

The central roles of salt (NaCl) and the kidneys in the pathogenesis of most forms of hypertension are well established.1, 2 The linkage between salt retention and blood pressure (BP) elevation is often referred to as whole body autoregulation.3, 4 Surprisingly, however, the precise mechanisms that underlie this linkage (i.e., the signaling pathway) have escaped elucidation. Here, we examine the evidence that endogenous ouabain (EO), Na+ pumps (Na,K-ATPase) and the Na/Ca exchanger (NCX) are critical molecular mechanisms in this pathway.
Ca2+ and the Control of Myogenic Tone
At constant cardiac output (CO), mean arterial BP ≈ CO × TPR (where TPR = total peripheral vascular resistance).5 In most (chronic) hypertension, in humans and animals, the CO is relatively normal, and the high BP is maintained by an elevated TPR.1, 4 TPR is controlled dynamically by vasoconstriction/dilation in small “resistance” arteries which exhibit tonic constriction (“tone”). This can be studied in isolated, cannulated small arteries which develop spontaneous (myogenic) tone, MT,6 under constant or increasing intralumenal pressure. Indeed, the level of tone in isolated arteries “is often comparable to that observed in the same vessels in vivo”,6 and may even be used to predict BP changes7 (see below).
MT is triggered by Ca2+ entry, primarily through voltage-gated Ca2+ channels in arterial smooth muscle (ASM) cells,6 and contraction is activated by the rise in cytosolic Ca2+ concentration, [Ca2+]CYT.8 In salt-dependent hypertension, the enhanced vasoconstriction, and increased tone and TPR are, at least in part, functional and reversible phenomena.9 Numerous mechanisms contribute to the regulation of myocyte [Ca2+]CYT and vasoconstriction, but the plasma membrane (PM) NCX provides an unique, direct link between Na+ and [Ca2+]CYT and, thus, Ca2+ signaling and contraction in ASM cells.10 NCX-mediated Ca2+ transport is governed by the Na+ electrochemical gradient generated by the PM Na+ pump.
We proposed that an endogenous Na+ pump inhibitor, i.e., a ouabain-like compound, with vasotonic action might be secreted in response to salt retention.11 In other words, this substance might be a missing hormonal link between salt retention, and the increased TPR and hypertension. Conservation of the high affinity ouabain binding site amino acid sequence in mammalian evolution (see below) implies that there must be an endogenous ligand for this site. Partial Na+ pump inhibition by the endogenous inhibitor should promote the net gain of Ca2+ via the myocyte NCX, and thereby augment Ca2+ signaling and vasoconstriction.10, 11
Endogenous Ouabain
These ideas triggered an intense international search for the postulated endogenous Na+ pump inhibitor, a ligand for the pump’s ouabain/digoxin binding site, that might mediate the vascular response. In 1991, our group purified EO from human plasma; the substance was identified as ouabain by mass spectroscopy.12 It is now possible to quantify EO by liquid chromatography-tandem mass spectroscopy (LC-MS-MS) starting from small (1 ml) samples of human or animal plasma.13 The LC-MS-MS spectra from human and rodent plasma extracts exhibit a major product ion at a mass:charge ratio (m/z) of 445 (Supplementary Figures 1-3; please see http://hyper.ahajournals.org); this is the lithiated aglycone of EO (i.e., lithiated ouabagenin). The possibility that EO might be the 11β isomer of ouabain14 is excluded because the 11-epimers of ouabain have different chromatographic behavior.15
Rat adrenal cortex is highly enriched with EO, and human and cow adrenals also contain very high levels.12 Bilateral adrenalectomy causes a large decline in rat plasma EO, while treatment of uni-nephrectomized rats with DOCA (deoxycorticosterone acetate) + salt increases plasma EO more than 10-fold, and significantly elevates BP.12 These data imply that EO is primarily an adrenocortical hormone, although it may also be synthesized in, and secreted by, the hypothalamus.16
Studies of humans and intact animals, and of adrenocortical cell cultures, reveal that EO is synthesized in the adrenal cortex, and that its synthesis and secretion are stimulated by adrenocorticotropic hormone (ACTH)12, 17-19 In humans19 and animals,18 ACTH-induced hypertension is associated with elevation of EO. Indeed, a preliminary report indicates that certain rare adrenocortical tumors, which are associated with severe hypertension, may produce prodigious amounts of EO.20
About 50% of humans with untreated essential hypertension and a majority of patients with adrenocortical adenomas and hypertension have significantly elevated plasma EO; moreover, BP correlates directly with plasma EO.21 Even in normal human subjects, a high salt diet elevates plasma EO,22 and a 10 min infusion of low dose ouabain increases vascular resistance and elevates BP for >60 min.23
Plasma EO levels are elevated in several rodent salt-sensitive hypertension models,12, 24, 25 and chronic administration of low dose ouabain to normal rodents usually induces hypertension in 1-3 weeks.26, 27 Furthermore, sub-pressor doses of ouabain and DOCA act synergistically to induce hypertension.28 Ouabain-induced BP elevation in rodents is counteracted by the ouabain antagonist, PST-2238 (“Rostafuroxin”).29 Also, in ACTH,18, 30 DOCA+salt,31 or reduced renal mass25 hypertension, Digibind (digoxin-selective Fab fragments that bind ouabain with high affinity)32 lowers BP.
Interestingly, low-dose ouabain increases TPR in dogs, but doesn’t raise BP, presumably because heart rate and CO are markedly reduced.33 Ouabain also doesn’t induce hypertension in sheep34 or in mineralocorticoid-resistant35 Long-Evans rats.36 These apparent exceptions may, however, yield novel information to help clarify the relationship between EO and hypertension.
Many of the findings cited above provide strong evidence that circulating EO has a key role in the pathogenesis of salt-sensitive hypertension. Other studies suggest, however, that brain, not plasma, EO,16 or even marinobufagenin,37 may be important.
Surprisingly, digoxin, another cardiotonic steroid and Na,K-ATPase inhibitor, does not induce hypertension in rodents.26, 38 Also, Digitalis glycosides do not elevate BP in patients treated for congestive heart failure or cardiac arrhythmias.39 Remarkably, digoxin actually lowers BP in ouabain-hypertensive rats26, 38 and in many patients with essential hypertension.40 Thus, Strophanthus glycosides such as ouabain may interact differently with Na+ pumps than do the structurally distinct Digitalis glycosides. Moreover, many observations now indicate that EO is a growth hormone, and that it may participate in a variety of kinase-mediated and other signaling pathways independent of its effects on Na+ pump-mediated Na+ transport.41, 42 EO may therefore contribute to vascular remodeling and target organ damage in hypertension. Clearly, there is much that we do not yet understand about the physiology and pharmacology of these agents.
