL1), which has recently been shown to effectively reduce CUL1-based SCF activity (31). SiRNA-mediated knock-down of CUL1 led to centriole multiplication with an increase of cells showing multiple daughter centrioles at single maternal centrioles (Fig. 2A) from 0.6% in controls to 8.5% in CUL1-depleted cells (p≤0.05; Fig. 2B, left panel). Centriole multiplication was also detected in cells transiently transfected with DN-CUL1 with an increase from 1.3% in controls to 12% in DN-CUL1-transfected cells (p≤0.05; Fig. 2B, right panel).
Figure 2. Inhibition of CUL1 causes centriole multiplication.

(A) Fluorescence microscopic analysis of U-2 OS/centrin-GFP cells transfected with either control siRNA duplexes (siControl) or siRNA targeting CUL1 (siCUL1) for 72 h. A DsRED-encoding plasmid was used as transfection marker. Nuclei stained with DAPI. Arrowheads in bottom insert indicate centriole multiplication with three daughter centrioles at a single mother. Scale bar indicates 10 μm.
(B) Quantification of centriole multiplication (>4 centrioles total, >1 daughter at a single mother) in U-2 OS/centrin-GFP cells transfected with either control (siControl) or CUL1 (siCUL1) siRNA duplexes for 72 h (left panel) or ectopic expression of empty vector control or DN-CUL1 for 48 h (right panel). Mean and standard error of three independent experiments with at least 100 cells counted per experiment are shown. Asterisk indicates statistically significant differences (p≤0.05; Student’s t test for independent samples).
(C) Immunoblot analysis of whole cell extracts from U-2 OS/centrin-GFP cells transiently transfected with siRNA duplexes targeting CUL1 (siCUL1) or control siRNA (siControl) for the indicated time intervals (left panels) or ectopic expression of empty vector control or DN-CUL1 for 48 h (right panel). Immunoblots for CUL1, cyclin E and CDK2 are shown. DN-CUL1 was detected using an OctA-Probe/Flag® antibody (Santa Cruz). Immunoblot for Actin shows protein loading.
CUL1 is involved in the degradation of a number of critical cell cycle regulators including cyclin E (32, 33). In line with this notion, we detected an increase of cyclin E protein expression in U-2 OS/centrin-GFP cells transfected with either siRNA against CUL1 or DN-CUL1 (Fig. 2C).
These results suggest that cyclin E may contribute to centriole multiplication induced by inhibition of CUL1. However, when cyclin E together with its kinase subunit CDK2 was overexpressed in U-2 OS/centrin-GFP cells, no significant increase of cells with centriole multiplication was detected (Fig. 3A). Only when cyclin E/CDK2 complexes were co-expressed with increasing amounts of the centrosomal kinase PLK4, a significant increase of cells with centriole multiplication was detected (Fig. 3A, B). Co-expression of PLK4 together with cyclin E/CDK2 consistently led to a higher frequency of centriole multiplication than overexpression of PLK4 alone (Fig. 3A).
Figure 3. Ectopic expression of cyclin E/CDK2 does not stimulate centriole multiplication but causes.

aberrant PLK4 recruitment to maternal centrioles.
(A) Quantification of the proportion of cells with centriole multiplication (>4 centrioles, >1 daughter per maternal centriole) after transient transfection with empty vectors (controls) or either cyclin E/CDK2 alone (0 μg PLK4 plasmid DNA) or increasing amounts of PLK4 alone (grey bars) or a combination of cyclin E/CDK2 with increasing amounts of PLK4 plasmid DNA (black bars). Asterisks indicate statistically significant differences (p≤0.05 at 0.5 μg and p≤0.005 at 2 μg PLK4 plasmid DNA).
(B) Fluorescence microscopic analysis of U-2 OS/centrin-GFP cells after transient transfection with empty vector (control) or cyclin E, CDK2 and PLK4. Note centriole multiplication in the right panel.
(C) Immunofluorescence microscopic analysis of U-2 OS/centrin-GFP cells for endogenous PLK4 expression after transient transfection with either empty vector (control) or cyclin E/CDK2. Arrows indicate an aberrant daughter centriole that co-localizes with PLK4 at the maternal centriole. Arrowheads indicate an aberrant PLK4 signal at a maternal centriole without a detectable centrin-positive daughter.
