

Abstract
Our previous studies have shown that very low-density lipoprotein receptor (VLDLR) is a negative regulator of the Wnt pathway. The present study showed that VLDLR gene knockout (Vldlr−/−) mice displayed impaired cone ERG responses at early ages. Immunostaining of mid-wavelength cones showed significantly decreased cone densities in the retina and shortened cone outer segments in Vldlr−/− mice. At older ages, Vldlr−/− mice displayed declined rod ERG responses, decreased layers of photoreceptor nuclei, reduced rhodopsin levels and decreased levels of 11-cis retinal, the chromophore of visual pigments. As shown by fluorescein angiography and permeability assay, Vldlr−/− mice had severe retinal vascular leakage. ZO-1, a tight junction protein, was down-regulated in Vldlr−/− mouse retinae, further supporting the impaired blood-retinal barrier. Double staining of pericytes and endothelial cells in retinal sections revealed that neovasculature in Vldlr−/− mice lacks pericyte coverage, suggesting impaired maturation of retinal vasculature in Vldlr−/− mice. Staining of adherent leukocytes in the retinal vasculature revealed significant leukostasis in Vldlr−/− mice. Moreover, Vldlr−/− mice displayed up-regulated expression of multiple pro-inflammatory factors and activated NF-κB and HIF-1α, key regulators of inflammation. These findings suggest that deficiency of VLDLR leads to retinal degeneration and inflammation.
Keywords: Wnt pathway, LRP, inflammation, angiogenesis, retinal degeneration, age-related macular degeneration
INTRODUCTION
Age-related macular degeneration (AMD) is the leading cause of blindness in the developed countries. The prevalence of AMD increases exponentially with age (Ambati et al., 2003; Nowak, 2006). AMD is a chronic and progressive retinal degenerative disease. At early stages, AMD is characterized by generation of drusen in the macular area and hyper- or hypo-pigmentation of the RPE (Bylsma, 2005; Provis et al., 2005). At late stages, AMD appears in two major forms, the dry AMD and wet AMD. The dry AMD is characterized by geographic atrophy, cone degeneration and loss of cone functions. In the wet AMD, abnormal neovasculature, originated from the choroid, forms either beneath the retinal pigment epithelium (RPE) or in the subretinal space after penetration through Bruch’s membrane, forming choroidal neovascularization (CNV). The abnormal neovasculature has high permeability, which can lead to macular edema and macrophage infiltration. CNV is a major vision threatening complication of AMD (Provis et al., 2005).
AMD is a complex disorder involving multiple genes and risk factors. Increased oxidative stress and inflammation in the retina are believed to play a pathogenic role in AMD (Ambati et al., 2003; Gong et al., 2001; Schlingemann, 2004). The recent breakthrough in genetic studies has shown that complement factor H plays a critical role in the pathogenesis of AMD, further supporting the pathogenic role of inflammation in AMD (Edwards et al., 2005; Klein et al., 2005; Nozaki et al., 2006). Over-expression of pro-inflammatory factors such as VEGF and intercellular adhesion molecule-1 (ICAM-1) has been shown to play a key role in retinal inflammation and CNV. VEGF functions not only as an angiogenic factor, but also as a pro-inflammatory and permeability factor (Croll et al., 2004; Dvorak, 2000; Ishida et al., 2003; Usui et al., 2004; Vinores et al., 2001). ICAM-1 is a major adhesion molecule leading to leukostasis in the retina (Miyamoto et al., 2000). Additionally, patients with severe wet AMD have several characteristics of chronic inflammatory diseases, such as elevated levels of tumor necrosis factor-α (TNF-α), soluble ICAM-1, and circulating vascular cell adhesion molecule-1 (VCAM-1), which attributes to manifestation of local edema (Bok, 2005).
The pathway mediating inflammation in AMD has not been defined. A recent study of genetic linkage and allelic association has identified three genes, the very low-density lipoprotein receptor (VLDLR), VEGF and low-density lipoprotein receptor-related protein-6 (LRP6), with significant linkage or association to AMD (Haines et al., 2006). LRP6 showed linkage in the family-based dataset and association in the case-control dataset. VEGF showed evidence of linkage and demonstrated significant independent allelic association in both the family-based and case-control datasets. VLDLR showed evidence of association in both the family-based and case-control datasets. Therefore, LRP6, VEGF and VLDLR have been suggested to play roles in the development of AMD (Haines et al., 2006). Our recent studies showed that LRP6 and its down-stream Wnt signaling pathway may play an important role in AMD (Chen et al., 2007).
