

Abstract
B-1 cells reside predominantly within the coelomic cavities, tonsils, Peyer’s patches, spleen (a minor fraction – ∼5%) and are absent in the lymph nodes. They are the primary sources of natural IgM in the body. B-1 cells express polyreactive B cell receptors (BCR) that cross react with self antigens and thus implicated in auto-immune disorders. Previously, we reported that peritoneal B-1 cells are deficient in CD19-mediated intracellular signals leading to Ca2+ mobilization. Here, we find that splenic B-1 cells, like peritoneal B-1 cells, are defective in Ca2+ release upon B cell activation by co-cross-linking BCR and CD19. In the absence of extracellular sources of Ca2+, intracellular Ca2+ flux is similar between B-1 and B-2 cells. Moreover, the intracellular component of Ca2+ release in both subsets of B cells is mostly PI3K dependent. BCR and CD19 co-cross-linking activates Akt, a key mediator of survival and proliferation signals downstream of PI3K in splenic B-2 cells. Splenic B-1 cells, on the other hand, do not phosphorylate Akt (S473) upon similar treatment. Furthermore, BCR + CD19 cross-linking induced phosphorylation of JNK is much reduced in splenic B-1 cells. In contrast, B-1 cells exhibited increased levels of constitutively active pLyn which appears to have an inhibitory role. The CD19 induced Ca2+ response and BCR induced proliferation response were restored by a partial inhibition of pLyn with Src kinase specific inhibitors. These findings suggest a defect in CD19 mediated signals in both peritoneal and splenic B-1 B lymphocytes, which is in part, due to higher levels of constitutively active Lyn.
Keywords: mouse B-1 cells, B cell receptor, CD19 signaling
INTRODUCTION
B-1 cells are distinguished from B-2 cells based on their anatomical localization, cell surface phenotype, self-renewal capacity, B cell receptor signal generation, and contribution to natural circulating serum IgM (Berland and Wortis, 2002; Hayakawa et al., 1985; Hayakawa et al., 1984; Hayakawa et al., 1986). B-1 cells are localized in the coelomic cavities (peritoneal and pleural), Peyer’s patches, tonsils, and spleen (~5% of splenic B cells) and absent in lymph nodes. B cell activation initiates a cascade of intracellular signaling events leading to cell proliferation, differentiation and secretion of the antibody or apoptosis. Binding of antigen or its surrogate, anti-IgM leads to clonal expansion in the follicular (FO) or the ‘conventional’ B-2 B cells. Unlike B-2 cells, B-1 cells are hyporesponsive to BCR cross-linking and share some similarities with anergic cells (Hippen et al., 2000). Paradoxically, B-1 cell numbers are increased in several murine models of autoimmune disorders, such as NZB, MRL/lpr and motheaten mice, as well as in humans with lupus or rheumatoid arthritis (Burastero et al., 1988; Dauphinee et al., 1988; Hayakawa et al., 1983; Mohan et al., 1998; Sidman et al., 1986). In blood, rheumatoid factor producing B cells were found to be predominantly B-1 type (Haas et al., 2005; Hardy et al., 1987).
B-1 cells are further subdivided into B-1a and B-1b subtypes as the former but not the latter express CD5 (Stall et al., 1992). Bikah et al. have established that CD5 antigen on B-1a cells inhibits signaling through the B cell receptor (Bikah et al., 1996). Sen et al. showed that CD5 down regulates BCR signaling by recruiting SHP-1 (Src homology 2 (SH2) domain containing protein tyrosine phosphatase-1) into the BCR complex (Sen et al., 1999). More recently, Dal Porto et al. showed that CD5 may induce activation of Lck which may in turn inhibit BCR signaling in B-1 cells (Dal Porto et al., 2004). This, however, is controversial since Frances et al. showed that B-1 cells do no express Lck (Frances et al., 2005). We have shown that FACS sorted peritoneal B-1a and B-1b B cells are equally defective in BCR induced proliferative response (Sen et al., 2002). B-1a and B-1b B cells collaborated in immunity to S. pneumoniae by respectively contributing to innate and adaptive immune responses (Haas et al., 2005). Since B-1b cells do not express CD5, the basis of BCR signaling defect is unclear. Recently, it has been shown that B-1b B cells may be primarily responsible for IgM memory cells, as they were expanded preferentially in a murine model of relapsing fever (Alugupalli et al., 2004). B-1b B cells have thus gained attention as crucial players of cell mediated antibody responses independent of T cell help (Alugupalli, 2008). Recent description of IL-10 producing splenic CD1dhi CD5+ B cells in mice with a regulatory role reinforces the importance of B-1 B cells in T-cell mediated immunity (Yanaba et al., 2008). These regulatory B cells (Breg) are proposed to suppress activation and differentiation of CD4+, CD8+, NKT and other immune cell types thereby demanding caution in B cell depletion therapeutics as it may hinder maintenance of tolerance (Mauri and Ehrenstein, 2008).
The B cell restricted glycoprotein CD19 in concert with CD21/CR2 and CD81/TAPA-1 forms a co-receptor complex and aids in BCR function as a positive regulator of B cell signaling by lowering the threshold for B cell activation (Carter and Fearon, 1992). Activation of CD19 is dependent upon Lyn-mediated phosphorylation of CD19 cytoplasmic domain (Fujimoto et al., 2001). There are 9 conserved tyrosine residues on CD19 cytoplasmic tail that upon phosphorylation allow recruitment of adaptor molecules such as Grb2, Sos, Vav and activation of PLCγ, Fyn, Lyn and PI3K (Wang et al., 2002). These molecules are responsible for downstream signaling events leading to calcium (Ca2+) mobilization, mitogen activated protein kinase (MAPK) activation and induction of transcription factors. We had previously reported that peritoneal B-1a and B-1b B cells are defective in CD19-dependent signaling events and speculated on the possible candidates that are differently regulated in B-1 versus B-2 cells (Sen et al., 2002).
Recently it has been proposed that splenic B-1 cells are distinct from peritoneal B-1 cells since the latter but not the former express constitutively activated form of STAT-3 (Fischer et al., 2001). Moreover, peritoneal but not splenic B-1 cells responded to stimulation with PMA alone. Hence we attempted to perform a comprehensive study of CD19 signaling in B-1a and B-1b B cells from both peritoneal and spleen of wild type mice. Additionally, we utilized splenic B-1 cells from VH12 transgenic mice to determine the biochemical basis of CD19 dependent signaling in B-1 cells (Arnold et al., 1994). We show that the positive signaling role of CD19 is defective in all B-1 cell subsets (B-1a and B-1b from both spleen and peritoneum) examined. Biochemically, this resulted in a lack of activation of JNK and Akt, key enzymes required for B cell survival and proliferation. Here, we demonstrate that B-1 cells have elevated levels of constitutively active Lyn and that it plays a role in the negative regulation of BCR and CD19 signaling.
MATERIALS AND METHODS
Mice
8–16 week old female C57BL/6 mice were obtained from the Jackson Laboratories (Bar Harbor, MA). VH12 transgenic mice in the C.B-17 background were described by Arnold et al. and were kept in heterozygous state by crossing to C.B-17 mice (Arnold et al., 1994). The PCR screened negative littermates were used as control mice.
Purification of B cells
3–4 month old female C57BL/6 mice were used to obtain peritoneal cells by peritoneal lavage with Hank’s balanced salt solution. For in vitro proliferation assays with peritoneal and splenic B cells, macrophages were removed by short-term plastic adherence and T cells were depleted using an antibody cocktail with anti-Thy 1.2, anti-CD4 (L3T4) and anti-CD8 (Lyt2) followed by rabbit complement, as described earlier (Sen et al., 2002). Anti-B220/CD45RB~FITC, anti-CD23~PE or anti-CD43~FITC (BD Pharmingen, San Jose, CA) were used to identify and sort B-2 (B220+ CD43−) and B-1 (B220+ CD43+) cells from splenic populations of C57BL/6 or VH12 transgenic mice (Wells et al., 1994).
In vitro cell proliferation assay
Splenic B-2 or B-1 and peritoneal B-1 cells were cultured in IF-12 medium (with 10% FBS). Cells plated at 2.5 × 105 cells/well were stimulated with 50 μg/ml polyclonal goat anti-IgM F(ab′)2 (ICN/MP Biomedicals, Irvine, CA), 1 μg/ml LPS (Sigma, St. Louis, MO) and 1:1000 anti-CD40 (1C10) ascites (Heath et al. 1994). Cultures were pulsed with 3[H] Thymidine (DuPont/NEN) on day 2 for 4 hours, harvested (Packard, Meriden, CT) and the incorporated radioactivity was measured using a Matrix 96 β-counter (Packard, Downers Grove, IL). Results are represented as mean + SE of triplicate cultures.