(D) Quantification of the percentage of U-2 OS/centrin-GFP or BJ/TERT/centrin-GFP cells with aberrant (two or more) PLK4 dots at maternal centriole following transfection with empty vector (control) or cyclin E/CDK2.
We therefore asked next whether cyclin E/CDK2 complexes can alter the expression of PLK4 at centrioles. To test this idea, U-2 OS/centrin-GFP cells were transiently transfected with cyclin E and CDK2 followed by immunofluorescence microscopy for endogenous PLK4 protein (Fig. 3C). In empty vector-transfected control cells, disengaged centrioles or duplicated centrioles were associated with a single dot of endogenous PLK4 that typically localized to the site of daughter centriole synthesis at the wall of the maternal centriole (Fig. 3C - control). In cells ectopically expressing cyclin E/CDK2, an increased proportion of cells with two or more dots of endogenous PLK4 at single mothers was detected (Fig. 3C - cyclin E/CDK2). These aberrant PLK4 dots were occasionally found to precede the formation of centrin-containing procentrioles (Fig. 3C, bottom panels), which is in line with previous results suggesting that PLK4 functions early during daughter centriole synthesis (12). Aberrant PLK4 dots at maternal centrioles were detected in 28 out of 52 cells (53.8%) transfected with cyclin E/CDK2 in comparison to 10 out of 51 cells (19.6%) transfected with empty vector (Fig. 3D). Overexpression of cyclin E/CDK2 also triggered an aberrant expression of endogenous PLK4 in non-transformed human cells with 14 out of 28 BJ/TERT/centrin-GFP fibroblasts transfected with cyclin E/CDK2 showing aberrant dots of endogenous PLK4 at maternal centrioles (50%) in comparison to 7 out of 28 empty vector-transfected cells (25%; Fig. 3D).
Taken together, these results suggest that the centriole multiplication induced by inhibition of CUL1 cannot solely be explained by an accumulation of cyclin E but depends also on increased levels of PLK4.
PLK4 is degraded by the proteasome
Our findings that cyclin E/CDK2 complexes cause an aberrant recruitment of PLK4 to maternal centrioles but do not lead to centriole multiplication unless PLK4 is overexpressed suggest that PLK4 levels are rate-limiting for excessive daughter centriole assembly. To explore how PLK4 protein expression may be regulated, we first analyzed U-2 OS/centrin-GFP cells ectopically expressing PLK4 and treated with the proteasome inhibitor Z-L3VS, which readily stimulates centriole multiplication (8). Immunoblot analyses revealed a stabilization of ectopically expressed PLK4, together with cyclin E, in Z-L3VS-trated cells (Fig. 4A). In addition, Z-L3VS treatment led to an excessive amount of endogenous PLK4 at maternal centrioles. (Fig. 4B). These results indicate that PLK4 protein levels are, at least in part, regulated by the ubiquitin-proteasome machinery. Furthermore, the effectiveness of Z-L3VS to stimulate centriole multiplication may be based on the fact that both, cyclin E and PLK4, are stabilized.
Figure 4. PLK4 is degraded by the proteasome.
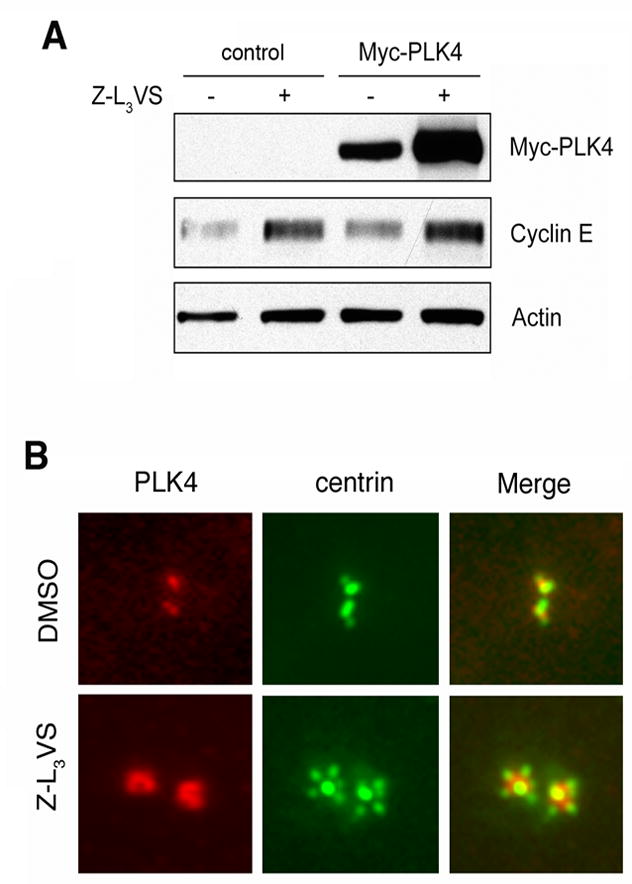
(A) Immunoblot analysis of whole cell extracts from U-2 OS/centrin-GFP cells transiently transfected with either empty vector (control) or PLK4 and treated with 1 μM of the proteasome inhibitor Z-L3VS or 0.1% DMSO at 24 h after transfection for an additional 24 h.
