

Abstract
Akt plays a role in protecting macrophages from apoptosis induced by some oxysterols Previously we observed enhanced degradation of Akt in P388D1 moncocyte/macrophages following treatment with 25-hydroxycholesterol (25-OH) or 7-ketocholesterol (7-KC). In the present report we examine the role of the ubiquitin proteasomal pathway in this process. We show that treatment with 25-OH or 7-KC results in the accumulation of poly-ubiquitinated Akt, an effect that is enhanced by co-treatment with the proteasome inhibitor MG-132. Modification of Akt by the addition of a Gly-Ala repeat (GAr), a domain known to block ubiquitin-dependent targeting of proteins to the proteasome, resulted in a chimeric protein that is resistant to turn-over induced by 25-OH or 7-KC and provides protection from apoptosis induced by these oxysterols. These results uncover a new aspect of oxysterol regulation of Akt in macrophages; oxysterol-stimulated poly-ubiquitination of Akt and degradation by the proteasomal pathway.
Keywords: oxysterols, 7-ketocholesterol, 25-hydroxycholesterol, Akt, apoptosis, ubiquitination, proteasome, Gly-Ala repeat
Introduction
Although the role of the phosphorylation mediated regulation of Akt/PKB activity by PI3-kinase [1] and PP2A [2, 3] has been extensively studied, the regulation of Akt levels in cells has been less well analyzed. One such process is regulation of the proteolytic degradation of Akt. Akt can undergo both caspase-dependent [4] and independent degradation [5] in response to proapoptotic signals. However, there is evidence that of involvement of the proteasome in Akt degradation [6, 7], as well. In some instances, proteasomal degradation of Akt, accompanied by ubiquitination takes place subsequent to caspase-6 cleavage [8]. It is also noteworthy that ubiquitin-independent proteasomal degradation can be observed for a number of proteins [9] and there is some evidence that Akt can be degraded without ubiquitination [10]. Perhaps the most widely studied pathway of degradation of Akt, is that induced by Hsp90 inhibitors, such as geldanomycin and 17-allylaminogeldanomycin, which are in clinical trials as anti-cancer drugs [11]. These compounds promote proteasomal degradation of Akt, and other proteins, by a caspase-independent, ubiquitination-dependent pathway[12, 13]. Thus, there are non-proteosomal and proteosomal pathways that regulate cellular Akt levels.
We previously reported that the oxysterols, 25-hydroxycholesterol (25-OH) and 7-ketocholesterol (7-KC), induce apoptosis in a monocyte/macrophage cell line, P388D1 cells, by a process involving Akt degradation and that a proteasome inhibitor blocked the oxysterol-induced Akt degradation[14]. This process is of potential clinical interest since oxysterol-induced apoptosis has been widely reported for vascular cells with potential pro and anti-atherogenic effects depending on the stage of plaque development and the tissue affected[15, 16].
To further explore the mechanism by which 7-KC and 25-OH induce Akt degradation we have attempted to create a proteasome-resistant Akt. The Gly-Ala repeat (GAr) domain of the Epstein-Barr virus (EBV) nuclear antigen-1 (EBNA-1) has been reported[17] to be a modulator of proteasomal-dependent proteolysis. The GAr interferes with the proteasomal processing of EBNA-1, prolonging its half-life and abrogating the presentation of EBNA-1 epitopes to the major histocompatibility complex class I-restricted CD8+ T-lymphocytes [18, 19]. The GAr acts as a cis-acting transferable element on different proteasome substrates, providing an attractive tool for regulating the proteolysis of many substrates of potential interest [20, 21]. In the current study, a myr-Akt-GAr chimera was constructed and stably expressed in P388D1 cells in order to gain further understanding of oxysterol regulation of Akt activity by degradation. Since Akt activity mediates many important biological processes and is an emerging target in cancer chemotherapy, gaining further understanding of regulation of Akt activity by degradation could illuminate novel regulation of many downstream effectors of Akt.
Materials and methods
Construction of the myr-Akt-GAr Chimera
A plasmid expressing a Myc-His-tagged, myristoylation-activated murine Akt1 (myr-Akt1) was purchased from Upstate Biotechnology, Inc (Charlottesville, VA). An Xba1 site located at 935 bp in myr-Akt1 was removed by conserved site-directed mutagenesis of a serine codon (TCT→TCC). Then a pair of synthetic oligonucleotides encoding a 29 aa sequence of the EBV EBNA1 GAr domain and containing Xba1 overhangs (underlined) (5′-CTAGAGCTGGAGCAGGCGGTGGAGCAGGTGCTGGAGGTGCAGGTGGAGCAGGCGGTGCAGGAGCAGGTGGTGCAGGTGCTGGAGGTGGAGCAGGTT-3′ and 5′ -CTAGAACCTGCTCCACCTCCAGCACCTGCACCACCTGCTCCTGCACCGCCTGCTC CACCTGCACCTCCAGCACCTGCTCCACCGCCTGCTCCAGCT-3′) were annealed and ligated into the Xba1 site at nucleotide 2461. The resulting plasmid, myr-Akt-GAr, encodes a protein that is identical to myr-Akt1 except for the addition of the 29 a.a. GAr domain between the C-terminus of Akt and the myc-his tag. The construct was confirmed by sequencing using upstream primer 5′ -GCAGCACCGGTTCTTTGC- 3′ and downstream primer 5′ –GAAGGGCCCTCTAGAACCTGC- 3′.
