

Abstract
Multiple pro-inflammatory mediators contribute to cardiac dysfunction caused by bacterial lipopolysaccharide (LPS). The rapid TNF-α response is likely involved in the induction of down-stream myocardial depressant factors. Studies by our laboratory and others indicate an important role for ICAM-1 in endotoxemic cardiac dysfunction through leukocyte-independent mechanisms. The purpose of this study was to determine: whether ICAM-1 knockout improves cardiac function during endotoxemia, and whether TLR4 and TNF- α regulate LPS-induced myocardial ICAM-1 expression.
Methods and results
Mice were treated with E. coli LPS (0.5 mg/kg iv). Myocardial ICAM-1 levels were analyzed by immunoblotting, and left ventricular developed pressure (LVDP) was assessed by the Langendorff technique. In wild-type mice, peak ICAM-1 levels were observed at 4 h when myocardial contractility was depressed. Myocardial contractility was improved following LPS in mice lacking functional TLR4, TNF-α or ICAM-1. TLR4 mutation abolished ICAM-1 expression with abrogation of precedent TNF-α response. Similarly, TNF-α knockout reduced myocardial ICAM-1 level following LPS treatment.
Conclusions
ICAM-1 contributes to the mechanism of endotoxemic cardiac dysfunction. TNF-α is involved in the regulation of myocardial ICAM-1 expression by TLR4.
Keywords: lipopolysaccharide, innate immunity, myocardium
1. Introduction
Bacterial lipopolysaccharide (LPS) induces cardiac contractile dysfunction that is associated with the production of pro-inflammatory mediators including TNF-α and ICAM-1 [1,2]. While TNF-α and ICAM-1 have been found to be myocardial depressant factors [2–4], their relative contribution to endotoxemic cardiac dysfunction and their interaction in this cardiac disorder are unclear. Toll-like receptor 4 (TLR4) plays a central role in LPS signaling and regulates LPS-induced production of multiple pro-inflammatory mediators [5]. The regulatory role of TLR4 in the induction of myocardial ICAM-1 during endotoxemia remains to be determined.
TNF-α is an early cytokine involved in the multifactorial mechanisms of endotoxemic cardiac dysfunction. TNF-α is rapidly expressed in the myocardium following exposure to LPS [1], and is also known to have a depressive effect on cardiac myocytes in vitro [6]. However, there is a time lag between peak myocardial TNF-α level and the occurrence of contractile dysfunction in an animal model of endotoxemia [1]. Thus, TNF-α may depress myocardial contractility via the induction of other factors. We have observed that LPS induces IL-18, a cardio-depressant factor, through a TNF-α dependent mechanism [7]. Therefore, it is likely that TNF-α induces multiple factors which can subsequently depress myocardial contractility.
Adhesion molecules play important roles in leukocyte transmigration and infiltration at sites of inflammation [8]. Indeed, ICAM-1 has been shown to contribute to post-ischemic myocardial injury through neutrophil-dependent mechanisms, and neutrophil depletion attenuates LPS-induced liver and lung injury [9]. However, lung neutrophil accumulation following LPS or hemorrhagic shock is not attenuated in ICAM-1 KO mice, suggesting that ICAM-1 may not be an important factor for neutrophil accumulation in lungs in endotoxemia and hemorrhagic shock and that ICAM-1 contributes to lung injury in these situations through neutrophil-independent mechanisms [10]. In this regard, our previous study demonstrates that neutrophil depletion has no influence on endotoxemic cardiac dysfunction although neutralization of ICAM-1 attenuates myocardial depression in endotoxemia [2]. Several studies have found that ligation of ICAM-1 activates cellular signaling mechanisms [11–13]. Recent studies [3,14] suggest that ICAM-1 regulates myocardial function and plays an important role in endotoxemic myocardial depression, likely through its direct effect on myocyte contractility. While LPS induces myocardial expression of ICAM-1 [2], it is not clear whether TLR4 regulates myocardial ICAM-1 expression and whether TNF-α plays a role in the induction of myocardial ICAM-1.
We tested the hypothesis that TNF-α links TLR4 signaling to the expression of myocardial depressant factors in endotoxemia. The purposes of this study were to determine: 1) the contribution of ICAM-1 to cardiac dysfunction, 2) the regulatory role of TLR4 in myocardial ICAM-1 expression, and 3) the role of TNF-α as an upstream factor in the induction of ICAM-1 in endotoxemia.