(B) Immunofluorescence microscopic analysis or U-2 OS/centrin-GFP cells treated with 0.1% DMSO or 1 μM Z-L3VS for 48 h and stained for PLK4. Note centriole multiplication together with an excessive amount of PLK4 at maternal centrioles in Z-L3VS-treated cells
Cyclin E/CDK2 can destabilize PLK4
To explore why cyclin E/CDK2 can only promote centriole multiplication in the presence of excessive amounts of PLK4, PLK4 protein levels were determined by immunoblotting after ectopic expression of Myc-tagged PLK4 alone or in combination with cyclin E and/or CDK2 (Fig. 5). Surprisingly, we detected a reduction of PLK4 protein levels in the presence of ectopically expressed cyclin E or cyclin E/CDK2 (Fig. 5A). Similar results were obtained when we analyzed whole cell extracts obtained from cells transfected with cyclin E/CDK2 and increasing amounts of PLK4 (Supplementary Material) and these findings could also be reproduced in HeLa cells (not shown).
Figure 5. Cyclin E/CDK2 reduce PLK4 protein stability in a PLK4 kinase activity-dependent manner.

(A) Immunoblot analysis of whole cell extracts from U-2 OS/centrin-GFP cells following transient transfection (48 h) with empty vector (control), Myc-tagged PLK4, Myc-tagged kinase-inactive mutant PLK4 D154A, cyclin E and/or CDK2. Note the decreased protein levels of wild-type PLK4 upon co-transfection with cyclin E or cyclin E/CDK2. No such reduction was detected in cells transfected with catalytically inactive mutant PLK4 D154A. Immunoblots for Actin are shown to demonstrate protein loading (A-C).
(B) Immunoblot analysis of whole cell extracts from U-2 OS/centrin-GFP cells after transient transfection (48 h) with PLK4 alone or PLK4 and cyclin E/CDK2 and treatment with 30 μg/ml cycloheximide (CHX) for the indicated time intervals. Note the decreased PLK4 protein stability in the presence of ectopically expressed cyclin E/CDK2.
(C) Immunoblot analysis of whole cell extracts from U-2 OS/centrin-GFP cells transiently transfected (24 h) with empty vector (control) or PLK4 alone or PLK4 in combination with cyclin E/CDK2 and treatment with 1 μM of the CDK inhibitor indirubin-3′-oxime for 24 h (IO). Note the stabilization of PLK4 protein in IO- treated cells in comparison to controls (0.1% DMSO).
In contrast to wild-type PLK4, protein levels of catalytically inactive mutant PLK4 D154A (14) were not significantly decreased in the presence of ectopically expressed cyclin E/CDK2 (Fig. 5A). Since PLK4 D154A mutant has no apparent defect in its localization to centrioles (14), we interpret this finding as evidence that the degradation of PLK4 depends on its kinase activity. Similar results have been reported for other protein kinases such as SRC (34). The decrease of PLK4 protein levels in the presence of cyclin E/CDK2 was due to decreased protein stability as shown by a cycloheximide block experiment (Fig. 5B). Furthermore, inhibition of CDK activity using the small molecule inhibitor indirubin-3′-oxime (IO) led to a stabilization of PLK4 protein underscoring the CDK dependency of this process (Fig. 5C).
