

Abstract
Remodeling of central excitatory synapses is crucial for synapse maturation and plasticity, and contributes to neurodevelopmental and psychiatric disorders. Remodeling of dendritic spines and the associated synapses has been postulated to require the coordination of adhesion with spine morphology and stability; however, the molecular mechanisms that functionally link adhesion molecules with regulators of dendritic spine morphology are mostly unknown. Here, we report that spine size and N-cadherin content are tightly coordinated. In rat mature cortical pyramidal neurons, N-cadherin-dependent adhesion modulates the morphology of existing spines by recruiting the Rac1 guanine-nucleotide exchange factor kalirin-7 to synapses through the scaffolding protein AF-6/afadin. In pyramidal neurons, N-cadherin, AF-6, and kalirin-7 colocalize at synapses and participate in the same multiprotein complexes. N-cadherin clustering promotes the reciprocal interaction and recruitment of N-cadherin, AF-6, and kalirin-7, increasing the content of Rac1 and in spines and PAK (p21-activated kinase) phosphorylation. N-cadherin-dependent spine enlargement requires AF-6 and kalirin-7 function. Conversely, disruption of N-cadherin leads to thin, long spines, with reduced Rac1 contact, caused by uncoupling of N-cadherin, AF-6, and kalirin-7 from each other. By dynamically linking N-cadherin with a regulator of spine plasticity, this pathway allows synaptic adhesion molecules to rapidly coordinate spine remodeling associated with synapse maturation and plasticity. This study hence identifies a novel mechanism whereby cadherins, a major class of synaptic adhesion molecules, signal to the actin cytoskeleton to control the morphology of dendritic spines, and outlines a mechanism that underlies the coordination of synaptic adhesion with spine morphology.
Keywords: Rac1, GluR1, postsynaptic density, synaptic plasticity, cytoskeleton, synapse
Introduction
Remodeling of existing dendritic spines plays crucial roles in synapse maturation and plasticity (Yuste and Bonhoeffer, 2001). Conversely, aberrant spine morphogenesis is associated with mental retardation (Fiala et al., 2002), psychiatric disorders including schizophrenia (Glantz and Lewis, 2001; Fiala et al., 2002), and addiction (Robinson and Kolb, 1999). Synaptic maturation and plasticity entail changes in multiple processes, including spine morphology, transsynaptic adhesion, and glutamate receptor content, which have recently been postulated to be coordinately regulated (Luscher et al., 2000; Kasai et al., 2003). Accordingly, in vivo imaging studies revealed that, in the mammalian cortex, spine stability is well correlated with spine shape: thin spines are very dynamic, whereas large spines are stable (Trachtenberg et al., 2002). However, the molecular mechanisms that accomplish the coordination of adhesion and morphology in spines are not known.
Changes in synaptic adhesion, which occur in parallel with spine remodeling, contribute to synapse maturation and plasticity (Tang et al., 1998; Bozdagi et al., 2000; Huntley et al., 2002). Cadherins are a major class of adhesion molecules (Wheelock and Johnson, 2003) that play crucial roles in nervous system development and physiology (Bamji, 2005). Cadherins and associated proteins control spine morphology and stability: reduced cadherin or α-N-catenin function cause thin and more motile spines, whereas α-N-catenin overexpression results in larger spine heads and increased spine number because of reduced spine turnover (Togashi et al., 2002; Abe et al., 2004). Cadherins also play important roles in synaptic plasticity: synaptic activity regulates N-cadherin clustering and α- and β-catenin abundance in spines (Bozdagi et al., 2000; Tanaka et al., 2000; Murase et al., 2002; Abe et al., 2004), whereas N-cadherin adhesion is important for long-term potentiation (LTP) (Tang et al., 1998; Bozdagi et al., 2000) and memory (Schrick et al., 2007).
Cadherin clustering and signaling to the actin cytoskeleton are essential for adhesion. Signaling to the cytoplasm is accomplished by interactions of cadherins with cytoplasmic proteins including catenins, which in turn are thought to regulate Rho GTPases and subsequent actin rearrangements (Bamji, 2005). Rho GTPases are central regulators of actin dynamics and control spine morphology (Nakayama et al., 2000). Rac1 activation induces spine formation and enlargement; Rac1 inhibition produces thin and long spines (Tashiro and Yuste, 2004). However, the mechanisms whereby cadherins regulate GTPases are not known. We hypothesized that this may be accomplished through synaptic guanine-nucleotide exchange factors (GEFs), direct activators of Rho GTPases (Schmidt and Hall, 2002). Kalirin-7 is a neuron-specific Rac1-GEF concentrated in dendritic spines, where it activates Rac1 and regulates spine morphogenesis (Penzes et al., 2001, 2003; Xie et al., 2007). The link between cadherins and kalirin-7 may be provided by the scaffolding protein AF-6/afadin, which interacted with kalirin-7 in a yeast two-hybrid screen (Penzes et al., 2001), but is enriched in cadherin adhesion junctions through interaction with α-catenin and nectin (Mandai et al., 1997; Pokutta et al., 2002). In neurons, AF-6 is present in synapses (Buchert et al., 1999; Xie et al., 2005) and puncta adherentia (Nishioka et al., 2000), and controls spine morphogenesis in cortical pyramidal neurons (Xie et al., 2005).
To understand the mechanisms that allow synaptic adhesion molecules to control spine remodeling, which may also underlie the coordination of spine adhesion, structure, and stability, we investigated the roles of AF-6, kalirin-7, and Rac1 in N-cadherin-dependent spine remodeling.
Materials and Methods
Reagents.
The plasmid encoding N-cadherin was a gift from Dr. David R. Colman (Montreal Neurological Institute, Montreal, Quebec, Canada); myc-kalirin-7 and myc-L-AF-6 were described previously (Penzes et al., 2001; Xie et al., 2005). Myc-kal7-ΔGEF was generated by the deletion of the region between amino acids 1284 and 1484 in the myc-kalirin-7 plasmid; Rap-CA and AF-6-PDZ* was described by Xie et al. (2005). Antibodies were as follows: green fluorescent protein (GFP), postsynaptic density-95 (PSD-95), and GluR1-C-terminal polyclonal antibodies were generated in the laboratory of Dr. Richard L. Huganir, N-cadherin polyclonal (recognizing the C terminus of N-cadherin, but also E and R cadherins) was a gift from Dr. David Colman (Montreal Neurological Institute, Montreal, Quebec, Canada), phospho-p21-activated kinase (PAK) antibody was a gift from Dr. Jonathan Chernoff (Fox Chase Cancer Center, Philadelphia, PA). Kalirin antibodies were described by Penzes et al. (2001). The following antibodies were purchased: AF-6 polyclonal (Sigma-Aldrich); PSD-95 and Rac1 monoclonal (Millipore); N-cadherin monoclonal (recognizing the N terminus) (Sigma-Aldrich); N-cadherin goat polyclonal (Santa Cruz); α-N-catenin monoclonal and myc monoclonal (University of Iowa Hybridoma Bank); GFP monoclonal (Millipore Bioscience Research Reagents); and Rap1 polyclonal (Santa Cruz).
Neuronal cultures.
Dissociated cultures of primary cortical neurons were prepared from embryonic day 17 Sprague Dawley rat embryos, cultured in Neurobasal medium with B27 (Invitrogen) for 3–4 weeks (Xie et al., 2005), and transfected with plasmids (1–10 μg of DNA) using Lipofectamine 2000 (Invitrogen). Mature neuron cultures [days in vitro (div) 24∼28] were used in all experiments unless otherwise indicated. Rats were used in accordance with Animal Care and Use Committee institutional and national guidelines and regulations under approved protocols.
Quantitative analysis of spine morphologies.
Healthy pyramidal neurons were imaged with a confocal microscope (Zeiss LSM5 Pascal). Confocal images of single- and double-stained neurons were obtained with a Ziess LSM5 Pascal confocal microscope. To determine the lateral resolution of our confocal microscope, we imaged fluorescent microspheres (Duke Scientific) of known diameters, mounted in ProLong antifade solution, using the 63× oil immersion objective (numerical aperture, 1.4). Quantification indicated that the lateral point spread function was smaller than 0.5 μm, and that areas equal to or larger than 0.21 μm2 were accurately measured. Therefore, we did not include structures in our analysis that had an area of <0.25 μm2, because these structures are approaching the limit of the resolution of our microscope. It should also be noted that measurements of small spine features, such as thin spine necks, could be overestimated in our measurements. An anti-GFP antibody was used to circumvent potential unevenness of GFP diffusion in spines. Images of neurons were taken using the 63× oil immersion objective as z-series of three to eight images, averaged four times, taken at 0.37 μm intervals, 1024 × 1024 pixel resolution at a scan speed of 8 s per section. The acquisition parameters were kept the same for all scans. Two-dimensional maximum projection reconstructions of images, morphometric analysis, and quantification were done using MetaMorph software (Molecular Devices). Synapses formed onto healthy neurons with pyramidal morphologies were imaged as described above. Cultures that were directly compared were stained simultaneously and imaged with the same acquisition parameters. For each condition, five to six neurons each from at least three separate experiments were used, and two dendrites from each neuron (∼75–100 μm per dendrite) were analyzed. Experiments were done blind to conditions and on sister cultures. To examine the morphologies of dendritic spines, individual spines on dendrites were manually traced, and the area, maximum length, and head width of each spine was measured by MetaMorph. Student's unpaired t tests were used to determine the statistical significance of differences between two groups; one-way ANOVA were used to compare three or more groups, followed by Tukey-b post hoc for multiple comparisons. Statistical analyses were performed in Excel, SPSS, or GraphPad Prism.
Immunostaining and quantitative immunofluorescence.
Changes in clustering were quantified using immunofluorescence on neurons fixed with 3.7% formaldehyde (for GFP-expressing neurons) or −20°C methanol (for synaptic proteins) and visualized with specific antibodies. Fixed neurons were permeabilized and blocked simultaneously in PBS containing 2% normal goat serum and 0.2% Triton X-100 for 1 h at room temperature. Primary antibodies were added in PBS containing 2% normal goat serum for 2 h at room temperature or overnight at 4°C, followed by three washes (10 min each) in PBS. Secondary antibodies, anti-rabbit-FITC (Vector), anti-mouse-FITC (Vector), anti-rabbit-rhodamine (Pierce), anti-mouse-rhodamine (Pierce), anti-goat-Alexa 514 (Invitrogen), were incubated for 1 h at room temperature in 2% normal goat serum in PBS. An additional three washes (5 min each) were performed before coverslips were mounted using ProLong antifade reagent (Invitrogen). Healthy neurons with pyramidal morphologies were imaged as described above. Cultures that were directly compared were stained simultaneously and imaged with the same acquisition parameters. For each condition, five to six neurons each from three separate experiments were analyzed. Experiments were done blind and on sister cultures. For fluorescence intensity measurements, the background corresponding to areas without cells were subtracted to generate a “background-subtracted” image. Images were then thresholded equally to include clusters with intensity at least twofold above the adjacent dendrite. To measure the intensity of clusters in spines or colocalized with another signal, regions around spines were generated based on GFP fluorescence or cluster immunofluorescence in one channel. These regions were then transferred into the target channel, and only fluorescence integrated intensities within the colocalizing regions were measured.