Cell culture, Transfection and Stable cell line selection
P388D1 cells were maintained in a humidified atmosphere of 5% CO2 and 95% air at 37°C, and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells were transfected with the plasmid expressing Myr-Akt-GAr chimera using Lipofectamine Plus (Invitrogen) according to the manufacturer's instructions. Stable clones (Myr-Akt1-GAr P388D1) were selected by serial dilution in medium containing 1 mg/ml G418 for 7 days, followed by 14 days incubation with 0.25 mg/ml G418. G418 resistant clones were screened by immunoblotting using anti-Myc (Upstate Biotechnology, Inc.) and anti-Akt1/2(N-19) antibodies (Santa Cruz Biotechnology, Santa Cruz, CA).
Cell Lysis, Immunopreciptitation and Immunoblotting
Cells were collected by centrifugation at 1,000 × g for 5 minutes, rinsed by suspending in ice-cold PBS and collected by centrifugation again. Cell pellets were suspended in modified RIPA buffer containing 50 mM Tris (pH 7.4), 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA supplemented with protease inhibitors (Pierce). Lysates were incubated on ice for 10 min, and centrifuged at 14,000 × g for 10 min to remove the insoluble debris and protein concentration was measured by micro-BCA assay (Pierce). Standard procedures were applied for immunoprecipitation of Akt using anti-Akt1/2(N-19). Proteins were separated by SDA-PAGE on 4-12% NuPAGE gels (Invitrogen) and transferred to PVDF membranes (Immobilon-P, Millipore). The blots were probed using antibodies specific for the protein of interest and the HRP-conjugated secondary antibodies. Ubiquitinated conjugates were detected by immunoblotting with an anti-Ubiquitin antibody (Boston Biochem, Cambridge, MA). Visualization was achieved by SuperSignal® West Pico chemiluminescent kit (Pierce).
Caspase-3 Assay
Cells were seeded at 1 × 106 / well in 12-well culture plates, and treated with oxysterols, purchased from Steraloids (Wilton, NH), or an equivalent volume of solvent (ethanol). Following an incubation period, the cells were collected, washed in ice-cold PBS, and resuspended in lysis buffer (10 mM Tris (pH 7.5), 130 mM NaCl, 1% Triton X-100, 10 mM sodium Pi, and 10 mM sodium PPi). Cell lysates were cleared by centrifugation at 14,000 × g at 4 °C for 20 min and assayed for protein concentration using a micro-BCA kit (Pierce) and for caspase-3 activity as follows. Equal volumes were incubated at 37 °C for 2.5 h in 20 mM Hepes, pH 7.5, 10% glycerol, and 2 mM dithiothreitol containing 5 μM caspase-3 substrate Ac-DEVD-AFC in the presence and absence of 100 nM Ac-DEVD-CHO, a caspase-3 specific inhibitor, added 30 min prior to the addition of the substrate. The released AFC was measured using a spectrofluorometer (FluroMax 3, Jobin-Yvon Inc.) equipped with a microplate reader at an excitation wavelength of 400 nm and an emission wavelength of 505 nm. Net caspase-3 activity was determined by subtracting the relative fluorescent light units (RFLU) obtained in the presence of the inhibitor from the RFLU obtained in the absence of the inhibitor, and then normalized to the protein content of the sample. Treatments were performed in triplicate and the data presented as the mean induction ± S.D.
Akt Kinase Assay
Akt activity was measured using an Akt kinase assay kit from Cell Signaling Technology, Inc. following the manufacturer's directions. Essentially, an immobilized Akt monoclonal antibody was used to selectively immunoprecipitate Akt from cell lysates. The immunoprecipitated Akt was then incubated with a substrate, glycogen synthase kinase-3 fusion protein (GSK-3), in kinase buffer in the presence of ATP. The phosphorylation of glycogen synthase kinase-3 (Ser21/9) was then measured by immunoblotting using a phospho-GSK-3 (Ser21/9) antibody.
Pulse-Chase Labeling and Immunoprecipitation of Akt
Cells were grown in Met/Cys deficient DMEM medium supplemented with 5% FBS for 30 min and then pulsed with Tran35S-label (250 μCi/ml) for 1 h, washed and then chased for 30, 60, 90 and 240 min in medium supplemented with 3 mM Met/Cys and either 10 μg/ml 7-KC or 25-OH or an equivalent amount of vehicle (ethanol). Cells were lysed in RIPA buffer and subjected to immunoprecipitation using goat polyclonal Akt1/2 antibody (Santa Cruz Biotechnology, Inc.) and Seize® coated plate immunoprecipitation kits (Pierce) according to the manufacturer's instructions. The amount of radiolabeled Akt in the precipitates was determined by SDS-PAGE and phosphorimaging.