Membrane Microdomains: a Structural Basis for Ouabain’s Action
Na+ pumps are αβ heterodimers. The catalytic subunit, α, contains the Na+, K+, MgATP and ouabain binding sites, and is phosphorylated during each pump cycle.43 β is essential for pump function; it stabilizes the α subunit conformation and chaperones the αβ complex to the PM.43, 44 In some tissues, a third subunit, γ, may help to regulate Na+ pump activity.44 There are four mammalian α subunit isoforms (α1-α4); they are products of different genes, but have nearly 90% sequence identity, different expression patterns45 and different kinetics46, and they are differently regulated.43, 47 All cells express Na+ pumps with an α1 subunit and Na+ pumps with another α isoform.43, 45 Skeletal, cardiac and smooth muscles, for example, express Na+ pumps with an α2 subunit as well as pumps with an α1; most neurons express α1 and α3.48 Renal epithelia express predominantly (>90-95%) Na+ pumps with α1, which mediate the final step in net transepithelial Na+ reabsorption.47
The functions of the different α subunit isoforms were elucidated by the discovery that, in a variety of cell types, Na+ pumps with an α2 or α3 subunit are confined to PM microdomains situated adjacent to “junctional” sarco-/endoplasmic reticulum (jS/ER) (Figure 2).45 Here, these Na+ pumps co-localize with NCX, which are confined to the same PM microdomains.45 Na+ pumps with an α1 subunit are more widely distributed in the PM, but are apparently excluded from these microdomains.49 Importantly, the PM microdomains are separated by only 12-20 nm from the jS/ER,50 and these structures form a functional unit, termed the “PLasmERosome”.51 The volume of cytosol in the junctional space (J) between the PM and jS/ER of a single PLasmERosome is only on the order of 10-19 to 10-18 liters,51 and diffusion of Na+ and Ca2+ between this space and bulk cytosol is restricted. Thus, standing Na+ and Ca2+ concentration gradients between these compartments and bulk cytosol can be maintained.52-54
Figure 2.
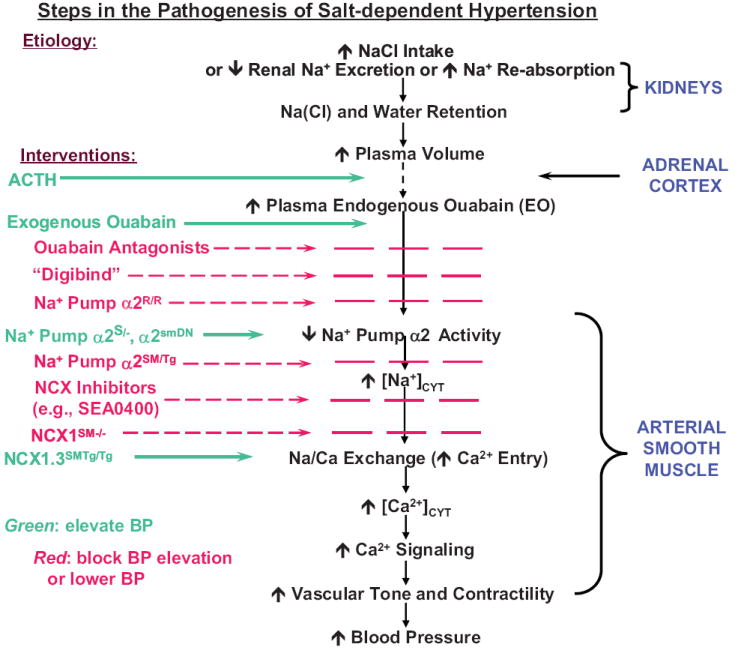
Proposed sequence of steps in the pathogenesis of salt-dependent hypertension. The “interventions,” listed at the left, indicate some of the pharmacologic and genetic manipulations that were used to test the proposed sequence, as discussed in this review. Genotype nomenclature for genetically-engineered mice is given in the text and in Figure 3 and 4 legends. Interventions shown in green increase traffic through the pathway and promote BP elevation; those shown in red block traffic through the pathway and prevent BP elevation or lower BP. Modified from Ref. 7.
Differences in Na+ pump α subunit isoform kinetics play a critical role in PLasmERosome function. The rodent α1 isoform has unusually low affinity for ouabain (KOuabain > 100 μM, vs < 0.05 μM in humans),55 so that nanomolar ouabain inhibits only the α2 Na+ pumps in rodent arterial myocyte PLasmERosomes.7 Even in humans, however, where α1 Na+ pumps have high affinity for ouabain, partial inhibition of Na+ pumps by nanomolar ouabain will raise [Na+] in the junctional space much more than in bulk cytosol. The reason is that the affinity of α2 Na+ pumps for Na+ is much lower (KNa ≈ 22 mM) than is the affinity of α1 Na+ pumps (KNa ≈ 12 mM).46
The widespread distribution of α1 Na+ pumps implies that they have a “housekeeping” function: they control, primarily, [Na+] in bulk cytosol. In contrast, the pumps with an α2 (in smooth muscle, for example) or α3 catalytic subunit regulate local [Na+] in the junctional space. Thus, these α2/α3 Na+ pumps control the local Na+ electrochemical gradient that influences Ca2+ transport by NCX. This organizational arrangement (Figure 1) uniquely links cell Ca2+ to Na+ metabolism. The transporters in the PLasmERosome regulate not only [Ca2+] in the junctional space, but S/ER Ca2+ stores and global Ca2+ signaling in the cells as well.51 Modulation of α2 Na+ pumps in arterial myocyte PLasmERosomes by EO can therefore influence arterial tone and BP. Below, we summarize recent studies in which genetic engineering and pharmacological manipulation of mouse Na+ pumps and NCX (Figure 2) have been used to examine the roles of these transporters in the long-term control of BP.
Figure 1.
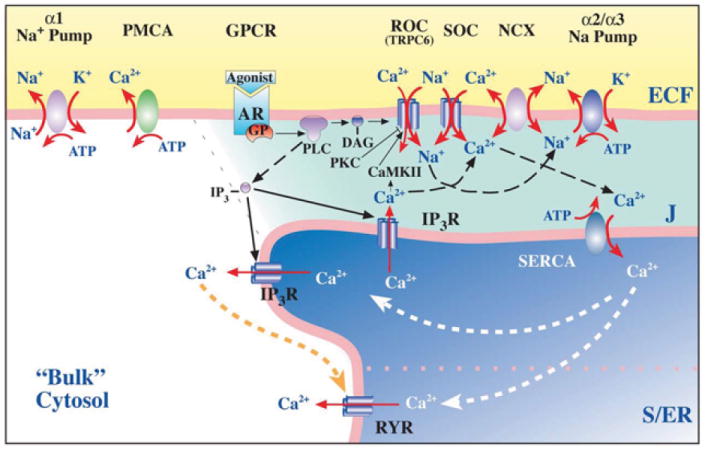
Model of the plasma membrane-junctional sarco-/endoplasmic reticulum (PM-jS/ER) region, the PLasmERosome, showing the location of key transport proteins involved in local control of jS/ER Ca2+ stores and Ca2+ signaling. The PLasmERosome consists of a PM microdomain, the adjacent jS/ER, and the intervening ‘diffusion-restricted’ junctional space (J). The PM microdomain contains α2 (in smooth muscle) or α3 Na+ pumps, NCX, and receptor and store-operated channels (ROCs and SOCs, composed of various transient receptor potential channel proteins or TRPCs). The jS/ER membrane contains SR Ca2+ pumps (SERCA), inositol trisphosphate receptors (IP3R) and ryanodine receptors (RYR). Other regions of the PM contain α1 Na+ pumps and Ca 2+ pumps (PMCA), as well as agonist receptors (ARs, which are G-protein coupled receptors, or GPCRs). Activation of GPCRs and release of G-proteins (GPs) stimulates phospholipase C (PLC) to produce IP3 and diacylglycerol (DAG). DAG may activate ROCs directly. Na+ may enter locally, through ROCs, SOCs or stretch-activated channels (not shown) to promote Ca2+ entry via NCX. Shading indicates relative Na+ and/or Ca2+ concentrations. (Reprinted with permission).52
Downstream Effector Mechanisms
α2 Na+ Pumps
As already noted, chronic administration of exogenous ouabain induces hypertension in rodents. The questions we now address are: How does ouabain (or EO) elevate BP? Is it due to inhibition of smooth muscle α2 Na+ pumps, as implied above?
