

Summary
The analysis of patient blood transcriptional profiles offers a means to investigate immunological mechanisms relevant to human diseases on a genome-wide scale. In addition, such studies provide a basis for the discovery of clinically-relevant biomarker signatures. We designed a strategy for microarray analysis that is based on the identification of transcriptional modules formed by genes coordinately expressed in multiple disease datasets. Mapping changes in gene expression at the module-level generated disease-specific transcriptional fingerprints which provide a stable framework for the visualization and functional interpretation of microarray data. These transcriptional modules were used as a basis for the selection of biomarkers and the development of a multivariate transcriptional indicator of disease progression in patients with systemic lupus erythematosus. Thus, this work describes the implementation and application of a methodology designed to support systems-scale analysis of the human immune system in translational research settings.
Introduction
Patient-based microarray transcriptional studies aim to discover biomarkers, and to identify novel biological knowledge that will unravel mechanisms of disease pathogenesis. However, these goals are met with considerable challenges. The use of gene expression microarrays in clinical research has led to the establishment of biomarker signatures, both from the analysis of tumor tissues (Alizadeh et al., 2000; Bittner et al., 2000; Golub et al., 1999), and blood samples (Allantaz et al., 2007; Baechler et al., 2003; Bennett et al., 2003; Burczynski et al., 2005; Chaussabel et al., 2005; Cobb et al., 2005; Kaizer et al., 2007; Ramilo et al., 2007; Thach et al., 2005). Yet, questions have been raised regarding the value of this approach for the discovery of stable disease markers (Michiels et al., 2005). Among the concerns are the fact that results of microarray analyses are prone to include noise (i.e. false positive results – (Ioannidis, 2005)) and do not compare well between laboratories and/or platforms (Bammler et al., 2005; Hyatt et al., 2006; Irizarry et al., 2005; Jarvinen et al., 2004; Larkin et al., 2005; Shi et al., 2006).
Leveraging patient transcriptional profiles as a means to identify relevant immunological mechanisms is also proving to be a challenge. In fact both microarray and patient-based studies are considered by immunologists as fundamentally descriptive: in the case of microarray studies because the results of system-wide screens do not conform to the reductionist knowledge discovery model prevailing in the field (Benoist et al., 2006); in the case of patient-based studies because the means of testing hypotheses in order to prove a mechanism are de facto very limited (Steinman and Mellman, 2004). Yet, carrying out such studies in patients, and at a systems level is necessary to advance immunological knowledge accumulated from the study of model organisms (Benoist et al., 2006; Steinman and Mellman, 2004).
Indeed, as technology platforms for systems-wide analysis become more sophisticated and more accessible than ever before, it is essential to continue exploring novel strategies for the exploitation of large-scale data. Among those, approaches to uncover the modular organization and function of transcriptional systems have already shown promise (Mootha et al., 2003; Rhodes et al., 2005; Segal et al., 2004); reviewed in (Segal et al., 2005). Indeed, such analyses can transform our perception of large scale transcriptional studies beyond the level of individual genes or lists of genes.
The present work describes the implementation of a unique approach for the analysis of blood microarray transcriptional profiles based on a modular data mining strategy. We showed that this approach could improve our understanding of disease pathogenesis and provide a basis for the selection of clinically-relevant transcriptional biomarkers.
Results
Construction of peripheral blood mononuclear cell (PBMC) transcriptional modules
Comparing transcriptional profiles of two or more study groups generates long lists of differentially expressed genes. Because of the large number of comparisons performed (usually >10,000), these results are permissive to noise, which in turn can affect biomarker discovery and data interpretation (Ioannidis, 2005; Michiels et al., 2005).
In order to circumvent these hurdles we focused the analysis on small sets of coordinately expressed transcripts. Indeed, the probability for multiple transcripts to follow a complex pattern of expression across dozens or hundreds of conditions only by chance is low, and such sets of genes should therefore constitute coherent and biologically meaningful transcriptional units. Thus, we designed an algorithm for constructing sets of coordinately expressed transcripts (i.e. modules) from PBMC microarray profiles generated from a wide range of diseases. This stable modular framework was then used as a basis for the analysis of separate PBMC dataset.
The algorithm used for the construction of transcriptional modules is described in detail in the methods section (Supplementary Figure 1). Briefly, the first step of the module construction process analyzes expression patterns of transcripts across samples for individual diseases: sets of coordinately expressed transcripts were identified using an unsupervised clustering algorithm; in this case, the GeneSpring Version 7.1 (Agilent) implementation of the K-Means algorithm (k=30). All transcripts detected in at least one sample were used as input; no screening for differential expression was performed. The second step of the module construction process analyzed the “clustering behavior” of transcripts across diseases, taking into account the possibility that genes may co-cluster in some diseases but not others. Also, in our example the transcripts that clustered together across all 8 diseases were grouped to form a set of modules (round 1 of selection) and the stringency of the analysis was then decreased gradually to identify transcripts that belong to similar K-means cluster in only a subset of diseases (round 2: 7 out of 8 diseases & round 3: 6 out of 8 diseases). This analysis of gene cluster membership across diseases relates to “graph theory” which is used in the mathematics and computer science fields to model pairwise relations between objects (Biggs, 1986). It is important to note that the module selection process is “data-driven” and does not involve manual selection of genes by the investigator.
We implemented the module construction strategy described above using as input a total of 239 peripheral blood mononuclear cell (PBMC) samples obtained from individuals with one of the following conditions: systemic juvenile idiopathic arthritis (n=47), systemic lupus erythematosus (n=40), type I diabetes (n=20), metastatic melanoma (n=39), acute infections (Escherichia coli (n=22), Staphylococcus aureus (n=18), Influenza A (n=16)), or liver transplant recipients undergoing immunosuppressive therapy (n=37). Transcriptional profiles were generated using Affymetrix U133A and U133B GeneChips (>44,000 probesets). A total of 4742 transcripts distributed among 28 sets were selected upon running the module construction algorithm described above (Supplementary Figure 1; a complete list is provided in Supplementary Table 1). Each module is assigned a unique identifier indicating the round and order of selection (i.e. M3.1 is the first module identified in the third round of selection).
The stringency of this algorithm was tested statistically by implementing the same module construction procedure after randomization of the original dataset. This process was repeated two hundred times without a single module being identified (See supplementary experimental procedures for details). Therefore, the analysis of gene cluster membership across multiple diseases provided a stringent means to identify PBMC transcriptional modules.
The next step consists in characterizing each module functionally. Keyword occurrence in PubMed abstracts associated with the genes within each module were analyzed by literature profiling (Chaussabel and Sher, 2002). Differences in patterns of keyword occurrence across modules were observed as illustrated in Supplementary Figure 2, and functional associations were identified for each of the 28 PBMC transcriptional modules (Table 1). Out of 28 PBMC modules, 14 could be clearly linked with pathways or cell types involved in immune processes (as detailed below). Functional associations were also observed in the remaining 14 modules. M2.5, for example, includes genes encoding immune-related molecules - CD40, CD80, CXCL12, IFNA5, IL4R - as well as cytoskeleton-related molecules - Myosin, Dedicator of Cytokenesis, Syndecan 2, Plexin C1, Distrobrevin). (See Table 1 for details). Thus transcriptional modules form coherent transcriptional and functional units.
Table 1.