2. Materials and methods
2.1 Animals and treatment
Male mice, body weight 20–25 grams, were used in this study. Mutant (ICAM-1 KO, TNF-α KO, C.C3H-Tlr4 Lps-d) mice and wild-type controls (C57BL/6 for ICAM-1 KO, B6.129 for TNF-α KO and BALB/cJ for C.C3H-Tlr4 Lps-d, respectively) were obtained from Jackson Laboratory (Bar Harbor, ME). Animals were quarantined and maintained on a standard pellet diet for a week before initiation of the experiments. All animal experiments were approved by the Animal Care and Research Committee, University of Colorado Health Sciences Center. Animals received humane care in compliance with the “Guide for the Care and Use of Laboratory Animals” [DHEW Publication No. (NIH) 85-23, revised 1985, Office of Science and Health Reports, DRR/NIH, Bethesda, MD 20205].
We have reported that a single sublethal dose of LPS induces cardiac dysfunction in rats and mice at 4 h after administration [1,7]. The mouse model of endotoxemia utilized in this study was developed by intravenous administration of E. coli LPS (serotype 055:B5, 0.5 mg/kg). Control animals receive an equal volume of normal saline. Animals were sacrificed at 1, 2 and 4 h. After anesthetization and heparinization (60 mg/kg of pentobarbital sodium and 2000 units/kg of heparin, ip), the chest was opened. Blood was collected from the right atrium, and plasma was separated by centrifugation for 10 min at 5,000 ×g at 4°C, and was stored at −70°C before cytokine assays. Heart was isolated, flushed with normal saline and frozen in liquid nitrogen for cytokine assays or immunoblotting of ICAM-1. For assessment of myocardial function, hearts were isolated from additional animals and perfused on a Langendorff apparatus.
2.2 Isolated heart perfusion
Isolated heart perfusion was performed by a modified isovolumetric Langendorff technique as described elsewhere [2,7]. Hearts were excised into cold modified Krebs-Henseleit solution s(in mM: 11 glucose, 119 NaCl, 1.2 CaCl2, 4.7 KCl, 25 NaHCO3, 1.18 KH2PO4 and 1.17 MgSO4). The aorta was cannulated with a 20G needle, and the heart was perfused within 2 min after isolation. Hearts were perfused in the isovolumetric mode (70 mmHg) with the modified Krebs-Henseleit solution which was saturated with 92.5% O2-7.5% CO2 to achieve a PO2 of 400–450 mmHg and a PCO2 of 35–40 mmHg. Myocardial temperature was maintained by placing the heart in a jacketed tissue chamber that was kept at 37°C by circulating warm water. A thin latex balloon was inserted through the left atrium into the left ventricle, and the balloon was filled with water to achieve a left ventricular end-diastolic pressure of 8 to 15 mmHg. Pacing wires were fixed to the right atrium, and the heart was paced at 5.0 Hz (400 beats/min) during perfusion. After 20 min of equilibration, left ventricular developed pressure (LVDP) and its first derivatives (+dP/dt and −dP/dt) were continuously recorded for 20 min using a computerized pressure amplifier/digitizer (Maclab 8, AD Instrument, Cupertino, CA).
2.3 TNF-α assay
Myocardial homogenate was prepared in 8 volumes of homogenate buffer [0.1 M sodium phosphate (pH 7.5) containing 0.1% Triton X100, 2 mM EGTA, 1.0 mM benzamidine and 1.0 mM phenylmethylsulfonyl fluoride]. Centrifugation was carried out at 4°C, 1000 ×g for 15 min with an Eppendorf centrifuge (model 5417R, Brinkmann Instruments, Inc., Westbury, NY). The supernatant was collected, and protein concentrations were analyzed. TNF-α levels in plasma and in myocardial homogenate were measured using a mouse ELISA kit as described previously [15].
2.4 Immunoblotting of ICAM-1
Myocardial homogenate is mixed with an equal volume of sample buffer (100 mM Tris-HCl, pH 6.8, 2% SDS, 0.02% bromophenol blue and 10% glycerol). Size fraction of crude proteins is performed by electrophoresis as previously described [Shames, 1998 #1789]. Crude protein (20 μg) is fractioned on 4–20% acrylamide gradient gel and transferred onto a nitrocellulose membrane. The membrane is incubated in PBS containing 5% non-fat dry milk for 45 min to block non-specific binding. The membrane is subsequently incubated with a rabbit polyclonal antibody against ICAM-1 (purchased from Serotec, 1:1000 dilution with PBS containing 0.05% Tween 20 and 5% dry milk) for 60 min and with peroxidase-labeled goat anti-rabbit IgG (1:5000 dilution with PBS containing 0.05% Tween 20 and 5% dry milk) for 45 min. After a thorough wash with PBS, protein band is developed using enhanced chemiluminescence (ECL) technique. The membrane is then exposed to Kodak X-Omat film. Densitometry is performed using a computerized densitometer (Molecular Dynamics, Sunnyvale, CA).