If circulating ouabain (or EO) elevates BP by inhibiting arterial myocyte α2 Na+ pumps, reduced expression of α2 Na+ pumps should have a similar effect. Therefore, we studied mice with a null mutation in either the α1 or α2 Na+ pump.56 Heterozygous (α1+/- and α2+/-), but not homozygous, null mutants survive, and they express ~50% of the normal complement of α1 or α2 pump protein, respectively, in ASM.7 Isolated mesenteric small arteries from the α2+/-, but not α1+/- mice, exhibit augmented myogenic reactivity in response to step-wise increases in intralumenal pressure, and significantly elevated myogenic tone (MT) when pressurized to 70 mm Hg.7 Nanomolar ouabain also augments myogenic reactivity and increases MT with an EC50 of ~1.3 nM.7 Consistent with these effects in isolated arteries, α2+/-, but not α1+/-, mice have significantly elevated BP (Figure 3).7 Moreover, the α2+/- mice are “salt-sensitive”: a high salt diet increases BP much more in these mice than in their wild type (WT) littermates (Figure 3).
Figure 3.
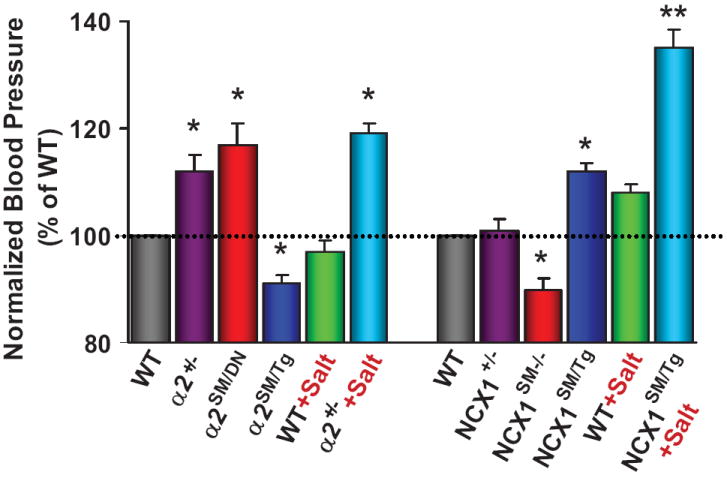
Relative blood pressures of mice with genetically-engineered α2 Na+ pumps and NCX1. The data from several sources, are normalized to the BPs of the respective control wild type (WT) mice. Mice with a null mutation in one α2 Na+ pump allele (α2+/-)7 or smooth muscle-specific α2 knockdown (α2SM/DN) (Song, Chen, Kotlikoff and Blaustein, unpublished; see Figure 4 in Supplementary Data), or increased smooth muscle-specific NCX1 overexpression (NCX1SM/Tg),66 had significantly elevated BP. A high salt diet augmented the elevated BP in α2+/- mice (4% NaCl × 2 weeks) and NCX1SM/Tg mice (8% NaCl + 1% NaCl in tap water × 4 weeks). Smooth muscle-specific overexpression of α2 Na+ pumps (α2SM/Tg)59 or Cre-recombinase knockdown of NCX1 (NCX1SM-/-)70 significantly reduced BP. * = P < 0.05, ** = P < 0.01 vs WT or the respective genotypes on a normal (0.5%) salt diet.
The α2+/- mice are “global” single allele null mutants, but it is important to determine if the effects are the result of reduced α2 Na+ pump activity/expression in ASM. Recently, we found that expression of a short N-terminal segment of the α2 Na+ pump is dominant negative (DN) for expression of full-length α2 pumps.57 Therefore, we generated mice (α2SM/DN) that express the α2 N-terminal segment with a smooth muscle (SM)-specific myosin heavy chain promoter.58 These mice exhibit greatly reduced α2 Na+ pump expression in artery smooth muscle (Supplementary Figure 4; please see http://hyper.ahajournals.org) and elevated BP (Figure 3).
In a complimentary approach, α1 and α2 Na+ pumps were overexpressed, independently, in mice, under the control of an α-actin smooth muscle-specific promoter.59 The mice that overexpressed α2, but not those that overexpressed α1, Na+ pumps had, on average, significantly reduced BP compared to WT mice (Figure 3).
The roles of ouabain/EO and α2 Na+ pumps in elevating BP was also examined in two other ways. One type of study utilized Rostafuroxin, a derivative of digitoxigenin,60 that antagonizes the inhibitory action of ouabain on Na,K-ATPase.29 In isolated arteries, Rostafuroxin counteracted the augmentation of MT by nanomolar ouabain, but not the (ouabain-independent) augmenting effect of reduced α2 expression on MT.7 Rostafuroxin also lowered BP in ouabain-induced hypertension29 and in about 30% of humans with essential hypertension.29
As an alternative, in a knock-in study, two amino acids in the α2 Na+ pump ouabain binding site were mutated to reduce, markedly, the α2 pump’s affinity for ouabain.18, 48 Mice that expressed ouabain-resistant α2 pumps (α2R/R) were resistant to ACTH-induced hypertension (Figure 4)18 as well as to ouabain-induced hypertension.48 Moreover, Digibind prevented the ouabain-induced elevation of BP in the wild type mice.48 Clearly, ACTH-induced hypertension depends upon the existence of a high-affinity cardiotonic steroid binding site on the α2 Na+ pump, and upon a water-soluble ligand that binds to this site. The plasma level of this ligand (presumably EO) was increased by ACTH and, like ouabain,32 bound to Digibind with high affinity.48
Figure 4.

Effects of ACTH on blood pressure in mice with high (normal) and low ouabain affinity α2 Na+ pumps. ACTH (500 μg/kg s.c. every 8 hr × 5 days) elevated BP in wild type (WT) mice, but not in mutant mice expressing α2 Na+ pumps with low affinity for ouabain (α2R/R).18 Treatment with Digibind (30 μg/kg daily, 2 hr before BP measurement), which binds ouabain with high affinity,32 but not control IgGγ, prevented the ACTH-induced elevation of BP in WT mice. ** = P < 0.1 for the pairings indicated. These re-graphed BP data were obtained by tail cuff,18 but comparable results were recently obtained by telemetry (Lingrel and colleagues, unpublished).