In 2003, VLDLR gene knockout (Vldlr−/−) mice were found to develop subretinal NV (Heckenlively et al., 2003). Our recent studies demonstrated that Vldlr−/− mice develop CNV which is induced by the activation of the Wnt pathway and VEGF over-expression (Chen et al., 2007). Recent studies showed that neovascularization in Vldlr−/− mice recapitulates features of retinal angiomatous proliferation in human, a subtype of AMD (Chen et al., 2007; Hu et al., 2008; Li et al., 2007). The present study provided evidence showing that Vldlr−/− mice manifest most pathological features of AMD such as cone degeneration and loss of cone function, retinal inflammation and vascular leakage.
MATERIALS AND METHODS
Animals
Animals were kept in a 12-hour light–dark cycle with an ambient light intensity of 85 ± 18 lux. Vldlr−/− mice were genotyped as described previously (Chen et al., 2007). Vldlr−/− mice and wild-type (wt) C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, MA). Care, use and treatment of the animals were in strict agreement with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Western blot analysis
The eyes were enucleated, and the corneas and lenses removed. The eyecups from each mouse were combined and homogenized, and protein concentration measured by the Bradford method. The same amount (50 µg) of total protein from each mouse was resolved by SDS-PAGE and electrotransferred onto a polyvinylidene fluoride (PVDF) membrane, as described previously. The membrane was blotted with primary antibodies and followed by secondary antibodies. The signal was detected using the enhanced chemiluminescence (ECL) system (GE Healthcare, Piscataway, NJ).
Histologic analysis
The mice were scarified and immediately perfused with fixative consisting of 2.5% gluteraldehyde and 2% formaldehyde in 0.1 M cacodylate buffer with 0.08 M CaCl2. The eyes were removed, bisected along the vertical meridian, postfixed in osmium tetroxide, and embedded in an Epon-Araldite mixture. Sections of the entire retina along the optic nerve were cut 1-µm in thickness and stained with toluidine blue. The thickness of the outer nuclear layer (ONL) was measured as described (Komeima et al., 2007) and the mean ONL thickness obtained from six locations of 25%, 50%, and 75% of the distance between the superior and inferior poles and the optic nerve.
Immunohistochemistry
Frozen sections (4 µm) were cut, air dried and washed in phosphate-buffered saline (PBS), blocked with 20% goat serum, 0.1% Triton X-100 and 1% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO) in PBS. The sections were incubated with primary antibodies overnight at 4°C. The sections were rinsed several times with PBS and incubated with secondary antibodies for 30 min. After this, the slides were rinsed in PBS and the nucleus was counter-stained with 4',6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, St. Louis, MO). The sections were then mounted in the antifade medium (Vectashield; Vector Laboratories, Burlingame, CA) and viewed on a laser scanning confocal microscope (model LSM 510; Carl Zeiss Meditec, Inc., Jena, Germany).
The dilutions of primary antibodies were: 1:200 for the rabbit anti-NF-κB antibody (Abcam, Cambridge, MA), 1:300 for the rat anti mouse anti-CD31 (BDpharmingen, San Jose, CA) antibody, 1:1000 for mouse anti-α-smooth muscle actin (SMA) antibody (Sigma, St. Louis, MO) and 1:1000 for the mouse anti-HIF-1α antibody (Abcam, Cambridge, MA).
The secondary antibodies were FITC (or Texas Red)-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Laboratory, Inc., West Grove, PA), Fluorescein anti-rat IgG with mouse adsorbed (Vector Laboratories, Burlingame, CA) (or Texas Red-conjugated goat anti-rat IgG, Invitrogen, Carlsbad, CA ) and Texas Red-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratory, Inc., West Grove, PA) at the dilution of 1:200.
Electroretinogram (ERG) recoding
To determine if the photoreceptor function is impaired in Vldlr−/− mice, the photopic and scotopic ERG were recorded in Vldlr−/− mice at ages of 3, 4, 5, 8, 10 and 32 weeks. Mice were dark adapted for at least 12 h. The mice were anesthetized, and pupils were dilated with topical application of 2.5% phenylephrine and 1% tropicamide. ERG responses were recorded with a silver chloride needle electrode placed on the surface of the cornea after 1% tetracaine topical anesthesia. A reference electrode was positioned at the nasal fornix, and a ground electrode on the foot. The duration of light stimulation was 10 mSec. The band pass was set at 0.3 to 500 Hz. Fourteen responses were recorded and averaged, with flash intervals of 20 sec. For quantitative analysis, the B-wave amplitude was measured between A-and B-wave peaks. The ERG waveforms of both eyes in the same animal were simultaneously recorded and compared as the right-to-left–eye ratio of B wave amplitude.