2.5 Statistical analysis
Data are presented as mean ± standard error (SE). ANOVA with a post hoc Bonferroni/Dunn test was performed to analyze differences between experimental groups. Statistical significance was accepted within 95% confidence limit.
3. Results
3.1 The relationship of ICAM-1 expression with endotoxemic myocardial depression
Immunoblotting detected an increased myocardial ICAM-1 level at 1 h following administration of LPS, and myocardial ICAM-1 level peaked at 4 h (Figure 1). ICAM-1 was undetectable at 4 h following LPS treatment in the myocardium of ICAM-1 KO mice (Figure 1). Peak ICAM-1 expression in the wild-type mice correlated with myocardial depression: both LVDP and +dP/dt were attenuated at 4 h, but not at 1 and 2 h, following administration of LPS (Figure 2).
Figure 1. Time course of myocardial ICAM-1 expression.

Myocardial samples were collected from wild-type (C57BL/6) mice at 0, 1, 2 and 4 h following administration of LPS or saline for analysis of ICAM-1 levels by immunoblotting. A. A representative gel shows that ICAM-1 level increased at 1, 2 and 4 h following LPS treatment in wild-type mice, and ICAM-1 was not detectable at 4 h after LPS treatment in ICAM-1 KO mice. B. Densitometric data show peak ICAM-1 level at 4 h after LPS treatment. n = 4 in each group.
Figure 2. Cardiac dysfunction during endotoxemia.

Wild-type mice were treated with E. coli LPS (serotype 055:B5, 0.5 mg/kg ip) or normal saline (0.2 ml ip). Hearts were isolated at 1, 2 and 4 h after treatment, and left ventricular developed pressure (LVDP, A) and +dP/dt (B) were assessed by Langendorff. Cardiac Dysfunction occurs at 4 h after LPS treatment. Data are expressed as mean ± SE. n = 6 in each group; *P<0.05 vs. saline control.
We utilized a knockout approach to determine the contribution of ICAM-1 to endotoxemic cardiac dysfunction. Myocardial contractile function was improved in mice lacking the ability to express ICAM-1. LVED was 46.6 ± 4.6 mmHg at 4 h after LPS treatment in ICAM-1 KO mice as compared to 25.1 ± 2.3 mmHg in LPS-treated wild-type mice (Figure 3A). Similarly, ICAM-1 KO has significantly improved +dP/dt at 4 h after administration of LPS (Figure 3B). It is noteworthy that ICAM-1 KO improved cardiac function to a level comparable to that observed in TLR4-defective mice or TNF-α KO mice.
Figure 3. The roles of ICAM-1, TLR4 and TNF-α in endotoxemic cardiac dysfunction.