Results and discussion
25-hydroxycholesterol and 7-ketocholesterol up-regulate the ubiquitination of Akt which targets it for degradation via the proteasome
A previous report[14] from our laboratory indicated that 25-OH and 7-KC could produce a reduction in the level of total Akt in P388D1 cells by enhancing its rate of degradation. We were able to show that a proteasome inhibitor blocked enhanced degradation of Akt in response to 25-OH treatment which is consistent with our hypothesis that these oxysterols regulate degradation of Akt via the proteasome. Proteasomal degradation of proteins can be either ubiquitination dependent or independent[9]. To assess any role for ubiquitination in oxysterol-regulated degradation of Akt we determined the effect of 25-OH and 7-KC on the ubiquitination of Akt. To do this, Akt was immunoprecipitated from oxysterol-treated P388D1 cells and then assessed for derivatization with ubiquitin by immunoblotting with a ubiquitin antibody. The study was limited to 25-OH and 7-KC as, among the oxysterols examined; 7-betahydroxycholesterol, 7-ketocholesterol, 24-hydroxycholesterol, 25-hydroxycholesterol and 27-hydroxycholesterol, only 25-OH and 7-KC displayed a significant ability to induce apoptosis in P388D1 cells as determined by induction of caspase 3 activity (Figure 1). The results demonstrate that treatment of P388D1 cells with 25-OH or 7-KC produces ubiquitination of Akt (Figure 2). The accumulation of ubiquitinated Akt is considerably enhanced by co-treatment with the proteasome inhibitor MG-132 (Figure 2B). These findings are consistent with the hypothesis that stimulation of Akt degradation by 25-OH and 7-KC is via ubiquitination-dependent targeting of Akt to the proteasome.
Fig.1.
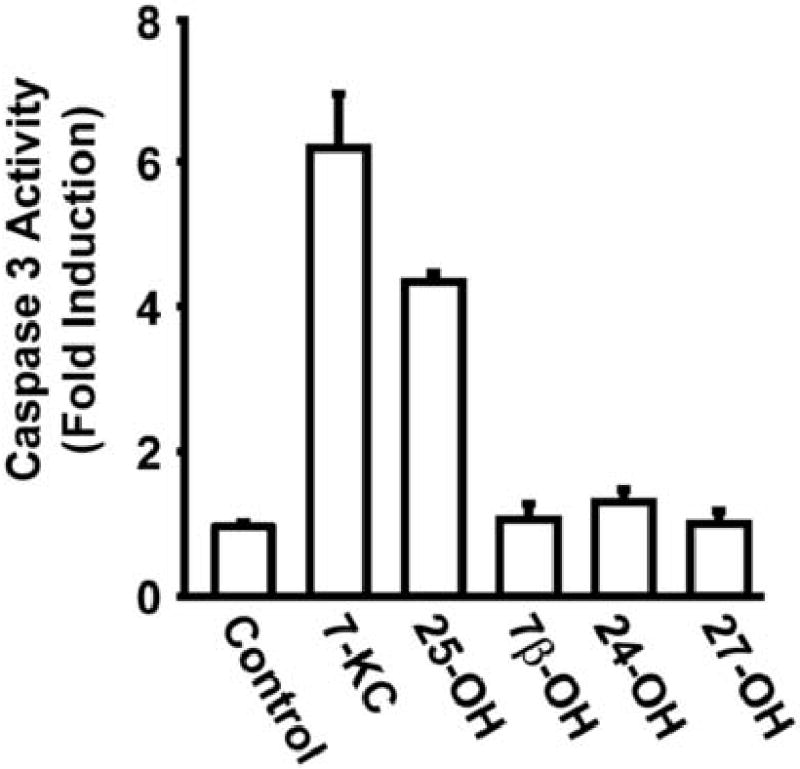
Activation of apoptosis by oxysterols in P388D1 monocyte/macrophages. P388D1 cells were culture for 30 h in the presence of (15 μg/ml) 7-betahydroxycholesterol (7β–OH), 7-ketocholesterol (7-KC), 24-hydroxycholesterol (24-OH), 25-hydroxycholesterol (25-OH) or 27-hydroxycholesterol (27-OH) as indicated in the figure. Caspase 3 activity in total cell lysates was determined as describe in Materials and methods. Only 7-KC and 25-OH treatment resulted in significant induction of caspase -3 activity in P388D1 cells.
Fig. 2.