To measure individual cluster immunofluorescence integrated intensities, total gray values of each cluster were measured automatically. For total integrated intensities/dendritic segment length, regions were outlined along dendrites and total gray values of signals colocalized with a reference signal within these regions were measured and normalized to dendritic length.
N-cadherin activating and blocking treatments.
To stimulate adhesion by engaging N-cadherins, we incubated neurons for 5 h with recombinant Fc-N-cadherin (Fc-Ncad) protein chimera between the extracellular domain of N-cadherin (amino acids 1–724) and the Fc region of human IgG (R&D Systems) preclustered with anti-human Fc antibody (goat) (Lambert et al., 2000). To block N-cadherin-mediated adhesion, we incubated neurons for 5 h with GC-4 interfering antibody (Sigma-Aldrich), which binds to the N-terminal extracellular domain of N-cadherin and disrupts N-cadherin mediated adhesion (Meyer et al., 1992).
Coimmunoprecipitation.
For coimmunoprecipitation, HEK293 cells were harvested in RIPA buffer (in mm: 150 NaCl, 10 Tris-HCl, pH 7.2, 5 EDTA, 0.1% SDS, 1% Triton X-100, 1% deoxycholate, plus inhibitors). Lysates were then sonicated, cleared by centrifugation at 14,000 × g for 10 min, and precleared with 30 μl of protein A-Sepharose for 60 min at 4°C. Supernatants were incubated with 3–5 μl of antibody for 2–4 h; 60 μl of protein A-Sepharose was added for 1 h at 4°C, after which they were washed three times with 1 ml of RIPA. Coimmunoprecipitations from cortical pyramidal neurons were performed essentially as described above. All samples were boiled for 5 min at 95°C after addition of Laemmli buffer and analyzed by SDS-PAGE and Western blotting.
Subcellular fractionation and detergent extractions.
Rat forebrain homogenates were fractionated by centrifugation to yield soluble (S2) and particulate (P2) synaptosomal fractions using standard techniques (Penzes et al., 2000). P2 fraction was then extracted with 1% Triton X-100 (TX), 1% SDS (SDS), 1% deoxycholate (Doc), or 1% RIPA, for 30 min on ice, followed by centrifugation. Supernatant (S) and pellet (P) fractions were analyzed by SDS-PAGE and Western blotting.
Statistics.
All data represent means ± SEM. Student's unpaired t test was used to determine the statistical significance of the differences between two groups. One-way ANOVA followed by post hoc Tukey's multiple-comparison test was used to determine the statistical significance of the differences among multiple groups (GraphPad Prism). Differences were deemed significant when p < 0.05.
Results
N-cadherin-mediated adhesion controls spine morphology and GluR1 content
Recent studies reported that interfering with N-cadherin, β-catenin, p120 catenin, and α-N-catenin functions affected spine morphology in hippocampal neurons (Murase et al., 2002; Togashi et al., 2002; Abe et al., 2004; Elia et al., 2006) providing support for the hypothesis that synaptic adhesion and spine size are coordinated (Kasai et al., 2003). Indeed, we observed a close correlation between dendritic spine sizes and N-cadherin contents in mature (div 28) cortical pyramidal neurons (Fig. 1a,b), further establishing a relationship between adhesion and spine morphology. N-cadherin was concentrated in clusters along dendrites where it colocalized with PSD-95, indicating enrichment in excitatory synapses (supplemental Fig. S1a, available at www.jneurosci.org as supplemental material), and thus is well positioned to control adhesion-dependent spine remodeling in these neurons. To determine whether N-cadherin-mediated adhesion affected spine size, we activated or inhibited N-cadherin adhesion in pyramidal neurons (Fig. 1c,d). To mimic enhanced adhesion, we incubated mature (div 28) cortical pyramidal neurons with recombinant Fc-N-cadherin preclustered with an anti-Fc antibody (Lambert et al., 2000) to engage N-cadherins. To disrupt N-cadherin adhesion, we incubated neurons with GC-4 interfering antibody, which binds to the N-terminal extracellular domain (Meyer et al., 1992). Treatment with preclustered Fc-N-cadherin (clust FcNcad) resulted in the enlargement of existing spines (areas: control, 0.88 ± 0.02; clust FcNcad, 1.69 ± 0.05 μm2) and substantial increase of N-cadherin immunofluorescence inside spines [N-cadherin integrated intensities: control, 1.93 ± 0.22; clust FcNcad, 8.33 ± 1.39 arbitrary units (a.u.)], without affecting spine length and linear densities; whereas GC-4 (interf Ab) resulted in longer spines (length: control, 1.20 ± 0.04; interf Ab, 2.42 ± 0.05 μm) and slightly elevated linear spine density (Fig. 1d) (control, 5.40 ± 0.43; interf Ab, 7.40 ± 0.67 spines/10 μm). Whereas interference did not cause an apparent reduction in average spine area, analysis of the spine length/breadth ratios revealed that this was attributable to the simultaneous elongation of spines, which occluded the changes in areas (as reflected by higher length/breadth ratios) (Fig. 1e,f). In summary, N-cadherin activation caused enlargement of existing spines, whereas interference with N-cadherin dimerization caused remodeling of spines into thin, long shapes (Fig. 1f). IgG alone, unclustered Fc-Ncadherin (data not shown), or clustered ephrinB1 (supplemental Fig. S2a,b, available at www.jneurosci.org as supplemental material) had no detectable effect. As expected, clustered Fc-N-cadherin induced formation of large N-cadherin clusters localized in spines (Fig. 1c, right, arrows), whereas GC-4 resulted in N-cadherin clusters in the dendritic shaft (arrowheads) (although no significant reduction in spine heads), indicating that endogenous N-cadherin was affected by these reagents (Fig. 1d). The N-cadherin-mediated spine enlargement observed in this study is independent of the ephrinB–EphB receptor pathway identified previously, in that ephrinB-induced spine morphogenesis was only seen in young hippocampal but not in mature cortical neurons (Penzes et al., 2003). We confirmed these results by treating mature cortical neurons with clustered ephrinB: no change in spine size, length, or density was observed (supplemental Fig. S2a,b, available at www.jneurosci.org as supplemental material). Furthermore, treatment with clust FcNcad in neurons expressing a kinase-dead mutant of the EphB receptor still produced spine enlargements concurrent to those in treated untransfected cells (supplemental Fig. S2a,b, available at www.jneurosci.org as supplemental material).
Figure 1.
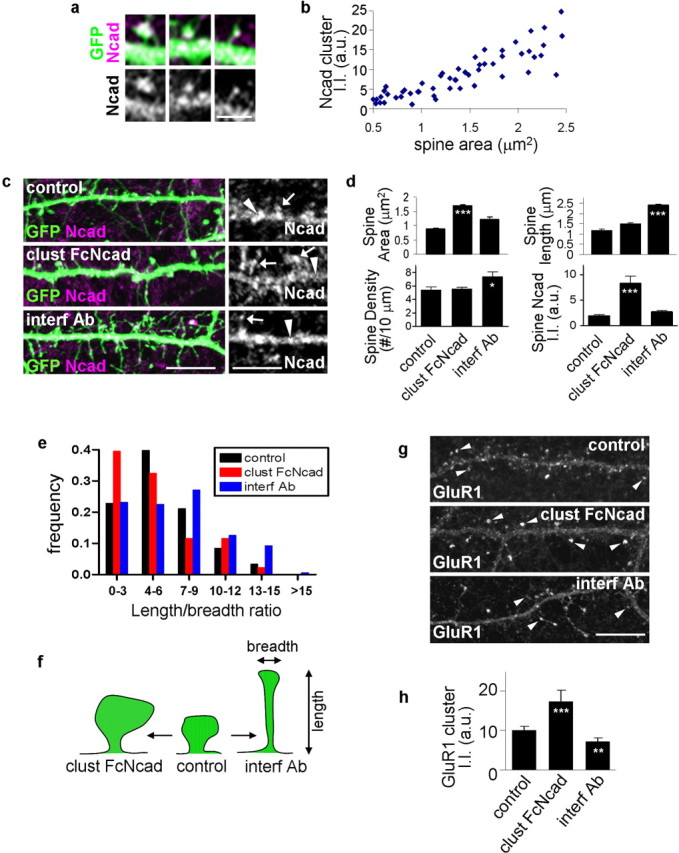
Activation and inhibition of N-cadherin adhesion result in spine remodeling. a, Correlation of spine size with N-cadherin content. High-magnification images of individual spines on GFP-expressing cortical neurons (div 28) immunostained for GFP and N-cadherin. N-cadherin clusters are present in dendritic spines. b, Quantification of the relationship between spine size and N-cadherin content in spines. Spine size (area) versus N-cadherin immunofluorescence integrated intensity was plotted for individual spines; N-cadherin content increases with spine size (correlation coefficient, R2 = 0.89). c, Activation or inhibition of N-cadherin adhesion induces spine remodeling. Cortical neurons (div 28) in medium-density culture expressing GFP were treated for 5 h with preclustered Fc-N-cadherin (clust FcNcad), interfering antibody GC4 (interf Ab), or left untreated (control). Right, N-cadherin clusters on dendrites of these neurons; arrows indicate clusters in spines, and arrowheads point to clusters in the dendritic shaft. d, Quantification of spine area, length, linear density, and N-cadherin integrated intensity within spines, from c: activation of N-cadherin adhesion results in spine enlargement and greater amounts of N-cadherin in spines (***p < 0.001 compared with control), whereas inactivation results in spine elongation (***p < 0.001 compared with control) and increased spine density (*p < 0.05 compared with control). e, Frequency distribution of the spine length/breadth ratio: activation of N-cadherin adhesion shifts the length/breadth distribution toward lower values, corresponding to larger, stubby spines; whereas inactivation of N-cadherin adhesion shifts the length/breadth distribution toward higher values, corresponding to longer and thinner spines. e, Schematic diagram showing spines morphological changes on activation or inhibition of N-cadherin adhesion. g, Activation or inhibition of N-cadherin adhesion affects GluR1 content (arrowheads) in spines. h, Quantification of GluR1 cluster integrated intensity in g: N-cadherin activation results in more GluR1 in clusters, inactivation results in less GluR1 in clusters (***p < 0.001, **p < 0.01 compared with control). Error bars indicate SEM. Scale bars: a, 2.5 μm; c, g, 10 μm.