Analysis of endogenous retinoids in the mouse eyecup
The eyecups (including the retina and RPE) from each mouse were combined and homogenized in 200 µL of PBS in a glass minigrinder. Retinoids were extracted under dim red light following a published procedure (Moiseyev et al., 2005). After the addition of 300 µL cold methanol and 60 µL 1 M hydroxylamine in 0.2 M sodium phosphate buffer (pH 7.0), the resultant suspension was thoroughly mixed (vortexed) for 30 sec, and 300 µL of dichloromethane were added and vortexed for another 30 sec. The solution was centrifuged at 10,000 ×g for 5 min. The lower organic layer was collected with a Pasteur pipette. The residual suspension was extracted once more with an equal volume of dichloromethane. The combined organic layer was evaporated with oxygen-free argon. The residue was dissolved in 200 µL of HPLC mobile phase and applied to the HPLC column. The HPLC separation of retinoids and peak analyses were performed as described (Moiseyev et al., 2005).
Vascular permeability assay
Retinal vascular permeability was quantified by measuring FITC-albumin leakage from blood vessels into the retina following a documented method (Lip et al., 2001) with modifications. Animals were anesthetized and FITC-labeled albumin (Sigma, St. Louis, MO) injected through the femoral vein (10 mg/kg body weight) under microscopic inspection. After the injection, the animals were kept on a warm pad for 2 h to ensure the complete circulation of FITC-labeled albumin. Then the chest cavity was opened, and blood collected through the right atrium. The mice were perfused via the left ventricle with PBS (pH 7.4), which were pre-warmed to 37°C to prevent vasoconstriction. Immediately after perfusion, the eyes were enucleated, and the retinae carefully dissected under an operating microscope. The fluorescein-albumin was extracted by sonication and centrifugation. The fluoresce intensity of FITC-albumin from the supernatant and serum was measured at excitation wavelength of 485 nm and emission wavelength of 530 nm. Retinal protein levels were measured by A280. FITC-albumin levels in the retina were normalized by serum fluoresce density and total retinal protein concentrations.
Leukostasis assay
The assay was performed following a documented protocol (Ishida et al., 2003). Briefly, anesthetized mice were perfused with PBS to remove non-adherent leukocytes in vessels. The adherent leukocytes in the vasculature were stained by perfusion with a Cy3-conjugated antibody specific for CD45, and vascular endothelial cells stained by FITC-conjugated concanavalin-A (Con-A, 40 µg/ml). The retinae were then flat-mounted and adherent leukocytes in the vasculature were counted under a fluorescent microscope.
ELISA for TNF-α and soluble ICAM-1
The eyecups or retinae were homogenized and centrifuged at 3000 rpm for 3 min. The total protein concentration in the supernatant was measured using the bicinchoninic acid protein assay reagent kit (Pierce). TNF-α and soluble ICAM-1 protein levels were measured using ELISA kits (R&D Systems Inc. Minneapolis, MN) according to the manufacturer's instructions and normalized by total protein concentrations in the retina.
Statistical analysis
All of the quantitative data were analyzed and compared between the wt and Vldlr−/− mice using unpaired Student’s t tests. Statistical significance was set at p<0.05.
RESULTS
Impaired cone functions and cone degeneration in Vldlr−/− mice at early ages
To determine if the photoreceptor function is impaired in Vldlr−/− mice, the photopic and scotopic ERG were recorded in Vldlr−/− mice at different ages and age-matched wt mice. Compared with age-matched wt mice, Vldlr−/− mice exhibited a significantly suppressed a wave in photopic ERG starting at 10 weeks, to a level approximately 40% of that in age-matched wt mice (P<0.05, Fig. 1 A–D), suggesting a compromised cone functions. In contrast, the scotopic ERG did not show significant change until age 32 weeks in Vldlr−/− mice. At all the ages analyzed, the b wave amplitudes of both scotopic and photopic ERG were not significantly changed in the Vldlr−/− mice (Fig. 1C&D).
Figure 1. Declined cone ERG responses and decreased cone densities in Vldlr−/− mice.