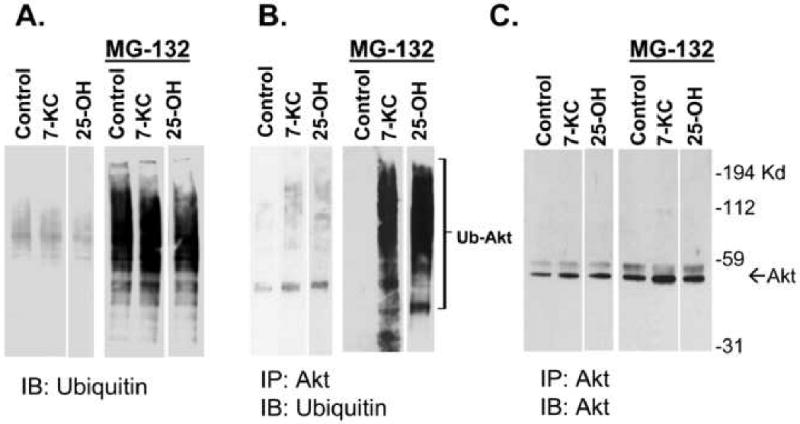
Stimulation of Akt ubiquitination by 7-ketocholesterol and 25-hydroxycholesterol in P388D1 macrophages. (A) Total cell lysates prepared from P388D1 cells treated with 7-KC or 25-OH (10 μg/ml) in the presence and absence of a proteasome inhibitor, MG-132 (30 μM), were subjected to immunoblotting with an anti-ubiquitin antibody. (B) To detect ubiquitinated Akt, Akt was immunoprecipitated and subjected to immunoblot analysis using ubiquitin antibody. Treatment with the proteasome inhibitor resulted in a large increase in ubiquitinated Akt. (C) Stripping and reprobing of the blot shown in (B) with an Akt antibody reveals that equivalent amounts of immunoprecipitated Akt were loaded on the gel. The arrow and brackets indicate the location of Akt and ubiquitinated Akt protein bands on the blots.
25-hydroxycholesterol and 7-ketocholesterol do not activate degradation of a proteasome- resistant mutant of Akt
Since ubiquitination can signal other events besides proteasomal degradation[22], we sought another approach to testing the hypothesis that these oxysterols induce proteasome degradation of Akt. Studies of other proteins degraded via the proteasome, subsequent to ubiquitination, have indicated that a GAr domain can block ubiquitination-dependent targeting to the proteasome. The effect of a GAr is much like that of a proteasome inhibitor and acts to suppress degradation of the ubiquitinated protein. However, it has the advantage over proteasome inhibitors of only targeting the protein of interest thus reducing the possibility of non-specific effects associated with proteasome inhibitors. Since Akt is ubiquitinated in response to oxysterol treatment, we predicted that Akt modified with a GAr would be resistant to down-regulation by oxysterols if the ubiquitination was targeting degradation via the proteasome. Since one of our eventual goals in this study is to develop an oxysterol-resistant Akt we chose as our base construct a constitutively activated (myristoylated) Akt (myr-Akt) which we modified to contain a carboxyl terminal GAr domain as described in Methods. We have previously reported that wild type Akt and myr-Akt levels decline in cells treated with 25-OH or 7-KC [14]. To examine the effect of oxysterols on myr-Akt-GAr chimera, both wild type P388D1 cells and P388D1 cells stably expressing myr-Akt-GAr (myr-Akt-GAr P388D1) were treated with10 μg/ml 7-KC for various periods of time. The level of Akt in wild-type cells, as well as the endogenous wild-type Akt levels in myr-Akt-GAr P388D1 cells, decreased following treatment with 7-KC. However, myr-Akt-GAr levels were increased by 7-KC treatment compared (Figure 3A).
Fig. 3.
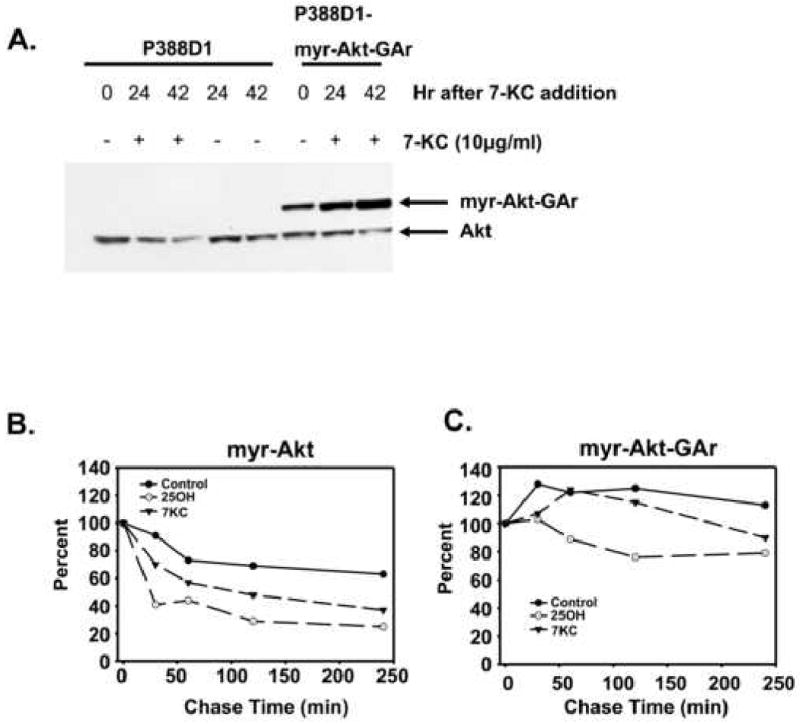
Addition of a GA repeat to Akt prevents its regulated degradation by 7-ketocholesterol and 25-hydroxycholesterol. myr-Akt-GAr is myc tagged and constitutively activated by myristoylation. (A) Wild-type P388D1 cells or P388D1 cells expressing myr-Akt-GAr were treated with 7-ketocholesterol for various periods of time as indicated and cell lysates were examined for Akt levels by immunoblotting with an Akt antibody. (B) P388D1 cells expressing myc tagged myristoylated Akt or (C) myc-tagged myristoylated Akt-GAr were pulsed with S35 methionine and chased with unlabeled methionine for various periods of time in the presence and absence of 7-KC or 25-OH. The ecotopic Akt was immunoprecipitated with an antibody specific for the myc epitope.