Spine size and AMPA receptor content are thought to be tightly coordinated in that larger spines, as those induced by N-cadherin activation, have more AMPA receptors, resulting in stronger and more stable synapses (Kasai et al., 2003). Recent studies reported that N-cadherin interacts with AMPA receptors (Dunah et al., 2005; Nuriya and Huganir, 2006), suggesting that cadherin-mediated adhesion may regulate spine AMPA receptors. We tested this hypothesis by stimulating or blocking N-cadherin adhesion and examining the content of GluR1 subunit of AMPA receptors in spines (Fig. 1g,h). N-cadherin engagement resulted in more GluR1 in clusters, whereas N-cadherin inactivation resulted in less GluR1 in clusters (integrated intensities/cluster: control, 10.0 ± 1.0; clust FcNcad, 17.3 ± 2.9; interf Ab, 7.0 ± 0.4 a.u.). These experiments indicate that synaptic adhesion strength actively controls spine morphology and AMPA receptor content, consistent with the requirement for N-cadherin adhesion in LTP (Tang et al., 1998; Bozdagi et al., 2000).
AF-6 colocalizes and interacts with N-cadherin in neurons
The interaction of the cytoplasmic tail of N-cadherin with cytoplasmic proteins, including α-N-catenin, β-catenin, and p120-catenin, are important for N-cadherin-dependent spine morphogenesis (Murase et al., 2002; Togashi et al., 2002; Abe et al., 2004; Elia et al., 2006). However, the detailed mechanism whereby N-cadherin controls spine remodeling is not known. Because the protein AF-6 is a component of the cadherin-based adhesion complexes in epithelial cells (Pokutta et al., 2002) and controls spine remodeling in cultured neurons (Xie et al., 2005), we reasoned that it may also control adhesion-dependent spine remodeling. Therefore, we examined the colocalization and interaction of AF-6 with components of the N-cadherin complexes in cultured pyramidal neurons and rat forebrain. Consistent with previous reports (Nishioka et al., 2000; Mizoguchi et al., 2002), we found that in young neurons (div 7) AF-6 was enriched in contact sites between neurites, where it partially colocalized with N-cadherin (supplemental Fig. S1b, available at www.jneurosci.org as supplemental material). In developing neurons (div 14), AF-6 and N-cadherin partially colocalized in clusters along dendrites, suggesting a role for this interaction in synapse maturation (supplemental Fig. S1c, available at www.jneurosci.org as supplemental material). In mature (div 24–28) neurons, AF-6 colocalized with N-cadherin at the somatic plasma membrane, in clusters along dendrites, and in spines (Fig. 2a). Similarly, AF-6 partially colocalized with α-N-catenin, its direct interacting partner in the N-cadherin complex (Fig. 2b, arrows). As revealed by colocalization with PSD-95, a large fraction of AF-6 clusters in dendrites were at excitatory synapses (supplemental Fig. S1d,e, available at www.jneurosci.org as supplemental material). Together, these data support a role for AF-6 in N-cadherin-dependent adhesion events in neurons at different stages of development. Although a large fraction of AF-6 is present in spines, AF-6 is also associated with adhesion complexes at other sites, such as soma and dendritic shafts, where it may participate in adhesion events with specific functions. To confirm in vivo the interaction between AF-6 and N-cadherin-associated protein complexes, we performed coimmunoprecipitation from rat forebrain homogenates. An AF-6 antibody specifically coprecipitated N-cadherin, and an N-cadherin antibody specifically coprecipitated AF-6 (Fig. 2c), suggesting that AF-6 and N-cadherin participate in the same protein complexes. Together with their colocalization, this indicates that AF-6 interacts with components of the N-cadherin-based adhesion complexes in neurons.
Figure 2.
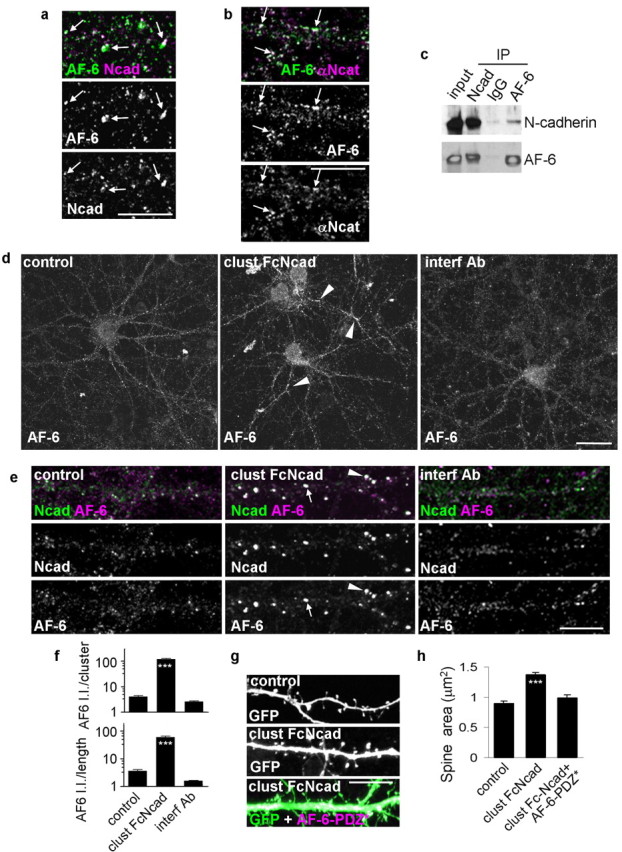
Activation and inhibition of N-cadherin adhesion result in AF-6 redistribution that underlies spine remodeling. a, AF-6 and N-cadherin colocalize in spines (arrows) in mature neurons (div 24–28). b, AF-6 and α-N-catenin colocalize at dendrite plasma membrane and in spines (arrows). c, AF-6 and N-cadherin coimmunoprecipitate from rat forebrain homogenate. d, N-cadherin activation or inhibition affects AF-6 localization in dendrites. The arrowheads indicate AF-6 clusters. e, Activation of N-cadherin adhesion induces formation of larger clusters of AF-6 along dendritic shafts (arrows) and in spines (arrowheads). f, Integrated intensities of AF-6 clusters that are colocalized with N-cadherin; quantification done for individual clusters and for all clusters within a segment of dendrite normalized to dendritic length. ***p < 0.001 compared with control and interf Ab. g, Spine enlargement induced by N-cadherin activation is blocked by expression of a mutant AF-6 with a nonfunctional PDZ domain (AF-6-PDZ*). h, Quantification of spine areas in g. ***p < 0.001 compared with control or clust FcNcad plus AF-6-PDZ*. Error bars indicate SEM. Scale bars: a, b, e, g, 10 μm; d, 30 μm.
AF-6 is regulated by N-cadherin-mediated adhesion and is required for cadherin-dependent spine enlargement
If AF-6 was a functional component of adhesion complexes, activation of N-cadherins would likely affect its subcellular localization. Our results revealed that N-cadherin activation induced the formation of larger clusters of endogenous AF-6 along dendrites (Fig. 2d, arrowheads) and in spines (Fig. 2e, arrowheads). Furthermore, AF-6 recruitment to N-cadherin clusters is greatly enhanced by N-cadherin activation (Fig. 2f), as evidenced by an increase in N-cadherin-colocalized AF-6 cluster immunofluorescence (integrated intensity/cluster: control, 4.14 ± 0.39; clust FcNcad, 120.90 ± 5.70; interf Ab, 2.62 ± 0.17 a.u.). Similar changes were found when total integrated intensity of colocalized AF-6 clusters along a segment of dendrite were measured and normalized to dendritic length (control, 3.64 ± 0.43; clust FcNcad, 60.41 ± 5.27; interf Ab, 1.59 ± 0.08 a.u.). Disruption of N-cadherin adhesion did not affect AF-6 clusters.
We previously showed that AF-6 controls spine morphogenesis, and overexpression of AF-6 induced formation of spines with diverse morphologies, including spines with large heads (arrowheads) as well as thin/long spines (arrows) (supplemental Fig. S1f, available at www.jneurosci.org as supplemental material), whereas deletion of AF-6 N-terminal Rap-association domains also resulted in large spines (Xie et al., 2005). Conversely, a mutation in the postsynaptic density-95/Drosophila disc large tumor suppressor/zonula occludens-1 (PDZ) domain of AF-6 (AF-6-PDZ*) eliminated the ability of AF-6 to induce larger spines. This suggests that AF-6 plays a role in spine enlargement caused by N-cadherin activation. To test this, we incubated neurons expressing GFP and AF-6-PDZ* with clustered Fc-N-cadherin (Fig. 2g,h). AF-6-PDZ* abolished spine enlargement induced by clustered Fc-N-cadherin (spine areas: control, 0.89 ± 0.04; clust FcNcad, 1.37 ± 0.05; clust FcNcad plus AF-6-PDZ*, 0.98 ± 0.06 μm2), supporting the requirement for AF-6 in N-cadherin adhesion-induced spine enlargement. Spine length and density were not affected by AF-6-PDZ* expression (data not shown). The effects on spine morphology were not caused by altered surface expression of N-cadherin, because in neurons expressing AF-6-PDZ*, surface N-cadherin clusters were equally accessible to “decoration” by clustered Fc-Ncadherin as in nontransfected control neurons (supplemental Fig. S1g, available at www.jneurosci.org as supplemental material). These experiments indicate that N-cadherin-dependent recruitment of AF-6 molecules to synapses is crucial for adhesion-dependent spine enlargement, and that interactions mediated by the PDZ domain of AF-6 play a key role in these processes.