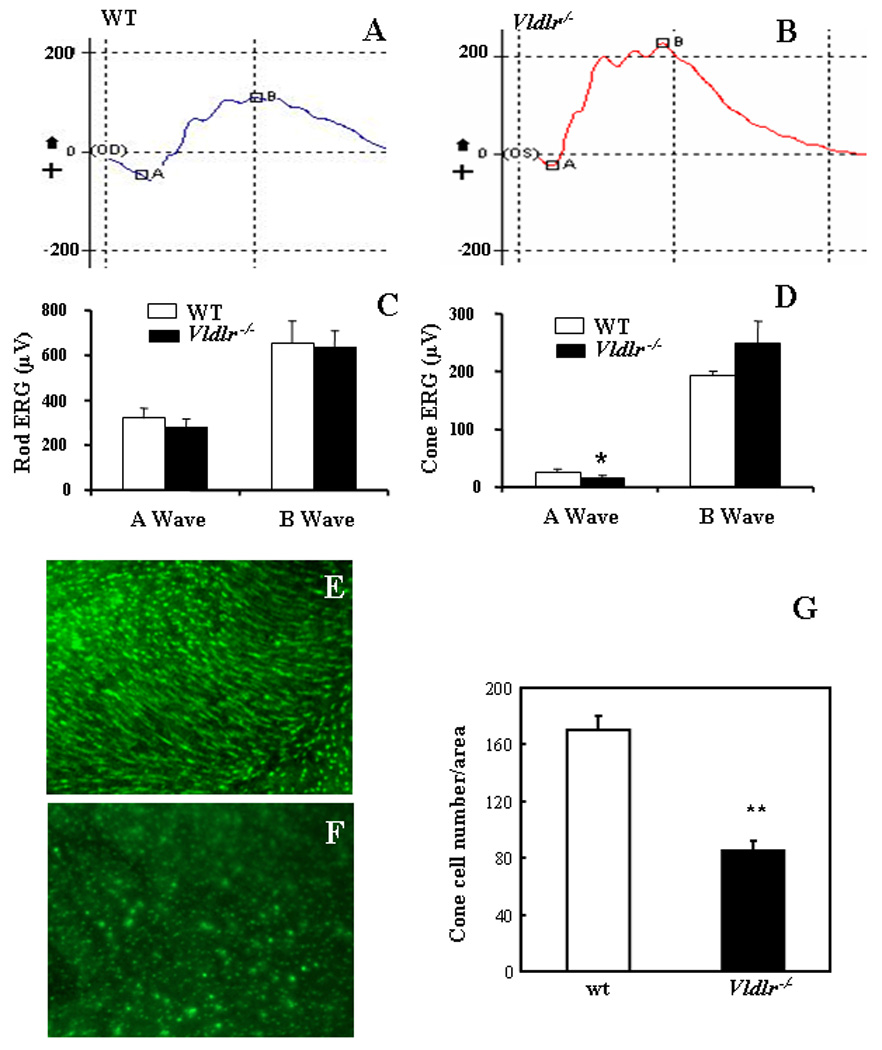
(A&B) Representative photopic ERG waveforms from wt (A) and Vldlr−/− (B) mice showed an apparently declined a wave in Vldlr−/− mice. (C&D) Amplitudes of a and b waves (mean±SD) showing that there is no significant difference in a wave or b wave in scotopic ERG between wt and Vldlr−/− mice (C), while the a wave amplitude in the photopic ERG was significantly lower in Vldlr−/− mice at age of 10 weeks than in age-matched wt mice (mean±SD, n=5) (D). (E&F) Whole-mounted retinae from wt and Vldlr−/− mice at age of 3 months were stained with an antibody specific for the mid-wavelength (MWL) cone opsin. The MWL cone density in the central retina of Vldlr−/− mice (F) was apparently lower, compared to that in the same region of age-matched wt mice (E). The cone outer segments were shorter in Vldlr−/− mice than in wt mice (E&F). (G) The cone density was quantified and presented as number of cones per 100×100 µm area in the retina (mean±SD, n=8). *P<0.05; ** P<0.01.
To further determine the morphology and density of cone photoreceptors in Vldlr−/− mice, the flat-mounted retinae were stained with an antibody specific for MWL cone opsin which is located predominantly in the outer segment. In the central retina, the cone density was apparently decreased in Vldlr−/− mice at age of 4 months, compared to that in the same area in wt mice at the same age (Fig. 1E&F). To quantify the cone density, the stained cones were counted in 10 random 100×100 µm areas per retina and presented as cones per area of (Fig. 1G). In addition to the reduced cone density, the cone outer segment was also apparently shortened in Vldlr−/− mice, compared to that in wt mice, suggesting cone degeneration at the early age in Vldlr−/− mice (Fig. 1E&F).
Progressive retinal degeneration in Vldlr−/− mice
Plastic retinal cross sections were stained with toluidine blue to compare the thickness of the outer nuclear layer (ONL). At age of 3 months, the ONL was apparently thinner in Vldlr−/− mice than that in age-matched wt mice (Fig. 2 A&B). To quantify the loss of photoreceptors, the thickness of the ONL were measured on cross sections and averaged. The results showed that Vldlr−/− mice had significantly decreased thickness of ONL than that in wt (P<0.01, n=7, Fig. 2C).
Figure 2. Retinal degeneration, disturbed retinoid profile and decreased rhodopsin levels in Vldlr−/− mice.