Clone B cells are P388D1 cells stably expressing myr-Akt, the parent construct of myr-Akt-GAr [14]. We performed pulse-chase experiments to examine the degradation rates of myr-Akt in clone B cells and myr-Akt-GAr in P388D1 after treatment with 7-KC or 25-OH. The result showed that both 7-KC and 25-OH enhanced the rate of myr-Akt degradation (Figure 3B) but not myr-Akt-GAr (Figure 3C). These results indicate that these oxysterols reduce Akt levels in P388D1 cells, at least in part, by upregulating Akt degradation through the ubiquitination-proteasome pathway.
Myr-Akt-GAr is active and protects cells from oxysterol-induced apoptosis
Since regulation of Akt reflects activity as well as expression, the functional significance of myr-Akt-GAr expression was assessed at the level of kinase activity. Clone B and myr-Akt-GAr P388D1 cells were treated with 10 μg/ml 7-KC and cell lysates were assayed for Akt kinase activity using GSK-3 as a substrate (Figure 4A). Both forms of myr-Akt were active. An increase in GSK-3 phosphorylation by myr-Akt-GAr P388D1 cell lysates was observed indicating increased activity as well as the increased level of myr-Akt-GAr in response to 7-KC treatment seen in Figure 3A. Therefore, the myr-Akt-GAr chimera retained the functional properties of wild type Akt. As expected, Akt activity was not detectable in wild type cells, even in the absence of oxysterol treatment. Consistent with the increased Akt kinase activity, immunoblotting using an antibody specific for phospho-Akt (Ser473) showed an increased level of phospho-Akt, the active form of Akt, in myr-Akt-GAr P388D1 cells following treatment with 7-KC (Figure 4B).
Fig. 4.
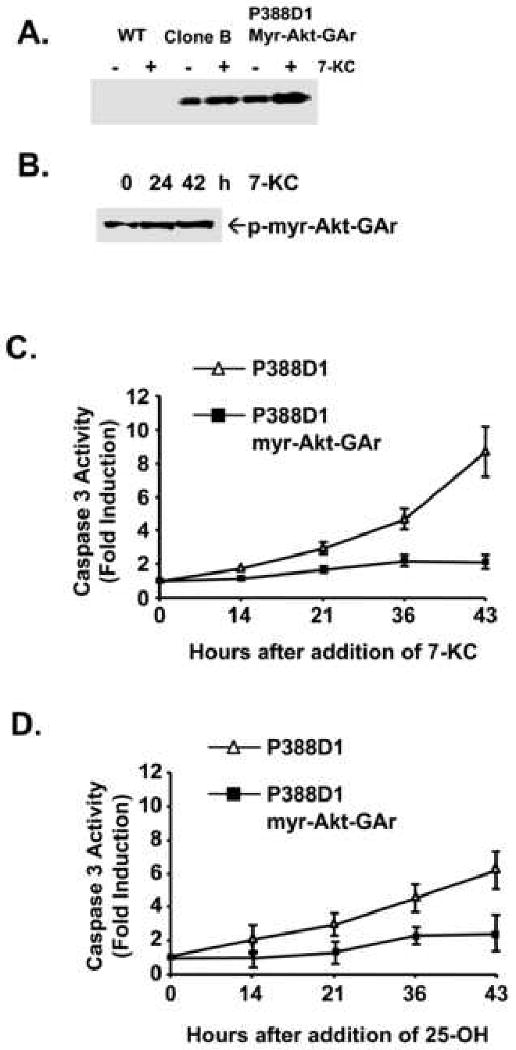
myr-Akt-GAr is biochemically active. (A) Akt activity in wild type P388D1 cells, clone B, and myr-Akt-GAr cells after treatment with 10 μg/ml 7-KC for 16 hours. The Akt kinase activity was assayed using GSK-3 as a substrate for phosphorylation as described in the Materials and Methods. The phospho-GSK-3 formed is visualized by immunoblotting with antibody specific for phospho-GSK-3 (Ser21/9). (B) Activation of myr-Akt-GAr as determined by immunoblotting with a phospho-Akt(ser473) antibody. (C and D) The myr-Akt-GAr construct confers sustained resistance to 7-KC or 25-OH (15 μg/ml) induced apoptosis as determined by measurement of caspase 3 activity.