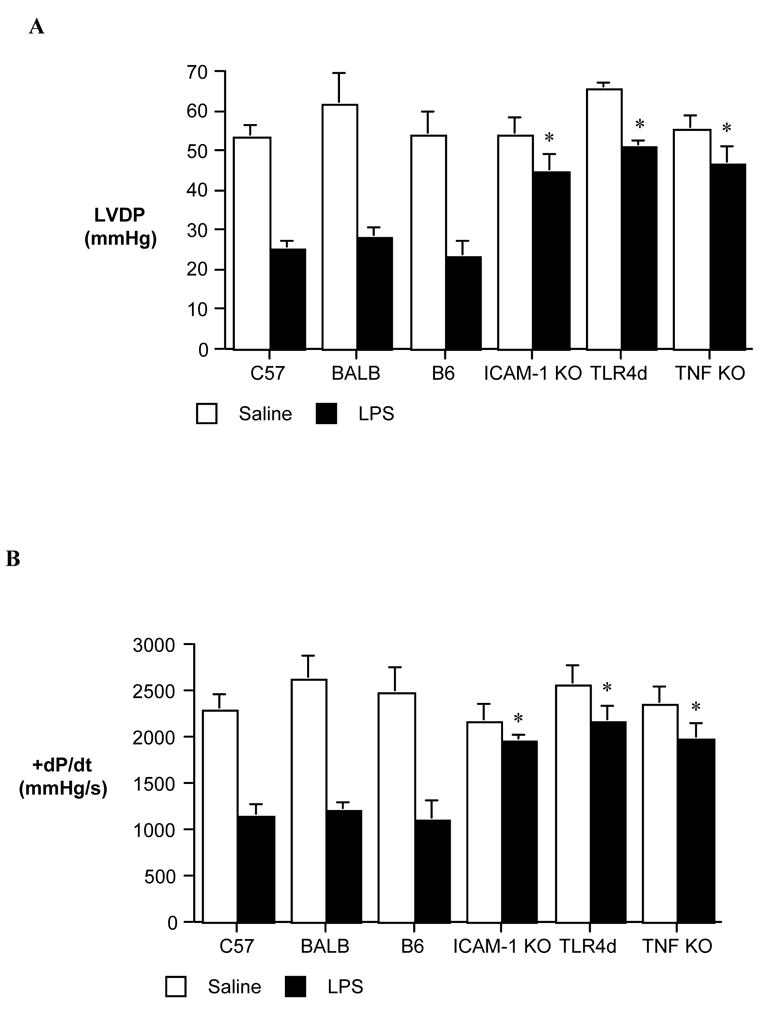
Hearts were isolated at 4 h after LPS or normal saline treatment from wild-type mice and mutant mice, and left ventricular developed pressure (LVDP, A) and +dP/dt (B) were assessed by Langendorff. In comparison to that in wild-type mice, myocardial contractile function was improved following LPS treatment in mice lacking ICAM-1, functional TLR4 or TNF-α. Data are expressed as mean ± SE. n = 6 in each group; * P<0.05 vs. LPS-treated wild-type group.
3.2 The roles of TLR4 and TNF-α in the induction of ICAM-1 during endotoxemia
To determine the regulatory role of TLR4 in myocardial expression of ICAM-1, we employed TLR4 mutant mice and examined their myocardial ICAM-1 levels following LPS treatment. While myocardial ICAM-1 level increased by 5.6 and 9.8 folds respectively at 2 and 4 h following LPS treatment in wild-type mice, there was no myocardial ICAM-1 increase in TLR4 mutant mice at either 2 or 4 h after administration of LPS (Figure 4). Similarly, LPS-induced TNF-α production was abrogated in both the plasma and myocardial tissue of TLR4 mutant mice (54.7 ± 5.1 pg/ml and 2.1 ± 0.5 pg/mg, respectively, in the plasma and the myocardium of TLR4 mutant mice vs. 1941 ± 62.5 pg/ml and 13.6 ± 1.2 pg/mg in the wild-type, both p<0.01).
Figure 4. The effect of TLR4 mutation on myocardial ICAM-1 levels.
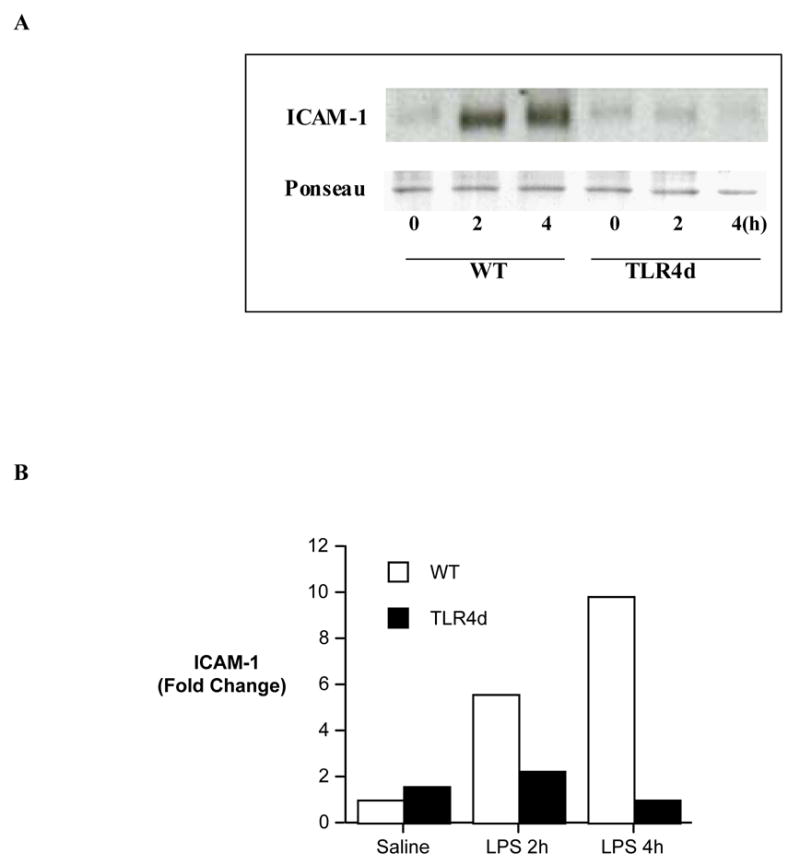
Myocardial samples were collected from wild-type (BALB/cJ) mice and TLR4 defective mutant mice at 0, 2 and 4 h following LPS treatment for analysis of ICAM-1 levels. A. A representative gel of 2 separate experiments shows that TLR4 mutation abrogates the induction of myocardial ICAM-1 by LPS. B. Densitometric data confirm the absence of ICAM-1 induction at both 2 and 4 h after LPS treatment in TLR4 defect mice.