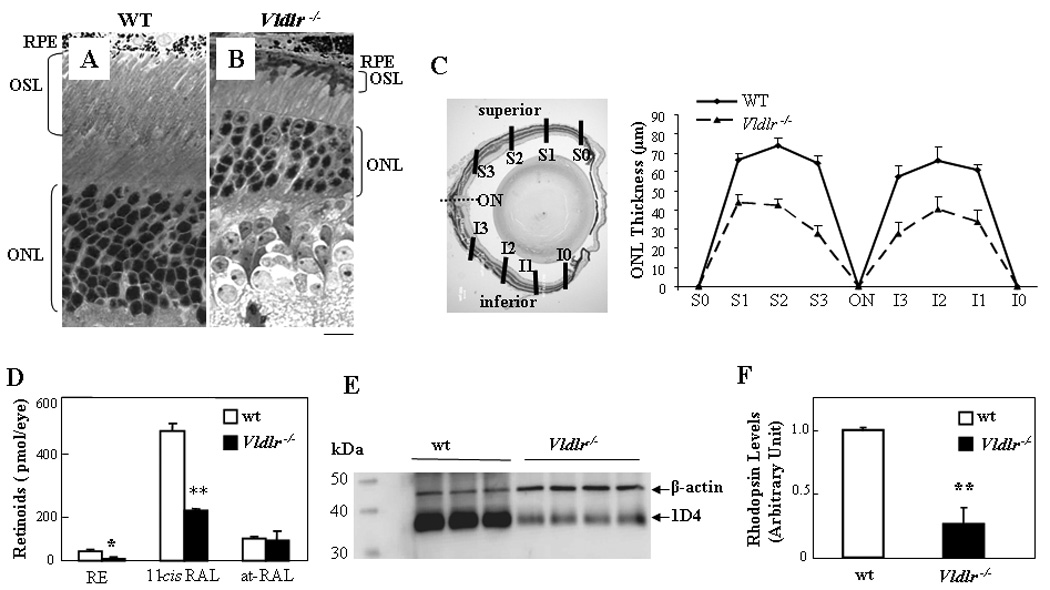
(A&B) Representative retinal cross sections from wt (A) and Vldlr−/− (B) mice at age of 3 months stained with toluidine blue. Note that an apparently thinner ONL in Vldlr−/− mice. Scale bar, 20 µm. (C) The thickness of the ONL were measured at six locations of each retina, at 25% (S3 or I3), 50% (S2 or I2), and 75% (S1 or I1) of the distance between the superior pole (S) or inferior pole (I) and the optic nerve (ON) and averaged (mean±SD, n=7, **P<0.01). (D) Eyecups containing RPE and retina were dissected from dark-adapted wt and Vldlr−/− mice at age of 8 months. Retinoids were extracted from the retina and RPE and analyzed by HPLC. Each form of retinoids was identified by its characteristic elution time and comparison with respective standards, and the amounts quantified (mean±SD, n=4). The amounts of 11-cis retinal (11-cis RAL) and total retinyl ester (RE), but not the all-trans retinal (at-RAL) were significantly lower in Vldlr−/− eyecups than that in wt (**P<0.001 and *P<0.05, respectively). (E) Western blot analysis of rhodopsin at age of 8 months. The same amount (5 µg) of retinal protein from each mouse was blotted with the 1D4 antibody and an antibody specific for β-actin (each lane represents an individual mouse). Each lane represents an individual mouse. (F) Rhodopsin levels normalized by β-actin levels as quantified by densitometry were significantly decreased in Vldlr−/− mice, to approximate 20% of that in wt mice (mean±SD, n=4, **P<0.01).
Disturbed retinoid visual cycle and decreased rhodopsin levels in Vldlr−/− mice
To further confirm the photoreceptor degeneration, we have measured visual pigment levels. Since 11-cis retinal is the chromorphore for both the rod and cone visual pigments, we compared the abundance of each form of retinoid in the retina and RPE of Vldlr−/− mice with those in the age-matched wt mice. Endogenous retinoids were extracted from the eyecups of dark-adapted mice and analyzed by HPLC as described previously (Moiseyev et al., 2005). As quantified by HPLC analysis, the Vldlr−/− mouse retina and RPE contained significantly lower amounts of 11-cis retinal and retinyl esters compared to the wt mice at the same age (Fig. 2D), suggesting decreased visual pigments in Vldlr−/− mice. In contrast, all-trans retinal levels were not significantly changed in Vldlr−/− mice.
To evaluate the extent of rod degeneration in late ages, rhodopsin levels in the retina were compared between the wt and Vldlr−/− mice at age of 8 months by Western blot analysis using 1D4, a monoclonal antibody against rhodopsin. The rhodopsin levels after normalization by β-actin levels were approximately 5 folds lower in Vldlr−/− mice than in age-matched wt mice (Fig. 2E&F).