Calcium release analysis
10 × 106 splenic cells were loaded with 1 μM Indo-1 AM (Molecular Probes Inc., Eugene, OR) at 37°C for 30 min. Cells were then stained with anti-B220~PE and anti-CD43~FITC on ice for 30 min along with biotinylated goat anti-mouse IgM F (ab′)2 (MP Biomedicals, Irvine, CA) and anti-CD19 (1D3) antibodies, washed and resuspended in insulin and progesterone free IF-12 media (10% FBS). Two minutes before analysis, the cells were resuspended in prewarmed media (6 mM EGTA containing media was used for intracellular Ca2+ release study) and a baseline calcium release was measured for 30 s. Then, avidin (Sigma, St. Louis, MO) was added to the cell suspension and calcium levels were measured for 5 min. The ratio of fluorescence (405/545 nm) of the gated B-2 and B-1 cells was determined using the MoFlo cytometer (Dako Cytomation Inc., Fort Collins, CO) and analyzed using the Summit software program. An increase in the ratio of violet to blue fluorescence indicates an increase in Ca2+ concentration in the cell. Cells from peritoneal lavage were treated similar to splenic cells. They were, however, stained with α-B220~CY, α-Mac-1~PE and α-CD5~FITC for distinguishing B-1a, B-1b and B-2 subsets. To determine the importance of PI3K, B cells were pre-incubated with Wortmannin for 25 min at room temperature. 6 mM EGTA containing media was used to determine the effect of CD19 signaling on intracellular calcium release. 2 μM Thapsigargin (Molecular Probes Inc., Eugene, OR) was added after obtaining a 30 s Indo-1 baseline. Experiments with PP1 and PP2 (Biomol International, L.P., Plymouth Meeting, PA) were performed by addition of the inhibitor to cells at room temperature for 20–40 min prior to 37°C pre-warming for 2 min.
B cell stimulation and Immunoblotting
5–10 × 106 splenic B cells were initially allowed to rest for 3–4 h in serum free IF-12 at 37°C. Appropriate stimulations were performed for indicated periods of time while incubating in a 37°C water bath. Reactions were stopped with addition of cold PBS, pH 7.2, containing 10mM NaF and 1mM Na3VO4. After two washes with stop buffer, cell pellet was lysed in 1% NP-40 cell lysis buffer containing 10 μg/ml Aprotinin, 10 mM PMSF, 1 mM Leupeptin for 30 min on ice. Lysates were cleared of nuclear debris by centrifugation at 10,000 rpm for 10 min at 4°C. Wherever indicated, 2 μM PP1 was added to cell suspension and incubated at 37° C for 30 min prior to addition of stimulating antibodies.
Proteins were denatured by boiling for 5–10 min in Laemmli’s reducing sample buffer and separated on 10% SDS-PAGE gels. Separated proteins were transferred to PVDF membrane (Immobilon-P transfer membrane, Millipore Corporation, Billerica, MA). Membranes were blocked with 1% BSA (for phospho-antibodies) or 5% milk in TBS containing 0.05% Tween-20 for an hour at room temperature. The blots were probed with primary (1 hr or overnight) and secondary (HRP conjugated) antibodies (1hr at room temperature) followed by developing in SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockfold, IL) for a minute. Protein bands were visualized on the Kodak Image Station 200RT (Eastman Kodak Company, New Haven, CT) and quantified according to the manufacturer’s software. Alternatively, the membrane was exposed to BioMax XAR Film (Eastman Kodak Company, Rochester, NY) and the bands were quantified using Scion Image software. Fold induction of bands specified was calculated by dividing the mean band intensities of protein of interest to the appropriate loading control and normalizing the media control group as 1.0.
Reagents
Rabbit polyclonal antibodies to pJNK, pAkt (S473), Akt and pSrc (Y416) were purchased from Cell Signaling Technology (Beverly, MA); monoclonal anti-mouse β-actin was from Sigma-Aldrich (St. Louis, MO). Anti-JNK1, anti-SHP-1, anti-Lyn and secondary reagents were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA).
Statistics
Student’s t test was used to determine statistical significance of differences between various groups.
RESULTS
Splenic B-1 cells are hyporesponsive to BCR induced proliferation
B cell receptor cross-linking of splenic B-2 cells leads to clonal expansion as observed by the vigorous proliferation of these cells. Peritoneal B-1 cells, on the other hand, do not mount a proliferative response to anti-IgM stimulation (Bikah et al., 1996). Because of the recent studies on differential role of B-1a and B-1b B cells in immune responses to Borrelia hermsii and S. penumoniae and reported differences between splenic and peritoneal B-1 cells, we re-examined the ability of these subsets to proliferate when BCR is cross-linked with anti-IgM. As shown in Fig. 1A, FACS sorted peritoneal B-1a and B-1b B cells failed to proliferate while B-2 cells responded well to BCR cross-linking. Both subsets responded to LPS and CD40 stimulation (data not shown). Similarly, sort purified splenic B-1 cells from C57BL/6 mice behaved like the peritoneal B-1 cells and do not proliferate in response to anti-IgM stimulation (Fig. 1B). Splenic B-1 cells obtained from VH12 transgenic mice also behaved like those from a wild type strain and were unresponsive to BCR signals (Fig. 1C). B-1 cell response to other stimulants such as LPS or anti-CD40 is comparable to B-2 cells suggesting that the defect in B-1 cells is in the BCR initiated proliferative signals while the other pathways remain relatively intact. Previous reports have indicated that splenic B-1 cells do not resemble peritoneal B-1 cells in all properties (Fischer et al., 2001; Frances et al., 2006; Tumang et al., 2004). However, with respect to BCR induced growth response, both peritoneal and splenic B-1 cells were similarly unresponsive. Also the transgenic splenic B-1 cells were similar to peritoneal B-1 cells from wild type mice in BCR responses.
Figure 1. Peritoneal and splenic B-1 cells are hyporesponsive to BCR mediated proliferation.

(A) T cell-depleted splenic and FACS purified peritoneal B-1a and B-1b B cells from C57BL/6 mice were cultured at a cell density of 2.0 × 105 cells/well with anti-IgM F (ab′)2 or anti-CD40 for 48 hours with indicated mitogens. Shown are the mean proliferation + SE from triplicate set of wells.
(B) FACS purified splenic B-1 (B220+ CD43+) and B-2 (B220+ CD43−) cells were plated at 2.5 × 105 cells/well, treated with the stimulants at the indicated concentrations, cultured for 48 hours and measured for proliferative response by 3[H] Thymidine incorporation. The graph represents one of three independent assays.
(C) T cell-depleted VH12 transgenic splenic B-1 cells and negative control littermates were plated at a cell density of 2.5 × 105 cells/well and cultured for 48 hours as in (B). Graph represents one of three independent assays. * indicates p < 0.01 when comparing B-1 and B-2 cell responses.
Splenic B-1 cells are defective in BCR and CD19 induced Ca2+ mobilization
It has been previously established that CD5 expression on B-1 cell surface confers their inherent hypo-responsiveness to BCR signaling and lack of CD5 on peritoneal B-1 cells (CD5−/−) can restore the BCR responsiveness (Bikah et al., 1996). The fact that both B-1a (CD5+) and B-1b (CD5−) subtypes are deficient in anti-IgM induced proliferation indicates that apart from CD5, BCR signaling in B-1 cells may be regulated by some other cell surface molecule (Fig. 1A) (Sen et al., 2002). In this context, Sen et al. demonstrated that co-cross-linking of CD19 and BCR induces a synergistic increase in Ca2+ response and that both peritoneal B-1a and B-1b cells are deficient in a synergistic response in comparison to splenic B-2 cells. Recently, peritoneal B-2 cells were reported to be less responsive than splenic B-2 (Hastings et al., 2006). Therefore, we determined if the peritoneal B-1 cell response to BCR and CD19 cross-linking is less when compared to peritoneal B-2 response. Essentially three different peritoneal B cell subsets (B-2, B-1a, B-1b) in the same tube were tested for calcium mobilization by using B220, Mac-1 and CD5 markers to electronically gate the various B cell populations. We chose a concentration of anti-IgM (◆) which on its own induces a low Ca2+ response in peritoneal B-2 cells (Fig. 2A). In the peritoneal B-2 cells, this response was dramatically enhanced by co-ligation with CD19 (▲) similar to the studies of Sen et al. (Sen et al., 2002) with B2 cells. In both peritoneal B-1a (△) and B-1b (■) B cell populations, the Ca2+ response induced by BCR and CD19 was significantly reduced in comparison to the peritoneal B-2 cell response. Ligation of B cells with anti-CD19 alone causes a very low Ca2+ flux (⋄). Thus B-1 cell responses to CD19-BCR co-ligation were reduced whether they were compared to splenic B-2 (Sen et al.) or to peritoneal B-2 cells (Fig. 2A) (Sen et al., 2002).
Figure 2. BCR and CD19 induced Ca2+flux is defective in peritoneal B-1a, B-1b; and splenic B-1 cells.