These genetic engineering studies reveal that arterial myocyte α2 Na+ pumps mediate the effects of EO and play a role in the long-term regulation of BP. Genetically or pharmacologically reduced α2 activity elevates BP, whereas increased α2 activity lowers BP (Figure 2). The next question is: By what specific mechanism does the altered α2 Na+ pump activity influence BP? The answer appears to lie in Na/Ca exchange.
NCX Type-1
Na/Ca exchange uniquely and directly links Na+ to Ca2+ metabolism and is a distal regulator of cytosolic Ca2+. There are two classes of Na/Ca exchangers, those that co-transport K+ with Ca2+ (NCKX), and those that do not (NCX).61 The predominant exchanger in arterial myocytes is NCX, even though NCKX has also been detected,62 There are three mammalian NCX isoforms (NCX1-NCX3), each the product of a different gene.63 NCX1, which is expressed in ASM, has multiple splice variants; NCX1.3 is the main variant in arterial myocytes.64
Inhibition of Na+ pumps by nanomolar ouabain augments Ca2+ signaling without elevating bulk cytosolic Na+ in primary cultured rat arterial myocytes.51 Even knockout of α2 Na+ pumps in cultured cells (astrocytes) has only minimal effect on bulk cytosolic Na+, but a large effect on Ca2+ signaling.65 These findings are consistent with a functional linkage between α2 (but not α1) Na+ pumps and NCX1, and local reduction of the trans-PM Na+ gradient when α2 activity is reduced, as implied by the PLasmERosome model (Figure 1). Moreover, recent pharmacological and genetic engineering studies now reveal that NCX1 influences not only arterial myocyte Ca2+ metabolism, but long-term vascular tone and BP as well.
Mice in which NCX1 is overexpressed in smooth muscle with an α-actin promoter (NCX1SM/Tg) have elevated BP that is markedly increased by a high salt diet; i.e., the mice are “salt-sensitive” (Figure 3).66 The elevated BP in the NCX1 overexpressors on high dietary salt is abolished by SEA0400, a selective NCX1 inhibitor,67 but not if the overexpressed NCX1 contains a G833C mutation,66 which specifically abrogates the action of SEA0400.68
To perform the converse experiment, mice with floxed NCX1 (NCX1flx/flx)69 were crossed with mice containing a Cre recombinase gene under the control of the smooth muscle myosin heavy chain promoter58 to generate smooth muscle-specific NCX1 knockout (NCX1SM-/-) mice. NCX1SM-/- mice have abnormally low blood pressure (Figure 3), and isolated, pressurized small arteries from these mice have abnormally low MT.70 Indeed, SEA0400 also lowers BP by about 5-10 mm Hg in WT mice66 and reduces MT by about 10% in isolated arteries from these mice.7, 66 Thus, NCX1 activity apparently makes a modest, but direct, contribution to normal MT and BP. SEA0400 also attenuates the increased MT in arteries from α2+/- mice,7 indicating that NCX1 mediates effects distal to α2 Na+ pumps. The BP and MT data from α2+/- and NCX1SM-/- mice support the view that MT in isolated arteries is an in vitro reflection of BP6 and, most likely, TPR.
The mice with genetically engineered NCX1 demonstrate that this exchanger contributes to long-term BP regulation: increased NCX1 expression increases BP while knockout of NCX1 reduces BP (Figures 2 and 3). This conclusion is supported by the effects of NCX blockers in several rodent models of salt-dependent or ACTH-induced hypertension. In DOCA+salt hypertensive rats, spontaneously hypertensive rats (SHR) on a high salt diet, and Dahl salt-sensitive rats on high salt, SEA0400 markedly reduces BP.66 Also, KB-R7943, a less potent NCX blocker, prevents ACTH from elevating BP in mice.18 Moreover, although a null mutation in one NCX1 allele has negligible effect on BP (NCX1+/- in Figure 3) or MT,71 it prevents the induction of hypertension by DOCA+salt.66 Importantly, SEA0400 did not lower BP in several salt-independent rat hypertension models: SHR on a normal salt diet, stroke prone-SHR, and the renin-dependent two-kidney/one-clip rat.66 The implication is that NCX1 makes an important contribution to the pathogenesis of salt-dependent hypertension, but not to salt-independent hypertension.
“Kalzium? Ja, das ist Alles!” (Calcium is Everything: O. Loewi)
Arterial myocyte contraction depends, ultimately, upon the availability of cytosolic Ca2+,8 and the sensitivity of the contractile apparatus to that Ca2+.72 Furthermore, NCX1, under the control of the Na+ gradient generated by the adjacent α2 Na+ pumps, helps regulate myocyte Ca2+ homeostasis (Figure 1). For example, the nanomolar ouabain-induced increase in MT is associated with increased myocyte [Ca2+];7 conversely, reduction of MT by SEA0400 is associated with reduced myocyte [Ca2+] (Figure 5).66 Thus, it is apparent that α2 Na+ pumps and NCX1 are relatively distal mechanisms in the final common path that links salt to vasoconstriction and hypertension (Figure 2). Indeed, all upstream vasoconstrictor and vasodilator mechanisms (neural and humoral) must, inevitably, be influenced by the activity of these two transporters, because they regulate basal [Ca2+]CYT.
Figure 5.
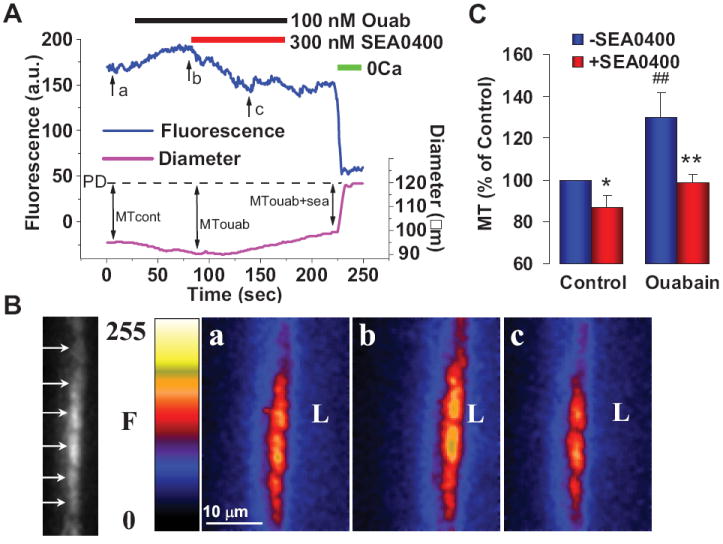
Effects of low dose ouabain and SEA0400 on [Ca2+]CYT and myogenic tone (MT) in mouse pressurized mesenteric small arteries. A. Simultaneous recording of fluorescence (F, in arbitrary units, a.u., a measure of [Ca2+]CYT) and external diameter in a representative fluo-4 loaded normal mouse artery pressurized to 70 mm Hg. Bars at the top indicate periods of exposure to 100 nM ouabain, 300 nM SEA0400 and 0Ca medium (to determine passive diameter, PD). B. Arrows in the black and white spinning disk confocal image at the left indicate fluorescence in individual myocytes of a longitudinal cross-section through one wall of the artery in A. Pseudocolor images of this artery wall were captured at the times indicated by arrows “a” (control MT), “b” (MT with ouabain) and “c” (MT with ouabain + SEA0400) in A; “L” is located in the artery lumen. C. Summary of normalized MT data from this and five other, similar experiments. * = P < 0.05, ## = P < 0.01 vs untreated control; ** = P < 0.01 vs ouabain alone. Corrected from Ref. 66.