The role of TNF-α in myocardial expression of ICAM-1 was evaluated in TNF-α KO mice. Significantly reduced myocardial ICAM-1 levels were found in TNF-α KO mice at 2 and 4 h (2.1 and 3.2 folds, respectively, vs. 7.1 and 11 folds in wild-type, Figure 5) after administration of LPS. Thus, TNF-α is involved in myocardial ICAM-1 expression during endotoxemia, and TNF-α appears to be an intermediate in the regulation of myocardial ICAM-1 expression by TLR4.
Figure 5. The effect of TNF-α knockout on myocardial ICAM-1 levels.
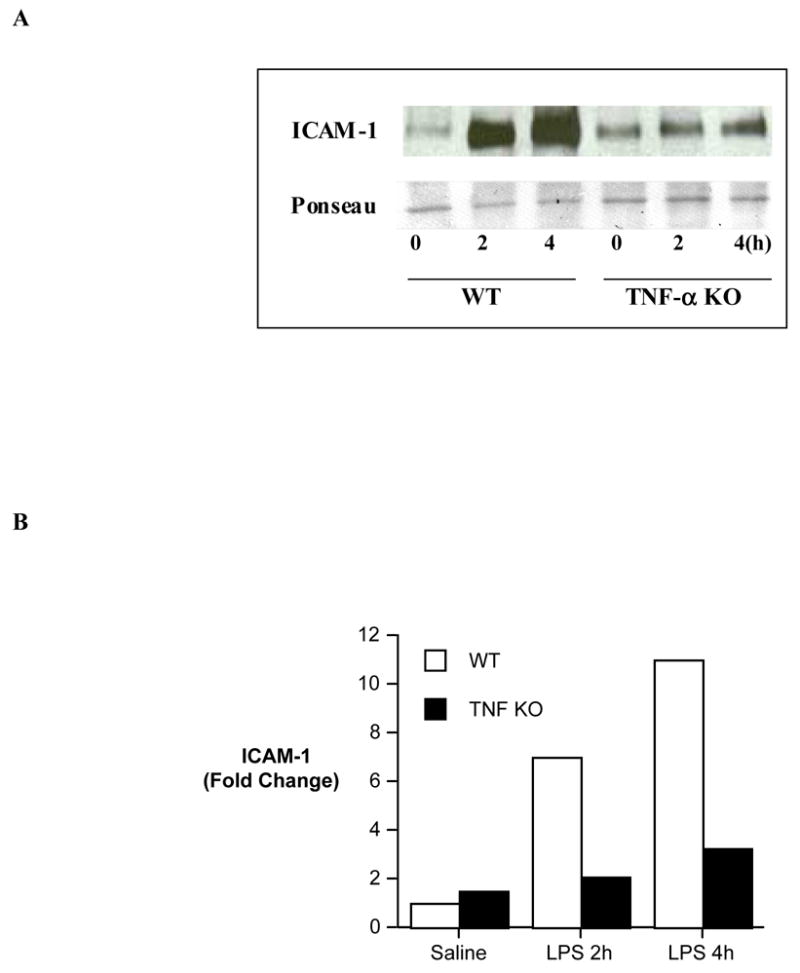
Myocardial samples were collected from wild-type (B6.129) mice and TNF-α knockoutmice at 0, 2 and 4 h following LPS treatment for analysis of ICAM-1 levels. A. A representative gel of 2 separate experiments shows that TNF-α KO attenuates the increase in myocardial ICAM-1. B. Densitometric analysis shows slightly increased, but markedly reduced in comparison to wild-type, myocardial ICAM-1 levels in TNF-α knockoutmice following LPS treatment.