Akt has been well characterized as an anti-apoptotic kinase that transduces survival signals in many cell types including macrophages[23-25]. To examine whether the expression of myr-Akt-GAr chimera protein could provide resistance to oxysterol-induced apoptosis, cells were treated with 15 μg/ml 7-KC or 25-OH for different periods of time and assayed for caspase-3 activity. Consistent with the increased kinase activity described above, only a slight increase in the induction of caspase-3 activity was detected in myr-Akt-GAr P388D1 cells following treatment with 7-KC or 25-OH (Figure 4C and 4D, respectively). In contrast, caspase-3 activity was significantly increased in wild type cells by treatment with 7-KC or 25-OH. These results demonstrate that 7-KC and 25-OH induce ubiquitin-dependent degradation of Akt in P388D1 cells. Furthermore, inhibition of this degradation, particularly with a constitutively active form of Akt, partially rescues P388D1 cells from induction of apoptosis by these oxysterols.
Oxysterols affect multiple pro- and anti- apoptotic signaling pathways in macrophages [26]. We partially characterized the steps in one of the oxysterol-induced pro-apoptotic pathways as involving a calcium-influx mediated activation of cytosolic phospholipase A2 resulting in the production of arachidonic acid which then is utilized as a substrate by acyl coenzyme A: cholesterol transferase (ACAT) to esterify the oxysterol [14, 27-29]. The subsequent steps in the pathway are unknown and currently under investigation.
Cannabinoids protect neuronal cells from apoptosis induced by a variety of insults by modulating Akt [30] and recent work from our laboratory showed macrophages lacking the cannabinoid type 2 receptor are resistant to 7-KC-induced apoptosis and 7-KC-induced dephosphorylation of Akt [31]. Combined with the result of the current study, these studies indicate that oxysterol regulation of Akt in macrophages occurs by multiple mechanisms and that the integration of the signals controlling these mechanisms is likely to be critical in determining the eventual fate of the macrophage.
A few studies have investigated the effects of oxLDL, and/or 7-ketocholesterol, on derivatization of proteins in other vascular cell types, smooth muscle cells (SMC) and endothelial cells [32-34]. In endothelial cells, OxLDL induces ubiquitination, as well as derivatizations with 4-hydroxy-2-nonenal (4-HNE), of cellular proteins [32]. In vascular SMC, 7-KC induces immunoreactivity for 4-HNE and enhanced ubiquitination of cellular proteins [34]. In these studies, treatment with a proteasome inhibitor was demonstrated to potentiate the toxic effects of oxLDL or 7-KC [32, 34]. In contrast, our results (Fig 4C and 4D) demonstrate that expression of a degradation-resistant Akt protects P388D1 cells from oxysterol-induced apoptosis. This difference may be the result of cell specific responses to proteasome inhibition, or, more likely, reflect the difference between selectively inhibiting the degradation of a strong survival signal, AKt, versus generalized inhibition of all proteasomal targeted proteins within the cell. The endothelial isoform of nitric oxide synthase (eNOS) is carbonylated and nitrated in endothelial cells after exposure to oxLDL and undergoes proteasomal degradation [33]. Whether oxysterols induce other forms of Akt derivatization in P388D1 cells is unknown and currently under investigation.
Alteration of the phosphorylation status of several proteins has been shown to create recognition sites for ubiquitin ligases [35], and phosphorylation/activation-induced ubiquitination and proteasomal degradation of other kinases has been suggested to be a mechanism preventing the inappropriate accumulation of activated kinases in cells[36, 37]. Akt has several sequences known to affect its activity or stability [38]. In the current study, 7-KC treatment did not stimulate Akt activity (Fig 4A) or Ser473 phosphorylation (Fig 4B) suggesting that increased Akt ubiquitination and degradation is not dependent upon Akt phosphorylation at Ser473 or Akt activation. Alternatively, the effect of oxysterol treatment might be to induce conformation changes which unmask or activate destabilizing sequences. PEST sequences mediate rapid turn over of proteins [39] and a putative PEST sequence in the N-terminal region of Akt as been identified [38]. Regardless of the mechanism(s) involved in destabilizing Akt, our results document a new aspect of oxysterol regulation of Akt, ubiquitination and targeted proteasomal degradation.
Since activated Akt is found in lesions and appears to play an important role in determining macrophage survival, we anticipate that the development of a degradation-resistant form of Akt, myr-Akt-GAr, could prove very useful in developing a further understanding of the functional significance of oxysterol-induced Akt degradation in atherosclerosis. As the phenotype produced by such a construct in whole animals would be dominant we expect that the development of transgenic mice expressing myr-Akt-GAr under the control of a macrophage specific promoter would be informative in studying the role of oxysterol-induced Akt degradation in lesion development and progression.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grant HL085137 (D.P.T).