An alternative suggestion is that activation of Rho/Rho kinase via the G12-G13-mediated G protein-coupled receptor pathway, which modulates the Ca2+ sensitivity of the contractile apparatus in ASM,72 is selective for salt-dependent hypertension.73 Those authors, however, studied only a salt-dependent (DOCA+salt) mouse model; they did not test whether the G12-G13 pathway also operates in salt-independent forms of hypertension.73 In fact, interference with the G12-G13 pathway, whether at the agonist receptor level,74 or at the level of Rho kinase,75 lowers BP in salt-independent models such as the stroke-prone spontaneously hypertensive rat74 and the NO synthase-inhibited rat.75 The G12-G13 pathway is, therefore, downstream, and distinct from the key salt-sensitive steps in Na+-dependent hypertension. Once salt-sensitive NCX1-mediated Ca2+ entry has occurred, the G12-G13 pathway helps modulate the increases in vascular tone and BP.
Endgame: Na/Ca Exchange, Ca2+ Entry and Myogenic Tone
In the heart, the main role of NCX is to extrude, during diastole, much of the Ca2+ that enters through voltage-gated channels (VGCs) during systole.76 Consequently, reduced cardiac NCX1 function as a result, for example, of α2 Na+ pump inhibition by cardiotonic steroids, is associated with Ca2+ gain and augmented signaling in cardiac myocytes. Therefore, it might at first seem surprising that ASM NCX1 contributes directly to vascular tone, and that reduced expression or pharmacological inhibition of NCX1 in arterial myocytes lowers [Ca2+]CYT and attenuates Ca2+ signaling (Figure 5). Indeed, Ca2+ entry via NCX has sometimes been called “reverse mode” exchange, implying, erroneously, that this is the backward or abnormal operation of the exchanger.77 NCX can transport Ca2+ in either direction across the PM,78 under the control of the local Na+ electrochemical gradient across the PM (Figure 1), and considerations of the electrical component of this gradient are of paramount importance. In the heart, the driving force on the exchanger, i.e., the difference between the prevailing membrane voltage, VM, and the NCX1 reversal potential, ENa/Ca,a which determines the direction of net Ca2+ movement, varies during the cardiac cycle. For example, the rapid membrane depolarization during the upstroke of the cardiac action potential rapidly switches NCX1 from the Ca2+ exit to Ca2+ entry mode, as the driving force, VM-ENa/Ca, becomes positive. Then, as VM repolarizes, VM-ENa/Ca again becomes negative and favors Ca2+ exit.78
A different situation exists in ASM, where changes in VM are normally quite slow and cells are often partially depolarized for very long periods of time.79 Here, intralumenal pressure in small arteries depolarizes the myocytes and activates dihydropyridine-sensitive L-type VGCs. Opening of stretch-activated non-selective cation channels80 may initiate the depolarization. This depolarization is insensitive to dihydropyridines: nifedipine blocks Ca2+ entry through L-type VGCs and reduces MT, but has little effect on the pressure-activated depolarization.79 The Na+ entry through stretch-activated channels and consequent depolarization, as well as the rise in [Ca2+]CYT,80 should also have another, previously unrecognized consequence: they should promote Ca2+ entry via NCX1 and thereby contribute to MT. The reason is that the exchanger is activated by cytosolic Ca2+,81 and the rise in cytosolic [Na+] and the depolarization augment the Ca2+ entry mode of NCX1 by increasing the driving force, VM-ENa/Ca. The implication is that both the L-type VGCs and NCX1 contribute to the maintenance of Ca2+ entry, elevated [Ca2+]CYT and arterial tone when the arteries are pressurized.
“In My End is My Beginning” (T.S Eliot)
In this review, we have explored some of the critical steps that link salt retention to the long-term increase in TPR and elevation of BP. Recent results, especially those from chemical analyses of human and rodent plasma samples, and from genetic engineering and pharmacological studies in rodents and rodent arteries, are summarized above. These studies give new insight into some of the molecular events that help regulate cytosolic Ca2+ and vascular tone. The data supply compelling evidence that EO, and smooth muscle α2 Na+ pumps and NCX1, are key mechanisms in the pathway that leads from salt retention to hypertension (Figure 2).
While these findings provide a framework, the story is far from complete. For example, a key area where knowledge is lacking is at the early steps between salt retention and the release of EO, as indicated by the broken vertical lines in Figure 2. Also, Coffman and colleagues recently demonstrated that the renal and extra-renal arteries make apparently independent (and equal) contributions to the long-term regulation of BP.82 But how the distal mechanisms, discussed above, affect the renal and extra-renal vasculature and renal function, and thereby contribute to BP control, is still unexplored. And, of course, a fundamental question is: What makes us salt-sensitive in the first place? Hopefully, the progress outlined above will clarify new directions for hypertension research to help resolve these issues.
Supplementary Material
Acknowledgments
Sources of Funding This work has been supported by National Institutes of Health grants HL-45215 and HL-78870 (to MPB), DK-65992 (to M.I.K), HL-28573 (to J.B.L.), HL-66062 (to J.B.L.), HL-48509 (to K.D.P.), HL-73094 (to W.G.W.), and HL-75584 (to JMH), a Japanese National Heart Institute KAKENHI grant on Priority Areas 20056030 (to T.I.), and an American Heart Association National Scientist Development Grant and an International Heart Association-Pfizer Award (to JZ).