| Module I.D. | Number of probe sets | Keyword selection | Assessment |
|---|---|---|---|
| M 1.1 | 76 | Ig, Immunoglobulin, Bone, Marrow, PreB, IgM,Mu. | Plasma cells. Includes genes coding for Immunoglobulin chains (e.g. IGHM, IGJ, IGLL1, IGKC, IGHD) and the plasma cell marker CD38. |
| M 1.2 | 130 | Platelet, Adhesion, Aggregation, Endothelial, Vascular | Platelets. Includes genes coding for platelet glycoproteins (ITGA2B, ITGB3, GP6, GP1A/B), and platelet-derived immune mediators such as PPPB (pro-platelet basic protein) and PF4 (platelet factor 4). |
| M 1.3 | 80 | Immunoreceptor, BCR, B-cell, IgG | B-cells. Includes genes coding for B-cell surface markers (CD72, CD79A/B, CD19, CD22) and other B-cell associated molecules: Early B-cell factor (EBF), B-cell linker (BLNK) and B lymphoid tyrosine kinase (BLK). |
| M 1.4 | 132 | Replication, Repression, Repair, CREB, Lymphoid, TNF-alpha | Undetermined. This set includes regulators and targets of cAMP signaling pathway (JUND, ATF4, CREM, PDE4, NR4A2, VIL2), as well as repressors of TNF-alpha mediated NF-KB activation (CYLD, ASK, TNFAIP3). |
| M 1.5 | 142 | Monocytes, Dendritic, MHC, Costimulatory, TLR4, MYD88 | Myeloid lineage. Includes molecules expressed by cells of the myeloid lineage (CD86, CD163, FCGR2A), some of which being involved in pathogen recognition (CD14, TLR2, MYD88). This set also includes TNF family members (TNFR2, BAFF). |
| M 1.6 | 141 | Zinc, Finger, P53, RAS | Undetermined. This set includes genes coding for signaling molecules, e.g. the zinc finger containing inhibitor of activated STAT (PIAS1 and PIAS2), or the nuclear factor of activated T-cells NFATC3. |
| M 1.7 | 129 | Ribosome, Translational, 40S, 60S, HLA | MHC/Ribosomal proteins. Almost exclusively formed by genes coding MHC class I molecules (HLA-A,B,C,G,E)+ Beta 2-microglobulin (B2M) or Ribosomal proteins (RPLs, RPSs). |
| M 1.8 | 154 | Metabolism, Biosynthesis, Replication, Helicase | Undetermined. Includes genes encoding metabolic enzymes (GLS, NSF1, NAT1) and factors involved in DNA replication (PURA, TERF2, EIF2S1). |
| M 2.1 | 95 | NK, Killer, Cytolytic, CD8, Cell-mediated, T-cell, CTL, IFN-g | Cytotoxic cells. Includes cytotoxic T-cells amd NK-cells surface markers (CD8A, CD2, CD160, NKG7, KLRs), cytolytic molecules (granzyme, perforin, granulysin), chemokines (CCL5, XCL1) and CTL/NK-cell associated molecules (CTSW). |
| M 2.2 | 49 | Granulocytes, Neutrophils, Defense, Myeloid, Marrow | Neutrophils. This set includes innate molecules that are found in neutrophil granules (Lactotransferrin: LTF, defensin: DEAF1, Bacterial Permeability Increasing protein: BPI, Cathelicidin antimicrobial protein: CAMP…). |
| M 2.3 | 148 | Erythrocytes, Red, Anemia, Globin, Hemoglobin | Erythrocytes. Includes hemoglobin genes (HGBs) and other erythrocyte-associated genes (erythrocytic alkirin: ANK1, Glycophorin C: GYPC, hydroxymethylbilane synthase: HMBS, erythroid associated factor: ERAF). |
| M 2.4 | 133 | Ribonucleoprotein, 60S, nucleolus, Assembly, Elongation | Ribosomal proteins. Including genes encoding ribosomal proteins (RPLs, RPSs), Eukaryotic Translation Elongation factor family members (EEFs) and Nucleolar proteins (NPM1, NOAL2, NAP1L1). |
| M 2.5 | 315 | Adenoma, Interstitial, Mesenchyme, Dendrite, Motor | Undetermined. This module includes genes encoding immune-related (CD40, CD80, CXCL12, IFNA5, IL4R) as well as cytoskeleton-related molecules (Myosin, Dedicator of Cytokenesis, Syndecan 2, Plexin C1, Distrobrevin). |
| M 2.6 | 165 | Granulocytes, Monocytes, Myeloid, ERK, Necrosis | Myeloid lineage. Includes genes expressed in myeloid lineage cells (IGTB2/CD18, Lymphotoxin beta receptor, Myeloid related proteins 8/14 Formyl peptide receptor 1), such as Monocytes and Neutrophils. |
| M 2.7 | 71 | No keywords extracted. | Undetermined. This module is largely composed of transcripts with no known function. Only 20 genes associated with literature, including a member of the chemokine-like factor superfamily (CKLFSF8). |
| M 2.8 | 141 | Lymphoma, T-cell, CD4, CD8, TCR, Thymus, Lymphoid, IL2 | T-cells. Includes T-cell surface markers (CD5, CD6, CD7, CD26, CD28, CD96) and molecules expressed by lymphoid lineage cells (lymphotoxin beta, IL2-inducible T-cell kinase, TCF7, T-cell differentiation protein mal, GATA3, STAT5B). |
| M 2.9 | 159 | ERK, Transactivation, Cytoskeletal, MAPK, JNK | Undetermined. Includes genes encoding molecules that associate to the cytoskeleton (Actin related protein 2,3, MAPK1, MAP3K1, RAB5A). Also present are T-cell expressed genes (FAS, ITGA4/CD49D, ZNF1A1). |
| M 2.10 | 106 | Myeloid, Macrophage, Dendritic, Inflammatory, Interleukin | Undetermined. Includes genes encoding for Immune-related cell surface molecules (CD36, CD86, LILRB), cytokines (IL15) and molecules involved in signaling pathways (FYB, TICAM2-Toll-like receptor pathway). |
| M 2.11 | 176 | Replication, Repress, RAS, Autophosphorylation, Oncogenic | Undetermined. Includes kinases (UHMK1, CSNK1G1, CDK6, WNK1, TAOK1, CALM2, PRKCI, ITPKB, SRPK2, STK17B, DYRK2, PIK3R1, STK4, CLK4, PKN2) and RAS family members (G3BP, RAB14, RASA2, RAP2A, KRAS). |
| M 3.1 | 122 | ISRE, Influenza, Antiviral, IFN-gamma, IFN-alpha, Interferon | Interferon-inducible. This set includes interferon-inducible genes: antiviral molecules (OAS1,2,3,L, GBP1, G1P2, EIF2AK2, PKR, MX1, PML), chemokines (CXCL10), signaling molecules (STAT1, STAT2, IRF7, ISGF3G). |
| M 3.2 | 322 | TGF-beta, TNF, Inflammatory, Apoptotic, Lipopolysaccharide | Inflammation I. Includes genes encoding molecules involved in inflammatory processes (e.g. IL8, ICAM1, C5R1, CD44, PLAUR, IL1A, CXCL16), and regulators of apoptosis (MCL1, FOXO3A, RARA, BCL3,6,2A1, GADD45B). |
| M 3.3 | 276 | Inflammatory, Defense, Lysosomal, Oxidative, LPS | Inflammation II. Includes molecules inducing or inducible by inflammation (IL18, ALOX5, ANPEP, AOAH, HMOX1, SERPINB1), as well as lysosomal enzymes (PPT1, CTSB, NEU1, ASAH1, LAMP2, CAST). |
| M 3.4 | 325 | Ligase, Kinase, KIP1, Ubiquitin, Chaperone | Undetermined. Includes protein phosphatases (PPP1R12A, PTPRC, PPP1CB, PPM1B) and phosphoinositide 3-kinase (PI3K) family members (PIK3CA, PIK32A, PIP5K3). |
| M 3.5 | 22 | No keyword extracted | Undetermined. Composed of only a small number of transcripts. Includes hemoglobin genes (HBA1, HBA2, HBB). |
| M 3.6 | 288 | Ribosomal, T-cell, Beta-catenin | Undetermined. This set includes mitochondrial ribosomal proteins (MRPLs, MRPs), mitochondrial elongations factors (GFM1,2), Sortin Nexins (SN1,6,14) as well as lysosomal ATPases (ATP6V1C). |
| M 3.7 | 301 | Spliceosome, Methylation, Ubiquitin | Undetermined. Includes genes encoding proteasome subunits (PSMA2, PSMB5,8); ubiquitin protein ligases HIP2, STUB1, as well as components of ubiqutin ligase complexes (SUGT1). |
| M 3.8 | 284 | CDC, TCR, CREB, Glycosylase | Undetermined. Includes genes encoding enzymes: aminomethyltransferase, arginyltransferase, asparagines synthetase, diacylglycerol kinase, inositol phosphatases, methyltransferases, helicases… |
| M 3.9 | 260 | Chromatin, Checkpoint, Replication, Transactivation | Undetermined. Includes genes encoding kinases (IBTK, PRKRIR, PRKDC, PRKCI) and phosphatases (e.g. PTPLB, PPP2CB/3CB, PTPRC, MTM1, MTMR2). |
Using modules to map transcriptional changes in health and disease
After identifying sets of coordinately expressed PBMC transcripts based on the analysis of patterns found in a wide range of diseases, we used this modular framework as a stable basis for analyzing individual PBMC datasets.
Modules were conceived as a stable framework for the analysis of data generated independently from the sets initially used for module construction. We analyzed PBMC microarray transcriptional profiles generated from 14 patients with acute Streptococcus pneumoniae infection and 10 age- and sex-matched healthy control subjects. This dataset was not used in the module selection process. Statistical comparisons between patient and healthy control groups were performed independently on a module-by-module basis (Mann-Whitney rank test, p<0.05). The transcriptional profiles of differentially expressed genes were then represented on a graph for individual modules (Figure 1A). The pie-chart indicates the proportion of differentially expressed transcripts for a given module (e.g. 49% of the 322 transcripts forming module M3.2 were overexpressed in patients with acute S. pneumoniae infection compared to healthy controls). As shown in Figure 1a, differentially expressed genes in each module were either predominantly overexpressed or predominantly underexpressed. This observation is notable because modules were not selected based on differences in expression between study groups but instead on clustering patterns.
Figure 1. Analysis of patient blood leukocyte transcriptional profiles.
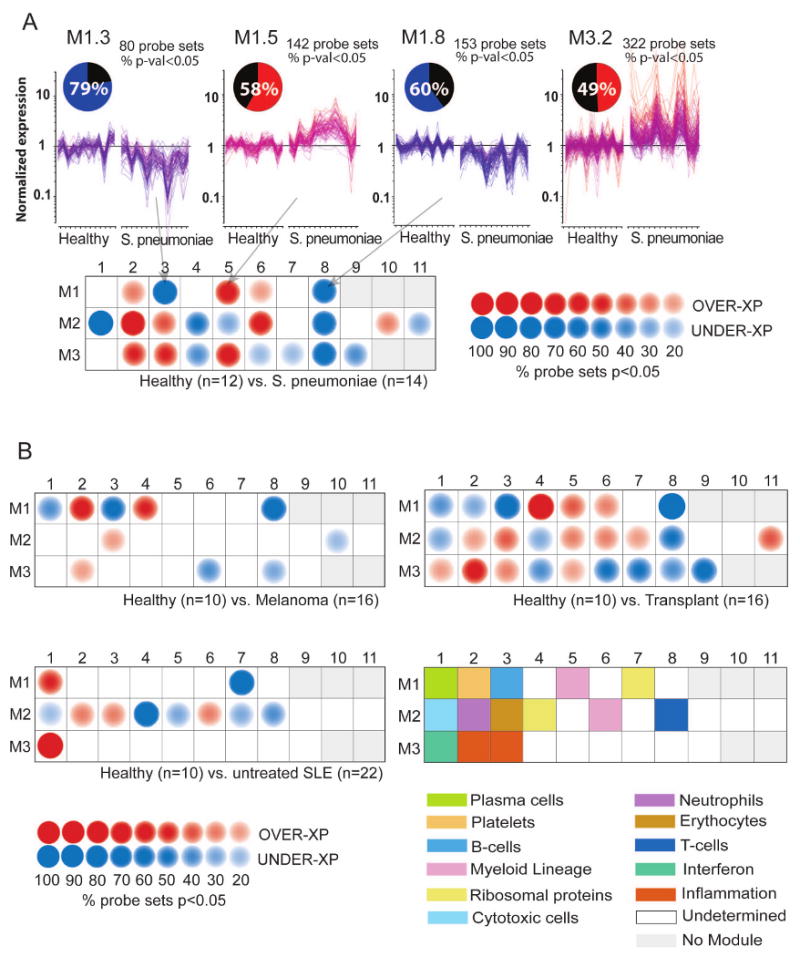
A) Module-level analysis: Gene expression from patients with acute S. pneumoniae infection and respective healthy volunteer PBMCs were compared (p<0.05, Mann-Whitney U test) in modules M1.3, M1.5, M1.8 and M3.2. Pie charts indicate the proportion of genes significantly changed for each module. Graphs represent transcriptional profiles of genes that were significantly changed. Each line shows levels of expression (y-axis) of a single transcript across multiple conditions (samples, x-axis). Expression is normalized to the median expression value of the control group. Results obtained for the 28 PBMC transcriptional modules are displayed on a grid. Coordinates indicate module IDs (e.g. M2.8 is row M2, column 8). Spots indicate proportion of genes significantly changed for each module in patient with S. pneumoniae infection as compared to healthy controls. Red: overexpressed, Blue: underexpressed. B) Disease Fingerprints: Three additional datasets were similarly processed. Profiles were obtained from patients with Systemic Lupus Erythematosus, Liver transplant recipients under pharamacological immunosuppression infection and patients with metastatic melanoma. Functional interpretation is indicated on a grid by a color code. Detailed functional module descriptions are in Table 1.