Taken together, these findings suggest cyclin E/CDK2 promote the aberrant recruitment of PLK4 to maternal centrioles followed by its degradation that depends on PLK4 kinase activity. This process may prevent the accumulation of active PLK4 at maternal centrioles at levels that may promote aberrant procentriole assembly. Our finding that additional dots of endogenous PLK4 at maternal centrioles are often smaller in size and weaker in signal intensity than normal endogenous PLK4 dots (Fig. 3C) may lend support to this notion.
CUL1 regulates PLK4 protein stability
We next asked whether CUL1 contributes to the regulation of PLK4 protein levels. Ectopic expression of DN-CUL1 caused an excess of endogenous PLK4 at maternal centrioles (Fig. 6A, left panels) that was phenotypically similar to PLK4 accumulation in response to Z-L3VS-associated inhibition of proteasomal PLK4 degradation. To assess this increase, the integrated density of PLK4 signals at maternal centrioles was quantified using ImageJ and expressed in arbitrary units (Fig. 6A, right panel). A 6.9-fold increase of endogenous PLK4 protein was detected in cells with centriole multiplication after transient transfection with DN-CUL1 after normalization for endogenous PLK4 expression in empty vector-transfected controls and formation of only a single daughter. Treatment of cells with the CDK inhibitor IO led only to a modest 1.3-fold increase of endogenous PLK4 in comparison to DMSO-treated control cells (Fig. 6A, right panel).
Figure 6. CUL1 regulates PLK4 protein stability.

(A) Immunofluorescence microscopic analysis of U-2 OS/centrin-GFP cells for endogenous PLK4 at 48 h after transfection of empty vector (control) or DN-CUL1 (left panels). Note centriole multiplication together with an excessive amount of endogenous PLK4 in DN-CUL1-transfected cells (bottom). Quantification of the fold-changes of PLK4 integrated signal intensities at maternal centrioles in U-2 OS/centrin-GFP cells transfected with empty vector or DN-CUL1 for 48 h or treated with 1 μM IO or 0.1% DMSO (control) for 48 h (right panel).
(B) Immunoblot analysis of whole cell extracts from U-2 OS/centrin-GFP cells after transient transfection (48 h) with empty vectors (control) or PLK4 in combination with either cyclin E/CDK2 alone or cyclin E/CDK2 and DN-CUL1. Note the increase of PLK4 protein in the presence of DN-CUL1 in the last lane.
(C) Immunoblot analysis of whole cell extracts from U-2 OS/centrin-GFP cells after transient transfection with empty vectors (control) or dominant-negative CUL1 (DN-CUL1) for 48 h followed by transfection with Myc-PLK4 for 24 h and treatment with 30 μg/ml cycloheximide (CHX) for the indicated time intervals. Note the increased protein stability of Myc-PLK4 in the presence of DN-CUL1 (6 h CHX, last lane).
(D) Tentative model of centriole multiplication triggered by oncogenic stimuli such as c-MYC, E2F-1 or HPV-16 E7. These stimuli are known to deregulate cyclin E/CDK2 complexes and we show here that this leads to an aberrant recruitment of PLK4 to maternal centrioles. However, degradation of enzymatically active PLK4 (and low baseline protein expression) by CUL1-based SCF E3 ubiquitin ligase activity prevents the formation of supernumerary daughter centrioles. Only when PLK4 is overexpressed or its CUL1-mediated degradation is altered, centriole multiplication occurs. Hence, it is likely that oncogene-induced centriole multiplication, for example by HPV-16 E7, also involves impairment of CUL1-mediated protein degradation through mechanisms that remain to be determined. Genetic alterations such as deletion of the CUL1 locus in certain human malignancies are likewise to promote centriole duplication.
We tested next the effects of DN-CUL1 on cyclin E/CDK2-induced PLK4 degradation. Using immunoblotting, we found that DN-CUL1 abrogates the reduction of PLK4 protein levels associated with ectopic expression of cyclin E/CDK2 (Fig. 6B). DN-CUL1 also increased the baseline protein stability of ectopically expressed PLK4 in the absence of overexpressed cyclin E/CDK2 (Fig. 6C). Depletion of CUL1 by siRNA led to similar results (not shown).
Taken together, these results suggest that CUL1 is involved in both cyclin E/CDK2-associated degradation of PLK4 and baseline PLK4 protein stability.