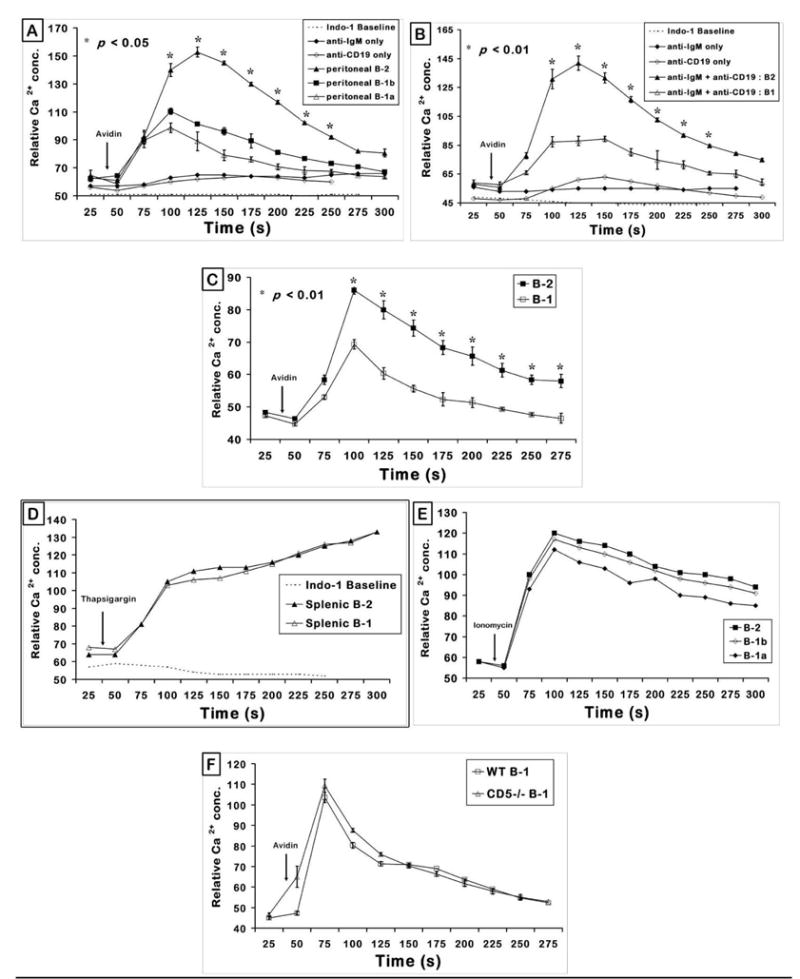
(A) Peritoneal cells (B220+) were gated on B-1a (Mac-1+, CD5+), B-1b (Mac-1+, CD5−) and B-2 (Mac-1−, CD5−) cells to obtain Ca2+ flux upon 0.5 μg/ml biotinylated anti-IgM + 2 μg/ml biotinylated anti-CD19 stimulation. Arrow indicates the time of addition of avidin to cross-link biotinylated anti-IgM and/or anti-CD19 antibodies. Graph represents mean of relative Ca2+ concentration from triplicate analysis + SE. p < 0.05 when comparing responses of peritoneal B-2 with B-1a or B-1b separately at each time point (seconds) indicated by the (*). Data shown are from a representative experiment repeated thrice.
(B) Splenic cells from C57BL/6 female mice were gated on B-1 (B220+ CD43+) and B-2 (B220+ CD43−) B cell subsets and the relative Ca2+ concentration plotted as a function of time. p < 0.05 (*) compares the response from splenic B-2 and B-1 cells. Graphs are representation of one of four independent experiments.
(C) Splenic cells from 4–6 month old VH12 transgenic mice were gated on B-1 and B-2 B cell populations as in panel B above and the relative Ca2+ concentration from triplicate set of tubes was plotted as a function of time. p value was < 0.01 (*) at the 75–200 second determinations. The graph is a representative of more than 6 independent experiments.
(D) Non-receptor induced Ca2+ flux in B-1 and B-2 cells: Thapsigargin (1 μM) was added to C57BL/6 mouse splenic cells 30 s after taking a baseline Ca2+ level (downward arrow). Relative Ca2+ concentration evoked by thapsigargin in splenic cells gated on B-1 (B220+ CD43+) and B-2 (B220+ CD43−) as a function of time is plotted.
(E) Peritoneal cells from C57BL/6 mice were treated with 1 μM Ionomycin after obtaining the 30 sec baseline calcium concentration. Response shown was the effect of non-receptor mediated generation of Ca2+ in the gated B-2, B-1b and B-1a sub-populations.
(F) Peritoneal cells from 3 month-old C57BL/6 (WT) or CD5−/− mice were gated on B-1 (B220+, Mac-1+) to obtain Ca2+ flux upon anti-IgM + anti-CD19 (both antibodies are biotinylated) stimulation. Arrow indicates the time of addition of avidin to cross-link biotinylated anti-IgM and anti-CD19 antibodies. Graph represents mean of relative Ca2+ concentration from triplicate analysis + SE. This is one of two independent experiments with similar results.
Because of the reported differences between splenic and peritoneal B cells we tested if splenic B-1 cells behave similarly to peritoneal B-1 cells with respect to this defect in CD19 induced Ca2+ response (Tumang et al., 2004). B-2 and B-1 cells from the same splenic cell population were marked by B220 and CD43 so that their Ca2+ response can be tested under identical stimulation conditions. Once again a low dose of biotinylated anti-IgM was used so that it induced a very small calcium flux which was dramatically enhanced when IgM and CD19 were co-crosslinked. The anti-CD19 antibody by itself had a minimal effect on the response. When gated on B-1 population (B220+ CD43+) from wild type splenic cells; the synergistic response obtained from BCR and CD19 co-cross-linking was again lower in magnitude (△) than that from the gated B-2 population (B220+ CD43−) in the same sample analyzed (▲) (Fig. 2B). The B-1 cells did not respond to anti-IgM (low dose) or anti-CD19 (optimal dose) when added separately. Transgenic splenic B-1 cells from VH12 mice, like the wild type strain, mounted a Ca2+ response to BCR-CD19 co-cross-linking that was comparably lower than B-2 cells from the same pool of cells (Fig. 2C).
In order to confirm that the splenic and peritoneal B-1 cells are deficient in mobilizing receptor mediated Ca2+ and not in non-receptor evoked Ca2+ flux; we stimulated splenic cells with thapsigargin. Thapsigargin is an inhibitor of sarcoplasmic endoplasmic reticulum calcium-ATPase (SERCA), a channel which functions to reuptake Ca2+ into the endoplasmic reticulum (ER) (Thastrup et al., 1990). Thapsigargin depletes ER Ca2+ stores independent of IP3 generation. Fig. 2D indicates that splenic B-1 cells, like splenic B-2 cells, are equally capable of mobilizing Ca2+ when treated with thapsigargin. Calcium ionophores such as Ionomycin and A23187 facilitate the direct transport of Ca2+ across the plasma membrane (Klaus et al., 1985). When peritoneal B cells were treated with 1 μM Ionomycin a similar calcium response was elicited in all subsets of peritoneal B cells (Fig. 2E). The differences between the three subpopulations were small and not statistically significant. Therefore, the Ca2+ flux defect that we observe in Figure 2 (A,B,C) from B-1 cells is specific to that generated via the BCR and CD19.
CD5 does not regulate CD19 induced calcium responses
Since CD5 has been shown to affect Ca2+ responses induced by BCR alone, we questioned if CD5 expression affected the BCR + CD19 induced response (Tarakhovsky et al., 1994). When gated on the B220+ Mac-1+ B-1 cells, the response from the CD5−/−peritoneal B-1 cells was of similar magnitude to that observed from the wild type cells (Fig. 2F). Thus the BCR + CD19 induced Ca2+ response of B-1 cells from wild type or CD5−/− mice was identical suggesting that the CD19 response is not regulated by CD5.
BCR and CD19 induced intracellular Ca2+ release in B-1 and B-2 subsets is similar and is PI3K dependent
The Ca2+ flux detected upon BCR and CD19 co-cross-linking in splenic B cells is constituted by intra- and extra-cellular components, which are almost not exclusive. BCR activation leads to phosphorylation of CD19 which allows recruitment of PI3K to CD19 (Y482, Y513) and subsequent activation of PI3K leads to generation of PI(3,4,5)P3 from PI(4,5)P2 (Wang et al., 2002). PIP3 is utilized by Btk to activate PLCγ which now generates second messenger molecules DAG and IP3 (Fujimoto et al., 2001). IP3 upon binding to IP3 receptors on ER membrane facilitates extrusion of Ca2+ from ER stores which generates the initial cytosolic pool of Ca2+. Depletion of ER Ca2+ stores triggers opening of TRPC (C-type transient receptor potential) class of ion channels (Ambudkar et al., 2007). The activated TRPCs allow further influx of Ca2+ from extracellular sources (Inada et al., 2006). In order to examine if the defect in CD19 induced Ca2+ is in the intra- or extra-cellular calcium mobilization, the response of B cell subsets in the absence of extracellular supply of Ca2+ was examined. Splenic B cells from BL/6 mice were resuspended in EGTA and stimulated via their BCR and CD19. Fig. 3A recapitulates the difference between B-1 and B-2 cells in calcium containing medium. Addition of EGTA not only greatly reduces the overall Ca2+ response but the difference in magnitude of response between B-1 and B-2 is minimal (Fig. 3B). This similarity in the intracellular Ca2+ release upon anti-IgM and anti-CD19 stimulation of B-1 and B-2 subsets is evident in the VH12 negative littermate (Fig. 3C) as well as VH12 transgenic mice (Fig. 3D) suggesting that the major reason for defective BCR + CD19 induced response is in the influx of extracellular Ca2+ into the cell.