To graphically represent global transcriptional changes, spots were aligned on a grid, with each position corresponding to a different module (Figure 1A). Spot intensity positively correlates with the proportion of differentially expressed transcripts, whereas spot color indicates the polarity of the change (red: overexpressed, blue: underexpressed). The resulting map represents molecular perturbations associated with a disease state. A blank grid would indicate that no significant differences exist between the disease and healthy baseline. Conversely, the presence of blue and red spots as in Figure 1A indicates that expression levels of sets of transcripts are increased or decreased in patients with acute S. pneumoniae infection in comparison to healthy controls. In addition, to facilitate data interpretation, modules' coordinates were associated to the functional categories which have been previously assigned (Figure 1B, Table 1). For instance, Modules M1.3 and M2.1, respectively associated with B-cells and cytotoxic cells, are under-expressed in the S. aureus group when compared to healthy, while modules M2.2 and M3.2, respectively associated with neutrophils and inflammation are over-expressed.
Thus, we showed that sets of transcriptional modules can be used as a reference for the analysis and interpretation of data generated independently from those used for module construction. Furthermore we have developed means to visualize transcriptional changes on a module-by-module basis, which in conjunction with functional annotations yield an interpretable representation of microarray results.
We next generated module maps for three additional groups of patients (22 Systemic Lupus Erythematosus (SLE), 16 metastatic melanoma, and 16 liver transplant recipients) compared to their respective control groups composed of 10 to 12 healthy donors who were matched for age and sex (Figure 1B, Supplementary Table 2). Results for M1.1 and M1.2 alone distinguished all four diseases (S. pneumoniae: M1.1 = no change, M1.2 = over-expressed; SLE: M1.1= over-expressed, M1.2 = no change; melanoma M1.1 = under-expressed, M1.2 = over-expressed; transplant: M1.1 = under-expressed, M1.2 = under-expressed). A number of genes in M3.2 (“inflammation”) were overexpressed in patients with melanoma, S. pneumoniae infection as well as transplant recipients, while genes in M3.1 (“interferon-inducible”) were overexpressed in patients with SLE and, to a lesser extent, in transplant recipients. M2.1 and M2.8 include, respectively, cytotoxic cell-related genes and T-cell transcripts, which are underexpressed in lymphopenic SLE patients and transplant recipients treated with immunosuppressive drugs. Overall, whereas these comparisons showed that modules can be shared between diseases (e.g. under-expression for M1.3 transcripts in both S. pneumoniae and Melanoma groups) global modular changes remained disease-specific.
Module maps provide a means to organize and reduce the dimension of complex data, and thereby to facilitate its interpretation. However useful, this oversimplified representation lacks at the same time the depth that systems-scale analyses are able to provide. Representing changes at the module-level with a red or blue spot for instance does not indicate which of the genes are significantly changed. Indeed, a spot of the same color in two different diseases may be attributed to two different subsets of genes belonging to the same module. The disconnect between gene-level and module-level data is especially apparent when the results are presented in a static format; i.e. on paper. Thus, we have developed an interactive web-interface allowing users to switch seamlessly between the module-level and gene-level (Supplementary Figure 3). Interactive module maps can be accessed at: www.biir.net/modules. In addition to the four datasets analyzed in the context of this manuscript we loaded on this tool third party datasets made publicly available by others (Burczynski et al., 2006). Mapping transcriptional changes in patients with Crohn's disease and ulcerative colitis highlighted similarity and differences between these diseases, with for instance a characteristic over-expression of transcripts linked to plasma cells (M1.1) in ulcerative colitis. This repository will be updated as more blood transcriptional data become available from our and other groups.
Using modules as a basis for the discovery of blood transcriptional biomarkers
Microarray gene expression data generated from blood not only provide valuable insights into mechanisms of disease pathogenesis but also constitute a promising source of biomarkers. The difficulty, however, lies in the extraction of indicators of potential clinical value from the vast amounts of data generated. We used modular transcriptional data as the foundation of our biomarker discovery strategy. This approach was implemented using a dataset generated from patients with SLE.
Microarray analyses have been carried out on peripheral blood mononuclear cells obtained from pediatric and adult SLE patients (Baechler et al., 2003; Bennett et al., 2003; Crow et al., 2003; Kirou et al., 2004). Using an earlier generation of Affymetrix arrays (∼12,600 probe sets), we identified a type I interferon (IFN) signature in all active pediatric patients (Bennett et al., 2003). This analysis also revealed the presence of neutrophil, immunoglobulin (Ig) and lymphopenic signatures that correlated with the presence of low density granulocytes, plasma cell precursors and a reduction in lymphocyte numbers in SLE blood, respectively (Bennett et al., 2003).
These findings were confirmed in the present study, with significant changes observed in modules M3.1, M2.2, M1.1 and M2.8 (interferon-inducible, neutrophils, plasma cells and T lymphocytes, respectively) for a new dataset generated from a cohort of 22 pediatric lupus patients sampled at the time of diagnosis and before initiation of treatment. Transcriptional changes were observed in 7 additional modules (M1.7, M2.1, M2.3, M2.4, M2.5, M2.6, and M2.7). Two of these modules, M1.7 and M2.4, included transcripts encoding ribosomal protein family members whose expression was recently found altered in acute infection and sepsis (Calvano et al., 2005; Thach et al., 2005). Furthermore, our unpublished observations have shown that in vitro exposure of purified human monocytes to interferon alpha results in a late downregulation of the transcripts forming these modules. In addition, marked changes in gene expression were also observed for modules M2.1 and M2.3 which include transcripts expressed in cytotoxic cells and erythrocytes, respectively. Interestingly, the pattern of change in M2.1, M2.2, M2.3 and M2.4 for the SLE group was well conserved across diseases. Indeed, increased expression for M2.2 & M2.3, and decreased expression for M2.1 and M2.4 was also observed in transplant recipients, as well as in patients with acute S. pneumoniae infections. This partial convergence is likely to reflect the existence of core transcriptional responses to disease or injury (e.g. inflammation).
The proposed biomarker selection strategy relies on modules for reducing highly dimensional microarray datasets in a step-wise manner (Figure 2). Starting from the full set of 28 modules only those for which a set minimum proportion of transcripts are significantly changed between the study groups are selected (Figure 2A; e.g. minimum proportion of differentially expressed transcripts at p<0.05 = 15% over-expressed or under-expressed transcripts; in the example given 11 SLE modules meet this criterion). This eliminates from the selection pool the modules registering fewer consistent changes that may be attributed to noise. The cutoffs used for gene selection can be adjusted to adapt the number of candidate markers that will be returned by this analysis.
Figure 2. Module-based biomarker selection strategy.
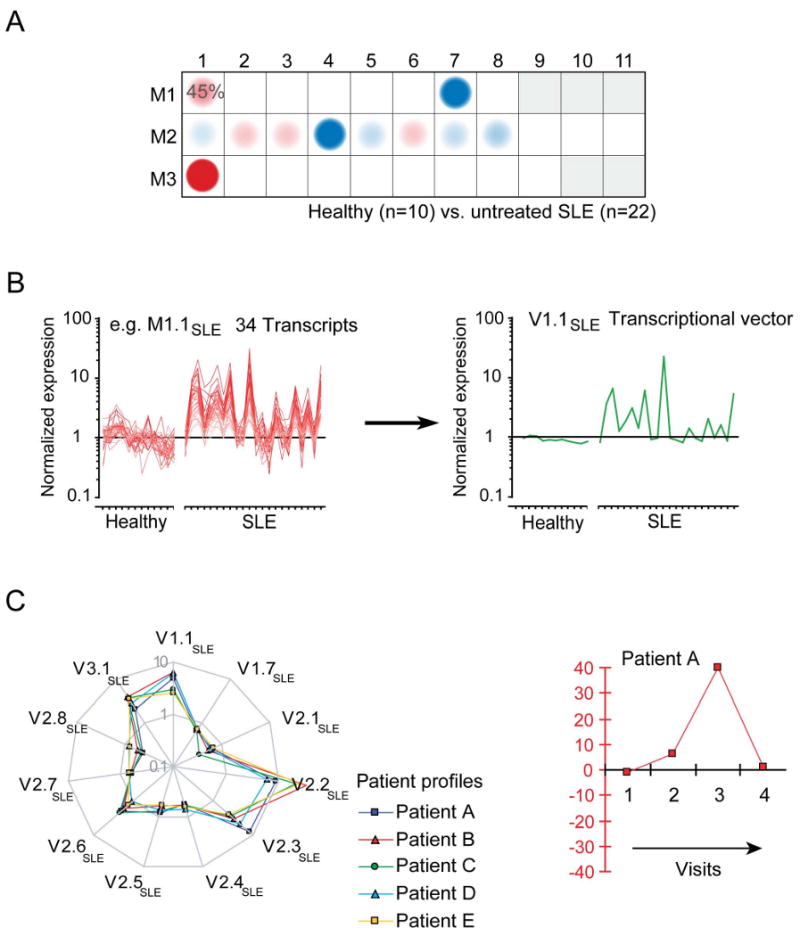
Modules are used as a starting point for the generation of biomarker signatures and progressive reduction of the dimension of microarray data: A) Mapping global transcriptional changes using a modular framework identified 11 modules for which at least 15% of the transcripts are significantly changed between controls and SLE. B) Transcriptional vectors were generated by averaging the normalized expression values of differentially expressed transcripts for each one of the 11 modules selected. C) Composite expression values are plotted as vectors on a “spider graph”. Each line represents a patient profile. Multivariate scores can be generated to recapitulate the changes registered by several transcriptional vectors and monitor changes in an individual patient over time.