Discussion
Results presented here show that the core SCF ubiquitin ligase component CUL1 is critical to tightly regulate PLK4 proteins levels in order to suppress aberrant centriole biogenesis at maternal centrioles.
Upregulation of PLK4 expression is a strong stimulus for centriole multiplication (8, 12, 15, 35). The fact that overexpression of cyclin E/CDK2 can aberrantly recruit PLK4 to maternal centrioles (Fig. 3C) provides mechanistic insight into how CDK2 and PLK4 may cooperate to stimulate excessive daughter centriole formation (8, 14). Our finding that overexpression of cyclin E/CDK2 leads to the degradation of PLK4 in a manner that depends on PLK4 kinase activity explains why overexpression of cyclin E/CKD2 alone is not sufficient to trigger centriole multiplication. The low endogenous levels of PLK4 together with the degradation of active PLK4 likely represent cellular failsafe mechanisms to limit normal centriole biogenesis to one per cell division cycle.
Several lines of evidence indicate that active protein kinases can be rapidly degraded by the ubiquitin-proteasome machinery whereas kinase-inactive mutants are more stable (34). Although the precise molecular mechanisms that render active kinases more susceptible to degradation are currently unknown, it has been suggested that the switch from an inactive to an active state is associated with a change in the conformation of the protein as well as phosphorylation events that promotes its ubiquitination and subsequent degradation. We will address this question for PLK4 in future experiments.
PLK4 has been shown to autophosphorylate (36) but no specific amino acid residues have been identified. It is known from other protein kinases e.g., CHK2 (37), that autophosphorylation is required for interaction with CUL1 and its degradation. It is hence possible that autophosphorylation of PLK4 plays a role in its degradation.
We were not able to unambiguously detect a direct interaction between CDK2 and PLK4 by co-immunoprecipitation (data not shown). This does not rule out a very weak and/or transient interaction. Our results are consistent with a model whereby high levels of cyclin E/CDK2 complexes cause an aberrant recruitment of PLK4 to centrioles through an indirect mechanism that remains to be identified. The fact that mutant PLK4 D154A is more stable than wild-type PLK4 in the presence of overexpressed cyclin E/CDK2 but has no centrosomal localization defect (14) suggests that it is the enzymatic activity of PLK4 and not only its centrosome association that triggers its degradation. One possibility is that cyclin E/CDK2 complexes phosphorylate a centrosomal protein that mediates the recruitment of PLK4 followed by its activation and subsequent degradation. It has been suggested, however, that the single Polo box of PLK4 reduces its ability to efficiently recognize phosphopeptides (38). PLK4 is protected from PEST-dependent proteolysis by the protein tyrosine kinase Tec (39). One possible mechanism hence may be that Tec activity is modulated by CDK2, which may then lead to an accumulation of active PLK4 at the centrosome. Tec contains a consensus S/T-PxK/R CDK2 phosphorylation motif and we will follow up on this interesting possibility in future experiments. Whether any of the centrosomal CDK2 targets such as nucleophosmin, CP110 or MPS1 play a role in this process remains to be determined (40–43).
The question whether normal activation of cyclin E/CDK2 complexes triggers PLK4 recruitment during normal centriole duplication also awaits further clarification. It is interesting to note, however, that CDK2 deficient MEFs cells have no detectable defects in centrosomal duplication (44). Oncogene-triggered centrosome overduplication, however, was clearly blocked in CDK2-deficient cells (44) suggesting that deregulation of CDK2, as frequently seen in tumors through increased cyclin E expression, may promote centriole overduplication by aberrantly recruiting PLK4. One prediction from our results is that such tumor cells may also harbor alterations of the PLK4 degradation machinery. Oncogenic stimuli that induce centriole overduplication may therefore not only function by deregulating cyclin E/CDK2 activity but also by impacting on CUL1-based SCF ubiquitin ligase activity (Fig. 6D).