Abbreviations
- 7-KC
7-ketocholesterol
- 25-OH
25-hydroxycholesterol
- GAr
glycine alanine repeat
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res. 1999;253:210–229. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- 2.Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24:186–191. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- 3.Resjo S, Goransson O, Harndahl L, Zolnierowicz S, Manganiello V, Degerman E. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell Signal. 2002;14:231–238. doi: 10.1016/s0898-6568(01)00238-8. [DOI] [PubMed] [Google Scholar]
- 4.Mann KK, Colombo M, Miller WH., Jr Arsenic trioxide decreases AKT protein in a caspase-dependent manner. Mol Cancer Ther. 2008;7:1680–1687. doi: 10.1158/1535-7163.MCT-07-2164. [DOI] [PubMed] [Google Scholar]
- 5.Martin D, Salinas M, Fujita N, Tsuruo T, Cuadrado A. Ceramide and reactive oxygen species generated by H2O2 induce caspase-3-independent degradation of Akt/protein kinase B. J Biol Chem. 2002;277:42943–42952. doi: 10.1074/jbc.M201070200. [DOI] [PubMed] [Google Scholar]
- 6.Yan D, Guo L, Wang Y. Requirement of dendritic Akt degradation by the ubiquitin-proteasome system for neuronal polarity. J Cell Biol. 2006;174:415–424. doi: 10.1083/jcb.200511028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan Y, Xie P, Zhang H, Guo S, Gu D, She M, Li H. Proteasome-dependent inactivation of Akt is essential for 12-O-tetradecanoylphorbol 13-acetate-induced apoptosis in vascular smooth muscle cells. Apoptosis. 2008;13:1401–1409. doi: 10.1007/s10495-008-0272-z. [DOI] [PubMed] [Google Scholar]
- 8.Medina EA, Afsari RR, Ravid T, Castillo SS, Erickson KL, Goldkorn T. Tumor necrosis factor-{alpha} decreases Akt protein levels in 3T3-L1 adipocytes via the caspase-dependent ubiquitination of Akt. Endocrinology. 2005;146:2726–2735. doi: 10.1210/en.2004-1074. [DOI] [PubMed] [Google Scholar]
- 9.Orlowski M, Wilk S. Ubiquitin-independent proteolytic functions of the proteasome. Arch Biochem Biophys. 2003;415:1–5. doi: 10.1016/s0003-9861(03)00197-8. [DOI] [PubMed] [Google Scholar]
- 10.Adachi M, Katsumura KR, Fujii K, Kobayashi S, Aoki H, Matsuzaki M. Proteasome-dependent decrease in Akt by growth factors in vascular smooth muscle cells. FEBS Lett. 2003;554:77–80. doi: 10.1016/s0014-5793(03)01109-8. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med. 2004;82:488–499. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- 12.Doong H, Rizzo K, Fang S, Kulpa V, Weissman AM, Kohn EC. CAIR-1/BAG-3 abrogates heat shock protein-70 chaperone complex-mediated protein degradation: accumulation of poly-ubiquitinated Hsp90 client proteins. J Biol Chem. 2003;278:28490–28500. doi: 10.1074/jbc.M209682200. [DOI] [PubMed] [Google Scholar]
- 13.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277:39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 14.Rusinol AE, Thewke D, Liu J, Freeman N, Panini SR, Sinensky MS. AKT/protein kinase B regulation of BCL family members during oxysterol-induced apoptosis. J Biol Chem. 2004;279:1392–1399. doi: 10.1074/jbc.M308619200. [DOI] [PubMed] [Google Scholar]
- 15.Colles SM, Maxson JM, Carlson SG, Chisolm GM. Oxidized LDL-induced injury and apoptosis in atherosclerosis. Potential roles for oxysterols. Trends Cardiovasc Med. 2001;11:131–138. doi: 10.1016/s1050-1738(01)00106-2. [DOI] [PubMed] [Google Scholar]
- 16.Panini SR, Sinensky MS. Mechanisms of oxysterol-induced apoptosis. Curr Opin Lipidol. 2001;12:529–533. doi: 10.1097/00041433-200110000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Levitskaya J, Coram M, Levitsky V, Imreh S, Steigerwald-Mullen PM, Klein G, Kurilla MG, Masucci MG. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature. 1995;375:685–688. doi: 10.1038/375685a0. [DOI] [PubMed] [Google Scholar]
- 18.Levitskaya J, Sharipo A, Leonchiks A, Ciechanover A, Masucci MG. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc Natl Acad Sci U S A. 1997;94:12616–12621. doi: 10.1073/pnas.94.23.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blake N, Lee S, Redchenko I, Thomas W, Steven N, Leese A, Steigerwald-Mullen P, Kurilla MG, Frappier L, Rickinson A. Human CD8+ T cell responses to EBV EBNA1: HLA class I presentation of the (Gly-Ala)-containing protein requires exogenous processing. Immunity. 1997;7:791–802. doi: 10.1016/s1074-7613(00)80397-0. [DOI] [PubMed] [Google Scholar]