Footnotes
For NCX1, which mediates the exchange of 3Na+ for 1Ca2+, ENa/Ca = 3ENa − 2ECa, where ENa and ECa are, respectively, the equilibrium potentials for Na+ and Ca2+ [ENa = (RT/F) ln ([Na]o/[Na]i) and ECa = (RT/2F) ln ([Ca]o/[Ca]i), and R, T and F are the gas constant, temperature (Kelvin) and Faraday’s number].78
Disclosures None
References
- 1.Kaplan NM. Kaplan’s Clinical Hypertension. 9. Philadelphia: Lippincott Williams & Wilkins; 2002. pp. 1–24.pp. 36–135. [Google Scholar]
- 2.Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev. 2005;85:679–715. doi: 10.1152/physrev.00056.2003. [DOI] [PubMed] [Google Scholar]
- 3.Guyton AC, Granger HJ, Coleman TG. Autoregulation of the total systemic circulation and its relation to control of cardiac output and arterial pressure. Circ Res. 1971;28(Suppl 1):93–97. [PubMed] [Google Scholar]
- 4.Cowley AWJ. Long-term regulation of arterial blood pressure. Physiol Rev. 1992;72:231–300. doi: 10.1152/physrev.1992.72.1.231. [DOI] [PubMed] [Google Scholar]
- 5.Berne RM, Levy MN. Cardiovascular Physiology. 8. St Louis, MO: Mosby; 2001. [Google Scholar]
- 6.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, Bianchi G, Ferrari P, Hamlyn JM, Iwamoto T, Lingrel JB, Matteson DR, Wier WG, Blaustein MP. Sodium pump α2 subunits control myogenic tone and blood pressure in mice. J Physiol. 2005;569:243–256. doi: 10.1113/jphysiol.2005.091801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruegg JC. Calcium in Muscle Contraction: Cellular and Molecular Physiology. 2. Springer-Verlag; 1992. pp. 201–238. [Google Scholar]
- 9.Schmidlin O, Sebastian AF, Morris RC., Jr What initiates the pressor effect of salt in salt-sensitive humans? Observations in normotensive blacks. Hypertension. 2007;49:1032–1039. doi: 10.1161/HYPERTENSIONAHA.106.084640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blaustein MP. Physiological effects of endogenous ouabain: control of intracellular Ca2+ stores and responsiveness. Am J Physiol Cell Physiol. 1993;264:C1367–C1387. doi: 10.1152/ajpcell.1993.264.6.C1367. [DOI] [PubMed] [Google Scholar]
- 11.Blaustein MP. Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am J Physiol Cell Physiol. 1977;232:C165–C173. doi: 10.1152/ajpcell.1977.232.5.C165. [DOI] [PubMed] [Google Scholar]
- 12.Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, Mathews WR, Ludens JH. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci U S A. 1991;88:6259–6263. doi: 10.1073/pnas.88.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pitzalis MV, Hamlyn JM, Messaggio E, Iacoviello M, Forleo C, Romito R, de Tommasi E, Rizzon P, Bianchi G, Manunta P. Independent and incremental prognostic value of endogenous ouabain in idiopathic dilated cardiomyopathy. Eur J Heart Fail. 2006;8:179–186. doi: 10.1016/j.ejheart.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 14.Mathews MR, DuCharme DW, Hamlyn JM, Harris DW, M F, Clark MA, Ludens JH. Mass spectral characterization of an endogenous digitalis-like factor from human plasma. Hypertension. 1991;17:930–935. doi: 10.1161/01.hyp.17.6.930. [DOI] [PubMed] [Google Scholar]
- 15.Hong B-C, Kim S, Kim T-S, Corey EJ. Synthesis and properties of several isomers of the cardioactive steroid ouabain. Tetrahedron Letters. 2006;47:2711–2715. [Google Scholar]
- 16.Huang BS, Amin MS, Leenen FH. The central role of the brain in salt-sensitive hypertension. Curr Opin Cardiol. 2006;21:295–304. doi: 10.1097/01.hco.0000231398.64362.94. [DOI] [PubMed] [Google Scholar]
- 17.Laredo J, Hamilton BP, Hamlyn JM. Ouabain is secreted by bovine adrenocortical cells. Endocrinology. 1994;135:794–797. doi: 10.1210/endo.135.2.8033829. [DOI] [PubMed] [Google Scholar]
- 18.Dostanic-Larson I, Van Huysse JW, Lorenz JN, Lingrel JB. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc Natl Acad Sci USA. 2005;102:15845–15850. doi: 10.1073/pnas.0507358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goto A, Yamada K, Hazama H, Uehara Y, Atarashi K, Hirata Y, Kimura K, Omata M. Ouabainlike compound in hypertension associated with ectopic corticotropin syndrome. Hypertension. 1996;28:421–425. doi: 10.1161/01.hyp.28.3.421. [DOI] [PubMed] [Google Scholar]
- 20.Evans G, Manunta P, Hamlyn JM, Hamilton BP, Gann DS. Ouabainoma: A new syndrome of endocrine hypertension caused by a ouabain-secreting cortical adenoma. Endocrine Society 75th Annual Meeting; Las Vegas. June 9-12, 1993; Abstracts:291. [Google Scholar]
- 21.Manunta P, Stella P, Rivera R, Ciurlino D, Cusi D, Ferrandi M, Hamlyn JM, Bianchi G. Left ventricular mass, stroke volume, and ouabain-like factor in essential hypertension. Hypertension. 1999;34:450–456. doi: 10.1161/01.hyp.34.3.450. [DOI] [PubMed] [Google Scholar]
- 22.Manunta P, Hamilton BP, Hamlyn JM. Salt intake and depletion increase circulating levels of endogenous ouabain in normal men. Am J Physiol Regul Integr Comp Physiol. 2006;290:R553–559. doi: 10.1152/ajpregu.00648.2005. [DOI] [PubMed] [Google Scholar]
- 23.Mason DT, Braunwald E. Studies on Digitalis.X. Effects of ouabain on forearm vascular resistance and venous tone in normal subjects and in patients in heart failure. J Clin Invest. 1964;43:532–543. doi: 10.1172/JCI104939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrandi M, Manunta P, Rivera R, Bianchi G, Ferrari P. Role of the ouabain-like factor and Na-K pump in rat and human genetic hypertension. Clin Exp Hypertens. 1998;20:629–639. doi: 10.3109/10641969809053241. [DOI] [PubMed] [Google Scholar]
- 25.Kaide J, Ura N, Torii T, Nakagawa M, Takada T, Shimamoto K. Effects of digoxin-specific antibody Fab fragment (Digibind) on blood pressure and renal water-sodium metabolism in 5/6 reduced renal mass hypertensive rats. Am J Hypertens. 1999;12:611–619. doi: 10.1016/s0895-7061(99)00029-1. [DOI] [PubMed] [Google Scholar]
- 26.Manunta P, Hamilton J, Rogowski AC, Hamilton BP, Hamlyn JM. Chronic hypertension induced by ouabain but not digoxin in the rat: antihypertensive effect of digoxin and digitoxin. Hypertens Res. 2000;23(Suppl):S77–85. doi: 10.1291/hypres.23.supplement_s77. [DOI] [PubMed] [Google Scholar]
- 27.Manunta P, Rogowski AC, Hamilton BP, Hamlyn JM. Ouabain-induced hypertension in the rat: relationships among plasma and tissue ouabain and blood pressure. J Hypertens. 1994;12:549–560. [PubMed] [Google Scholar]