PLK4 contains a consensus DSGxxS/T phosphodegron, which has been linked to SCFβ-TrCP-mediated degradation. Two recent studies in Drosophila cells have demonstrated a role of the SCFSlimb ubiquitin ligase in the regulation of SAK (PLK4) (15, 35). Slimb is the ortholog of the mammalian F-box protein β-TrCP and although knock-out of β-TrCP in mice has been found to lead to centrosome amplification (45), our own experiments did not show significant centriole multiplication after siRNA-mediated knock-down of β-TrCP in human U-2 OS cells (not shown). Our finding does not preclude a role of SCFβ-TrCP in PLK4 regulation in human cells but it raises the interesting possibility that there may be cell type specific differences in PLK4 regulation and/or that more than one F-box protein participates in SCF-mediated regulation of PLK4 stability in a spatio-temporally controlled manner. Given the need to tightly regulate PLK4 levels, it is likely that multiple mechanisms converge on PLK4 expression control and protein stability.
The CUL1 locus on chromosome 7q36 has been found to be recurrently deleted in a number of human malignancies including acute myeloid leukemias and myelodysplastic syndromes (46). Such malignancies are characterized by centrosome aberrations and genomic instability (47) and our results suggest that the activity of CUL1 to suppress centriole multiplication may play an important role in its function as a putative tumor suppressor (Fig. 6D). Another prominent example of centrosome amplification associated with the loss of a tumor that has E3 ubiquitin ligase activity is BRCA1 (48). It is likely that more proteins involved in restraining centriole overduplication through targeted degradation remain to be discovered.
Our findings provide a framework for future studies to dissect molecular interactions and posttranslational modifications that will add layers of complexity to the SCF/CUL1-based regulatory circuitry that restrains daughter centriole biogenesis at maternal centrioles.
Supplementary Material
Acknowledgments
We are grateful to Michel Bornens, Ole Gjoerup, Philip Leder, Jacqueline Lees, Laurent Meijer and Erich A. Nigg for sharing important reagents. This work was supported by Public Health Service grant NIH/NCI R01 CA112598 (to S. D.), the Susan G. Komen for the Cure Foundation (to S. D), R01 066980 (to K. M.), a Research Scholar Grant from the American Cancer Society (RSG-07-075-01, to S. D.), a grant from the Howard Hughes Medical Institute Undergraduate Research Program (to L. Z.), a Carnegie Mellon University Small Undergraduate Research Grant (to L. Z.), and, in part, by a grant from the Pennsylvania Department of Health. The Department specifically disclaims responsibility for any analyses, interpretations or conclusions.
References
- 1.Azimzadeh J, Bornens M. Structure and duplication of the centrosome. J Cell Sci. 2007;120(Pt 13):2139–42. doi: 10.1242/jcs.005231. [DOI] [PubMed] [Google Scholar]
- 2.Strnad P, Gonczy P. Mechanisms of procentriole formation. Trends Cell Biol. 2008;18(8):389–96. doi: 10.1016/j.tcb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Nigg EA. Centrosome aberrations: cause or consequence of cancer progression? Nature Rev Cancer. 2002;2:1–11. doi: 10.1038/nrc924. [DOI] [PubMed] [Google Scholar]
- 4.Duensing S, Munger K. Centrosome abnormalities, genomic instability and carcinogenic progression. Biochim Biophys Acta. 2001;2(8):M81–8. doi: 10.1016/s0304-419x(00)00025-1. [DOI] [PubMed] [Google Scholar]
- 5.Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer. 2007;7(12):911–24. doi: 10.1038/nrc2249. [DOI] [PubMed] [Google Scholar]
- 6.Lingle WL, Lukasiewicz K, Salisbury JL. Deregulation of the centrosome cycle and the origin of chromosomal instability in cancer. Adv Exp Med Biol. 2005;570:393–421. doi: 10.1007/1-4020-3764-3_14. [DOI] [PubMed] [Google Scholar]
- 7.Duensing A, Duensing S. Guilt by association? p53 and the development of aneuploidy in cancer. Biochem Biophys Res Commun. 2005;331(3):694–700. doi: 10.1016/j.bbrc.2005.03.157. [DOI] [PubMed] [Google Scholar]
- 8.Duensing A, Liu Y, Perdreau SA, Kleylein-Sohn J, Nigg EA, Duensing S. Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene. 2007;26(43):6280–8. doi: 10.1038/sj.onc.1210456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boveri T. The origin of malignant tumors. Baltimore: The Williams & Wilkins Company; 1929. [Google Scholar]