Retinal vascular leakage in Vldlr−/− mice
After infusion of FITC-BSA, the wt mice retina showed FITC-BSA only in the retinal vasculature, while Vldlr−/− mice showed apparent leakage of the FITC-BSA into the retina tissue parenchyma at both 5 min and 1 hr after the infusion of FITC-BSA (Fig. 3A–D). As shown by permeability assay with FITC-BSA as a tracer, vascular permeability in the Vldlr−/− retina was approximately 2-fold higher over that in the wt retina (Fig. 3E). Western blot analysis showed that retinal levels of ZO-1, a tight junction protein, were significantly decreased in Vldlr−/− mice (Fig. 3F), further supporting the breakdown of the blood-retinal barrier or increased vascular permeability in Vldlr−/− mice.
Figure 3. Retinal vascular leakage in Vldlr−/− mice.
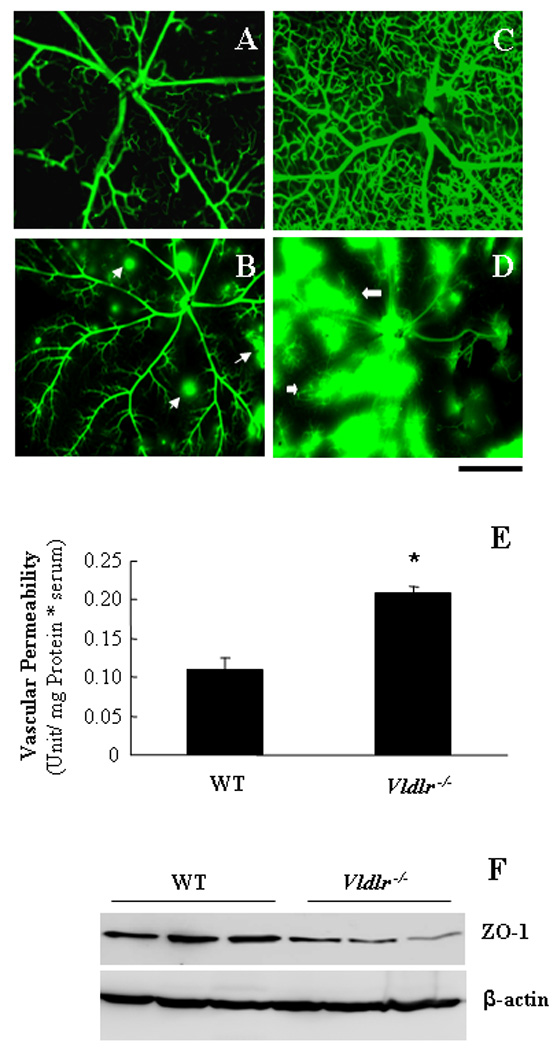
(A–D) Wt (A&C) and Vldlr−/− (B&D) mice at age of 6 weeks were infused with 10 mg/ml of FITC-BSA into ascending aorta. The retina was dissected 5 min (A&B) or 1 hr (C&D) following the FITC-BSA infusion, fixed immediately and flat-mounted. Scale bar, 50 µm. (E) Retinal vascular leakage was quantified by vascular permeability assay using FITC-BSA as a tracer (mean±SD, n=7). Note that Vldlr−/− mice had a significant increase of retinal vascular permeability, compared to the age-matched wt mice (*P<0.01). (F) Levels of ZO-1 in the retina were measured by Western blot analysis and normalized by β-actin, demonstrating decreased ZO-1 levels in the retina of Vldlr−/− mice age of 6 weeks. Each lane represents an individual mouse.
Vldlr gene knockout impairs the vascular maturity and integrity in the retina
To examine the integrity and maturity of the retinal vasculature, pericyte coverage of the capillaries in the retina and sub-retinal space was examined by double immunostaining of CD31 (endothelial marker) and SMA (pericyte marker). In the inner retina of wt mice (6 wks of age), CD31-positive endothelial cells were accompanied by SMA-positive pericytes, demonstrating maturity of retinal vasculature at this age. In contrast, the retinal and sub-retinal neovasculature in Vldlr−/− mice at the same age showed only CD31 staining but not the SMA signal, suggesting that endothelial cells are not covered by pericytes in the neovasculature in the sub-retinal space of Vldlr−/− mice (Fig. 4A&B).
Figure 4. Impaired vascular maturity and integrity in Vldlr−/− mice.