4. Discussion
4.1 Roles for TNF-α and ICAM-1 in myocardial depression
LPS induces the expression of TNF-α and ICAM-1, and peak TNF-α levels precede the onset of myocardial functional depression [1]. In the present study, peak myocardial ICAM-1 levels temporally correlate with cardiac dysfunction. Interestingly, myocardial contractile function was protected from depression in mice lacking this proinflammatory factor. These findings support the concept that endotoxemic cardiac dysfunction is mediated through the interaction of multiple factors, of which ICAM-1 is an important contributor.
ICAM-1 may depress myocardial contractility directly since the peak expression of this factor temporally correlates with myocardial depression. Indeed, we have found that neutralization of ICAM-1 with an antibody improves cardiac contractile function in endotoxemia [2]. Recent studies have identified ICAM-1 as a depressant factor, exerting its effects on the myocardium through both neutrophil-dependent and independent mechanisms [3,14]. However, recruitment of neutrophils via ICAM-1 may not account for its myocardial depressive effect in this endotoxemia model. Not only was a low neutrophil count observed in the myocardium in our study [2], but independent studies have found that neutrophil depletion failed to influence endotoxemic cardiac dysfunction [2,4]. Adhesion molecule ICAM-1 may have a novel role in myocardial depression, likely functioning as a direct depressant factor. Further elucidation of the mechanism by which ICAM-1 mediates myocardial depression might lead to new therapeutic approaches for myocardial protection in the setting of septic shock.
4.2 Regulation of myocardial ICAM-1 expression by TLR4
Toll-like receptors play important roles in the innate immunoresponse. First identified as putative homologues of the Drosophila Toll protein, several members of the Toll-like receptor family have now been identified in mammals [16]. Toll-like receptors transmit signals from the ectodomain, consisting of multiple leucine-rich repeats, to the cytoplasm via the TIR domain. This process has been best described between LPS and TLR4 [17]. In LPS-induced TLR4 signaling, the TIR domain uses a conserved signal transduction pathway requiring MyD88 [18]. MyD88 interacts with TLR4 through its own carboxy-terminal TIR domain. Through its amino-terminal death domain, MyD88 recruits IRAK4 which phosphorylates IRAK1 to propagate the pro-inflammatory signal, leading to the phosphorylation of the IKK complex and MAPKs, including p38 and JNK [18]. This phosphorylation event liberates bound NF-κB from the cytoplasm into the nucleus, resulting in the production of pro-inflammatory mediators.
TLR4 signaling has been shown to be involved in the expression of TNF-α and IL-1β [19] and contributes to myocardial depression in endotoxemic shock [19–22]. In the present study, we examined the expression of ICAM-1 in C.C3H-Tlr4 Lps-dmice that have an engineered mutation on the TIR domain, resulting in a TLR4 signaling defect [17]. We found that TLR4 mutation abolished ICAM-1 expression following LPS treatment. Thus, TLR4 mediates myocardial ICAM-1 expression during endotoxemia. The effects of TLR4 mutation on ICAM-1 expression correlated with an abrogation of LPS-induced TNF-α production. Moreover, peak TNF-α production preceded the expression of ICAM-1 [1]. TNF-α is likely an upstream factor which induces ICAM-1 expression.
4.3 TNF-α as an intermediary in the induction of ICAM-1
TNF-α has been demonstrated to be a myocardial depressant factor in a variety of animal models [1,23,24] and has been linked to heart failure in humans [25]. TNF-α expression is rapid and transient following LPS treatment. Both plasma and myocardial TNF-α levels peaked at 1 h and returned to baseline at 4 h at the same time that myocardial depression occurred [1]. While TNF-α can depress myocardial contractility in vitro [6], the time lag between TNF-α response and myocardial depression suggests that TNF-α is an indirect myocardial depressant factor in vivo. In this regard, TNF-α has been found to mediate the induction of IL-8 and IL-1β by IL-18 [26]. In this study, we observed that myocardial ICAM-1 levels were significantly reduced in TNF-α KO mice. Thus, both TLR4 mutation and TNF-α KO markedly attenuated LPS-induced myocardial ICAM-1 expression, suggesting that TNF-α signaling is involved in the regulation of myocardial ICAM-1 expression by TLR4. It should be noted that there is a low level of ICAM-1 induction by LPS in TNF-α knockout mice, particularly at 4h. Thus, a TNF-α-independent mechanism for myocardial ICAM-1 expression during endotoxemia does exist.