AF-6 interacts with the Rac1-GEF kalirin-7 in neurons
Previously, we demonstrated that AF-6 controls spine morphology; however, the downstream mechanisms by which AF-6 regulates spine morphogenesis remain unknown. Two splice variants of AF-6 are expressed in the brain: the long form (205 kDa) (L-AF-6) contains a C-terminal filamentous actin-binding domain that is not present in the short form (190 kDa) (s-AF-6). The presence of an F-actin binding domain in L-AF-6 suggested that AF-6 may directly cross-link actin filaments, implying L-AF-6 would control actin rearrangements differently to s-AF-6 because of the presence of this domain. Contrary to this prediction, we found that AF-6 did not affect F-actin distribution, demonstrated by the lack of F-actin redistribution in COS7 cells transfected with either L- or s-AF-6 (Fig. 3a). Both AF-6 forms contain a PDZ domain. The inactivation of its PDZ domain reduced its ability to induce spine morphogenesis (Xie et al., 2005) (Fig. 2g,h), suggesting that proteins interacting with this domain are important. To further dissect the downstream signaling mechanisms by which AF-6 regulates adhesion-dependent spine morphogenesis, we focused on proteins interacting with its PDZ domain. Using a yeast two-hybrid screen, AF-6 was found to interact with the C terminus of kalirin-7, a brain-specific Rac1-GEF and a regulator of spine morphogenesis (Xie et al., 2007). Therefore, we reasoned that kalirin-7 may mediate the actions of AF-6 on spines. To confirm that these two proteins interacted in vivo, we performed coimmunoprecipitations from rat forebrain homogenates. Antibodies specific to AF-6 and kalirin coprecipitated each other, respectively (Fig. 3b). The importance of the PDZ-mediated interaction between AF-6 and kalirin-7 was further confirmed by coimmunoprecipitation from transfected HEK293 cells (Fig. 3c): whereas AF-6 with an intact PDZ domain interacted with kalirin-7, AF-6-PDZ*, which blocked adhesion-dependent spine morphogenesis, did not interact with kalirin-7. Furthermore, AF-6 and kalirin-7 colocalize in numerous dendritic spines (Fig. 3e, arrows).
Figure 3.
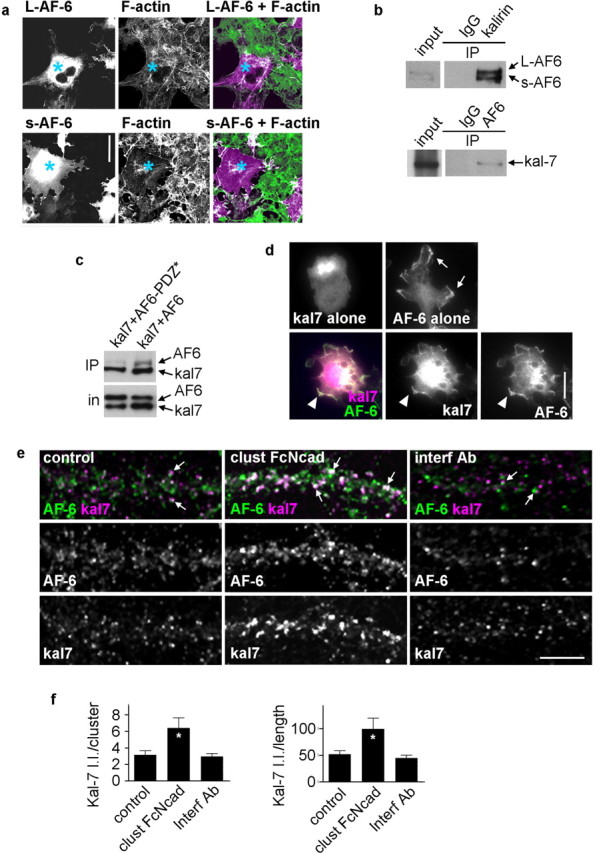
AF-6 interacts with kalirin-7 in neurons. a, Long (L-AF-6) and short (s-AF-6) isoforms do not induce actin filament (F-actin) rearrangements in COS7 cells. Cells expressing exogenous myc-L-AF-6 and myc-s-AF-6 proteins (blue asterisks) do not show increased FITC-phalloidin fluorescence compared with nonexpressing cells. Myc-AF-6 proteins were visualized using myc antibody, and actin filaments were visualized with FITC-phalloidin. b, AF-6 and kalirin-7 coimmunoprecipitate from rat cerebral cortex homogenates; both L- and s-AF-6 interact with kalirin-7; an IgG negative control does not immunoprecipitate either protein. c, AF-6 and kalirin-7 interact directly, as shown by coimmunoprecipitation of exogenous proteins expressed in HEK293 cells. This interaction is dependent on a functional PDZ domain of AF-6, because it is abolished by a point mutation in this domain (AF-6-PDZ*). Immunoprecipitation was done with kalirin antibody and Western blot detection with a myc antibody. d, AF-6 recruits kalirin-7 to the plasma membrane in HEK293 cells: whereas kalirin-7 alone is diffuse in the cytosol and AF-6 is partially associated with the plasma membrane, when coexpressed, AF-6 and kalirin-7 partially colocalize at the plasma membrane. e, Activation of N-cadherin adhesion affects the localization of kalirin-7 in dendrites (arrows, colocalized AF-6 and kalirin-7 clusters). f, Integrated intensities of kalirin-7 clusters that are colocalized with AF-6 in e; quantification done for individual clusters and for all clusters within a segment of dendrite normalized to dendritic length. *p < 0.05 compared with control and interf Ab. Error bars indicate SEM. Scale bars, 10 μm.
The requirement of a PDZ-mediated interaction for AF-6-dependent spine morphogenesis suggested that AF-6 may recruit kalirin-7 to specific sites on the plasma membrane in dendrites and thereby control spine remodeling. Indeed, in transfected hEK293 cells, whereas kalirin-7 alone was diffusely distributed in the cytoplasm and AF-6 alone was partially associated with the plasma membrane (arrows), AF-6 recruited kalirin-7 to the plasma membrane (arrowheads) (Fig. 3d). Because activation of N-cadherin adhesion modulated the clustering of AF-6 in dendrites, this may have also modulated the localization of kalirin-7. Thus, we examined the N-cadherin adhesion-dependent recruitment of endogenous kalirin-7 to AF-6 puncta in dendrites (Fig. 3e,f). N-cadherin engagement increased kalirin-7 recruitment to AF-6 puncta: colocalized kalirin-7 cluster integrated intensity (control, 3.15 ± 0.52; clust FcNcad, 6.41 ± 1.24; interf Ab, 2.95 ± 0.39 a.u.); total integrated intensity of colocalized kalirin-7 clusters on a segment of dendrite normalized to dendritic length (control, 51.7 ± 7.03; clust FcNcad, 99.6 ± 20.4; interf Ab, 44.9 ± 5.78 a.u.).
AF-6 recruits kalirin-7 to spines
Anchoring by the AF-6 PDZ domain may be a mechanism to recruit kalirin-7 to adhesion sites on the dendritic and synaptic plasma membrane. To test this hypothesis, we expressed kalirin-7 with or without AF-6 or AF-6-PDZ* in cortical neurons and examined kalirin-7 clusters. Coexpression of AF-6 increased the integrated intensity of kalirin-7 puncta, whereas coexpression with AF-6-PDZ* resulted in reduced integrated intensity of kalirin-7 clusters (Fig. 4a,b) (kal7 alone, 12.37 ± 1.0; kal7 plus AF-6, 19.6 ± 1.6; kal7 plus AF-6-PDZ*, 9.8 ± 0.5, kal7 cluster intensities).
Figure 4.
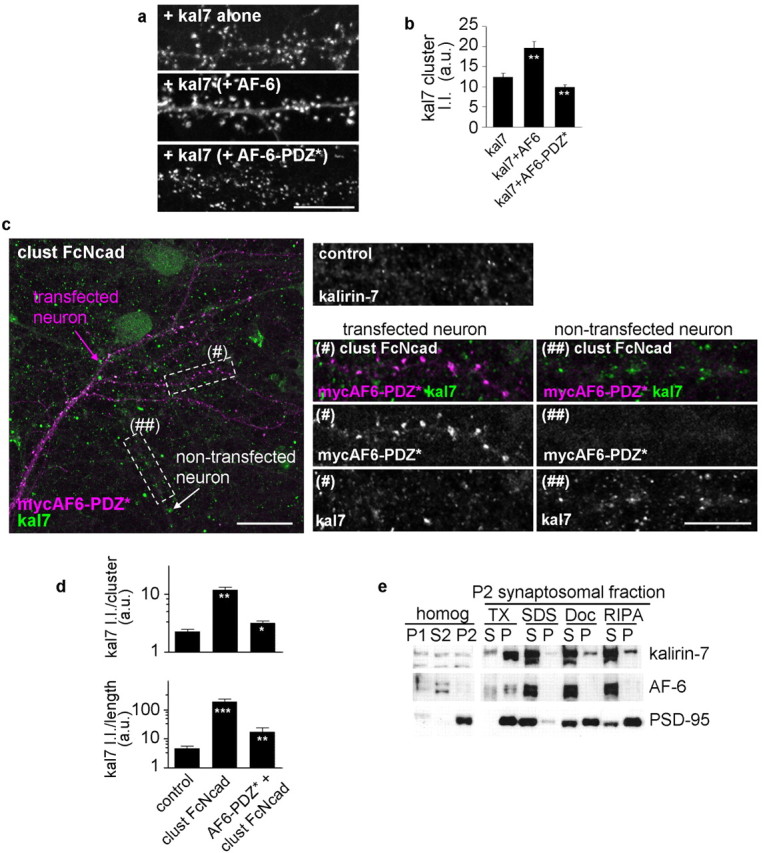
AF-6 PDZ domain mediates interaction with kalirin-7 in neurons. a, In cortical neurons, exogenous kalirin-7 is targeted to punctate structures along dendrites, coexpression of AF-6 enhanced kalirin-7 clustering into puncta; coexpression with myc-AF-6-PDZ* reduced kalirin-7 clustering. b, Quantification of a: kalirin-7 integrated intensity per cluster (**p < 0.01 compared with control). c, Functional PDZ domain in AF-6 is necessary for N-cadherin activation-dependent recruitment of kalirin-7. Neurons transfected with myc-AF6-PDZ* were treated with clust FcNcad or untreated (control) and immunostained for kalirin-7 (green) and myc (purple). Insets show endogenous kalirin-7 puncta in dendrites of a control neuron and in clust FcNcad-treated neurons expressing myc-AF6-PDZ* (#) or nontransfected (##). The insets are from magnified regions denoted with dashed white lines. d, Integrated intensities of kalirin-7 clusters in c; quantification done for individual clusters and for all clusters within a segment of dendrite normalized to dendritic length; myc-AF-6-PDZ* prevents spine kalirin-7 recruitment by N-cadherin adhesion (***p < 0.001 compared with control; **p < 0.01 compared with clust FcNcad). e, Differential association of AF-6, kalirin-7, and PSD-95 with the synaptic cytoskeleton. P2 synaptosomal fraction was solubilized in buffers with detergents of different strengths, and soluble (S) or insoluble, particulate (P) fractions were separated by centrifugation. Presence in the S fraction indicates solubility in that detergent, whereas presence in P indicates association with the insoluble proteins. Error bars indicate SEM. Scale bars: a, c, insets, 10 μm; c, 20 μm.