- 10.Tsou MF, Stearns T. Mechanism limiting centrosome duplication to once per cell cycle. Nature. 2006 doi: 10.1038/nature04985. [DOI] [PubMed] [Google Scholar]
- 11.Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, et al. SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol. 2005;15(24):2199–207. doi: 10.1016/j.cub.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 12.Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA. Plk4-induced centriole biogenesis in human cells. Dev Cell. 2007;13(2):190–202. doi: 10.1016/j.devcel.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Strnad P, Leidel S, Vinogradova T, Euteneuer U, Khodjakov A, Gonczy P. Regulated HsSAS-6 Levels Ensure Formation of a Single Procentriole per Centriole during the Centrosome Duplication Cycle. Dev Cell. 2007;13(2):203–13. doi: 10.1016/j.devcel.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. 2005;7(11):1140–6. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- 15.Cunha-Ferreira I, Rodrigues-Martins A, Bento I, et al. The SCF/Slimb ubiquitin ligase limits centrosome amplification through degradation of SAK/PLK4. Curr Biol. 2009;19(1):43–9. doi: 10.1016/j.cub.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 16.Vidwans SJ, Wong ML, O’Farrell PH. Anomalous centriole configurations are detected in Drosophila wing disc cells upon cdk1 inactivation. J Cell Sci. 2003;116:137–43. doi: 10.1242/jcs.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson RG, Brenner RM. The formation of basal bodies (centrioles) in the Rhesus monkey oviduct. J Cell Biol. 1971;50(1):10–34. doi: 10.1083/jcb.50.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dirksen ER. Centriole morphogenesis in developing ciliated epithelium of the mouse oviduct. J Cell Biol. 1971;51(1):286–302. doi: 10.1083/jcb.51.1.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freed E, Lacey KR, Huie P, et al. Components of an SCF ubiquitin ligase localize to the centrosome and regulate the centrosome duplication cycle. Genes Dev. 1999;13(17):2242–57. doi: 10.1101/gad.13.17.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fabunmi RP, Wigley WC, Thomas PJ, DeMartino GN. Activity and regulation of the centrosome-associated proteasome. J Biol Chem. 2000;275(1):409–13. doi: 10.1074/jbc.275.1.409. [DOI] [PubMed] [Google Scholar]
- 21.Wigley WC, Fabunmi RP, Lee MG, et al. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol. 1999;145(3):481–90. doi: 10.1083/jcb.145.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gstaiger M, Marti A, Krek W. Association of human SCF Skp2 subunit p19 Skp1 with interphase centrosomes and mitotic spindle poles. Exp Cell Res. 1999;247:554–62. doi: 10.1006/excr.1999.4386. [DOI] [PubMed] [Google Scholar]
- 23.Piva R, Liu J, Chiarle R, Podda A, Pagano M, Inghirami G. In vivo interference with Skp1 function leads to genetic instability and neoplastic transformation. Mol Cell Biol. 2002;22(23):8375–87. doi: 10.1128/MCB.22.23.8375-8387.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piel M, Meyer P, Khodjakov A, Rieder CL, Bornens M. The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells. J Cell Biol. 2000;149(2):317–30. doi: 10.1083/jcb.149.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70(6):993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- 26.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262(5142):2050–4. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 27.Jin J, Ang XL, Shirogane T, Harper JW. Identification of substrates for F-box proteins. Methods Enzymol. 2005;2005(399):287–309. doi: 10.1016/S0076-6879(05)99020-4. [DOI] [PubMed] [Google Scholar]
- 28.Furukawa M, Zhang Y, McCarville J, Ohta T, Xiong Y. The CUL1 C-terminal sequence and ROC1 are required for efficient nuclear accumulation, NEDD8 modification, and ubiquitin ligase activity of CUL1. Mol Cell Biol. 2000;20(21):8185–97. doi: 10.1128/mcb.20.21.8185-8197.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guarguaglini G, Duncan PI, Stierhof YD, Holmstrom T, Duensing S, Nigg EA. The Forkhead-associated Domain Protein Cep170 Interacts with Polo-like Kinase 1 and Serves as a Marker for Mature Centrioles. Mol Biol Cell. 2005;16(3):1095–107. doi: 10.1091/mbc.E04-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duensing S, Duensing A, Crum CP, Munger K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001;61(6):2356–60. [PubMed] [Google Scholar]