5. Conclusion
The results of this study demonstrate that: 1) ICAM-1 contributes to endotoxemic cardiac dysfunction, 2) myocardial ICAM-1 expression during endotoxemia is regulated by TLR4 and 3) TNF-α is an upstream factor for ICAM-1 induction.
Acknowledgments
This work was supported in part by National Institutes of Health grants GM49222 and HL079051.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Meng X, Ao L, Meldrum DR, Cain B, Shames BD, Selzman CH, et al. TNF-α and myocardial depression in endotoxemic rats: temporal discordance of an obligatory relationship. Am J Physiol. 1998;275:R502–8. doi: 10.1152/ajpregu.1998.275.2.R502. [DOI] [PubMed] [Google Scholar]
- 2.Raeburn CD, Calkins CM, Zimmerman MA, Song Y, Ao L, Banerjee A, et al. ICAM-1 and VCAM-1 mediate endotoxemic myocardial dysfunction independent of neutrophil accumulation. Am J Physiol Regul Integr Comp Physiol. 2002;283:R477–86. doi: 10.1152/ajpregu.00034.2002. [DOI] [PubMed] [Google Scholar]
- 3.Davani EY, Dorscheid DR, Lee CH, vanBreemen C, Walley KR. Novel regulatory mechanism of cardiomyocyte contractility involving ICAM-1 and the cytoskeleton. Am J Physiol Heart Circ Physiol. 2004;287:H1013–22. doi: 10.1152/ajpheart.01177.2003. [DOI] [PubMed] [Google Scholar]
- 4.Tavener SA, Kubes P. Cellular and molecular mechanisms underlying LPS-associated myocyte impairment. Am J Physiol Heart Circ Physiol. 2006;290:H800–6. doi: 10.1152/ajpheart.00701.2005. [DOI] [PubMed] [Google Scholar]
- 5.Baumgarten G, Knuefermann P, Schuhmacher G, Vervolgyi V, vonRappard J, Dreiner U, et al. Toll-like receptor 4, nitric oxide, and myocardial depression in endotoxemia. Shock. 2006;25:43–9. doi: 10.1097/01.shk.0000196498.57306.a6. [DOI] [PubMed] [Google Scholar]
- 6.Cain BS, Meldrum DR, Dinarello CA, Meng X, Joo K, Banerjee A, et al. TNF-α and IL-1β synergistically depress human myocardial function. Crit Care Med. 1999;27:1309–18. doi: 10.1097/00003246-199907000-00018. [DOI] [PubMed] [Google Scholar]
- 7.Raeburn C, Dinarello CA, Zimmerman MA, Calkins C, Pomerantz BJ, McIntyre RC, et al. Neutralization of IL-18 attenuates lipopolysaccharide-induced myocardial dysfunction. Am J Physiol Heart Circ Physiol. 2002;283:H650–7. doi: 10.1152/ajpheart.00043.2002. [DOI] [PubMed] [Google Scholar]
- 8.Albelda SM, Smith CW, Ward PA. Adhesion molecules and inflammatory injury. FASEB J. 1994;8:504–12. [PubMed] [Google Scholar]
- 9.Hewett JA, Schultze AE, VanCise S, Roth RA. Neutrophil depletion protects against liver injury from bacterial endotoxin. Lab Invest. 1992;66:347–61. [PubMed] [Google Scholar]
- 10.Song Y, Ao L, Calkins CM, Raeburn CD, Harken AH, Meng X. Differential cardiopulmonary recruitment of neutrophils during hemorrhagic shock: a role for ICAM-1? Shock. 2001;16:444–8. doi: 10.1097/00024382-200116060-00007. [DOI] [PubMed] [Google Scholar]
- 11.Lorenzon P, Vacile E, Nardon E, Ferrero E, Harlan JM, Tedesco F, et al. Endothelial cell E- and P-selectin and vascular cell adhesion molecule-1 function as signaling receptors. J Cell Biol. 1998;142:1381–91. doi: 10.1083/jcb.142.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clayton A, Evans RA, Pettit E, Hallett M, Williams JD, Steadman R. Cellular activation through ligation of intercellular adhesion molecule-1. J Cell Sci. 1998;111:443–53. doi: 10.1242/jcs.111.4.443. [DOI] [PubMed] [Google Scholar]