To test whether an intact PDZ domain of AF-6 was required for N-cadherin-dependent kalirin-7 recruitment, we examined the clustering of endogenous kalirin-7 in presence or absence of overexpressed AF-6-PDZ* (Fig. 4c,d). In dendrites of neurons expressing AF-6-PDZ* [Fig. 4c, (#)], N-cadherin engagement failed to induce kalirin-7 clustering; whereas in dendrites of neurons not expressing AF-6-PDZ* [Fig. 4c, (##)], kalirin-7 was clustered on N-cadherin engagement (kalirin-7 cluster integrated intensity: control, 2.25 ± 0.22; clust FcNcad, 11.81 ± 1.43; AF-6-PDZ* plus clust FcNcad, 3.17 ± 0.29; total kalirin-7 integrated intensity on a segment of dendrite normalized to dendritic length: control, 4.64 ± 1.07; clust FcNcad, 194.80 ± 48.33; interf Ab, 17.55 ± 6.98 a.u.). These data indicate that AF-6 may regulate spine morphogenesis by recruiting kalirin-7 to synapses through a PDZ domain-dependent interaction.
Because kalirin-7 also interacted with PSD-95 (Penzes et al., 2001), we compared the relative strength of the association of AF-6 versus PSD-95 versus kalirin-7 with cytoskeletal and PSD elements, as an indirect measure of the mobility of a certain kalirin-7 pool, specifically associated with one or the other scaffolding protein (Fig. 4e). One possibility is that different scaffolding proteins that bind kalirin-7 may have different degrees of mobility within the PSD, therefore leading to more stable versus more dynamic pools of kalirin in spines. Although both AF-6 and PSD-95 cofractionate with kalirin-7 in the crude synaptosomal fraction, the strength of the association of AF-6 versus PSD-95 with cytoskeletal and PSD is different. AF-6 can be solubilized with a weaker detergent (Triton X-100, deoxycholate, RIPA buffer), along with a fraction of kalirin-7, indicating a more mobile pool of protein. These results suggest that a larger fraction of kalirin-7 is loosely associated with PSD, possibly through interaction with proteins such as AF-6, indicating a higher mobility within spine. In contrast, PSD-95 along with another fraction of kalirin-7 can only be solubilized by strong detergents (SDS) but is left in the pellet by (deoxycholate, RIPA buffer), indicating a pool of kalirin-7 stably associated with PSD, likely by interaction with PSD-95.
Kalirin-7 interacts with N-cadherin-containing adhesion complexes in neurons
Rho GTPase activation is thought to play a key role in N-cadherin-dependent spine remodeling (Bamji, 2005; Elia et al., 2006). In particular, Rac1 activation induces spine enlargement similar to N-cadherin activation (Nakayama et al., 2000), whereas its inhibition causes remodeling into thin and long morphologies (Tashiro and Yuste, 2004). AF-6, by linking the N-cadherin complex with the Rac1-GEF kalirin-7, may provide adhesion complexes the ability to rapidly control Rac1-dependent spine remodeling. We reasoned that a signaling pathway that includes N-cadherin/AF-6/kalirin-7 may control adhesion-dependent spine remodeling. We first examined the colocalization of kalirin-7 with components of the N-cadherin adhesion complexes in dendrites. Indeed, kalirin-7 colocalized with α-N-catenin (Fig. 5a,b) and N-cadherin (Fig. 5c) in clusters on dendrites and in many spines (arrows). Because AF-6 interacts with both kalirin-7 and N-cadherin-associated proteins, a subset of AF-6 molecules may interact with both kalirin-7 and components of the N-cadherin complex. To test this possibility in a cellular system, HEK293 cells were transfected with all three proteins simultaneously (Fig. 5d); N-cadherin, AF-6, and kalirin-7 were observed to partially colocalize at the plasma membrane (Fig. 5d).
Figure 5.
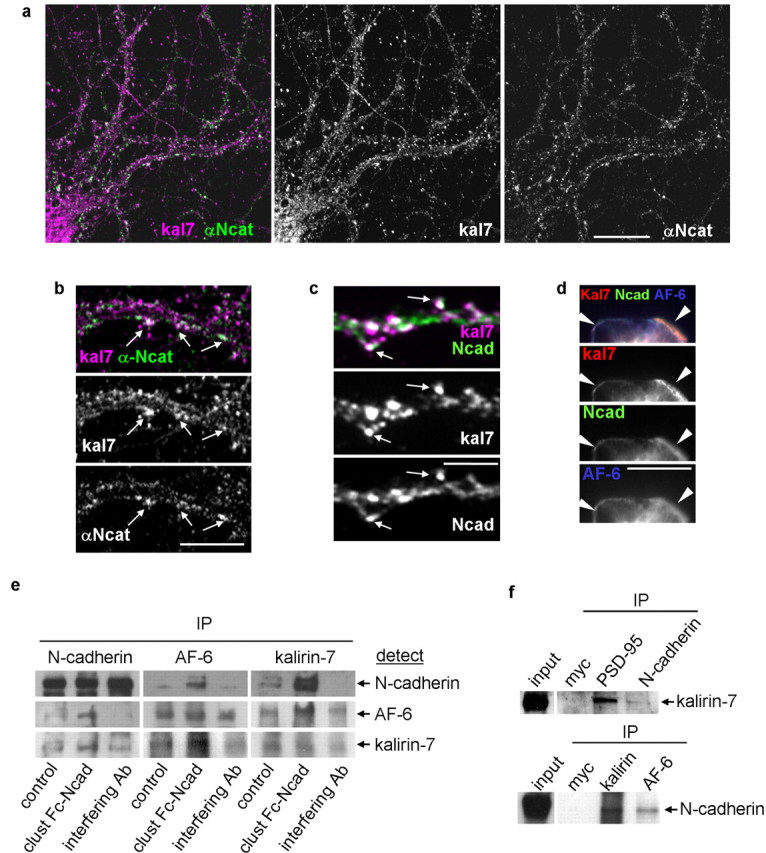
Kalirin-7 colocalizes and interacts with N-cadherin adhesion complexes. a, Kalirin-7 partially colocalizes with α-N-catenin. b, Colocalization of N-catenin and kalirin-7 in spine-like structures (arrows). c, Kalirin-7 partially colocalizes with N-cadherin in dendrites (arrows). d, Exogenous kalirin-7, AF-6, and N-cadherin colocalize at the plasma membrane of HEK293 cells. e, Activation or inhibition of N-cadherin adhesion modulates the interactions among AF-6, kalirin-7, and N-cadherin in neurons. Coimmunoprecipitation was done with N-cadherin, AF-6, or kalirin-7 antibodies, respectively; all immunoblots were probed for N-cadherin, AF-6, and kalirin-7. N-cadherin activation causes increased interaction among N-cadherin, AF-6, and kalirin-7; inhibition of N-cadherin reduces interaction among N-cadherin, AF-6, and kalirin-7. f, N-cadherin and kalirin-7 coimmunoprecipitate from rat forebrain homogenates. Scale bars: a, 25 μm; b, 10 μm; c, d, 5 μm.
Together, these experiments suggested that, in neurons, kalirin-7 participates in N-cadherin containing multiprotein complexes through interaction with AF-6. In addition, because AF-6 clustering is modulated by N-cadherin adhesion, the reciprocal interaction among kalirin-7, AF-6, and N-cadherin in such multiprotein complexes may also be modulated by N-cadherin adhesion. To test this, we stimulated or inhibited N-cadherin in neurons and examined the interactions among kalirin-7, AF-6, and N-cadherin through coimmunoprecipitation (Fig. 5e). All three proteins were found to interact with one another, demonstrating the existence of protein complexes that contain all three proteins: N-cadherin, AF-6, and kalirin-7. In addition, N-cadherin engagement enhanced the reciprocal interaction of N-cadherin, AF-6, and kalirin-7, whereas N-cadherin interference reduced the extent of coimmunoprecipitation between these proteins. These results demonstrate that N-cadherin adhesion modulates the extent of interaction among N-cadherin, AF-6, and kalirin-7 in neurons. We further confirmed the interaction of kalirin-7 with N-cadherin in vivo and compared it with its interaction with the synaptic scaffolding protein PSD-95. Although coimmunoprecipitation from rat forebrain homogenate confirmed that kalirin-7 interacted with N-cadherin (Fig. 5f), as expected the interaction between kalirin-7 with PSD-95 was comparatively stronger. This suggests that the kalirin-7/N-cadherin interaction is indirect and potentially mediated by AF-6.
N-cadherin adhesion modulates Rac1 and PAK signaling in dendrites
Because N-cadherin activation modulates AF-6 clustering in addition to the interaction among N-cadherin, AF6, and kalirin-7, it may also modulate the localization of kalirin-7 in dendrites. Indeed, N-cadherin engagement enhanced kalirin-7 recruitment to N-cadherin clusters (Fig. 6a,b), indicated by increased integrated intensities of colocalized kalirin-7 clusters (Fig. 6a, arrows) (control, 1.55 ± 0.11; clust FcNcad, 9.08 ± 1.46; interf Ab, 1.55 ± 0.20 a.u.); similar changes were found in the total colocalized kalirin-7 integrated intensity on a segment of dendrite normalized to dendritic length (control, 18.49 ± 1.72; clust FcNcad, 114.40 ± 16.93; interf Ab, 21.12 ± 2.67 a.u.). We also compared the intensities of exogenous kalirin-7 clusters (Fig. 6c). In control neurons overexpressed kalirin-7 was concentrated in small puncta along the dendrites. Coexpression with N-cadherin resulted in clusters containing more kalirin-7, without affecting the number of clusters, suggesting that overexpressed N-cadherin recruited more kalirin-7 to preexisting clusters (kalirin-7 cluster integrated intensities: kal7 alone, 12.37 ± 0.10; kal7 plus Ncad, 18.96 ± 0.14 a.u.; p < 0.001).
Figure 6.