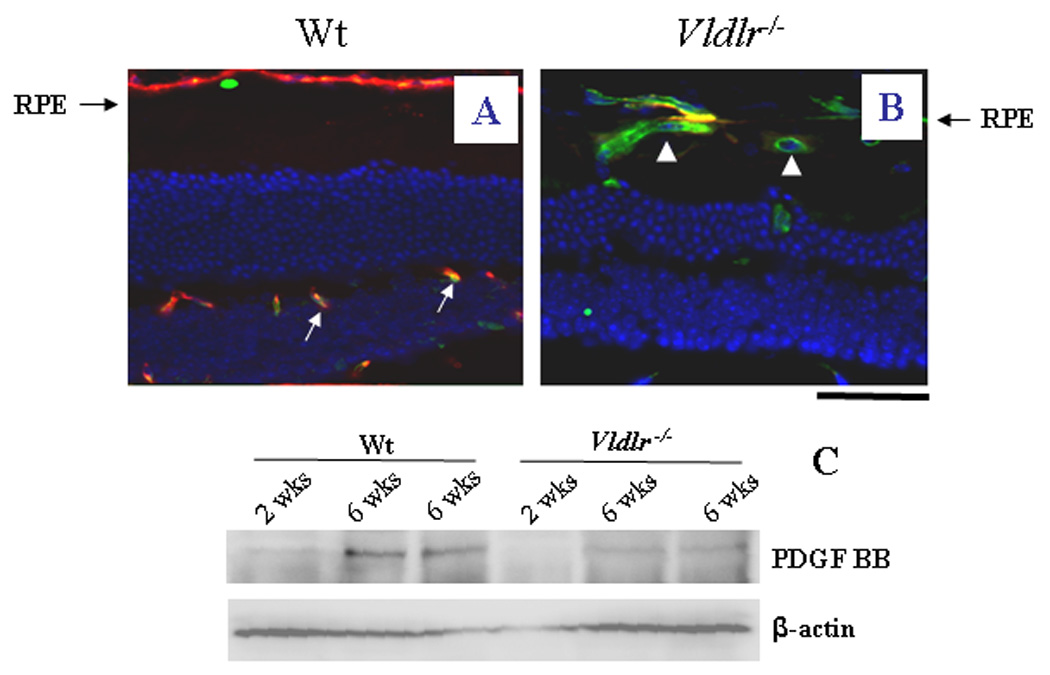
(A&B) Cross eye sections from wt (A) and Vldlr−/− (B) mice at age of 6 weeks were double stained with an antibody specific for CD31, an endothelial cell marker (green) and an antibody for SMA, a pericyte marker (red). In the inner retinal layer of wt mice, endothelial cells are covered by pericytes (orange color in merged image in A), while in the subretinal vasculature in the Vldlr−/− retina, most endothelial cells are not accompanied by pericytes (indicated by arrows in B). Scale bar, 100 µm. (C) PDGF levels in the retina were measured by Western blot analysis in wt and Vldlr−/− mice (2 and 6 wks of age) and normalized by β-actin levels, demonstrating the lack of PDGF in the Vldlr−/− retina at both 2 and 6 wks of age (each lane represents an individual mouse).
The premature vasculature was further investigated by Western blot analysis of platelet-derived growth factor–BB (PDGF- BB), a survival factor for retinal pericytes. At 2 weeks of age, both wt and Vldlr−/− mice showed nearly undetectable levels of PDGF, which correlates with the lack of pericyte coverage in the retina. At 6 weeks of age, PDGF levels in the retina were significantly increased in wt mice, correlating with maturation of retinal vasculature in wt mice. In contrast, PDGF levels remained at low levels in Vldlr−/− mice, similar to that of 2 weeks of age, suggesting that knockout of the VLDLR delays the maturation of blood vessels in the retina (Fig. 4C).
Leukostasis in Vldlr−/− mice
As retinal inflammation is a characteristic feature of AMD, we have examined the adherent leukocytes in the retinal microvasculature (leukostasis) in Vldlr−/− mice. The retinal leukostasis was examined by double staining of vascular endothelium and adherent leukocytes with FITC-conjugated Con-A and a Cy3-labeled antibody against CD45, a leukocyte marker, respectively. After the circulating leukocytes were removed by perfusion, the adherent leukocytes were visualized in the flat-mounted retina under a fluorescent microscope. Multiple adherent leukocytes were observed in the retinal vasculature of Vldlr−/− mice but not in the wt mice (Fig. 5A&B). Quantification of leukocytes showed that adherent leukocytes were increased approximately 10-fold in the Vldlr−/− retina, over that in wt mice (P<0.001, Fig. 5C). Western blot analysis showed that levels of CD45 were significantly elevated in the retina of Vldlr−/− mice after perfusion (Fig. 5D), providing further evidence of increased leukocytes in the Vldlr−/− retina.
Figure 5. Leukostasis in the Vldlr−/− retina.