- 28.Sekihara H, Yazaki Y, Kojima T. Ouabain as an amplifier of mineralocorticoid-induced hypertension. Endocrinology. 1992;131:3077–3082. doi: 10.1210/endo.131.6.1446641. [DOI] [PubMed] [Google Scholar]
- 29.Ferrari P, Ferrandi M, Valentini G, Bianchi G. Rostafuroxin: an ouabain antagonist that corrects renal and vascular Na+-K+- ATPase alterations in ouabain and adducin-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2006;290:R529–535. doi: 10.1152/ajpregu.00518.2005. [DOI] [PubMed] [Google Scholar]
- 30.Li M, Wen C, Whitworth JA. Hemodynamic effects of the Fab fragment of digoxin antibody (digibind) in corticotropin (ACTH)-induced hypertension. Am J Hypertens. 1997;10:332–336. doi: 10.1016/s0895-7061(96)00318-4. [DOI] [PubMed] [Google Scholar]
- 31.Krep H, Price DA, Soszynski P, Tao QF, Graves SW, Hollenberg NK. Volume sensitive hypertension and the digoxin-like factor. Reversal by a Fab directed against digoxin in DOCA-salt hypertensive rats. Am J Hypertens. 1995;8:921–927. doi: 10.1016/0895-7061(95)00181-N. [DOI] [PubMed] [Google Scholar]
- 32.Pullen MA, Brooks DP, Edwards RM. Characterization of the neutralizing activity of digoxin-specific Fab toward ouabain-like steroids. J Pharmacol Exp Ther. 2004;310:319–325. doi: 10.1124/jpet.104.065250. [DOI] [PubMed] [Google Scholar]
- 33.Hildebrandt DA, Montani J-P, Heath BJ, Granger JP. Chronic ouabain (QUA) infusion alters systemic hemodynamics in normal dogs. Faseb J. 1999;9:A297. [Google Scholar]
- 34.Pidgeon GB, Lewis LK, Yandle TG, Richards AM, Nicholls MG. Endogenous ouabain, sodium balance and blood pressure. J Hypertens. 1996;14:169–171. doi: 10.1097/00004872-199602000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Rowland NE. NaCl appetite in two strains of rat reported to be resistant to mineralocorticoid-induced hypertension. Physiol Behav. 1998;64:49–56. doi: 10.1016/s0031-9384(98)00018-3. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Tempini A, Schnyder B, Montani JP. Regulation of blood pressure during long-term ouabain infusion in Long-Evans rats. Am J Hypertens. 1999;12:423–426. doi: 10.1016/s0895-7061(98)00250-7. [DOI] [PubMed] [Google Scholar]
- 37.Anderson DE, Fedorova OV, Morrell CH, Longo DL, Kashkin VA, Metzler JD, Bagrov AY, Lakatta EG. Endogenous sodium pump inhibitors and age-associated increases in salt sensitivity of blood pressure in normotensives. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1248–1254. doi: 10.1152/ajpregu.00782.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang BS, Kudlac M, Kumarathasan R, Leenen FH. Digoxin prevents ouabain and high salt intake-induced hypertension in rats with sinoaortic denervation. Hypertension. 1999;34:733–738. doi: 10.1161/01.hyp.34.4.733. [DOI] [PubMed] [Google Scholar]
- 39.Gheorghiade M, van Veldhuisen DJ, Colucci WS. Contemporary use of digoxin in the management of cardiovascular disorders. Circulation. 2006;113:2556–2564. doi: 10.1161/CIRCULATIONAHA.105.560110. [DOI] [PubMed] [Google Scholar]
- 40.Abarquez RF., Jr Digitalis in the treatment of hypertension. A prelminary report. Acta Med Philipp. 1967;3:161–170. [PubMed] [Google Scholar]
- 41.Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am J Cardiovasc Drugs. 2007;7:173–189. doi: 10.2165/00129784-200707030-00004. [DOI] [PubMed] [Google Scholar]
- 42.Li Z, Xie Z. The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflugers Arch. 2008 doi: 10.1007/s00424-008-0470-0. in press. [DOI] [PubMed] [Google Scholar]
- 43.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol Renal Physiol. 1998;275:F633–F650. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- 44.Geering K. Functional roles of Na,K-ATPase subunits. Curr Opin Nephrol Hypertens. 2008;17:526–532. doi: 10.1097/MNH.0b013e3283036cbf. [DOI] [PubMed] [Google Scholar]
- 45.Juhaszova M, Blaustein MP. Distinct distribution of different Na+ pump alpha subunit isoforms in plasmalemma. Physiological implications. Ann N Y Acad Sci. 1997;834:524–536. doi: 10.1111/j.1749-6632.1997.tb52310.x. [DOI] [PubMed] [Google Scholar]
- 46.Zahler R, Zhang ZT, Manor M, Boron WF. Sodium kinetics of Na,K-ATPase alpha isoforms in intact transfected HeLa cells. J Gen Physiol. 1997;110:201–213. doi: 10.1085/jgp.110.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Summa V, Camargo SM, Bauch C, Zecevic M, Verrey F. Isoform specificity of human Na+, K+-ATPase localization and aldosterone regulation in mouse kidney cells. J Physiol. 2004;555:355–364. doi: 10.1113/jphysiol.2003.054270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dostanic I, Paul RJ, Lorenz JN, Theriault S, Van Huysse JW, Lingrel JB. The alpha2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol. 2005;288:H477–485. doi: 10.1152/ajpheart.00083.2004. [DOI] [PubMed] [Google Scholar]
- 49.Lee MY, Song H, Nakai J, Ohkura M, Kotlikoff MI, Kinsey SP, Golovina VA, Blaustein MP. Local subplasma membrane Ca2+ signals detected by a tethered Ca2+ sensor. Proc Natl Acad Sci U S A. 2006;103:13232–13237. doi: 10.1073/pnas.0605757103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Somlyo AV, Franzini-Armstrong C. New views of smooth muscle structure using freezing, deep-etching and rotary shadowing. Experientia. 1985;41:841–856. doi: 10.1007/BF01970000. [DOI] [PubMed] [Google Scholar]
- 51.Arnon A, Hamlyn JM, Blaustein MP. Ouabain augments Ca2+ transients in arterial smooth muscle without raising cytosolic Na+ Am J Physiol Heart Circ Physiol. 2000;279:H679–H691. doi: 10.1152/ajpheart.2000.279.2.H679. [DOI] [PubMed] [Google Scholar]
- 52.Blaustein MP, Wier WG. Local sodium, global reach: filling the gap between salt and hypertension. Circ Res. 2007;101:959–961. doi: 10.1161/CIRCRESAHA.107.164459. [DOI] [PubMed] [Google Scholar]
- 53.Poburko D, Liao CH, Lemos VS, Lin E, Maruyama Y, Cole WC, van Breemen C. Transient receptor potential channel 6 mediated, localized cytosolic [Na+] transients drive Na+/Ca2+ exchanger mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ Res. 2007;101:1030–1038. doi: 10.1161/CIRCRESAHA.107.155531. [DOI] [PubMed] [Google Scholar]
- 54.Rembold CM, Chen XL. The buffer barrier hypothesis, [Ca2+]i homogeneity, and sarcoplasmic reticulum function in swine carotid artery. J Physiol. 1998;513:477–492. doi: 10.1111/j.1469-7793.1998.477bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Brien WJ, Lingrel JB, Wallick ET. Ouabain binding kinetics of the rat alpha two and alpha three isoforms of the sodium-potassium adenosine triphosphate. Arch Biochem Biophys. 1994;310:32–39. doi: 10.1006/abbi.1994.1136. [DOI] [PubMed] [Google Scholar]