We next generated composite values for each sample. The arithmetic average of normalized expression values across significantly over-expressed or under-expressed genes selected from each module was calculated (Figure 2B). Each resulting “transcriptional vector” recapitulates the expression of a given module (or select set of genes within a module) in a given patient. A spider graph connects all the vector values obtained for each patient (Figure 2C). This is in contrast with the module maps defined earlier, which display the frequency of significant changes for an entire patient cohort module-by-module.
SLE patient profiles are linked to disease activity
Transcriptional vectors were derived for the entire cohort of 22 untreated pediatric SLE patients using the set of 11 SLE modules detailed above (the 628 differentially expressed genes distributed among those 11 vectors are listed in Supplementary Table 3). On Figure 3A each line represents the expression profile of one patient; the thicker line shows the average expression for the patients forming this group. The values are normalized per-gene using the median expression value of healthy and are represented on a logarithmic scale. Figure 3B displays the expression pattern characteristic of healthy volunteers. Differences between the healthy and SLE groups were statistically significant for each of the modules (p<0.01 Mann-Whitney U test). Patient profiles were also generated for an independent set of 31 children with SLE treated orally with steroids and/or cytotoxic drugs and/or hydroxychloroquine (Figure 3C – Supplementary Table 4). Interestingly, average profiles for both treated and untreated patient cohorts were almost superimposable (Figure 3D – no significant difference at p<0.01; V2.2 p=0.04; Mann Whitney U test). However, patient selection in both groups was such that they presented similar disease activity as measured by the clinical index SLEDAI (SLE disease activity index – untreated patients average=11.5 ± 7.9; treated patients=9.4 ± 6.4, Student's t-Test p=0.3).
Figure 3. SLE transcriptional vectors.
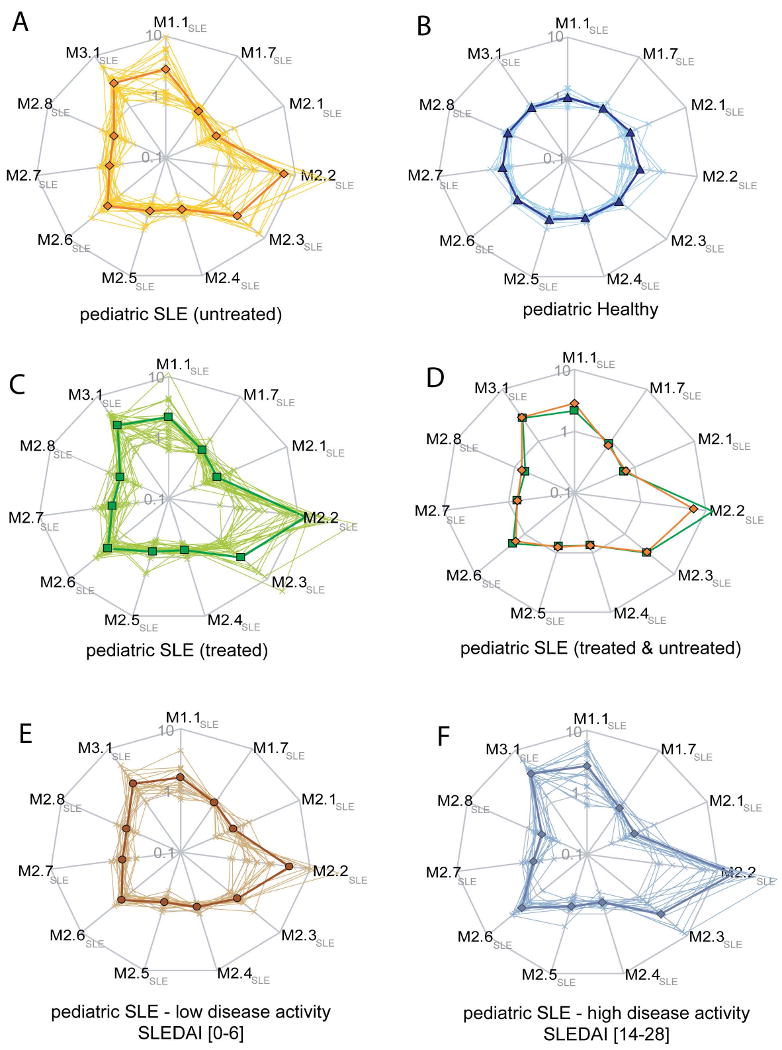
A) Composite transcriptional vectors identified from a pediatric SLE patient population sampled prior to the initiation of therapy. Each line on the radar plot represents a patient profile (logarithmic scale). Values are normalized per-gene using the median expression value of healthy. The thicker line represents the average normalized expression profile for this group of patients. Profiles generated for healthy volunteers B) and an independent cohort of pediatric SLE patients under treatment C). Averaged normalized expression profiles for treated (green) and untreated (orange) SLE patients cohorts D). Patient profiles were plotted on the same vectors on the basis of clinical activity (SLEDAI), regardless of treatment. E) Patients with low disease activity (SLEDAI from 0 to 6). F) Patients with high disease activity (SLEDAI from 14 to 28).
In order to investigate a possible link between SLE activity and patient transcriptional profiles we stratified samples based solely on SLEDAI scores. Samples from patients with mild disease activity (SLEDAI [0-6]) presented a profile closer to that of healthy subjects (Figure 3E); whereas patients with high disease activity (SLEDAI [14-28]) presented an exacerbated profile (Figure 3F – comparison of mild vs. high disease activity: V1.7, V2.2, V2.3, V2.4, V2.8 & V3.1: p<0.01; V1.1 p=0.07; V2.1 p=0.06; V2.5 p=0.8; V2.6 p=0.02; V2.7 p=0.08; Mann Whitney U test).
These results suggest that composite transcriptional vectors identified in SLE patients are associated with disease severity and have potential value as biomarkers.
Generating multivariate transcriptional scores as indicators of SLE disease progression
SLE is a heterogeneous multisystemic disease presenting a wide range of clinical and laboratory abnormalities. Objectively assessing disease activity across patients or longitudinally in individual patients can therefore be challenging. At least 6 composite measures of SLE global disease activity have been developed (Bae et al., 2001; Bencivelli et al., 1992; Bombardier et al., 1992; Hay et al., 1993; Liang et al., 1989; Petri et al., 1999) and have been used to assess disease progression during clinical trials. These measures, however, rely on a series of clinical and laboratory findings and are cumbersome to obtain. The SLEDAI, one of the simplest measures, considers 24 different attributes that need to be obtained at every clinic visit. Additionally, given the heterogeneous nature of the clinical disease, not all SLE manifestations are computed within these measures, making the overall assessment of the patient sometimes difficult. Thus, establishment of an objective disease activity index would be beneficial. We engaged to assess whether such activity index could be generated from blood leukocyte microarray transcriptional data.
The analysis of pediatric SLE patient profiles carried out above showed a link between transcriptional vectors and clinical disease manifestations. We have previously found that expression of individual genes, or gene signatures (such as interferon-inducible genes) could be affected by treatment (Bennett et al., 2003). Therefore, our aim was to maximize the number of transcriptional signatures used as a basis for the generation of a clinical indicator of disease activity. We computed correlations between composite expression values for individual dimensions (transcriptional vectors) and the clinical activity index (SLEDAI) for each of the patients in our untreated cohort (Figure 4A). We found that two dimensions (corresponding to V2.2 - “neutrophil” - and V3.1 - “interferon-inducible” - modules) correlated positively with disease activity, whereas dimensions corresponding to V1.7, V2.4 and V2.8 (“ribosomal proteins” and “T cells”) correlated negatively. We next verified that differences in expression observed for a selection of transcripts that belong to these five modules could be confirmed using real-time PCR. Two transcripts from M1.7, M2.2, M2.4, M2.8 and M3.1 were tested in 10 healthy controls and 25 patients. Differences in expression were significant in 9 out of the 10 transcripts tested (CCDC72, ELA2, MPO, FBL, EEF1D, IL23A, SIGLEC1 p<0.001; GATA3, MX1 p<0.05; GLTSCR2 p=0.8). Only one of the transcripts tested, GLTSCR2, did not display the expected difference between control and SLE groups. This degree of concordance is consistent with rates reported in the literature (Bosotti et al., 2007). The discrepancy could be attributed to differences in probe selection for the respective assays. Expression values obtained by real-time PCR for the 9 differentially expressed transcripts were also significantly correlated with microarray data (Supplementary Figures 4 & 5).
Figure 4. SLE transcriptional vectors correlate with disease activity.