- 31.Yen HC, Elledge SJ. Identification of SCF ubiquitin ligase substrates by global protein stability profiling. Science. 2008;322(5903):923–9. doi: 10.1126/science.1160462. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Penfold S, Tang X, et al. Deletion of the Cul1 gene in mice causes arrest in early embryogenesis and accumulation of cyclin E. Curr Biol. 1999;9(20):1191–4. doi: 10.1016/S0960-9822(00)80024-X. [DOI] [PubMed] [Google Scholar]
- 33.Dealy MJ, Nguyen KV, Lo J, et al. Loss of Cul1 results in early embryonic lethality and dysregulation of cyclin E. Nat Genet. 1999;23(2):245–8. doi: 10.1038/13886. [DOI] [PubMed] [Google Scholar]
- 34.Harris KF, Shoji I, Cooper EM, Kumar S, Oda H, Howley PM. Ubiquitin-mediated degradation of active Src tyrosine kinase. Proc Natl Acad Sci U S A. 1999;96(24):13738–43. doi: 10.1073/pnas.96.24.13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rogers GC, Rusan NM, Roberts DM, Peifer M, Rogers SL. The SCF Slimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. J Cell Biol. 2009;184(2):225–39. doi: 10.1083/jcb.200808049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leung GC, Ho CS, Blasutig IM, Murphy JM, Sicheri F. Determination of the Plk4/Sak consensus phosphorylation motif using peptide spots arrays. FEBS Lett. 2007;581(1):77–83. doi: 10.1016/j.febslet.2006.11.080. [DOI] [PubMed] [Google Scholar]
- 37.Lovly CM, Yan L, Ryan CE, Takada S, Piwnica-Worms H. Regulation of Chk2 ubiquitination and signaling through autophosphorylation of serine 379. Mol Cell Biol. 2008;28(19):5874–85. doi: 10.1128/MCB.00821-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van de Weerdt BC, Medema RH. Polo-like kinases: a team in control of the division. Cell Cycle. 2006;5(8):853–64. doi: 10.4161/cc.5.8.2692. [DOI] [PubMed] [Google Scholar]
- 39.Yamashita Y, Kajigaya S, Yoshida K, et al. Sak serine-threonine kinase acts as an effector of Tec tyrosine kinase. J Biol Chem. 2001;276(42):39012–20. doi: 10.1074/jbc.M106249200. [DOI] [PubMed] [Google Scholar]
- 40.Okuda M, Horn HF, Tarapore P, et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell. 2000;103(1):127–40. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 41.Fisk HA, Winey M. The mouse Mps1p-like kinase regulates centrosome duplication. Cell. 2001;106(1):95–104. doi: 10.1016/s0092-8674(01)00411-1. [DOI] [PubMed] [Google Scholar]
- 42.Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD. CP110, a cell-cycle-dependent cdk substrate, regulates centrosome duplication in human cells. Dev Cell. 2002;3:339–50. doi: 10.1016/s1534-5807(02)00258-7. [DOI] [PubMed] [Google Scholar]
- 43.Kasbek C, Yang CH, Yusof AM, Chapman HM, Winey M, Fisk HA. Preventing the degradation of mps1 at centrosomes is sufficient to cause centrosome reduplication in human cells. Mol Biol Cell. 2007;18(11):4457–69. doi: 10.1091/mbc.E07-03-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duensing A, Liu Y, Tseng M, Malumbres M, Barbacid M, Duensing S. Cyclin-dependent kinase 2 is dispensable for normal centrosome duplication but required for oncogene-induced centrosome overduplication. Oncogene. 2006;25:2943–9. doi: 10.1038/sj.onc.1209310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guardavaccaro D, Kudo Y, Boulaire J, et al. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev Cell. 2003;4(6):799–812. doi: 10.1016/s1534-5807(03)00154-0. [DOI] [PubMed] [Google Scholar]
- 46.Dunbar AJ, Gondek LP, O’Keefe CL, et al. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68(24):10349–57. doi: 10.1158/0008-5472.CAN-08-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kramer A, Neben K, Ho AD. Centrosome aberrations in hematological malignancies. Cell Biol Int. 2005;29(5):375–83. doi: 10.1016/j.cellbi.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 48.Parvin JD. The BRCA1-dependent ubiquitin ligase, gamma-tubulin, and centrosomes. Environ Mol Mutagen. 2009 doi: 10.1002/em.20475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.