- 20.Sharipo A, Imreh M, Leonchiks A, Imreh S, Masucci MG. A minimal glycine-alanine repeat prevents the interaction of ubiquitinated I kappaB alpha with the proteasome: a new mechanism for selective inhibition of proteolysis. Nat Med. 1998;4:939–944. doi: 10.1038/nm0898-939. [DOI] [PubMed] [Google Scholar]
- 21.Zhang M, Coffino P. Repeat sequence of Epstein-Barr virus-encoded nuclear antigen 1 protein interrupts proteasome substrate processing. J Biol Chem. 2004;279:8635–8641. doi: 10.1074/jbc.M310449200. [DOI] [PubMed] [Google Scholar]
- 22.Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- 23.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10:262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 25.Liu H, Perlman H, Pagliari LJ, Pope RM. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages. Role of Mcl-1, independent of nuclear factor (NF)-kappaB, Bad, or caspase activation. J Exp Med. 2001;194:113–126. doi: 10.1084/jem.194.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berthier A, Lemaire-Ewing S, Prunet C, Montange T, Vejux A, Pais de Barros JP, Monier S, Gambert P, Lizard G, Neel D. 7-Ketocholesterol-induced apoptosis. Involvement of several pro-apoptotic but also anti-apoptotic calcium-dependent transduction pathways. Febs J. 2005;272:3093–3104. doi: 10.1111/j.1742-4658.2005.04723.x. [DOI] [PubMed] [Google Scholar]
- 27.Panini SR, Yang L, Rusinol AE, Sinensky MS, Bonventre JV, Leslie CC. Arachidonate metabolism and the signaling pathway of induction of apoptosis by oxidized LDL/oxysterol. J Lipid Res. 2001;42:1678–1686. [PubMed] [Google Scholar]
- 28.Rusinol AE, Yang L, Thewke D, Panini SR, Kramer MF, Sinensky MS. Isolation of a somatic cell mutant resistant to the induction of apoptosis by oxidized low density lipoprotein. J Biol Chem. 2000;275:7296–7303. doi: 10.1074/jbc.275.10.7296. [DOI] [PubMed] [Google Scholar]
- 29.Freeman NE, Rusinol AE, Linton M, Hachey DL, Fazio S, Sinensky MS, Thewke D. Acyl-coenzyme A:cholesterol acyltransferase promotes oxidized LDL/oxysterol-induced apoptosis in macrophages. J Lipid Res. 2005;46:1933–1943. doi: 10.1194/jlr.M500101-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molina-Holgado E, Vela JM, Arevalo-Martin A, Almazan G, Molina-Holgado F, Borrell J, Guaza C. Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J Neurosci. 2002;22:9742–9753. doi: 10.1523/JNEUROSCI.22-22-09742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freeman-Anderson NE, Pickle TG, Netherland CD, Bales A, Buckley NE, Thewke DP. Cannabinoid (CB2) receptor deficiency reduces the susceptibility of macrophages to oxidized LDL/oxysterol-induced apoptosis. J Lipid Res. 2008;49:2338–2346. doi: 10.1194/jlr.M800105-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vieira O, Escargueil-Blanc I, Jurgens G, Borner C, Almeida L, Salvayre R, Negre-Salvayre A. Oxidized LDLs alter the activity of the ubiquitin-proteasome pathway: potential role in oxidized LDL-induced apoptosis. Faseb J. 2000;14:532–542. doi: 10.1096/fasebj.14.3.532. [DOI] [PubMed] [Google Scholar]
- 33.Steffen Y, Jung T, Klotz LO, Schewe T, Grune T, Sies H. Protein modification elicited by oxidized low-density lipoprotein (LDL) in endothelial cells: protection by (-)-epicatechin. Free Radic Biol Med. 2007;42:955–970. doi: 10.1016/j.freeradbiomed.2006.12.024. [DOI] [PubMed] [Google Scholar]
- 34.Martinet W, De Bie M, Schrijvers DM, De Meyer GR, Herman AG, Kockx MM. 7-ketocholesterol induces protein ubiquitination, myelin figure formation, and light chain 3 processing in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:2296–2301. doi: 10.1161/01.ATV.0000146266.65820.a1. [DOI] [PubMed] [Google Scholar]
- 35.Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, Hung MC. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27:4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu Z, Liu D, Hornia A, Devonish W, Pagano M, Foster DA. Activation of protein kinase C triggers its ubiquitination and degradation. Mol Cell Biol. 1998;18:839–845. doi: 10.1128/mcb.18.2.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 38.Lee JH, Shin SH, Kang S, Lee YS, Bae S. A novel activation-induced suicidal degradation mechanism for Akt by selenium. Int J Mol Med. 2008;21:91–97. [PubMed] [Google Scholar]
- 39.Garcia-Alai MM, Gallo M, Salame M, Wetzler DE, McBride AA, Paci M, Cicero DO, de Prat-Gay G. Molecular basis for phosphorylation-dependent, PEST-mediated protein turnover. Structure. 2006;14:309–319. doi: 10.1016/j.str.2005.11.012. [DOI] [PubMed] [Google Scholar]