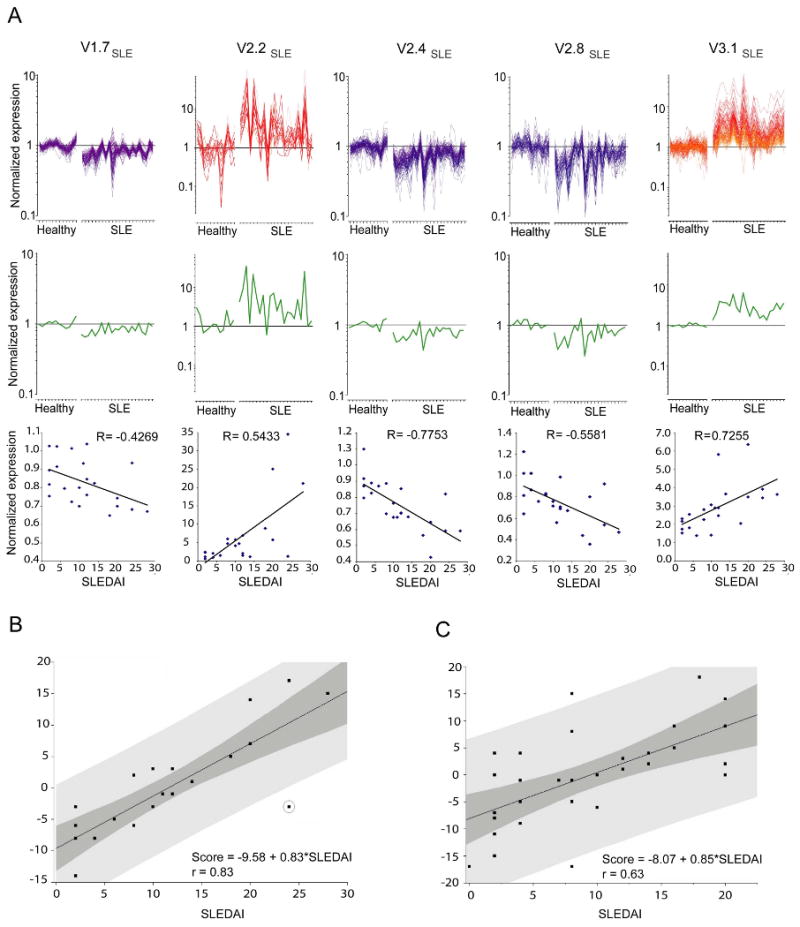
A) Expression profiles of genes forming vectors V1.7SLE, V2.2SLE, V2.4SLE, V2.8SLE and V3.1SLE correlating with a clinical SLE disease activity index (SLEDAI). Graphs represent expression level of individual transcripts forming each of the vectors in 12 healthy individuals and 21 untreated pediatric SLE patients. Average expression values across transcripts forming each vector are shown on the graph in yellow. Correlations between averaged vector expression values and SLEDAI are shown below (Spearman correlation). B) Multivariate scores were obtained for 22 untreated pediatric SLE patients by linear regression analysis of multivariate transcriptional U-scores (y axis) for vector V1.7SLE, V2.2SLE, V2.4SLE, V2.8SLE, V3.1 SLE, and SLEDAI (x axis). The light shaded area indicates the 95% limits confidence limits for individual predicted values. The dark shaded area indicates the 95% limits confidence limits for the slope and intercept. C) The same analysis applied to 31 pediatric SLE patients receiving different combinations of therapy.
A non-parametric method for analyzing multivariate ordinal data was used to score the patients based on these five dimensions (Spangler et al., 2004; Wittkowski et al., 2004). The advantage of this approach is that there is no need for additional assumptions and validations. Once available knowledge has been incorporated by making the initial transformations the proposed scores are valid by construction, as long as each variable increases or decreases with the unobservable latent factor. Thus, no empirical evaluation is needed. Because no assumptions are made regarding the functional form of the relationship, U- scores are scale independent.
U-scores were obtained for all patients in the untreated cohort (n=22). A polarity of 1 was attributed to vectors correlating positively with disease activity (i.e. Neutrophil: V2.2, Interferon: V3.1). The polarity of vectors correlating inversely with disease activity was set to -1 (T cells: V2.8, and Ribosomal proteins: V1.7 and V2.4). This allowed the ranking of all patients within this group. U-scores have positive (most severe disease) or negative values (less severe disease) reflecting the rank of each sample vs. the other patients forming this cohort. The association between the multivariate “transcriptional scores” and SLEDAI was assessed using linear regression and was determined to be statistically significant (Figure 4B; r=0.83, df=1, t=6.66, and p-value<.0001). The correlation achieved by this score was superior to that of its individual components. Using the same process, correlation between “Transcriptional score” and SLEDAI was examined for the treated pediatric SLE patient cohort (n=31) and was found to be statistically significant as well (Figure 4C; r=0.63, df=1, t=4.40, and p-value=0.0001).
Thus, distinct immunological signatures associated with the pathogenesis of SLE have been reduced to a unique multivariate score correlating with disease activity.
Multivariate transcriptional scores are used to monitor disease progression in patients with SLE
Lupus disease flares can lead to irreversible worsening of the status of the patient. We tested the relevance of the multivariate transcriptional score for the longitudinal monitoring of disease activity a cohort of 20 pediatric SLE patients (two to four time points/patient, intervals between each time point varied from one month to 18 months). Half of the patients had been included in our cross-sectional analysis before they were enrolled in this longitudinal study.
During the follow up period, the SLEDAI fluctuated in 10 patients whereas it remained constant in the other 10 (Figure 5). Parallel trends were observed between transcriptional U-scores and SLEDAI longitudinal measures in a majority of patients. The positive association between SLEDAI and transcriptional scores was verified statistically using a linear regression model. The estimated model was: transcriptional score = 18.13 + 1.26 (SLEDAI) - 0.03 (Days). The overall model was statistically significant (df=1, chi-sq=28.44, and p-value<.0001), as was the association between the SLEDAI and transcriptional scores (df=28, t=2.41, and p-value=0.0229). For every one unit increase in SLEDAI score the transcriptional score increases by 1.3. Overall SLEDAI index and transcriptional scores reflected similar activities according to their respective scales in all but 6 patients (SLE31, SLE78, SLE125, SLE130, SLE135 and SLE 99) in whom the transcriptional U-scores were disproportionately high compared to SLEDAI index (SLEDAI values are positive while multivariate U-scores can be positive or negative). One of the patients with the highest discrepancy (SLE78) was diagnosed during the follow-up period with a life-threatening complication (pulmonary hypertension) which is not computed within the SLEDAI. Thus, severity of disease was more accurately assessed by the transcriptional score. Disease flaring and subsequent recovery was detected in one patient (SLE31) upon longitudinal follow up using both SLEDAI and transcriptional score. Interestingly, however, the amplitude of change observed in the case of the transcriptional U-score appears not only to be much greater (0 to 40 vs. 6 to 10 for SLEDAI), but an increase could already be detected at the second time point, 2 months before the worsening of the clinical condition of this patient was detected by SLEDAI. Thus, these data illustrate the potential value of microarray data and the multivariate transcriptional scores derived from it for the longitudinal follow up of disease progression in patients with complex multisystemic diseases like SLE.
Figure 5. Longitudinal disease monitoring with a multivariate disease activity score.
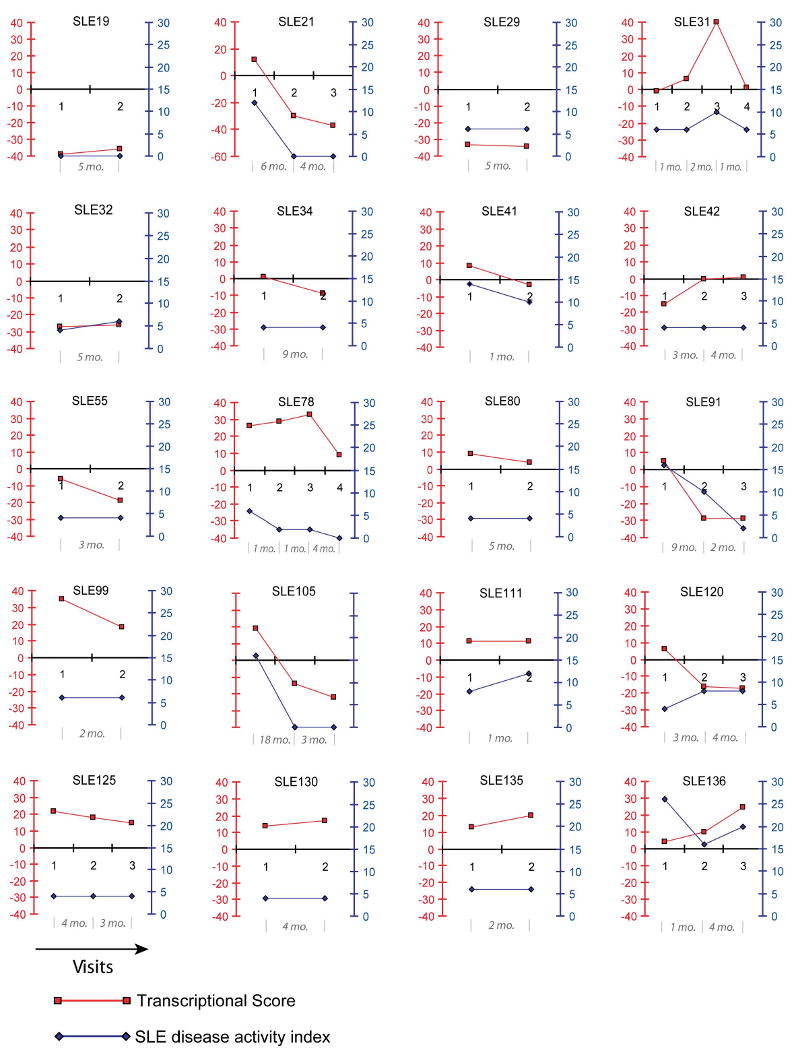
SLEDAI index (blue, right y axis) and transcriptional U-scores (red, left y axis) of pediatric patients (identified by an SLE ID) over time (x axis). Time elapsed between sampling is indicated in months.