Figure 3. Effect of chelating extracellular calcium with EGTA on B cells stimulated with BCR and CD19.

(A and B) BCR and CD19 induced intracellular Ca2+ response appears to be similar in both splenic B-1 and B-2 cells. C57BL/6 spleen cells were treated as in Fig. 2 and gated on splenic B-1 (B220+ CD43+) and B-2 cells (B220+ CD43−) (Panel A). To obtain intracellular Ca2+ flux, cells were resuspended in media containing 6 mM EGTA to chelate extracellular Ca2+ available in the media (Panel B). Data shown is representative of one out of four independent experiments. Graph is plotted with data of mean + SE from triplicate samples. p < 0.01 compares the B-2 cell response to the B-1 cell response at each time point indicated.
(C and D) Biotinylated anti-IgM and biotinylated anti-CD19 co-cross-linking induced intracellular Ca2+ flux in VH12 negative littermate control mice is similar in splenic B-1 and B-2 cells. Splenic cells from VH12 negative littermate control mice (panel C) and transgenic mice (panel D) were treated similar to wild type splenic cells as in (panel A) and intracellular Ca2+ flux obtained as described above. Data plotted is mean + SE from triplicate samples. p < 0.01 comparing the response from B-2 cells with and without EGTA.
Majority of the Ca2+ flux seen in B-2 cells upon BCR + CD19 stimulation is through PI3K activity. In order to assess if this is the case in B-1 cells; splenic B cells were pre-incubated at room temperature for 25 min with a PI3K inhibitor, wortmannin (Fig. 4). The Ca2+ response of B-2 cells was significantly reduced by wortmannin. The small response of splenic B-1 cells to anti-IgM and anti-CD19 was also reduced by wortmannin.
Figure 4. The Ca2+flux generated by co-cross-linking BCR and CD19 on wild type splenic B cells is PI3K dependent in both B-1 and B-2 subsets.

Splenic B cells were treated with 0.5 μg/ml biotinylated anti-IgM and 2 μg/ml biotinylated anti-CD19 and 10 μg/ml avidin was added to crosslink IgM and CD19. 25 nM wortmannin was added at room temperature 25 min prior to analysis, as described in the Methods section. p < 0.05 comparing the response from B-2 cells without and with wortmannin. This is a representation of at least 3 independent experiments.
CD19 dependent Akt activation is deficient in splenic B-1 cells
Phosphorylation of a serine/threonine protein kinase Akt is a downstream event in PI3K signaling pathway (Otero et al., 2001). Akt activation serves as a survival and proliferative mechanism by its ability to activate the NFκB pathway. BCR stimulation leads to phosphorylation of Akt in B-2 but not in B-1 cells (Wong et al., 2002). CD19 by itself is capable of activating the serine/threonine protein kinase Akt in primary splenic B cells and is necessary to enhance the Akt activation following cross-linking of mIgM (Otero et al., 2001). We have found that in B-2 cells anti-IgM induced pAkt (S473) is further upregulated slightly when co-crosslinked with anti-CD19 (Fig. 5). Interestingly, in VH12 transgenic B-1 cells, pAkt induced by optimal dose of anti-IgM was reduced. Neither BCR ligation alone nor BCR and CD19 coligation failed to induce any detectable phosphorylation of Akt. This may explain why there is a low proliferative response to BCR cross-linking.
Figure 5. BCR and CD19 signaling in transgenic splenic B-1 cells fails to activate Akt/PK-B.

Purified splenic B cells from VH12 transgenic mice (VH12-Tg) and negative littermate controls (VH12-NL) were stimulated with indicated antibodies for 15 min at 37°C and immunoblotted for pAkt (at S473). Anti-IgM indicates the antibody without biotin and is used at 50 μg/ml. Anti-μ~bio and anti-CD19~bio indicate biotin conjugated antibodies and are respectively used at 0.5 and 2.0 μg/ml. 10 μg/ml avidin is used to cross-link IgM and CD19. This is a representative of at least 3 independent experiments.
JNK activation is lower in splenic B-1 cells
BCR stimulation leads to activation of stress-activated protein kinase (SAPK/JNK). BCR induced pJNK is greater and occurs quicker in B-2 than in B-1 cells (O’Rourke et al., 1998). Phosphorylation of CD19 (Y391, Y421) leads to recruitment of Vav and culminates in activation of JNK (Wang et al., 2002). Co-cross-linking of BCR and CD19 on splenic B-2 cells leads to further enhancement of BCR induced phosphorylation of JNK (Tooze et al., 1997). JNK exists in two isoforms, p54 and p46 as shown in the immunoblot. We are showing only the phosphorylation of p54 isoform since phosphorylation of p46 isoform was minimal under these conditions. In our system, we demonstrate that the level of pJNK in splenic B-2 cells upon anti-IgM and anti-CD19 co-cross-linking is higher than with the either antibody alone. In the VH12 splenic B cells, the JNK phosphorylation induced by BCR ligation was less than in the B-2 cells and this response was not enhanced by co-ligation of BCR and CD19 at the time point tested (Fig. 6). Stimulation with PMA indicates that the B-1 cells are responsive to other non-BCR induced JNK activation. This indicates that splenic B-1 cells are deficient in another CD19 dependent function, i.e. JNK activation.
Figure 6. Splenic B-1 cells fail to induce pJNK when stimulated via BCR and CD19.

Purified splenic B cells from the two mouse strains were stimulated for 15 min at 37°C, lysed and immunoblotted for pJNK and JNK1 as loading control. Immunoblot represents one of 3 independent stimulations. Anti-IgM, anti-μ~bio and anti-CD19~bio and avidin are used as explained in legend to Fig 5.
Peritoneal B-1 cells express pLyn constitutively
Lyn, a src family tyrosine kinase member, is known to regulate B cell activation in both positive and negative manner (Xu et al., 2005). BCR induced Lyn phosphorylation is essential for CD19 activation. To test the status of Lyn in peritoneal B-1 and splenic B-2 cells, we immunoprecipitated Lyn from lysates obtained from macrophage and T cell-depleted peritoneal B-1 cells and splenic B-2 cells. When they were probed with anti-p-SFK antibody that detects activated form of Lyn, we observed that Lyn is constitutively phosphorylated in peritoneal B-1 cells as compared to a lower level of pLyn in resting splenic B-2 cells (Fig. 7A). There was no significant increase in total Lyn in B-1 cells compared to B-2 cells. We, therefore, considered the possibility that Lyn plays a negative role in the context of peritoneal B-1 cells by its ability to phosphorylate inhibitory receptors, enzymes and adaptor molecules.
Figure 7. Peritoneal B-1 cells express high pLyn levels.

(A) Peritoneal B-1 and splenic B-2 cells from C57BL/6 mice were resuspended in serum free IF-12, rested for 30 min at 37°C and immunoprecipitated with anti-Lyn antibody and probed for pSFK and Lyn.
(B) Peritoneal B-1 and splenic B-2 cells from wild type C57BL/6 mice were stimulated with biotinylated anti-IgM and/or anti-CD19 + avidin and treated as in panel A above to obtain whole cell lysates and immunoblotted for detection SHP-1 (~60 kDa), membrane was stripped and re-probed for actin as gel loading control.
(C) Whole cell lysates obtained from similar treatment as in panel A were immuno-precipitated with anti-Lyn antibody. The blot was probed for SHP-1, stripped and reprobed for Lyn to correct for gel loading.
(D and E) CD5-SHP-1 association is important for the negative regulatory role of CD5 in B-1 B cells: Total splenocytes from 3–4 week old viable motheaten (me/mev) and negative littermate control mice were treated similar to the splenocytes from wild type C57BL/6 mice in Fig. 2B. Duplicate set of cell suspensions stained independently for B-1 (B220+ CD43+) and B-2 (B220+ CD43−) B cells were stimulated with avidin after 30 second relative calcium concentration baseline determination. This is one of three independent experiments. The responses from motheaten B-1 cells are shown in panel D and those from B-2 in motheaten compared to littermate control mice in panel E.