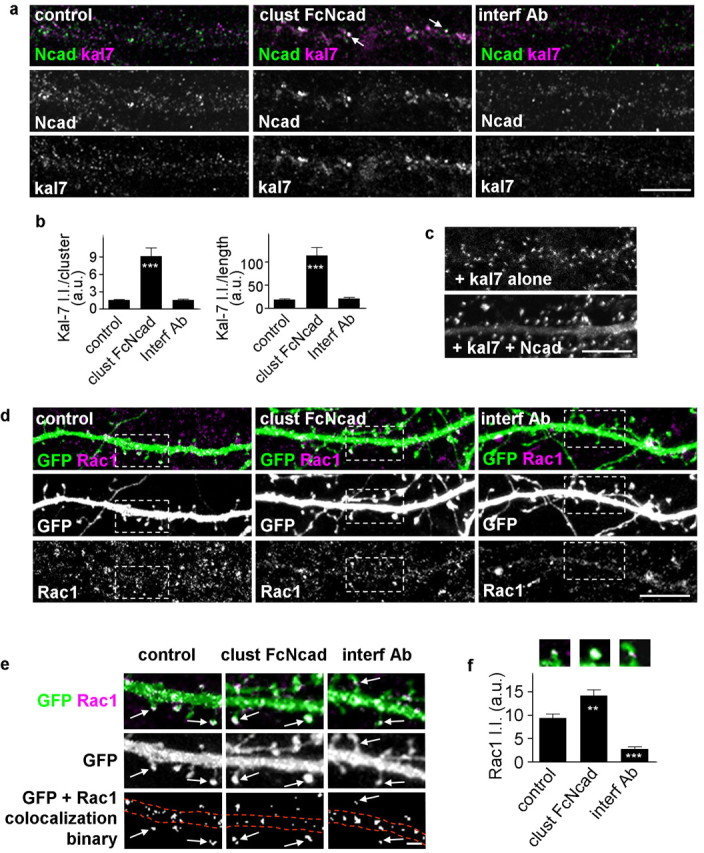
N-cadherin adhesion modulates kalirin-7 and Rac1 localization in dendrites and spines. a, N-cadherin activation affects the localization of kalirin-7 in dendrites; the arrows indicate colocalized N-cadherin and kalirin-7 clusters. b, Integrated intensities of kalirin-7 clusters that are colocalized with N-cadherin in a; quantification done for individual clusters and for all clusters within a segment of dendrite normalized to dendritic length. ***p < 0.001 compared with control and interf Ab. c, Coexpression of myc-kalirin-7 (kal7) with or without N-cadherin in neurons, immunostained for myc. Expression of kalirin-7 together with N-cadherin induces larger kalirin-7 cluster in dendritic spines. d, N-cadherin activation or inhibition modulates dendritic localization of Rac1. The dashed white boxes indicate areas magnified in the following panel. e, Magnified visualization of Rac1 in dendrites and spines of GFP-expressing neurons. Activation of N-cadherin increases Rac1 content in spines; inhibition of N-cadherin reduces Rac1 content in spines. Binary masks were generated for Rac1 puncta colocalized with GFP, to quantify the amount of Rac1 in spines. The arrows indicate Rac content in spine heads, and the dashed lines outline dendrites. f, Rac1 integrated intensities within spines were measured and normalized to spine sizes (**p < 0.01, ***p < 0.001 compared with control). Representative images of spines are indicated for each condition. Error bars indicate SEM. Scale bars: a, c, d, 10 μm; e, 1 μm.
Because N-cadherin activation or inhibition modulates the interaction among N-cadherin, AF6, and kalirin-7, it may also modulate the amount of Rac1 in spines. To test this, we measured the intensity of Rac1 immunofluorescence contained within dendritic spines (Fig. 6d–f). Rac1 intensity was normalized to spine sizes to eliminate the passive effect of changes in Rac1 levels caused by altered spine size (control, 9.37 ± 0.85; clust FcNcad, 14.18 ± 1.22; interf Ab, 2.76 ± 0.45 a.u.). In control neurons (Fig. 6d,e, left), small puncta of Rac1 immunoreactivity were detectable in dendrites and in spines (arrows); N-cadherin activation increased significantly the size of Rac1 puncta in spines (Fig. 6d,e, center). In contrast, interference with N-cadherin resulted in reduced Rac1 content in spines (Fig. 6d,e, right) and remodeling of spines into of thin, long shapes, consistent with the reported effects of reduced Rac1 activity on spine morphology: inhibition of Rac1 caused spine thinning and elongation (Tashiro and Yuste, 2004).
A major substrate of Rac1 is PAK. Binding of Rac1 causes activation and autophosphorylation of PAK, hence PAK phosphorylation can be used as a readout of Rac1 activation. We also shown that kalirin-7-induced spine morphogenesis is dependent on Rac1-mediated PAK activation (Xie et al., 2007). Therefore, to test whether N-cadherin engagement activated the Rac1–PAK pathway, and whether this was dependent on kalirin-7 GEF activity, we examined the phosphorylation of endogenous PAK (P-PAK) in neurons (Fig. 7a–c). N-cadherin engagement caused an increase in P-PAK immunoreactivity compared with untreated control neurons, indicating N-cadherin-dependent PAK phosphorylation. Expression of myc-kalirin-7-ΔGEF, a kalirin-7 truncation mutant lacking the GEF domain but containing an intact C terminus, reduced N-cadherin-dependent PAK phosphorylation (control, 3.24 ± 0.51; clust FcNcad, 12.55 ± 1.85; kal7-ΔGEF plus clust FcNcad, 5.71 ± 1.20 a.u.); this reduction was not seen in dendrites of neighboring nontransfected neurons (Fig. 7a–c).
Figure 7.

Kalirin-7 GEF activity is required for N-cadherin-dependent PAK phosphorylation. a, Cortical neuron cultures sparsely transfected with myc-kalirin-7ΔGEF (kal7-ΔGEF) were treated with clustered Fc-Ncad, and then immunostained for myc and endogenous phospho-Pak (P-PAK). N-cadherin adhesion enhances PAK phosphorylation in neurons not expressing kal7-ΔGEF (yellow outline); this enhanced PAK phosphorylation is prevented in neighboring neurons expressing kal7-ΔGEF (red outline). b, Magnified images of dendrites expressing kal7-ΔGEF or not transfected, cells immunostained as above. Untreated cells (control) display basal levels of P-PAK; activation of N-cadherin adhesion (clust FcNcad) results in increased P-PAK levels. Expression of kal7-ΔGEF prevents N-cadherin-mediated Pak phosphorylation. c, P-PAK integrated intensities per cluster in b (**p < 0.01 compared with control; *p < 0.05 compared with clust FcNcad). Error bars indicate SEM. Scale bars: a, b, 10 μm.
N-cadherin-dependent spine enlargement requires kalirin
Because kalirin-7 promotes spine enlargement, overexpression of kalirin-7 may prevent the remodeling of spines into thinner and longer shapes induced by interference with N-cadherin adhesion. We tested this hypothesis by treating neurons expressing GFP together with or without myc-kalirin-7 with the interfering antibody GC4, and comparing spine morphologies (Fig. 8a–c). Overexpression of kalirin-7 prevented the formation of long and thin spines induced by interference with N-cadherin adhesion (spine length: interf Ab vs control, p < 0.05; kal7 plus interf Ab vs control, no difference; spine area: kal7 plus interf Ab vs control, p < 0.05), demonstrating that kalirin-7 functions downstream of N-cadherin in this pathway (spine length: control, 1.77 ± 0.11; interf Ab, 2.47 ± 0.27; kal7 plus interf Ab, 2.02 ± 0.15 μm; spine area: control, 0.64 ± 0.06; interf Ab, 0.90 ± 0.11; kal7 plus interf Ab, 1.05 ± 0.15 μm2; spine linear density: control, 5.09 ± 0.36; interf Ab, 7.40 ± 0.67; kal7 plus interf Ab, 5.73 ± 0.38 per 10 μm).
Figure 8.

N-cadherin-dependent spine enlargement requires kalirin. a, Overexpression of myc-kalirin-7 (kal7 expressing) prevents the formation of long and thin spines induced by interference with N-cadherin adhesion (interf Ab); nontransfected neurons still form these spines. Cortical neurons were immunostained for GFP and myc. b, Magnified images of dendrites of neurons untreated (control), or treated with interfering antibody (interf Ab) with or without myc-kalirin-7 expression; cells immunostained as above. c, Quantification of spine lengths, areas, and linear densities (*p < 0.05 compared with control). d, Kalirin-7 knockdown by 3 d RNAi treatment prevents N-cadherin-induced spine enlargement; expression of an RNAi-insensitive kalirin-7 mutant rescues this effect (**p < 0.01 compared with control; ***p < 0.001 compared with kal7 RNAi or kal7 RNAi plus kal7 rescue). e, Overexpression of kal7-ΔGEF prevents spine enlargement induced by N-cadherin activation. Error bars indicate SEM. Scale bars: a, b, 10 μm; e, 5 μm.
Because kalirin has been shown to be crucial for spine morphogenesis induced by extracellular signals (Penzes et al., 2003; Xie et al., 2007), we tested whether it was required for N-cadherin-dependent spine enlargement. Using a kalirin-7-specific RNA interference (RNAi), previously characterized in detail (Xie et al., 2007), we knocked down kalirin expression in neurons. Although knockdown for 3 d does not cause significant effects on spines (Xie et al., 2007), it prevented N-cadherin-induced spine enlargement (Fig. 8d). This blocking of N-cadherin-induced spine enlargement was reversed in rescue experiments in which an RNAi-insensitive mutant of kalirin-7 was overexpressed (Fig. 8d) (spine area: control, 0.68 ± 0.06; clust FcNcad, 1.17 ± 0.14; clust FcNcad plus kal7 RNAi, 0.71 ± 0.04; clust FcNcad plus kal7 RNAi plus kal7 rescue, 1.08 ± 0.07 μm2), demonstrating that these effects were indeed attributable to kalirin-7 and not some offsite target of the characterized RNAi construct. To test whether the GEF domain of kalirin was also required for N-cadherin-dependent spine enlargement, neurons expressing GFP and myc-kal7-ΔGEF were treated with clustered Fc-N-cadherin or left untreated (Fig. 8e). Quantification of spine lengths and areas revealed that kal7-ΔGEF prevented spine enlargement induced by N-cadherin activation (spine area: control, 0.65 ± 0.03; clust FcNcad, 0.68 ± 0.04; p = 0.47; spine length: control, 1.92 ± 0.07; clust FcNcad, 1.81 ± 0.07; p = 0.25), without affecting spine linear densities (spine linear density: control, 7.52 ± 0.74; clust FcNcad, 7.54 ± 0.81; p = 0.98). Overexpression of kal7-ΔGEF did not affect the availability of N-cadherin on the dendritic surface, as shown by decorating transfected and nontransfected neurons with Fc-N-cadherin (supplemental Fig. S1h, available at www.jneurosci.org as supplemental material). Collectively, these data suggest that, in pyramidal neuronal dendrites, kalirin-7 colocalizes and interacts with N-cadherin-containing adhesion complexes. N-cadherin-mediated adhesion events recruit kalirin-7 and Rac1 to these synaptic adhesion sites, resulting in increased PAK phosphorylation. Furthermore, these results demonstrate crucial role for kalirin-7 in cadherin-dependent spine morphogenesis.