Composite transcriptional vectors are stable across laboratories and microarray platforms
To be truly viable as biomarkers, composite transcriptional vectors must prove reliable. Early on, poor reproducibility of microarray results obtained by different laboratories and across platforms raised suspicion about the validity of these results and remains a major concern (Bammler et al., 2005; Frantz, 2005; Ioannidis, 2005; Irizarry et al., 2005; Larkin et al., 2005; Michiels et al., 2005; Shi et al., 2006). We compared transcriptional profiles obtained using two commercial microarray platforms, Affymetrix and Illumina. PBMCs were isolated from four healthy volunteers and ten liver transplant recipients. Starting from the same source of total RNA, targets were generated independently and analyzed using Affymetrix U133 GeneChips (at the Baylor Institute for Immunology Research) and Illumina Human Ref8 BeadChips (at Illumina Inc.). Fundamental differences exist between the two microarray technologies (see Methods for details). Probe IDs provided by each manufacturer were converted into a common ID that was used for matching gene expression profiles. Overall the Affymetrix and Illumina profiles for M3.1 appear to be similar (Figure 6). However, correlations comparing both platforms performed for individual genes forming M3.1 resulted in a median R2 value of 0.36 (ranging from 0.17 and 0.55) In other modules such as M1.2 and M3.2 correlations observed at the level of individual genes were also poor (R2 median (range) = 0.13 (0.02-0.5) for genes forming M1.2; and 0.19 (0.06-0.4) for genes forming M3.2 – Figure 6). In order to compare overall modular expression pattern across the two platforms we derived for each module a composite transcriptional vector (averaging the values obtained for the genes forming each module). Remarkably, the module-level expression values thus derived from Affymetrix and Illumina data were highly comparable (Figure 6; transplant group Pearson correlation coefficient R2 = 0.83, 0.98 and 0.93, for M1.2, M3.1 and M3.2 respectively; p<0.0001 – in addition R2 values of M1.1=0.84; M1.3=0.95; M1.4=0.81; M1.5=0.74; M1.8=0.62; M2.1=0.98; M2.2=0.82; M2.3=0.99; M2.6=0.73; M2.8=0.83; M2.10=0.66; M3.3=0.65; M3.8=0.57; R2 values for other modules <0.5). Taken together, these results indicate that module-level composite expression data produce a more stable metric than individual gene expression values, thereby enhancing data reproducibility across microarray platforms. This property may be attributed to the stringent module selection process (transcripts must be co-expressed across many samples) and the fact that composite expression values are derived from multiple measurements (smoothing the imprecision observed at the level of individual probes).
Figure 6. Cross-microarray platform comparison.
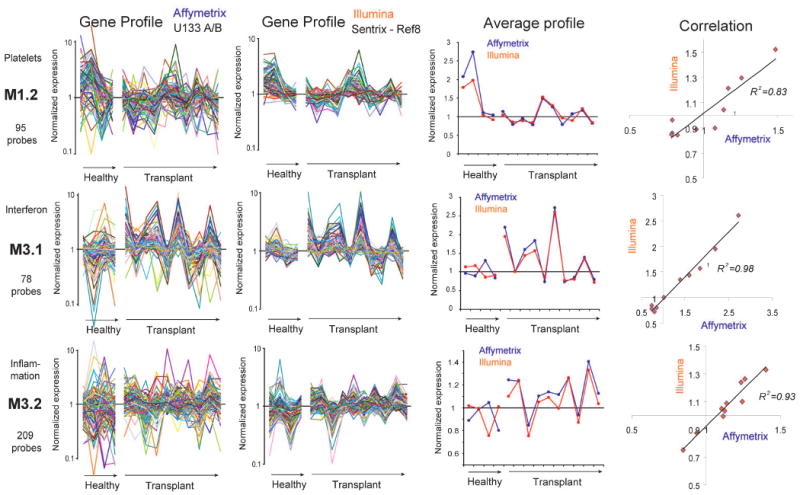
PBMC samples from healthy donors and liver transplant recipient analyzed on two different microarray platforms: Affymetrix U133A&B GeneChips and Illumina Sentrix Human Ref8 BeadChips. The same source of total RNA was used to independently prepare biotin-labeled cRNA targets. Expression is normalized to the median of measurements obtained across all samples. Averaged expression values of the genes in each module are shown for both Affymetrix and Illumina platforms.
Discussion
Patient blood transcriptional profiling studies generate large scale data that is difficult to exploit. Adopting a module-based data mining strategy can facilitate biomarker and biological knowledge discovery by focusing the analysis of microarray data on stable sets of transcripts selected on the basis of their clustering pattern across diseases.
The module construction strategy that we have designed takes advantage of the biological variability inherent to patient-based studies in order to identify the major transcriptional components of this system. A clustering algorithm teases apart the patterns emerging from the blood profiles obtained for different diseases. Once patterns have been identified for each disease the cluster membership of individual transcripts is compared. A module is formed of transcripts found to always belong to the same clusters across all diseases (8 out of 8 in our example). The stringency of this requirement is progressively relaxed during the subsequent rounds of selection so that modules are formed when transcripts fall in the same clusters in any combination of 7 (round 2) or combination of 6 diseases (round 3). This stepwise reduction of the stringency of filtering criteria accounts for the fact that transcripts may not be “turned on” in all diseases. Indeed, modules linked to interferon or inflammation (M3.1 and M3.2) were for instance not formed until the third round of selection. The validity of the transcriptional modules thus generated was verified by different approaches. Random permutations attested of the statistical validity of the module construction, while co-expression was confirmed in independent datasets (PBMC samples from healthy volunteers or patients with S. pneumoniae infection that were not used to identify modules), across laboratories and microarray platforms (Affymetrix vs. Illumina). Furthermore, as should be expected of transcriptional modules, literature profiling of genes forming each one of them revealed significant functional convergence, with half of the modules associated with clearly identifiable functional themes.
By including profiles from a wide range of diseases our goal was to identify a “universal” set of modules that could be used as a stable framework for subsequent analysis of any PBMC dataset. However, we nonetheless anticipate that adding more diseases to the selection pool will result in a refined partitioning of the modules already identified and will add modules to the existing set. Also, although the collection of genome-wide PBMC transcriptional profiles used for module selection is already extensive the identification of a definitive module set will require expending the scale of this analysis.
Reducing the dimension of microarray data makes it more amenable to interpretation. When confronted to such overwhelming amount of information it is necessary to reduce it to a manageable number of variables and to use visualization schemes as a means to facilitate the identification of patterns in the data, especially when performing comparisons across diseases. Furthermore, following functional interpretation, we found that the modules identified are linked to the two components driving differential gene expression in blood: changes in relative cellular abundance (e.g. B cell, cytotoxic cell modules), and gene regulation (e.g. inflammation, interferon). Thus, overlaying these functional annotations to the fixed module patterns further supports the interpretation of disease fingerprints. Inevitably, however, reducing microarray data to a small set of variables and broad functional categories can only offer an oversimplified view of the data, and interactive mining tools are therefore necessary to restore the unique depth perspective that systems scale data are able to provide.
We have also explored the use of transcriptional modules as a basis for biomarker discovery. By construction modules include only transcripts which co-clustered in at least 6 out of 8 diseases across many samples. The probability of this happening just by chance is very low. In fact we have run tests in which gene labels were permuted randomly in the different diseases and could not identify any modules. Also, using sets of transcriptional modules as a basis for biomarker discovery should help focus on biologically relevant transcripts. Another potential benefit of using modules as a framework for biomarker discovery is that it allows the reduction of the dimension of microarray data. Identifying a small set of clinically valuable markers from tens of thousands of candidates in a single analysis step is a considerable challenge, noise being again a major issue. However, when the data are first reduced from over 44,000 variables to about 5000 distributed in 28 modules, biomarker discovery becomes a much more manageable proposition. In the case of SLE, comparisons carried out on a module-by-module basis identified 11 sub-modules with a minimum of 15% of transcripts over- or under-expressed compared to healthy. Once the data are reduced to 11 composite values (or transcriptional vectors) it then becomes possible to summarize the results as one single multivariate score. Repeating measurements for multiple transcripts sharing the same pattern within a module also makes for a more robust measurement, which explains, at least in part, the level of correlation measured between data generated on two microarray platforms in two independent laboratories. Finally the fact that some of the modules can be associated to well-recognized biological pathways linked to disease pathogenesis will help in further asserting the credibility of biomarkers derived from such analysis.
Indeed, upon being identified SLE vectors were validated in an independent set of samples. Furthermore, multivariate resulting transcriptional scores were correlated to clinical disease activity indices in both cross-sectional and longitudinal sets of samples. Our data demonstrate that composite transcriptional vectors can be directly correlated to clinical disease activity in patients with lupus.
In conclusion, the modular analysis framework that we have generated could prove useful for the discovery of diagnostic or prognostic markers and provide the means to monitoring disease progression and response to treatment in other complex disease settings
Experimental Procedures
Patient information
Subjects were recruited at the Baylor University Medical Center at Dallas, Texas Scottish Rite Hospital and Children's Medical Center of Dallas. The study was approved by the Institutional Review Boards of UT Southwestern Medical Center, Texas Scottish Rite Hospital, and Baylor Health Care System, and informed consent was obtained from all patients (legal representatives and patients over 10 yr of age). Bacterial and viral infections were confirmed by standard bacterial cultures, direct fluorescent antigen testing, and viral cultures. Patients with infections were recruited once a confirmed microbiologic diagnosis was established. The clinical and demographic characteristics of SLE patients are summarized in Supplementary Table 4.
Processing of blood samples
Blood samples were collected in acid citrate dextrose or EDTA tubes (BD Vacutainer) and immediately delivered at room temperature to the Baylor Institute for Immunology Research, Dallas, TX, for processing. Peripheral blood mononuclear cells (PBMCs) were isolated via Ficoll gradient and immediately lysed in RLT reagent (Qiagen, Valencia, CA) with beta-mercaptoethanol (BME) and stored at −80°C prior to the RNA extraction step.