- 13.Lawson C, Ainsworth M, Yacoub M, Rose M. Ligation of ICAM-1 on endothelial cells leads to expression of VCAM-1 via a nuclear factor-κB-independent mechanism. J Immunol. 1999;162:2990–6. [PubMed] [Google Scholar]
- 14.Davani EY, Boyd JH, Dorscheid DR, Wang Y, Meredith A, Chau E, et al. Cardiac ICAM-1 mediates leukocyte-dependent decreased ventricular contractility in endotoxemic mice. Cardiovasc Res. 2006;72:134–42. doi: 10.1016/j.cardiores.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 15.Song Y, Ao L, Raeburn CD, Calkins CM, Abraham E, Harken AH, et al. A low level of TNF-alpha mediates hemorrhage-induced acute lung injury via p55 TNF receptor. Am J Physiol Lung Cell Mol Physiol. 2001;281:L677–84. doi: 10.1152/ajplung.2001.281.3.L677. [DOI] [PubMed] [Google Scholar]
- 16.Beutler B. Innate immune responses to microbial poisons: discovery and function of the Toll-like receptors. Ann Rev Pharmacol Toxicol. 2003;43:609–28. doi: 10.1146/annurev.pharmtox.43.100901.135729. [DOI] [PubMed] [Google Scholar]
- 17.Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 18.Janssens S, Beyaert R. A universal role for MyD88 in TLR/IL-mediated signaling. Trends Biochem Sci. 2002;27:474–82. doi: 10.1016/s0968-0004(02)02145-x. [DOI] [PubMed] [Google Scholar]
- 19.Baumgarten G, Knuefermann P, Nozaki N, Sivasubramanian N, Mann DL, Vallejo JG. In vivo expression of proinflammatory mediators in the adult heart after endotoxin administration: the role of toll-like receptor-4. J Infect Dis. 2001;183:1617–24. doi: 10.1086/320712. [DOI] [PubMed] [Google Scholar]
- 20.Nemoto S, Vallejo JG, Knuefermann P, Misra A, Defreitas G, Carabello BA, et al. Escherichia coli LPS-induced LV dysfunction: role of toll-like receptor-4 in the adult heart. Am J Physiol Heart Circ Physiol. 2002;282:H2316–H23. doi: 10.1152/ajpheart.00763.2001. [DOI] [PubMed] [Google Scholar]
- 21.Tavener SA, Long EM, Robbins SM, McRae KM, vanRemmen H, Kubes P. Immune cell Toll-like receptor 4 is required for cardiac myocyte impairment during endotoxemia. Circ Res. 2004;95:700–7. doi: 10.1161/01.RES.0000144175.70140.8c. [DOI] [PubMed] [Google Scholar]
- 22.Binck BW, Tsen MF, Islas M, White DJ, Schultz RA, Willis MS, et al. Bone marrow-derived cells contribute to contractile dysfunction in endotoxic shock. Am J Physiol Heart Circ Physiol. 2005;288:H577–H583. doi: 10.1152/ajpheart.00745.2004. [DOI] [PubMed] [Google Scholar]
- 23.Meldrum DR, Cain BS, Cleveland JC, Meng X, Ayala A, Banerjee A, et al. Adenosine decreases post-ischemic myocardial TNF-α: anti-inflammatory implications for preconditioning and transplantation. Immunology. 1997;92:472–7. doi: 10.1046/j.1365-2567.1997.00380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meldrum DR, Dinarello CA, Shames BD, Cleveland JC, Cain BS, Banerjee A, et al. Ischemic preconditioning decreases postischemic myocardial tumor necrosis factor-α production: potential ultimate effector mechanism of preconditioning. Circulation. 1998;98:II214–II9. [PubMed] [Google Scholar]
- 25.Henriksen PA, Newby DE. Therapeutic inhibition of tumor necrosis factor α in patients with heart failure: cooling an inflammed heart. Heart. 2003;89:14–8. doi: 10.1136/heart.89.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puren J, Fantuzzi G, Gu Y, Su MS, Dinarello CA. Interleukin-18 (IFNgamma-inducing factor) induces IL-8 and IL-1beta via TNFalpha production from non-CD14+ human blood mononuclear cells. J Clin Invest. 1998;101:711–21. doi: 10.1172/JCI1379. [DOI] [PMC free article] [PubMed] [Google Scholar]