Active Rap interferes with N-cadherin-dependent spine enlargement
AF-6 contains two Ras/Rap-association domains (RA) that preferentially bind activated Rap (Boettner et al., 2003). Interestingly, whereas overexpression of AF-6 induced formation of both thin spines and spines with large heads, deletion of the RA domains from AF-6, consistently induced spine head enlargement (Xie et al., 2005). This suggested that the RA domains of AF-6 may prevent or reduce the ability of AF-6 to induce spine enlargement. We previously showed that expression of constitutively active Rap1 (Rap1-CA) results in the localization of AF-6 at the dendritic plasma membrane and in the formation of thin and long spines. In contrast to this, expression of RapGAP, which inactivates Rap, resulted in AF-6 relocalization from the dendritic plasma membrane to the dendritic cytosol and in enlarged spines (Xie et al., 2005). Therefore, we hypothesized that the binding of active Rap to AF-6 may interfere with its ability to mediate N-cadherin-induced spine enlargement. Indeed, expression of Rap1-CA blocked spine head enlargement induced by N-cadherin activation (supplemental Fig. S3a,b, available at www.jneurosci.org as supplemental material), and resulted in thin spines (area: clust FcNcad, 1.69 ± 0.05; clust FcNcad plus Rap1-CA, 1.18 ± 0.04; p < 0.01; length: clust FcNcad, 1.41 ± 0.05; clust FcNcad plus Rap1-CA, 2.40 ± 0.03; p < 0.001). These spines were statistically undistinguishable from those caused by the expression of Rap-CA alone, but were longer and had larger areas than untreated controls (area: Rap1-CA, 1.25 ± 0.08; control, 0.88 ± 0.02; p < 0.01; length: Rap1-CA, 2.5 ± 0.03; control, 1.24 ± 0.04; p < 0.001); spine density was not affected by Rap1-CA. Expression of Rap1-CA did not reduce the amount of N-cadherin expressed on neuronal surface, as shown by unaffected binding of clustered N-cadherin to neurons (supplemental Fig. S1i, available at www.jneurosci.org as supplemental material). These experiments, together with our previous studies, demonstrate that active Rap interferes with AF-6 in mediating N-cadherin-induced spine enlargement.
Discussion
Dendritic spine remodeling is an important element of synapse maturation and plasticity. Previous studies have shown that increased N-cadherin adhesion or increased expression of N-cadherin and associated proteins induce spine enlargement (Togashi et al., 2002; Murase et al., 2002; Abe et al., 2004). These concurrent processes lead to increased synapse stability (Abe et al., 2004) and higher AMPA receptor content (Liao et al., 1999). Large spines are also more stable (Trachtenberg et al., 2002). Activity-dependent potentiation is also associated with simultaneous spine enlargement (Yuste and Bonhoeffer, 2001; Kasai et al., 2003) as well as recruitment and clustering of N-cadherin and associated proteins (Bozdagi et al., 2000; Murase et al., 2002). Conversely, disruption of N-cadherin or associated proteins caused formation of thin long spines and spine destabilization (Murase et al., 2002; Togashi et al., 2002; Abe et al., 2004). In all of these cases, synaptic adhesion, spine morphology, stability, and synaptic AMPA receptor content are coordinated (Kasai et al., 2003). However, the molecular mechanisms that coordinately regulate spine morphology and synaptic adhesion are not well understood. This coordination may be achieved by adhesion-dependent control of spine morphology, but little is known about the mechanisms whereby synaptic adhesion molecules such as N-cadherin modulate spine morphology.
Although N-cadherin adhesion has been shown to modulate the morphology of existing spines (Togashi et al., 2002; Abe et al., 2004), the mechanism underlying this process was not known. Our studies identify a novel mechanism that coordinates synaptic adhesion and spine morphology, accomplished by N-cadherin signaling to the Rac1-GEF kalirin-7, mediated by the scaffold protein AF-6, supporting the following model (Fig. 9). Increased transsynaptic N-cadherin clustering at spine heads, occurring during synapse maturation, stabilization, and potentiation, associated with increased adhesion, leads to the recruitment of AF-6 and kalirin-7 to adhesion sites in spines. Enrichment of kalirin-7 at in these spines enhances local activation of Rac1, causing enlargement of spines and increased stability of existing synaptic connections. Conversely, reduced N-cadherin-mediated adhesion, which occurs in less mature synapses, when synapses are dynamic and undergo extensive motility and remodeling (Lendvai et al., 2000), during synapse elimination (Mataga et al., 2004), experience-dependent plasticity of cortical circuits (Trachtenberg et al., 2002), or in long-term depression (Zhou et al., 2004), results in the dissociation of AF-6 and kalirin-7 from adhesion complexes, preventing the signal flow from N-cadherin to kalirin-7. This causes decreased Rac1 activation and content in spines, leading to formation of thin, long spines, which are less stable. Because N-cadherin-dependent adhesion is regulated during excitatory synapse development and in synaptic plasticity, this mechanism may underlie spiny synapse maturation and activity-dependent structural plasticity, and thus may contribute to experience-dependent plasticity and learning.
Figure 9.

Model of the role of N-cadherin and associated proteins, AF-6, and kalirin-7 in the dynamic coordination of synaptic adhesion and spine morphology. Clustering of N-cadherin at synapses during synapse maturation, stabilization, or potentiation recruits AF-6, which in turn recruits the Rac1-GEF kalirin-7. Kalirin-7 then activates Rac1 locally, to induce spine enlargement. These large spines are also rich in AMPA receptors. Conversely, weakening of N-cadherin adhesion during synapse destabilization, turnover, experience-dependent plasticity, or synaptic depression, results in the release of AF-6 and kalirin-7 from adhesion sites, leading to reduced local Rac1 activation, resulting in thinner and longer spines, with reduced AMPA receptor content.
A key role has been hypothesized for Rho GTPase activation in cadherin-dependent actin cytoskeletal rearrangements and spine remodeling (Bamji, 2005). However, until recently the mechanisms whereby N-cadherin controls Rho GTPase activation in spines has not been clear. A recent study has shown that p120-catenin controls the formation of new spines, reflected in spine and synapse density, through activation of Rac1 and inactivation of RhoA (Elia et al., 2006). In contrast, our results indicate that the N-cadherin/AF-6/kalirin-7 pathway may provide adhesion complexes the ability to rapidly control Rac1-dependent remodeling of existing spines. The two pathways may to function in tandem to control spine formation and density versus remodeling of existing spines. Kalirin-7, which is an enzyme, may provide an amplification step to this pathway: recruitment of one molecule of kalirin-7 may result in activation of many molecules of Rac1, leading to extensive and sustained changes in actin dynamics and spine morphology. In addition, direct participation of N-cadherin in multiprotein complexes with molecules that rapidly control actin dynamics, such as Rho-GEFs, offers adhesion events the ability to rapidly regulate spine morphology. This mechanism may underlie rapid activity-dependent coordinated changes in adhesion and spine structure during plasticity.
In several previous studies we showed that kalirin-7 is regulated by tyrosine or threonine phosphorylation (Penzes et al., 2003; Xie et al., 2007). However, in this study, we identify a novel mode of regulation of kalirin-7, mediated by its adhesion-dependent recruitment to synapses through its interaction with AF-6. Kalirin-7 targeting to spines depends on its C terminus (Penzes et al., 2001); however, the relative importance of proteins such as AF-6 or PSD-95 that interact with the kalirin-7 C-tail is not clear. The ability of AF-6 to bind kalirin-7 even in presence of PSD-95 in the brain, as shown by coimmunoprecipitation experiments, suggests that AF-6 links some kalirin-7 molecules to adhesion complexes even in the presence of PSD-95. AF-6 and PSD-95 may link kalirin-7 to two separate sets of upstream regulatory proteins, such as cadherin and EphB receptors, or NMDA receptors, respectively. Weaker association of AF-6 with the synaptic cytoskeleton relative to PSD-95 suggests that the AF-6/kalirin-7 complexes may be more dynamic, whereas the PSD-95/kalirin-7 complexes may be stably associated with the PSD.
Although AF-6 has been known to interact directly with α-catenin and nectins in adhesion junctions of epithelial cells (Pokutta et al., 2002; Honda et al., 2006), its role in synaptic adhesion was uncharacterized. In this study, we identified a mechanism linking synaptic adhesion with the remodeling of existing spines, and we show for the first time that AF-6 plays an important role in these processes. Our results place AF-6 in association with N-cadherin adhesion complexes in forebrain neuron synapses in culture and in vivo, demonstrate a role for N-cadherin in concentrating AF-6 in spines, and support a functional role for AF-6 in adhesion-dependent spine remodeling. Because AF-6 is rapidly recruited to spines on N-cadherin activation and because of its role in spine remodeling and its interactions with kalirin-7, it may provide adhesion complexes the ability to dynamically regulate the actin cytoskeleton, allowing synaptic adhesion events to rapidly control spine remodeling.
Previous studies have reported that, in different brain regions and at different stages of development, AF-6 is localized to different sites (Buchert et al., 1999; Nishioka et al., 2000; Mizoguchi et al., 2002; Honda et al., 2006). Our data show that AF-6 is enriched in most excitatory synapses in cultured neurons and PSD fractions of brain (Xie et al., 2005) and in spines of cortical pyramidal neurons. AF-6 also interacts with nectins, another class of Ca2+-independent adhesion molecules, raising the possibility that nectins may be upstream or downstream of AF-6 in regulating spine morphology. In cultured neurons, disruption of nectin-1-mediated adhesion resulted in a decrease in size and an increase in number of presynaptic puncta (Mizoguchi et al., 2002). Nectins have also been shown to mediate a specific interaction between axons and dendrites (Togashi et al., 2002). However, these results were not confirmed in nectin knock-out mice (Honda et al., 2006), suggesting that nectins play a minor role in controlling excitatory synapse morphology in cortical pyramidal neurons. However, nectins play a more important role in the formation and maintenance of hippocampal puncta adherentia between mossy fiber synapses and CA3 dendrites (Nishioka et al., 2000; Honda et al., 2006).