Microarray analysis
Total RNA was isolated using the RNeasy kit (Qiagen) according to the manufacturer's instructions and RNA integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA).
Affymetrix GeneChips
Target labeling was performed according to the manufacturer's standard protocol (Affymetrix Inc., Santa Clara, CA). Biotinylated cRNA targets were purified and subsequently hybridized to Affymetrix HG-U133A and U133B GeneChips (>44,000 probe sets). Arrays were scanned using an Affymetrix confocal laser scanner. Microarray Suite, Version 5.0 (MAS 5.0; Affymetrix) software was used to assess fluorescent hybridization signals, to normalize signals, and to evaluate signal detection calls. Normalization of signal values per chip was achieved using the MAS 5.0 global method of scaling to the target intensity value of 500 per GeneChip. A gene expression analysis software program, GeneSpring, Version 7.1 (Agilent), was used to perform statistical analysis and clustering.
Illumina BeadChips
Samples were processed and data acquired by Illumina Inc. (San Diego, CA). Targets were prepared using the Illumina RNA amplification kit (Ambion, Austin, TX). cRNA targets were hybridized to Sentrix HumanRef8 BeadChips (>25,000 probes), which were scanned on an Illumina BeadStation 500. Illumina's Beadstudio software was used to assess fluorescent hybridization signals.
Quantitative real-time PCR
Biotinylated cRNA prepared for microarray analysis was reverse transcribed into cDNA using the The High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster City CA). Real-time PCR was set up with Roche Probes Master reagents and Universal Probe Library hydrolysis probes. PCR reaction was performed on the LightCycler 480 (Roche Applied Science). Secondary derivative calculation data was collected and cross point values of target genes were normalized to two housekeeping genes (ARHGDIB and GUSB).
Module construction algorithm
Our goal was to extract from an extensive leukocyte microarray dataset groups of coordinately expressed transcripts spanning multiple diseases (i.e. identifying genes which expression is correlated across multiple samples). Although the initial steps of our approach produce clusters of coordinately transcribed genes in a similar manner as other groups, we refine the process by generating modules based on cluster membership across multiple independent microarray experiments: 1. parallel analyses were performed, grouping transcripts for 8 different disease datasets using the K-means clustering algorithm. 2. Transcripts that which are co-expressed in the context of several diseases were then identified (i.e. we examine cluster membership across multiple independent microarray experiments). Also, in the first round of selection we started by choosing transcripts with shared cluster membership for all 8 diseases. For the subsequent round of selection we accounted for the fact that the different diseases may produce different patterns, thus we decreased the level of stringency accordingly (i.e. allowing for one, or even two diseases to be dropped in the second, and the third rounds of selection, respectively). In summary this approach relies on the K-means clustering algorithm, and is tailored to capture transcriptional modules spanning multiple diseases, starting from a large number of transcripts. This module construction algorithm is described in detail in the supplementary experimental procedures section.
Multivariate U-scores
The detailed explanation of this method has been published recently (Wittkowski et al., 2004) and the required tools are available at http://Mustat.Rockefeller.edu. Briefly, scores were obtained by computing the average normalized expression levels for all transcripts within the modules that were identified as differentially expressed in SLE PBMCs.
Literature profiling
The literature profiling algorithm employed in this study has been previously described in detail (Chaussabel and Sher, 2002). This approach links genes sharing similar keywords. It uses hierarchical clustering to analyze patterns of term occurrence in literature abstracts.
Association between SLEDAI and Multivariate U-Scores
Linear regression was used in the cross sectional analyses to assess the association between the multivariate “transcriptional scores” and SLEDAI for the treated and untreated pediatric SLE patients. Results and figures were obtained using JMP statistical software (Version 7; SAS Institute). When assessing this association for the corresponding longitudinal data a linear mixed effect model with a random intercept was used to account for the repeated and unequally spaced observations. This modeling technique is well described in such texts as Verbeke and Molenberghs (Verbeke and Molenberghs, 2000) and Fitzmaurice et al. (Fitzmaurice et al., 2004). SAS statistical software (Version 9.1; SAS Institute) was used for this portion of the analysis.
Accession number for materials deposited in a public database
The microarray data used in this study has been deposited in NCBI's Gene Expression Omnibus (GEO) with the accession number GSE11907.
Supplementary Material
Supplementary Figure 1: Module construction algorithm. A) Data are generated in the context of a defined experimental system (e.g. ex vivo PBMCs). B) Transcriptional profiles are obtained for several experimental groups (e.g. G1-8). C) For each group, genes are distributed among x clusters (e.g. x = 30) based on similarity of expression profiles (using K-means clustering algorithm). D) The cluster distribution of each gene across the different experimental groups is recorded in a table and distribution patterns are matched. E) Modules are selected through an iterative process, starting with the largest set of genes distributed among the same cluster across all experimental groups (e.g. found in the same cluster for eight out of eight groups). The selection is expanded from this core reference pattern to include genes with 7/8, 6/8 and 5/8 matches. Once a module has been formed, the genes are withdrawn from the selection pool. The process is then repeated, starting with the second largest group of genes, progressively reducing levels of stringency.
Supplementary Figure 2: Modules form coherent functional units. Term occurrence levels in abstracts were computed for all the genes in M3.1, M1.5, M1.3 and M1.2 associated with at least ten publications (representing more than 26,000 abstracts). Keyword profiles were extracted for each module and a selection was used to generate this figure. Levels of keyword occurrence in abstracts are indicated by color scale, with yellow representing high occurrence. M3.1 (e.g. STAT1, CXCL10, OAS2, MX2) is associated with interferon, M1.5 (e.g. MYD88, CD86, TLR2, LILRB2, CD163) is associated with pathogen recognition molecules / myeloid lineage cells, M1.3 (e.g. CD19, CD22, CD72A, BLNK, PAX5) is associated with B-cells and M1.2 (e.g. ITGA2B, PF4, SELP, GP6) is associated with platelets.
Supplementary Figure 3: Screenshots from an interactive module mapping tool accessible online at www.biir.net/modules. Modules maps are used as a guide to explore gene-level data module-by-module, providing access to gene lists and gene profiles. This tool also gives users the opportunity to modify dynamically statistical cutoffs. Module maps obtained for the same dataset with p value cutoffs set to 0.05 (top) and 1 (no filter, bottom) are shown.
Supplementary Figure 4 & 5: Correlation of real-time PCR and microarray gene expression data. Two genes were selected from 5 transcriptional modules, M1.7, M2.2, M2.4, M2.8 and M3.1, for verification by real-time PCR. Transcript abundance was measured for 25 patients and 10 healthy controls. Real-time PCR and microarray expression values were normalized to the median of all 35 samples and natural-log transformed. The relationship between the real-time PCR and microarray expression values for each individual gene was assessed using linear regression. The estimated regression equation, Pearson correlation coefficients, and significance levels are shown on each graph. The light and dark shaded areas indicate the 95% limits confidence limits for individual predicted values and model parameters respectively. Healthy individuals are represented by hollow diamonds and the patients by darkened squares.
Acknowledgments
Supported by Baylor Health Care System Foundation, the Alliance for Lupus Research (VP), DANA foundation (AKP), Defense Advanced Research Planning Agency (JB), The National Institutes of Health (U19 AIO57234-02, P01 CA084512, R01 CA078846 and R01 I068842 to JB; Center for Lupus Research P50 AR054083 and R01 AR050770-01 to VP). JB holds the W.W. Caruth, Jr. Chair in Organ Transplantation Immunology. AKP holds the Michael A. Ramsay Chair for Cancer Immunology Research.
We thank our patients and their parents / guardians for agreeing to participate in the study. We thank Dr. Carson Harrod for editorial help, Quynh-Anh Nguyen for technical assistance, Gordon Hayward for IT support and Drs. Michael Ramsay and William Duncan for their continuous support.