The negative regulation of Lyn in B lymphocytes has been shown to involve recruitment of SHP-1 (Cornall et al., 1998). In order to determine if the increased levels of pLyn might be negatively regulating the BCR signal in B-1 cells via SHP-1, we first determined the level of total SHP-1 in lysates obtained from peritoneal B-1 cells before or after stimulation via BCR and CD19. As shown in Fig 7B, the total level of SHP-1 protein expressed in the peritoneal B-1 cells was not very different from the littermate splenic B-2 cells indicating that the cytoplasmic SHP-1 available for the phosphatase function of the enzyme is similar in B-1 and B-2 cells. The SHP-1 levels were not altered by BCR or CD19 stimulation but were always slightly lower upon co-cross-linking of CD19 and BCR, a phenomenon observed in both B-1 and B-2 cells. However, this does not necessarily mean all of the available SHP-1 is associated with pLyn. Lyn has been previously shown to bind the –SH2 domain of SHP-1 in activated B cells (Somani et al., 2001). When tested for enhanced Lyn/SHP-1 association in B cells, the lysates immunoprecipitated with anti-Lyn did not show differences in the association of SHP-1 in B-1 and B-2 cells suggesting that SHP-1 may not have a role in the negative regulatory function of Lyn under these experimental conditions (Fig. 7C). Consistent with this we found that B-1 cells from SHP-1 deficient motheaten mice exhibited similar defects in BCR + CD19 induced Ca2+ response when compared to B-2 cells from the normal littermate (Fig. 7D). Moreover, the splenic B-2 cell responses from motheaten mice are significantly better than those observed in the littermate control mice as has been previously reported (Fig. 7E). Recently Pao et al. have demonstrated that upon cross-linking BCR only, splenic B-1 cells (B220lo CD5+) with a B-cell specific deletion of ptpn6 gene exhibit greater increase in intracellular Ca2+ concentration than B-1 cells in littermate controls. Thus, they propose SHP-1 to be a negative regulator of BCR evoked Ca2+ responses in splenic and peritoneal CD5+ B-1a B cells (Pao et al., 2007).
Blocking Lyn activation partially rescues BCR and CD19 induced signaling defect in B-1 cells
We tested the putative negative regulatory role of pLyn in B-1 cells by determining if inhibition of Lyn can enable B-1 cells to overcome the BCR/CD19 signaling defects. Accordingly, we treated B-1 cells with a potent and selective src family tyrosine kinase (SFK) inhibitor, pyrazolopyrimidine PP1 (Hanke et al., 1996). Peritoneal cells from C57BL/6 mice were loaded with Indo-1, treated with anti-IgM and anti-CD19 and stained for B-1 specific markers (B220 and Mac-1). These cells were pre-incubated with various doses of PP1 at room temperature for 20 min, warmed to 37°C for 2 min and stimulated with avidin after taking a 30 s baseline (Fig. 8A). The B-1 cells that were not treated with PP1 showed a modest BCR + CD19 induced Ca2+ response (■), as shown in previous figures. Pre-treatment of cells with PP1 enabled the B-1 cells to overcome this Ca2+ defect and allowed them to flux cellular Ca2+ to a greater magnitude (△, ○). This enhancement of Ca2+ response was dose-dependent. Addition of PP1 alone or vehicle control (DMSO equivalent) did not elicit any measurable Ca2+ changes over the baseline (data not shown). Similar results were obtained with splenic B-1 cells (Fig. 8B). In order to test if this phenomenon of enhancement can be exhibited even by the CD5− B-1b subset, we have included the CD5 marker along with B220 and Mac-1 to monitor the generation of Ca2+ flux in all the three sub-populations of peritoneal B cells. Interestingly, pre-treatment of peritoneal cells with the SFK inhibitor PP1 enabled both B-1a and B-1b B cells to better elicit Ca2+ response to BCR + CD19 co-cross-linking (Fig. 8C). This indicates that there is a certain amount of pLyn required for B-1 cells to generate Ca2+ from the ER stores. If there are greater levels of pLyn than an optimal quantity, Lyn may potentially recruit negative regulators of B cell signaling and down modulate the Ca2+ response.
Figure 8. PP1 restores the BCR + CD19 signaling defect in B-1 cells.

(A) Peritoneal cells were loaded with Indo-1 and stained with α-B220 and α-Mac-1/CD11b as described in the Methods, treated with biotinylated anti-IgM and biotinylated anti-CD19; pre-incubated with PP1 at room temperature for 20 min prior to obtaining the 30 sec baseline. Avidin was added as indicated by the arrow. Graph represents one of three independent measurements.
(B) C57BL/6 splenocytes were Indo-1 loaded, stained with anti-B220~PE and anti-CD43~FITC, incubated with biotinylated anti-IgM F(ab′)2 and biotinylated anti-CD19, pre-treated with varying doses of PP1 for 20 min at room temperature, cell suspension warmed to 37°C, activated with avidin and calcium response monitored for 5 min. Shown were the responses of the gated splenic B-1 (B220+ CD43+) B cells at two doses of PP1 (μM).
(C) Cells from peritoneal lavage were treated as in (A), stained with α-B220~CY, α-Mac-1~PE, α-CD5~FITC, stimulated with biotinylated anti-IgM and biotinylated anti-CD19; pre-treated with PP1 at room temperature for 20 min prior to determining the 30 sec baseline calcium levels. Avidin was added as indicated by the arrow. Graph depicts the responses from gated B-1a (B220+ Mac-1+ CD5+) and B-1b (B220+ Mac-1+ CD5−) sub-populations.
(D) Peritoneal B-1 and VH12 transgenic splenic B cells (panel E) were treated with 50 μg/ml anti-IgM F (ab′)2 for 48 h in IF-12. The proliferative counts were determined as described in the Methods. Plot represents one of three independent in vitro proliferation assays. p < 0.005 compares the cpm at each of the PP1 doses to the 0 PP1 (μM) group i.e. anti-IgM alone group.
(F) Peritoneal B-1 cells were incubated with 2 μM PP1 for 30 min at 37°C. Cells were then treated with biotinylated antibodies to IgM and CD19 and stimulated with avidin for 10 min. The whole cell lysates were probed for pLyn (Y507), membrane was stripped and reprobed for total Lyn protein as loading control.
In order to investigate if PP1 induced restoration of calcium response can translate into recovery of a functional response, peritoneal B-1 cells were stimulated with anti-IgM in combination with increasing doses of PP1 for 48 hours in IF-12 medium. Fig. 8D shows that addition of PP1 to anti-IgM stimulated peritoneal B-1 cells enabled them to proliferate significantly to the BCR signal. Fig. 8E shows that the BCR induced proliferation response of splenic B-1 cells is also enhanced by PP1. As shown in Fig. 8F, PP1 indeed inhibited Lyn phosphorylation in unstimulated cells and in BCR+CD19 stimulated B-1 cells. Similar results were obtained with PP2, another SFK inhibitor both in Ca2+ experiments (Fig. 9A) and in proliferation response (Fig. 9B). Thus the high pLyn levels seen in the B-1 cells not only negatively regulate the initial signaling events such as Ca2+ generation (Fig. 8A), but also the proliferative response (Fig. 8D). Higher doses of PP1 and PP2 inhibited the BCR induced proliferation responses consistent with the need for optimal levels of Lyn for B cell activation.
Figure 9. Inhibition of SFKs by PP2 partially overcomes BCR + CD19 signaling defect in B-1 cells.

(A) Transgenic splenic B cells were treated similar to that in Fig. 8B. Cells were pre-treated with different doses of PP2 (μM) and the calcium response to BCR + CD19 co-cross-linking shown on the gated splenic B-1 cells.
(B) 2.5 × 105 VH12 transgenic splenic B-1 cells (T-cell depleted) were cultured for 48 hours in an in vitro proliferation assay. Cells were incubated at 37°C with anti-IgM and different concentrations of the pharmacological inhibitor PP2. Shown is a representative graph from one of two independent experiments.
DISCUSSION
Numerous reports have firmly established the positive signaling role of CD19 in aiding BCR induced responses both in vitro and in vivo. However, B cell activation has not been addressed in the context of CD19 function in B-1 B lymphocytes. We have demonstrated earlier that there is a deficiency in CD19 induced intracellular signals in the peritoneal B-1 cells (Sen et al., 2002). The current study examines the importance of CD19 in regulating BCR signaling defect in various B-1 cell subpopulations from normal wild type mice and in transgenic mice. Rothstein and colleagues have shown that splenic B-1 cells differ from peritoneal B-1 cells in certain functional properties and transcriptional programs (Fischer et al., 2001; Tumang et al., 2004). But, B-1 cells obtained from both these distinct anatomical locations are similar in one major aspect of B cell activation: they are hyporesponsive to B cell receptor induced growth signals. This result has enabled us to utilize splenic B-1 cells from wild type as well as VH12 transgenic mouse strains in order to understand the biochemical basis of BCR defect in B-1 cells (Arnold et al., 1994). We showed that B-1a and B-1b cells from peritoneum and spleen and from wild type and transgenic mice fail to proliferate in response to BCR ligation and exhibit reduced Ca2+ mobilization when BCR and CD19 are co-cross-linked. Many previous studies have been performed with B-1 cells from various BCR transgenic mice. Our study is unlike such reports in that it analyzes B-1 cells from various compartments in both normal and transgenic mice making the results representative of most B-cell subpopulations.