- 56.James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA, Lingrel JB. Identification of a specific role for the Na,K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol Cell. 1999;3:555–563. doi: 10.1016/s1097-2765(00)80349-4. [DOI] [PubMed] [Google Scholar]
- 57.Song H, Lee MY, Kinsey SP, Weber DJ, Blaustein MP. An N-terminal sequence targets and tethers Na+ pump α2 subunits to specialized plasma membrane microdomains. J Biol Chem. 2006;281:12929–12940. doi: 10.1074/jbc.M507450200. [DOI] [PubMed] [Google Scholar]
- 58.Xin HB, Deng KY, Rishniw M, Ji G, Kotlikoff MI. Smooth muscle expression of Cre recombinase and eGFP in transgenic mice. Physiol Genomics. 2002;10:211–215. doi: 10.1152/physiolgenomics.00054.2002. [DOI] [PubMed] [Google Scholar]
- 59.Pritchard TJ, Bullard DP, Lynch RM, Lorenz JN, Paul RJ. Transgenic mice expressing Na+-K+ ATPase in smooth muscle decreases blood pressure. Am J Physiol Heart Circ Physiol. 2007;293:H1172–1182. doi: 10.1152/ajpheart.00279.2007. [DOI] [PubMed] [Google Scholar]
- 60.Quadri L, Bianchi G, Cerri A, Fedrizzi G, Ferrari P, Gobbini M, Melloni P, Sputore S, Torri M. 17 beta-(3-furyl)-5 beta-androstane-3 beta, 14 beta, 17 alpha-triol (PST 2238). A very potent antihypertensive agent with a novel mechanism of action. J Med Chem. 1997;40:1561–1564. doi: 10.1021/jm970162e. [DOI] [PubMed] [Google Scholar]
- 61.Lytton J. Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J. 2007;406:365–382. doi: 10.1042/BJ20070619. [DOI] [PubMed] [Google Scholar]
- 62.Dong H, Jiang Y, Triggle CR, Li X, Lytton J. Novel role for K+-dependent Na+/Ca2+ exchangers in regulation of cytoplasmic free Ca2+ and contractility in arterial smooth muscle. Am J Physiol Heart Circ Physiol. 2006;291:H1226–H1235. doi: 10.1152/ajpheart.00196.2006. [DOI] [PubMed] [Google Scholar]
- 63.Quednau BD, Nicoll DA, Philipson KD. The sodium/calcium exchanger family-SLC8. Pflugers Arch. 2004;447:543–548. doi: 10.1007/s00424-003-1065-4. [DOI] [PubMed] [Google Scholar]
- 64.Nakasaki U, Iwamotot T, Hanada H, Imagawa T, Shigekawa M. Cloning of the rat aortic smooth muscle Na+/Ca2+ exchanger and tissue-specific expression of isoforms. J Biochem (Tokyo) 1993;114:528–534. doi: 10.1093/oxfordjournals.jbchem.a124211. [DOI] [PubMed] [Google Scholar]
- 65.Golovina VA, Song H, James PF, Lingrel JB, Blaustein MP. Na+ pump alpha 2-subunit expression modulates Ca2+ signaling. Am J Physiol Cell Physiol. 2003;284:C475–C486. doi: 10.1152/ajpcell.00383.2002. [DOI] [PubMed] [Google Scholar]
- 66.Iwamoto T, Kita S, Zhang J, Blaustein MP, Arai Y, Yoshida S, Wakimoto K, Komuro I, Katsuragi T. Salt-sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type-1 in vascular smooth muscle. Nature Med. 2004;10:1193–1199. doi: 10.1038/nm1118. [DOI] [PubMed] [Google Scholar]
- 67.Matsuda T, Arakawa N, Takuma K, Kishida Y, Kawasaki Y, Sakaue M, Takahashi K, Takahashi T, Suzuki T, Ota T, Hamano-Takahashi A, Onishi M, Tanaka Y, Kameo K, Baba A. SEA0400, a novel and selective inhibitor of the Na+-Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J Pharmacol Exp Ther. 2001;298:249–256. [PubMed] [Google Scholar]
- 68.Iwamoto T, Kita S, Uehara A, Imanaga I, M T, Baba A, Katsuragi T. Molecular determinants of Na+/Ca2+ exchange (NCX1) inhibition by SEA0400. J Biol Chem. 2004;279:7544–7553. doi: 10.1074/jbc.M310491200. [DOI] [PubMed] [Google Scholar]
- 69.Reuter H, Henderson SA, Han T, Ross RS, Goldhaber JI, Philipson KD. The Na+-Ca2+ exchanger is essential for the action of cardiac glycosides. Circ Res. 2002;90:305–308. doi: 10.1161/hh0302.104562. [DOI] [PubMed] [Google Scholar]
- 70.Zhang J, Ren C, Chen L, Raina H, Kinsey SP, Philipson KD, Kotlikoff MI, Matteson DR, Blaustein MP. How does smooth muscle specific NCX1 knockout cause reduced arterial contractility and low blood pressure? Hypertension. 2007;50(4):e110. [Google Scholar]
- 71.Zhang J, Chen L, Lingrel JB, Philipson KD, Blaustein MP. Arterial myocyte Na+ pumps and Na+/Ca2+ exchangers modulate Ca2+ signaling, contractility and long-term blood pressure. J Hypertens. 2006;24(Suppl 6):S61. [Google Scholar]
- 72.Maguire JJ, Davenport AP. Regulation of vascular reactivity by established and emerging GPCRs. Trends Pharmacol Sci. 2005;26:448–454. doi: 10.1016/j.tips.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 73.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 74.Wagner J, Drab M, Bohlender J, Amann K, Wienen W, Ganten D. Effects of AT1 receptor blockade on blood pressure and the renin-angiotensin system in spontaneously hypertensive rats of the stroke prone strain. Clin Exp Hypertens. 1998;20:205–221. doi: 10.3109/10641969809053215. [DOI] [PubMed] [Google Scholar]
- 75.Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, Isaka N, Hartshorne DJ, Nakano T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res. 2003;92:411–418. doi: 10.1161/01.RES.0000059987.90200.44. [DOI] [PubMed] [Google Scholar]
- 76.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 77.Noble D, Blaustein MP. Directionality in drug action on sodium-calcium exchange. Ann N Y Acad Sci. 2007;1099:540–543. doi: 10.1196/annals.1387.013. [DOI] [PubMed] [Google Scholar]
- 78.Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 79.Kotecha N, Hill MA. Myogenic contraction in rat skeletal muscle arterioles: smooth muscle membrane potential and Ca2+ signaling. Am J Physiol Heart Circ Physiol. 2005;289:H1326–1334. doi: 10.1152/ajpheart.00323.2005. [DOI] [PubMed] [Google Scholar]
- 80.Morita H, Honda A, Inoue R, Ito Y, Abe K, Nelson MT, Brayden JE. Membrane stretch-induced activation of a TRPM4-like nonselective cation channel in cerebral artery myocytes. J Pharmacol Sci. 2007;103:417–426. doi: 10.1254/jphs.fp0061332. [DOI] [PubMed] [Google Scholar]
- 81.Hilgemann DW, Collins A, Matsuoka S. Steady-state and dynamic properties of cardiac sodium-calcium exchange. Secondary modulation by cytoplasmic calcium and ATP. J Gen Physiol. 1992;100:933–961. doi: 10.1085/jgp.100.6.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, Spurney RF, Kim HS, Smithies O, Le TH, Coffman TM. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest. 2005;115:1092–1099. doi: 10.1172/JCI200523378. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.