References
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Allantaz F, Chaussabel D, Stichweh D, Bennett L, Allman W, Mejias A, Ardura M, Chung W, Wise C, Palucka K, et al. Blood leukocyte microarrays to diagnose systemic onset juvenile idiopathic arthritis and follow the response to IL-1 blockade. J Exp Med. 2007;204:2131–2144. doi: 10.1084/jem.20070070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae SC, Koh HK, Chang DK, Kim MH, Park JK, Kim SY. Reliability and validity of systemic lupus activity measure-revised (SLAM-R) for measuring clinical disease activity in systemic lupus erythematosus. Lupus. 2001;10:405–409. doi: 10.1191/096120301678646146. [DOI] [PubMed] [Google Scholar]
- Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bammler T, Beyer RP, Bhattacharya S, Boorman GA, Boyles A, Bradford BU, Bumgarner RE, Bushel PR, Chaturvedi K, Choi D, et al. Standardizing global gene expression analysis between laboratories and across platforms. Nat Methods. 2005;2:351–356. doi: 10.1038/nmeth754. [DOI] [PubMed] [Google Scholar]
- Bencivelli W, Vitali C, Isenberg DA, Smolen JS, Snaith ML, Sciuto M, Bombardieri S. Disease activity in systemic lupus erythematosus: report of the Consensus Study Group of the European Workshop for Rheumatology Research. III. Development of a computerised clinical chart and its application to the comparison of different indices of disease activity. The European Consensus Study Group for Disease Activity in SLE. Clin Exp Rheumatol. 1992;10:549–554. [PubMed] [Google Scholar]
- Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoist C, Germain RN, Mathis D. A plaidoyer for ‘systems immunology’. Immunol Rev. 2006;210:229–234. doi: 10.1111/j.0105-2896.2006.00374.x. [DOI] [PubMed] [Google Scholar]
- Biggs N, L E, Wilson R. Graph Theory. Oxford University Press; 1986. [Google Scholar]
- Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum. 1992;35:630–640. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- Bosotti R, Locatelli G, Healy S, Scacheri E, Sartori L, Mercurio C, Calogero R, Isacchi A. Cross platform microarray analysis for robust identification of differentially expressed genes. BMC Bioinformatics. 2007;8 1:S5. doi: 10.1186/1471-2105-8-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burczynski ME, Peterson RL, Twine NC, Zuberek KA, Brodeur BJ, Casciotti L, Maganti V, Reddy PS, Strahs A, Immermann F, et al. Molecular classification of Crohn's disease and ulcerative colitis patients using transcriptional profiles in peripheral blood mononuclear cells. J Mol Diagn. 2006;8:51–61. doi: 10.2353/jmoldx.2006.050079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burczynski ME, Twine NC, Dukart G, Marshall B, Hidalgo M, Stadler WM, Logan T, Dutcher J, Hudes G, Trepicchio WL, et al. Transcriptional profiles in peripheral blood mononuclear cells prognostic of clinical outcomes in patients with advanced renal cell carcinoma. Clin Cancer Res. 2005;11:1181–1189. [PubMed] [Google Scholar]
- Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- Chaussabel D, Allman W, Mejias A, Chung W, Bennett L, Ramilo O, Pascual V, Palucka AK, Banchereau J. Analysis of significance patterns identifies ubiquitous and disease-specific gene-expression signatures in patient peripheral blood leukocytes. Ann N Y Acad Sci. 2005;1062:146–154. doi: 10.1196/annals.1358.017. [DOI] [PubMed] [Google Scholar]
- Chaussabel D, Sher A. Mining microarray expression data by literature profiling. Genome Biol. 2002;3:RESEARCH0055. doi: 10.1186/gb-2002-3-10-research0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb JP, Mindrinos MN, Miller-Graziano C, Calvano SE, Baker HV, Xiao W, Laudanski K, Brownstein BH, Elson CM, Hayden DL, et al. Application of genome-wide expression analysis to human health and disease. Proc Natl Acad Sci U S A. 2005;102:4801–4806. doi: 10.1073/pnas.0409768102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. 2003;36:481–490. doi: 10.1080/08916930310001625952. [DOI] [PubMed] [Google Scholar]
- Fitzmaurice GM, Laird NM, Ware JH. Applied longitudinal analysis. Hoboken, N.J.: Wiley-Interscience; 2004. [Google Scholar]
- Frantz S. An array of problems. Nat Rev Drug Discov. 2005;4:362–363. doi: 10.1038/nrd1746. [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Hay EM, Bacon PA, Gordon C, Isenberg DA, Maddison P, Snaith ML, Symmons DP, Viner N, Zoma A. The BILAG index: a reliable and valid instrument for measuring clinical disease activity in systemic lupus erythematosus. Q J Med. 1993;86:447–458. [PubMed] [Google Scholar]
- Hyatt G, Melamed R, Park R, Seguritan R, Laplace C, Poirot L, Zucchelli S, Obst R, Matos M, Venanzi E, et al. Gene expression microarrays: glimpses of the immunological genome. Nat Immunol. 2006;7:686–691. doi: 10.1038/ni0706-686. [DOI] [PubMed] [Google Scholar]
- Ioannidis JP. Microarrays and molecular research: noise discovery? Lancet. 2005;365:454–455. doi: 10.1016/S0140-6736(05)17878-7. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Warren D, Spencer F, Kim IF, Biswal S, Frank BC, Gabrielson E, Garcia JG, Geoghegan J, Germino G, et al. Multiple-laboratory comparison of microarray platforms. Nat Methods. 2005;2:345–350. doi: 10.1038/nmeth756. [DOI] [PubMed] [Google Scholar]
- Jarvinen AK, Hautaniemi S, Edgren H, Auvinen P, Saarela J, Kallioniemi OP, Monni O. Are data from different gene expression microarray platforms comparable? Genomics. 2004;83:1164–1168. doi: 10.1016/j.ygeno.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Kaizer EC, Glaser CL, Chaussabel D, Banchereau J, Pascual V, White PC. Gene expression in peripheral blood mononuclear cells from children with diabetes. J Clin Endocrinol Metab. 2007;92:3705–3711. doi: 10.1210/jc.2007-0979. [DOI] [PubMed] [Google Scholar]
- Kirou KA, Lee C, George S, Louca K, Papagiannis IG, Peterson MG, Ly N, Woodward RN, Fry KE, Lau AY, et al. Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum. 2004;50:3958–3967. doi: 10.1002/art.20798. [DOI] [PubMed] [Google Scholar]
- Larkin JE, Frank BC, Gavras H, Sultana R, Quackenbush J. Independence and reproducibility across microarray platforms. Nat Methods. 2005;2:337–344. doi: 10.1038/nmeth757. [DOI] [PubMed] [Google Scholar]
- Liang MH, Socher SA, Larson MG, Schur PH. Reliability and validity of six systems for the clinical assessment of disease activity in systemic lupus erythematosus. Arthritis Rheum. 1989;32:1107–1118. doi: 10.1002/anr.1780320909. [DOI] [PubMed] [Google Scholar]
- Michiels S, Koscielny S, Hill C. Prediction of cancer outcome with microarrays: a multiple random validation strategy. Lancet. 2005;365:488–492. doi: 10.1016/S0140-6736(05)17866-0. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Petri M, Buyon J, Kim M. Classification and definition of major flares in SLE clinical trials. Lupus. 1999;8:685–691. doi: 10.1191/096120399680411281. [DOI] [PubMed] [Google Scholar]
- Ramilo O, Allman W, Chung W, Mejias A, Ardura M, Glaser C, Wittkowski KM, Piqueras B, Banchereau J, Palucka AK, Chaussabel D. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood. 2007;109:2066–2077. doi: 10.1182/blood-2006-02-002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Barrette TR, Ghosh D, Chinnaiyan AM. Mining for regulatory programs in the cancer transcriptome. Nat Genet. 2005;37:579–583. doi: 10.1038/ng1578. [DOI] [PubMed] [Google Scholar]
- Segal E, Friedman N, Kaminski N, Regev A, Koller D. From signatures to models: understanding cancer using microarrays. Nat Genet. 2005;37(Suppl):S38–45. doi: 10.1038/ng1561. [DOI] [PubMed] [Google Scholar]
- Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nat Genet. 2004;36:1090–1098. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY, et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler R, Wittkowski KM, Goddard NL, Avena NM, Hoebel BG, Leibowitz SF. Opiate-like effects of sugar on gene expression in reward areas of the rat brain. Brain Res Mol Brain Res. 2004;124:134–142. doi: 10.1016/j.molbrainres.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Mellman I. Immunotherapy: bewitched, bothered, and bewildered no more. Science. 2004;305:197–200. doi: 10.1126/science.1099688. [DOI] [PubMed] [Google Scholar]
- Thach DC, Agan BK, Olsen C, Diao J, Lin B, Gomez J, Jesse M, Jenkins M, Rowley R, Hanson E, et al. Surveillance of transcriptomes in basic military trainees with normal, febrile respiratory illness, and convalescent phenotypes. Genes Immun. 2005 doi: 10.1038/sj.gene.6364244. [DOI] [PubMed] [Google Scholar]
- Verbeke G, Molenberghs G. Linear mixed models for longitudinal data. New York, NY: Springer; 2000. [Google Scholar]
- Wittkowski KM, Lee E, Nussbaum R, Chamian FN, Krueger JG. Combining several ordinal measures in clinical studies. Stat Med. 2004;23:1579–1592. doi: 10.1002/sim.1778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Module construction algorithm. A) Data are generated in the context of a defined experimental system (e.g. ex vivo PBMCs). B) Transcriptional profiles are obtained for several experimental groups (e.g. G1-8). C) For each group, genes are distributed among x clusters (e.g. x = 30) based on similarity of expression profiles (using K-means clustering algorithm). D) The cluster distribution of each gene across the different experimental groups is recorded in a table and distribution patterns are matched. E) Modules are selected through an iterative process, starting with the largest set of genes distributed among the same cluster across all experimental groups (e.g. found in the same cluster for eight out of eight groups). The selection is expanded from this core reference pattern to include genes with 7/8, 6/8 and 5/8 matches. Once a module has been formed, the genes are withdrawn from the selection pool. The process is then repeated, starting with the second largest group of genes, progressively reducing levels of stringency.
Supplementary Figure 2: Modules form coherent functional units. Term occurrence levels in abstracts were computed for all the genes in M3.1, M1.5, M1.3 and M1.2 associated with at least ten publications (representing more than 26,000 abstracts). Keyword profiles were extracted for each module and a selection was used to generate this figure. Levels of keyword occurrence in abstracts are indicated by color scale, with yellow representing high occurrence. M3.1 (e.g. STAT1, CXCL10, OAS2, MX2) is associated with interferon, M1.5 (e.g. MYD88, CD86, TLR2, LILRB2, CD163) is associated with pathogen recognition molecules / myeloid lineage cells, M1.3 (e.g. CD19, CD22, CD72A, BLNK, PAX5) is associated with B-cells and M1.2 (e.g. ITGA2B, PF4, SELP, GP6) is associated with platelets.
Supplementary Figure 3: Screenshots from an interactive module mapping tool accessible online at www.biir.net/modules. Modules maps are used as a guide to explore gene-level data module-by-module, providing access to gene lists and gene profiles. This tool also gives users the opportunity to modify dynamically statistical cutoffs. Module maps obtained for the same dataset with p value cutoffs set to 0.05 (top) and 1 (no filter, bottom) are shown.
Supplementary Figure 4 & 5: Correlation of real-time PCR and microarray gene expression data. Two genes were selected from 5 transcriptional modules, M1.7, M2.2, M2.4, M2.8 and M3.1, for verification by real-time PCR. Transcript abundance was measured for 25 patients and 10 healthy controls. Real-time PCR and microarray expression values were normalized to the median of all 35 samples and natural-log transformed. The relationship between the real-time PCR and microarray expression values for each individual gene was assessed using linear regression. The estimated regression equation, Pearson correlation coefficients, and significance levels are shown on each graph. The light and dark shaded areas indicate the 95% limits confidence limits for individual predicted values and model parameters respectively. Healthy individuals are represented by hollow diamonds and the patients by darkened squares.