Our calcium mobilization experiments led to a very interesting finding that the defect lies in the differential levels of Ca2+ influx in B-1 and B-2 cells whereas the intracellular release is similar between the two populations (Fig. 3). This could possibly be due to comparably lowered activation status of the plasma membrane Ca2+ channels. STIM (stromal interaction molecule) proteins residing in the ER act as sensors to Ca2+ depletion from the ER stores (Spassova et al., 2006). Orai, the regulatory subunit of TRPC channels participates in the store-operated Ca2+ entry (SOCE) process (Inada et al., 2006). Disruption in one of more members of the TRPC channels has been shown to modulate the magnitude of Ca2+ flux in B cell line models (Mori et al., 2002).
Consistent with the notion that CD19-mediated signals are deficient in B-1 cells, BCR + CD19 induced pAkt was lower in B-1 cells which may have a role in the low response of B-1 cells to IgM induced in vitro proliferative signals (Fig. 5). Wong et al. have reported that B-1 cells are defective in BCR dependent MAPK activation, in particular ERK (Wong et al., 2002). We find that B-1 cells are also defective in BCR and CD19 induced JNK activation, a key MAPK enzyme involved in cell survival and proliferation thus possibly contributing to their reduced proliferative ability to IgM stimulation (Fig. 6) (Gururajan et al., 2005).
B-1b (CD5−) subset of B-1 cells have been shown by Dal Porto et al to be almost equally capable of mobilizing Ca2+ as do B-2 cells when triggered through the surface IgM (Dal Porto et al., 2004). We, however, have found B-1b B cells behave like B-1a B cells in eliciting reduced Ca2+ responses upon BCR and CD19 co-cross-linking when compared to splenic or peritoneal B-2 cells. Our findings about differences in B-1b and B-2 cells were performed in the same pool of cells in the same tube ruling out any artifacts. The difference between our results and those of Dal Porto et al. could be due to the fact that BCR response may be normal while CD19 response is defective. However, we find that BCR response is also defective in B-1b B cells. It was unclear how the assays were performed by Dal Porto et al. as they appear to compare peritoneal B-1b to splenic B-2 cells and the calcium profiles do not provide any information about the magnitude of response to cross-linking BCR alone. In our case, we find that both peritoneal B-1a (CD5+) and B-1b (CD5−) B cells cannot respond to anti-IgM initiated proliferative signals as well as peritoneal B-2 cells (Fig. 1). Additionally, these subsets of B-1 cells generate Ca2+ flux in response to BCR and CD19 co-cross-linking, the magnitude of which is drastically lower than either splenic or peritoneal B-2 cells when assayed in the same tube. Hence, CD5 alone may be insufficient to regulate CD19 + BCR responses in B-1 cells (Fig. 2). This is further strengthened by the observation that the BCR-CD19 response of B-1 cells is defective even in CD5−/− mice (Fig. 2F). Importantly, both splenic B-1 and peritoneal B-1 B cells were found to be similar in their defective response to BCR-CD19 cross-linking. This also indicates that the B-1 cells are deficient in CD19 initiated positive signaling events irrespective of their location. Previous studies suggested that CD5 dependent Lck activation may have a role in the reduced responses of B-1a cells but other investigators failed to find Lck expression in B-1 cells (Dal Porto et al., 2004; Frances et al., 2005).
Receptor proximal signaling events in B cell activation are highly crucial to determine the overall effector function of the B cell. Lyn forms an important link in BCR dependent CD19 activation and lyn−/− mice exhibit hyper-responsive phenotype culminating in auto-immunity (Chan et al., 1997). It has recently been discovered that Lyn phosphorylation can lead to dual effects of either upregulating or downmodulating B cell signaling (Xu et al., 2005). Since we observed high levels of pLyn in splenic and peritoneal B-1 cells both in resting as well as BCR + CD19 activated state in otherwise hyporesponsive B-1 cells; it is possible that pLyn phosphorylates the ITIM (immunoreceptor tyrosine-based inhibitory motif) of inhibitory receptors on B cell surface to a greater degree than activating proteins with ITAM (immunoreceptor tyrosine-based activation motif) domains. Alternatively, Lyn may directly phosphorylate phosphatases such as SHP-1, SHIP-1 and induce their activation, which in turn may inhibit BCR-induced downstream signaling intermediates.
Previous studies by Hashimoto et al. demonstrate that BCR cross-linking induced depolarization leading to a decrease in store-operated Ca2+ (SOC) influx and inhibition of store-operated calcium channels in DT40 B cells. This inhibition of SOC was abolished in Lyn-deficient but not in Btk, Syk, or SHIP-deficient cells indicating that Lyn plays a role in the inhibition of calcium influx (Hashimoto et al., 2001). This is consistent with our observation that Lyn activity is increased in B-1 cells and that they exhibit a defect in calcium infux upon BCR-CD19 cross-linking. Other proteins such as CD22, CD72, FcγRIIb, SHIP-1 and SHP-1 have also been implicated in regulating BCR initiated signaling pathways (Cornall et al., 1999; Nadler et al., 1997a). All these molecules are potential substrates for the hyper active Lyn in B-1 cells. The negative regulatory role of CD22 in BCR mediated calcium signaling has already been demonstrated in B cell lines (Nadler et al., 1997b). Lyn has been shown to be required for CD22 and CD72 inhibition (Smith, 1998) (Adachi et al., 2001). Lajaunias et al. found that CD22 does not regulate BCR responses in B-1 cells (Lajaunias et al., 2002). However, they did not look at the effect of CD22 on CD19 signaling. Although FcγRIIb is a well known negative regulator of BCR signaling, hyporesponsiveness of B-1 cells to BCR is thought to be independent of this receptor since similar results are obtained with intact and F(ab′)2 fragments of anti-BCR antibodies. Its role in CD19 signaling has to be investigated using F(ab′)2 fragments of anti-CD19 antibodies. A recently described novel B cell restricted lectin protein, Siglec-G, acts as an inhibitory receptor by diminishing α-IgM evoked Ca2+ response in B-1 B cells (Hoffmann et al., 2007). The mechanism of inhibition, however, remains to be determined. An analysis of the synergistic effect of BCR + CD19 co-cross-linking in B-1 vs. B-2 B cells in siglec−/− mice may lead to a better understanding of the inhibitory mechanism.
In our earlier studies we found that CD19 phosphorylation was similar in B-1 and B-2 cells stimulated via BCR at early time points but was less at late time points (30 minutes) (Sen et al., 2002). In this study we found an elevation in Lyn activity in B-1 cells even without any stimulation even though we did not examine Lyn activity at late time points after BCR signaling. The decrease in CD19 phosphorylation at late time points could be due to Lyn dependent activation of a phosphatase (whose identity is not known at this time) that dephosphorylates CD19 in B-1 but not B-2 cells. Alternatively, a negative regulator in B-1 cells is competing with CD19 for Lyn activity at later time points. Recently Pao et al. have demonstrated by using conditional B cell specific ptpn6 knock out mice that upon cross-linking BCR only, splenic B-1 cells (B220lo CD5+) exhibit greater increase in intracellular Ca2+ concentration than B-1 cells in littermate controls. Thus, they propose SHP-1 to be a negative regulator of BCR evoked Ca2+ responses in splenic and peritoneal CD5+ B-1a B cells (Pao et al., 2007).
Blocking Lyn activity by synthetic inhibitors PP1 or PP2 upregulated the anti-IgM induced proliferative response in B-1a and B-1b peritoneal B cells. At the same time, it also enhanced the ability of B-1 cells to flux Ca2+ to BCR + CD19 signaling. It, therefore, confirms the finding that high levels of pLyn in B-1 cells have a critical role in the unresponsiveness of this subset of B cells to BCR cross-linking. Reducing the phosphorylated levels of Lyn to a certain extent is able to restore the response in B-1 cells. Total inhibition of pLyn is also not desirable since Lyn activity is required for positive downstream signaling events in the B cell. Accordingly, higher doses of PP1/2 are inhibitory to B-1 cell function. Since the specificity of small molecule inhibition to Lyn is not absolute, role of Lyn has to be further confirmed with other approaches.
However, B-1 cells are known to be difficult to transfect. Presently, we are evaluating cell permeable peptide inhibition to block Lyn in B-1 cells, a technique successfully used by Mataraza et al. to demonstrate the importance of Cyclin D3 for B-1 cell proliferation (Mataraza et al., 2006).
The similar role of CD19 signaling in B-1a and B-1b B cell activation found in this study does not contradict the unique distribution and function of B-1a and B-1b B cells found in CD19 deficient and human CD19 transgenic mice as reported by Haas et al. (Haas et al., 2005). Thus CD19 signaling may have distinct roles in the development of B-1a and B-1b B cells which are thought to arise from distinct precursors in fetal liver and adult bone marrow (Dorshkind and Montecino-Rodriguez, 2007). Once these cell lineages have been generated they may have similar CD19 associated signaling pathways resulting in their hyporesponsiveness.