In this study, we show that active Rap1 prevents N-cadherin-induced spine enlargement. This novel role for Rap1 is likely mediated by AF-6, for several reasons. AF-6 promotes the formation of spines with large heads, similar to that induced by N-cadherin adhesion, whereas blocking N-cadherin adhesion results in formation of thin and elongated spines, similar to those caused by overexpression of activated Rap1 (Xie et al., 2005), suggesting that active Rap1 may interfere with the ability of N-cadherin to induce spine enlargement. Moreover, deletion of the N-terminal portion of AF-6, containing Rap-association domains, induced spine enlargement, suggesting that binding of active Rap1 to the RA domains of AF-6 was required for spine elongation. Indeed, we showed here that activated Rap1 blocked spine head enlargement caused by N-cadherin activation. Rap1 may interfere with N-cadherin/AF-6 downstream signaling in one of several possible ways. Binding of Rap1-GTP to AF-6 may affect its conformation and prevent it from interacting with N-cadherin-associated proteins. In addition, because active Rap1 recruits AF-6 to the plasma membrane through direct interaction between the two proteins, Rap1 may compete with the interaction of AF-6 with N-cadherin/α-N-catenin. Alternatively, Rap1 may sequester AF-6 away from adhesion junctions, preventing its interaction with N-cadherin/α-N-catenin or kalirin-7. Future studies are needed to investigate how the Rap1 and N-cadherin mediated upstream pathways interact to control AF-6 localization and function in neurons, because some studies suggested involvement of Rap1 in adhesion.
In summary, this study identifies a novel mechanism whereby N-cadherin, a major synaptic adhesion molecule, signals to the actin cytoskeleton to control the morphology of dendritic spines, and outlines a mechanism that may underlie the coordination of synaptic adhesion with spine morphology during synapse maturation and plasticity.
Footnotes
This work was supported by National Institutes of Health (NIH), National Alliance for Autism Research, and National Alliance for Research on Schizophrenia and Depression (P.P.), and NIH and Howard Hughes Medical Institute (R.L.H.). We thank Dr. Anjen Chenn (Northwestern University) for critically reading this manuscript and Dr. Anis Contractor (Northwestern University) for advice. We thank Kelly Jones for carefully editing this manuscript.
References
- Abe K, Chisaka O, Van Roy F, Takeichi M. Stability of dendritic spines and synaptic contacts is controlled by alpha N-catenin. Nat Neurosci. 2004;7:357–363. doi: 10.1038/nn1212. [DOI] [PubMed] [Google Scholar]
- Bamji SX. Cadherins: actin with the cytoskeleton to form synapses. Neuron. 2005;47:175–178. doi: 10.1016/j.neuron.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Boettner B, Harjes P, Ishimaru S, Heke M, Fan HQ, Qin Y, Van Aelst L, Gaul U. The AF-6 homolog canoe acts as a Rap1 effector during dorsal closure of the Drosophila embryo. Genetics. 2003;165:159–169. doi: 10.1093/genetics/165.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW. Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron. 2000;28:245–259. doi: 10.1016/s0896-6273(00)00100-8. [DOI] [PubMed] [Google Scholar]
- Buchert M, Schneider S, Meskenaite V, Adams MT, Canaani E, Baechi T, Moelling K, Hovens CM. The junction-associated protein AF-6 interacts and clusters with specific Eph receptor tyrosine kinases at specialized sites of cell-cell contact in the brain. J Cell Biol. 1999;144:361–371. doi: 10.1083/jcb.144.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Hueske E, Wyszynski M, Hoogenraad CC, Jaworski J, Pak DT, Simonetta A, Liu G, Sheng M. LAR receptor protein tyrosine phosphatases in the development and maintenance of excitatory synapses. Nat Neurosci. 2005;8:458–467. doi: 10.1038/nn1416. [DOI] [PubMed] [Google Scholar]
- Elia LP, Yamamoto M, Zang K, Reichardt LF. p120 catenin regulates dendritic spine and synapse development through Rho-family GTPases and cadherins. Neuron. 2006;51:43–56. doi: 10.1016/j.neuron.2006.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA. Dendritic spine density in schizophrenia and depression. Arch Gen Psychiatry. 2001;58:203. doi: 10.1001/archpsyc.58.2.203. [DOI] [PubMed] [Google Scholar]
- Honda T, Sakisaka T, Yamada T, Kumazawa N, Hoshino T, Kajita M, Kayahara T, Ishizaki H, Tanaka-Okamoto M, Mizoguchi A, Manabe T, Miyoshi J, Takai Y. Involvement of nectins in the formation of puncta adherentia junctions and the mossy fiber trajectory in the mouse hippocampus. Mol Cell Neurosci. 2006;31:315–325. doi: 10.1016/j.mcn.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Huntley GW, Benson DL, Colman DR. Structural remodeling of the synapse in response to physiological activity. Cell. 2002;108:1–4. doi: 10.1016/s0092-8674(01)00631-6. [DOI] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- Lambert M, Padilla F, Mege RM. Immobilized dimers of N-cadherin-Fc chimera mimic cadherin-mediated cell contact formation: contribution of both outside-in and inside-out signals. J Cell Sci. 2000;113:2207–2219. doi: 10.1242/jcs.113.12.2207. [DOI] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K. Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature. 2000;404:876–881. doi: 10.1038/35009107. [DOI] [PubMed] [Google Scholar]
- Liao D, Zhang X, O'Brien R, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat Neurosci. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- Luscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci. 2000;3:545–550. doi: 10.1038/75714. [DOI] [PubMed] [Google Scholar]
- Mandai K, Nakanishi H, Satoh A, Obaishi H, Wada M, Nishioka H, Itoh M, Mizoguchi A, Aoki T, Fujimoto T, Matsuda Y, Tsukita S, Takai Y. Afadin: a novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J Cell Biol. 1997;139:517–528. doi: 10.1083/jcb.139.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mataga N, Mizuguchi Y, Hensch TK. Experience-dependent pruning of dendritic spines in visual cortex by tissue plasminogen activator. Neuron. 2004;44:1031–1041. doi: 10.1016/j.neuron.2004.11.028. [DOI] [PubMed] [Google Scholar]
- Meyer RA, Laird DW, Revel JP, Johnson RG. Inhibition of gap junction and adherens junction assembly by connexin and A-CAM antibodies. J Cell Biol. 1992;119:179–189. doi: 10.1083/jcb.119.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi A, Nakanishi H, Kimura K, Matsubara K, Ozaki-Kuroda K, Katata T, Honda T, Kiyohara Y, Heo K, Higashi M, Tsutsumi T, Sonoda S, Ide C, Takai Y. Nectin: an adhesion molecule involved in formation of synapses. J Cell Biol. 2002;156:555–565. doi: 10.1083/jcb.200103113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase S, Mosser E, Schuman EM. Depolarization drives beta-Catenin into neuronal spines promoting changes in synaptic structure and function. Neuron. 2002;35:91–105. doi: 10.1016/s0896-6273(02)00764-x. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka H, Mizoguchi A, Nakanishi H, Mandai K, Takahashi K, Kimura K, Satoh-Moriya A, Takai Y. Localization of l-afadin at puncta adhaerentia-like junctions between the mossy fiber terminals and the dendritic trunks of pyramidal cells in the adult mouse hippocampus. J Comp Neurol. 2000;424:297–306. doi: 10.1002/1096-9861(20000821)424:2<297::aid-cne8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Nuriya M, Huganir RL. Regulation of AMPA receptor trafficking by N-cadherin. J Neurochem. 2006;97:652–661. doi: 10.1111/j.1471-4159.2006.03740.x. [DOI] [PubMed] [Google Scholar]
- Penzes P, Johnson RC, Alam MR, Kambampati V, Mains RE, Eipper BA. An isoform of kalirin, a brain-specific GDP/GTP exchange factor, is enriched in the postsynaptic density fraction. J Biol Chem. 2000;275:6395–6403. doi: 10.1074/jbc.275.9.6395. [DOI] [PubMed] [Google Scholar]
- Penzes P, Johnson RC, Sattler R, Zhang X, Huganir RL, Kambampati V, Mains RE, Eipper BA. The neuronal Rho-GEF Kalirin-7 interacts with PDZ domain-containing proteins and regulates dendritic morphogenesis. Neuron. 2001;29:229–242. doi: 10.1016/s0896-6273(01)00193-3. [DOI] [PubMed] [Google Scholar]
- Penzes P, Beeser A, Chernoff J, Schiller MR, Eipper BA, Mains RE, Huganir RL. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37:263–274. doi: 10.1016/s0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- Pokutta S, Drees F, Takai Y, Nelson WJ, Weis WI. Biochemical and structural definition of the l-afadin- and actin-binding sites of alpha-catenin. J Biol Chem. 2002;277:18868–18874. doi: 10.1074/jbc.M201463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci. 1999;11:1598–1604. doi: 10.1046/j.1460-9568.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- Schrick C, Fischer A, Srivastava DP, Tronson NC, Penzes P, Radulovic J. N-cadherin regulates cytoskeletally associated IQGAP1/ERK signaling and memory formation. Neuron. 2007;55:786–798. doi: 10.1016/j.neuron.2007.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Shan W, Phillips GR, Arndt K, Bozdagi O, Shapiro L, Huntley GW, Benson DL, Colman DR. Molecular modification of N-cadherin in response to synaptic activity. Neuron. 2000;25:93–107. doi: 10.1016/s0896-6273(00)80874-0. [DOI] [PubMed] [Google Scholar]
- Tang L, Hung CP, Schuman EM. A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron. 1998;20:1165–1175. doi: 10.1016/s0896-6273(00)80497-3. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- Wheelock MJ, Johnson KR. Cadherin-mediated cellular signaling. Curr Opin Cell Biol. 2003;15:509–514. doi: 10.1016/s0955-0674(03)00101-7. [DOI] [PubMed] [Google Scholar]
- Xie Z, Huganir RL, Penzes P. Activity-dependent dendritic spine structural plasticity is regulated by small GTPase Rap1 and its target AF-6. Neuron. 2005;48:605–618. doi: 10.1016/j.neuron.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56:640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]