Our findings contribute to a significant understanding of downstream signaling events that are responsible for BCR defect in B-1 cells, cells that are highly cross-reactive to auto-antigens. It still remains to be understood how such unresponsive B cells are capable of secreting abundant levels of serum IgM in the absence of intentional immunization. This is particularly intriguing because of the recent observations by Alugupalli et al. and Haas et al. that B-1b B cells are respond vigorously in making antibody responses upon challenge with Borrelia Hermsii and Streptococcus pneumoniae bacteria and that B-1b B cells are functional in the absence of CD19 (Alugupalli et al., 2004; Haas et al., 2005). Our proliferation results indicate that B-1a and B-1b B cells are equally capable of responding to LPS or T-dependent CD40 stimulation in vitro as do B-2 cells. This leads us to suggest that because of their auto-reactive receptor expression, the BCR signaling per se is maintained in a state of ‘anergy’. When activated via additional pathways, such as TLR and CD40, especially in the context of whole bacteria, the B-1 cells are able to overcome the B cell receptor unresponsiveness, as suggested by Alugupalli and Gerstein (Sindhava et al., manuscript in preparation) (Alugupalli and Gerstein, 2005).
In summary, we have shown that part of the reason for apparent anergic state of B-1a, B-1b B cells from spleen or peritoneum, or VH12 transgenic mice is due to a defect in CD19 signaling. For the first time, we have shown that the defective CD19 and BCR signaling is due to a constitutively high levels of activated Lyn and that the anergic state can be repaired by blocking pLyn.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi T, Wienands J, Wakabayashi C, Yakura H, Reth M, Tsubata T. SHP-1 requires inhibitory co-receptors to down-modulate B cell antigen receptor-mediated phosphorylation of cellular substrates. J Biol Chem. 2001;276:26648–26655. doi: 10.1074/jbc.M100997200. [DOI] [PubMed] [Google Scholar]
- Alugupalli KR. A distinct role for B1b lymphocytes in T cell-independent immunity. Curr Top Microbiol Immunol. 2008;319:105–30. doi: 10.1007/978-3-540-73900-5_5. [DOI] [PubMed] [Google Scholar]
- Alugupalli KR, Gerstein RM. Divide and conquer: division of labor by B-1 B cells. Immunity. 2005;23:1–2. doi: 10.1016/j.immuni.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T, Gerstein RM. B1b Lymphocytes Confer T Cell-Independent Long-Lasting Immunity. Immunity. 2004;21:379–390. doi: 10.1016/j.immuni.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Ambudkar IS, Ong HL, Liu X, Bandyopadhyay BC, Cheng KT. TRPC1: the link between functionally distinct store-operated calcium channels. Cell Calcium. 2007;42:213–23. doi: 10.1016/j.ceca.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Arnold LW, Pennell CA, McCray SK, Clarke SH. Development of B-1 cells: segregation of phosphatidyl choline-specific B cells to the B-1 population occurs after immunoglobulin gene expression. J Exp Med. 1994;179:1585–1595. doi: 10.1084/jem.179.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berland R, Wortis HH. Origins and functions of B-1 cells with notes on the role of CD5. Annu Rev Immunol. 2002;20:253–300. doi: 10.1146/annurev.immunol.20.100301.064833. [DOI] [PubMed] [Google Scholar]
- Bikah G, Carey J, Ciallella JR, Tarakhovsky A, Bondada S. CD5-mediated negative regulation of antigen receptor-induced growth signals in B-1 B cells. Science. 1996;274:1906–9. doi: 10.1126/science.274.5294.1906. [DOI] [PubMed] [Google Scholar]
- Burastero SE, Casali P, Wilder RL, Notkins AL. Monoreactive high affinity and polyreactive low affinity rheumatoid factors are produced by CD5+ B cells from patients with rheumatoid arthritis. J Exp Med. 1988;168:1979–92. doi: 10.1084/jem.168.6.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 1992;256:105–107. doi: 10.1126/science.1373518. [DOI] [PubMed] [Google Scholar]
- Chan VWF, Meng F, Soriano P, DeFranco AL, Lowell CA. Characterization of the B lymphocyte populations in lyn-deficient mice and the role of lyn in signal initiation and down-regulation. Immunity. 1997;7:69–71. doi: 10.1016/s1074-7613(00)80511-7. [DOI] [PubMed] [Google Scholar]
- Cornall RJ, Cyster JG, Hibbs ML, Dunn AR, Otipoby KL, Clark EA, Goodnow CC. Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity. 1998;8:497–508. doi: 10.1016/s1074-7613(00)80554-3. [DOI] [PubMed] [Google Scholar]
- Cornall RJ, Goodnow CC, Cyster JG. Regulation of B cell antigen receptor signaling by the Lyn/CD22/SHP1 pathway. Curr Top Microbiol Immunol. 1999;244:57–68. doi: 10.1007/978-3-642-58537-1_5. [DOI] [PubMed] [Google Scholar]
- Dal Porto JM, Burke K, Cambier JC. Regulation of BCR Signal Transduction in B-1 Cells Requires the Expression of the Src Family Kinase Lck. Immunity. 2004;21:443–453. doi: 10.1016/j.immuni.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Dauphinee M, Tovar Z, Talal N. B cells expressing CD5 are increased in Sjogren’s syndrome. Arthritis Rheum. 1988;31:642–7. doi: 10.1002/art.1780310509. [DOI] [PubMed] [Google Scholar]
- Dorshkind K, Montecino-Rodriguez E. Fetal B-cell lymphopoiesis and the emergence of B-1-cell potential. Nat Rev Immunol. 2007;7:213–9. doi: 10.1038/nri2019. [DOI] [PubMed] [Google Scholar]
- Fischer GM, Solt LA, Hastings WD, Yang K, Gerstein RM, Nikolajczyk BS, Clarke SH, Rothstein TL. Splenic and Peritoneal B-1 Cells Differ in Terms of Transcriptional and Proliferative Features That Separate Peritoneal B-1 from Splenic B-2 Cells. Cell Immunol. 2001;213:62–71. doi: 10.1006/cimm.2001.1860. [DOI] [PubMed] [Google Scholar]
- Frances R, Tumang JR, Kaku H, Gurdak SM, Rothstein TL. B-1 cells express transgelin 2: Unexpected lymphocyte expression of a smooth muscle protein identified by proteomic analysis of peritoneal B-1 cells. Mol Immunol. 2006 doi: 10.1016/j.molimm.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Frances R, Tumang JR, Rothstein TL. B-1 cells are deficient in Lck: defective B cell receptor signal transduction in B-1 cells occurs in the absence of elevated Lck expression. J Immunol. 2005;175:27–31. doi: 10.4049/jimmunol.175.1.27. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Poe JC, Hasegawa M, Tedder TF. CD19 Amplification of B Lymphocyte Ca2+ Responses. A ROLE FOR Lyn SEQUESTRATION IN EXTINGUISHING NEGATIVE REGULATION. J Biol Chem. 2001;276:44820–44827. doi: 10.1074/jbc.M107559200. [DOI] [PubMed] [Google Scholar]
- Gururajan M, Chui R, Karuppannan AK, Ke J, Jennings CD, Bondada S. c-Jun N-terminal kinase (JNK) is required for survival and proliferation of B-lymphoma cells. Blood. 2005;106:1382–91. doi: 10.1182/blood-2004-10-3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas KM, Poe JC, Steeber DA, Tedder TF. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23:7–18. doi: 10.1016/j.immuni.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Hardy RR, Hayakawa K, Shimizu M, Yamasaki K, Kishimoto T. Rheumatoid factor secretion from human Leu-1+ B cells. Science. 1987;236:81–3. doi: 10.1126/science.3105057. [DOI] [PubMed] [Google Scholar]
- Hashimoto A, Hirose K, Kurosaki T, Iino M. Negative Control of Store-Operated Ca2+ Influx by B Cell Receptor Cross-Linking. J Immunol. 2001;166:1003–1008. doi: 10.4049/jimmunol.166.2.1003. [DOI] [PubMed] [Google Scholar]
- Hastings WD, Tumang JR, Behrens TW, Rothstein TL. Peritoneal B-2 cells comprise a distinct B-2 cell population with B-1b-like characteristics. Eur J Immunol. 2006;36:1114–23. doi: 10.1002/eji.200535142. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Hardy RR, Herzenberg LA, Herzenberg LA. Progenitors for Ly-1 B cells are distinct from progenitors for other B cells. J Exp Med. 1985;161:1554–68. doi: 10.1084/jem.161.6.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Hardy RR, Honda M, Herzenberg LA, Steinberg AD, Herzenberg LA. Ly-1 B cells: functionally distinct lymphocytes that secrete IgM autoantibodies. Proc Natl Acad Sci U S A. 1984;81:2494–8. doi: 10.1073/pnas.81.8.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Hardy RR, Parks DR, Herzenberg LA. The “Ly-1 B” cell subpopulation in normal immunodefective, and autoimmune mice. Journal of experimental medicine. 1983;157:202–218. doi: 10.1084/jem.157.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath AW, Wu WW, Howard MC. Monoclonal antibodies to murine CD40 define two distinct functional epitopes. Eur J Immunol. 19994;24:1828–1834. doi: 10.1002/eji.1830240816. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Hardy RR, Stall AM, Herzenberg LA, Herzenberg LA. Immunoglobulin-bearing B cells reconstitute and maintain the murine Ly-1 B cell lineage. Eur J Immunol. 1986;16:1313–6. doi: 10.1002/eji.1830161021. [DOI] [PubMed] [Google Scholar]
- Hippen KL, Tze LE, Behrens TW. CD5 maintains tolerance in anergic B cells. J Exp Med. 2000;191:883–90. doi: 10.1084/jem.191.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Kerr S, Jellusova J, Zhang J, Weisel F, Wellmann U, Winkler TH, Kneitz B, Crocker PR, Nitschke L. Siglec-G is a B1 cell-inhibitory receptor that controls expansion and calcium signaling of the B1 cell population. Nat Immunol. 2007;8:695–704. doi: 10.1038/ni1480. [DOI] [PubMed] [Google Scholar]
- Inada H, Iida T, Tominaga M. Different expression patterns of TRP genes in murine B and T lymphocytes. Biochem Biophys Res Commun. 2006;350:762–7. doi: 10.1016/j.bbrc.2006.09.111. [DOI] [PubMed] [Google Scholar]
- Klaus GG, Bijsterbosch MK, Holman M. Activation and proliferation signals in mouse B cells. VII. Calcium ionophores are non-mitogenic polyclonal B-cell activators. Immunology. 1985;56:321–7. [PMC free article] [PubMed] [Google Scholar]
- Lajaunias F, Nitschke L, Moll T, Martinez-Soria E, Semac I, Chicheportiche Y, Parkhouse RM, Izui S. Differentially regulated expression and function of CD22 in activated B-1 and B-2 lymphocytes. J Immunol. 2002;168:6078–83. doi: 10.4049/jimmunol.168.12.6078. [DOI] [PubMed] [Google Scholar]
- Mataraza JM, Tumang JR, Gumina MR, Gurdak SM, Rothstein TL, Chiles TC. Disruption of cyclin D3 blocks proliferation of normal B-1a cells, but loss of cyclin D3 is compensated by cyclin D2 in cyclin D3-deficient mice. J Immunol. 2006;177:787–95. doi: 10.4049/jimmunol.177.2.787. [DOI] [PubMed] [Google Scholar]
- Mauri C, Ehrenstein MR. The “short” history of regulatory B cells. Trends in Immunology. 2008;29:34–40. doi: 10.1016/j.it.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Mohan C, Morel L, Yang P, Wakeland EK. Accumulation of splenic B1a cells with potent antigen-presenting capability in NZM2410 lupus-prone mice. Arthritis Rheum. 1998;41:1652–62. doi: 10.1002/1529-0131(199809)41:9<1652::AID-ART17>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Mori Y, Wakamori M, Miyakawa T, Hermosura M, Hara Y, Nishida M, Hirose K, Mizushima A, Kurosaki M, Mori E, Gotoh K, Okada T, Fleig A, Penner R, Iino M, Kurosaki T. Transient receptor potential 1 regulates capacitative Ca(2+) entry and Ca(2+) release from endoplasmic reticulum in B lymphocytes. J Exp Med. 2002;195:673–81. doi: 10.1084/jem.20011758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler MJS, Chen B, Anderson JS, Wortis HH, Neel BG. Protein-tyrosine phosphatase SHP-1 is dispensable for FcγRIIB- mediated inhibition of B cell antigen receptor activation. Journal of Biological Chemistry. 1997a;272:20038–43. doi: 10.1074/jbc.272.32.20038. [DOI] [PubMed] [Google Scholar]
- Nadler MJS, McLean PA, Neel BJ, Wortis HH. B cell antigen receptor-evoked calcium influx is enhanced in CD22-deficient B cell lines. J Immunol. 1997b;159:4233–4243. [PubMed] [Google Scholar]
- O’Rourke LM, Tooze R, Turner M, Sandoval DM, Carter RH, Tybulewicz VL, Fearon DT. CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity. 1998;8:635–45. doi: 10.1016/s1074-7613(00)80568-3. [DOI] [PubMed] [Google Scholar]
- Otero DC, Omori SA, Rickert RC. Cd19-dependent activation of Akt kinase in B-lymphocytes. J Biol Chem. 2001;276:1474–8. doi: 10.1074/jbc.M003918200. [DOI] [PubMed] [Google Scholar]
- Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, Thomas ML, Neel BG, Rajewsky K. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity. 2007;27:35–48. doi: 10.1016/j.immuni.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Sen G, Bikah G, Venkataraman C, Bondada S. Negative regulation of antigen receptor-mediated signaling by constitutive association of CD5 with the SHP-1 protein tyrosine phosphatase in B-1 B cells. Eur J Immunol. 1999;29:3319–28. doi: 10.1002/(SICI)1521-4141(199910)29:10<3319::AID-IMMU3319>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Sen G, Wu HJ, Bikah G, Venkataraman C, Robertson DA, Snow EC, Bondada S. Defective CD19-dependent signaling in B-1a and B-1b B lymphocyte subpopulations. Mol Immunol. 2002;39:57–68. doi: 10.1016/s0161-5890(02)00047-0. [DOI] [PubMed] [Google Scholar]
- Sidman CL, Shultz LD, Hardy RR, Hayakawa K, Herzenberg LA. Production of immunoglobulin isotypes by Ly-1+ B cells in viable motheaten and normal mice. Science. 1986;232:1423–5. doi: 10.1126/science.3487115. [DOI] [PubMed] [Google Scholar]
- Smith KGC, Tarlinton DM, Doody GM, Hibbs ML, Fearon DT. Inhibition of the B cell by CD22: A requirement for Lyn. J Exp Med. 1998;187:807–811. doi: 10.1084/jem.187.5.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somani A-K, Yuen K, Xu F, Zhang J, Branch DR, Siminovitch KA. The SH2 Domain Containing Tyrosine Phosphatase-1 Down-regulates Activation of Lyn and Lyn-induced Tyrosine Phosphorylation of the CD19 Receptor in B Cells. J Biol Chem. 2001;276:1938–1944. doi: 10.1074/jbc.M006820200. [DOI] [PubMed] [Google Scholar]
- Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc Natl Acad Sci U S A. 2006;103:4040–5. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stall AM, Adams S, Herzenberg LA, Kantor AB. Characteristics and development of the murine B-1b (Ly-1 B sister) cell population. Ann N Y Acad Sci. 1992;651:33–43. doi: 10.1111/j.1749-6632.1992.tb24591.x. [DOI] [PubMed] [Google Scholar]
- Tarakhovsky A, Muller W, Rajewsky K. Lymphocyte populations and immune responses in CD5-deficient mice. Eur J Immunol. 1994;24:1678–1684. doi: 10.1002/eji.1830240733. [DOI] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–70. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze RM, Doody GM, Fearon DT. Counterregulation by the coreceptors CD19 and CD22 of MAP kinase activation by membrane immunoglobulin. Immunity. 1997;7:59–67. doi: 10.1016/s1074-7613(00)80510-5. [DOI] [PubMed] [Google Scholar]
- Tumang JR, Hastings WD, Bai C, Rothstein TL. Peritoneal and splenic B-1 cells are separable by phenotypic, functional, and transcriptomic characteristics. Eur J Immunol. 2004;34:2158–67. doi: 10.1002/eji.200424819. [DOI] [PubMed] [Google Scholar]
- Wang Y, Brooks SR, Li X, Anzelon AN, Rickert RC, Carter RH. The physiologic role of CD19 cytoplasmic tyrosines. Immunity. 2002;17:501–14. doi: 10.1016/s1074-7613(02)00426-0. [DOI] [PubMed] [Google Scholar]
- Wells SM, Kantor AB, Stall AM. CD43 (S7) expression identifies peripheral B cell subsets. J Immunol. 1994;153:5503–15. [PubMed] [Google Scholar]
- Wong S-C, Chew W-K, Tan JE-L, Melendez AJ, Francis F, Lam K-P. Peritoneal CD5+ B-1 Cells Have Signaling Properties Similar to Tolerant B Cells. J Biol Chem. 2002;277:30707–30715. doi: 10.1074/jbc.M202460200. [DOI] [PubMed] [Google Scholar]
- Xu Y, Harder KW, Huntington ND, Hibbs ML, Tarlinton DM. Lyn tyrosine kinase: accentuating the positive and the negative. Immunity. 2005;22:9–18. doi: 10.1016/j.immuni.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A Regulatory B Cell Subset with a Unique CD1dhiCD5+ Phenotype Controls T Cell-Dependent Inflammatory Responses. Immunity. 2008;28:639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]