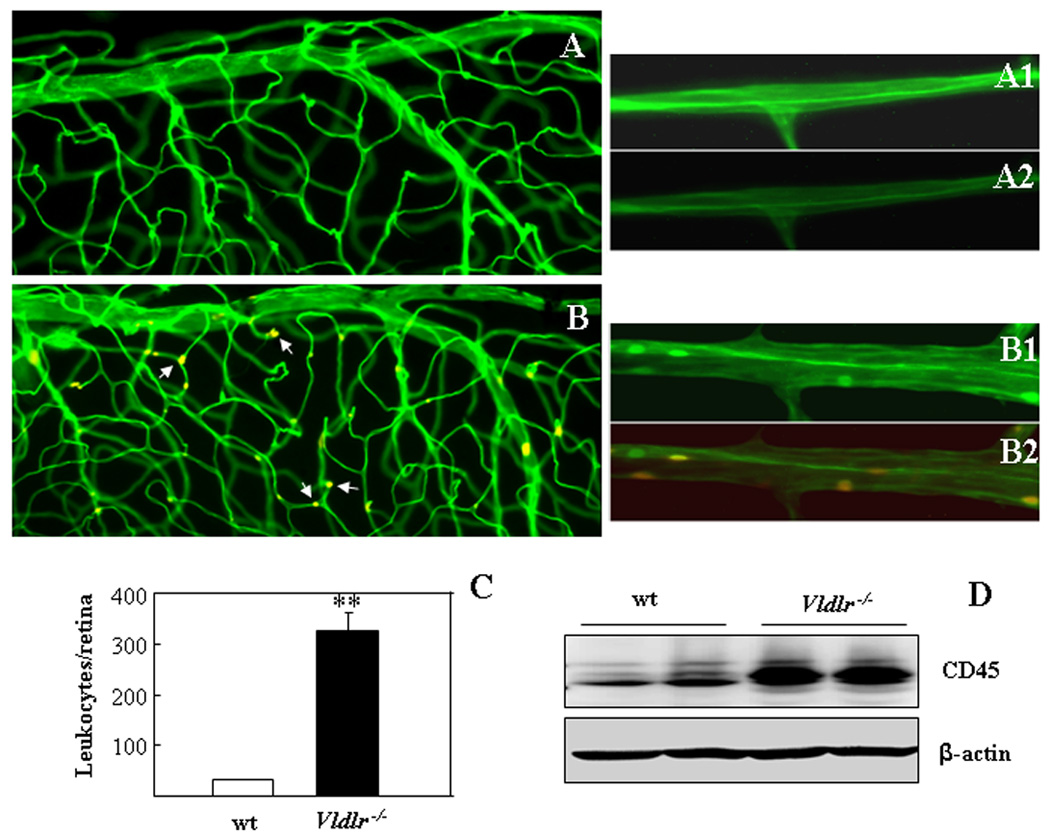
(A–C) Retinal vascular endothelium and adherent leukocytes were double stained with FITC-conjugated Con-A (green) and Cy3-conjugated CD45 (red) in 2 month-old wt (A) and Vldlr−/− (B) mice after the circulating leukocytes were removed by perfusion. The retinae were then flat-mounted, and the adherent leukocytes visualized under fluorescent microscope. Multiple leukocytes adherent to endothelium of retinal vasculature (yellow color in merge image as indicated by arrows) were observed in the Vldlr−/− retina (B,B1,B2) but not in the wt retina (A,A1,A2). (A1, B1) images of Con-A staining only; (A2, B2) superimposed images of the Con-A and CD45 double staining. Scale bar, 50 µm. (C) Adherent leukocytes were counted in the whole retinae of wt and Vldlr−/− mice (mean±SD, n=7), showing significantly increased adherent leukocytes in the Vldlr−/− retina (**P<0.01). (D) Leukocytes in the retina were measured by CD45 levels using Western blot analysis in wt and Vldlr−/− mice (6 wks of age) after perfusion and normalized by β-actin levels. Each lane represents an individual mouse.
Elevated pro-inflammatory factor levels in Vldlr−/− mice
As leukostasis is known to be mediated by ICAM-1, soluble ICAM-1 levels in the retina and RPE were quantified by ELISA, which showed that soluble ICAM-1 levels in Vldlr−/− eyecups were increased by approximate 2-fold over that in wt mice (Fig. 6A). Similarly, other inflammatory factors including TNF-α, endothelial nitric oxide synthase (eNOS) and cyclooxygenase-2 (COX2) were significantly up-regulated in the retina and RPE of Vldlr−/− mice, as measured by ELISA and Western blot analysis (Fig. 6B&C).
Figure 6. Elevated levels of pro-inflammatory factors in the Vldlr−/− retina.
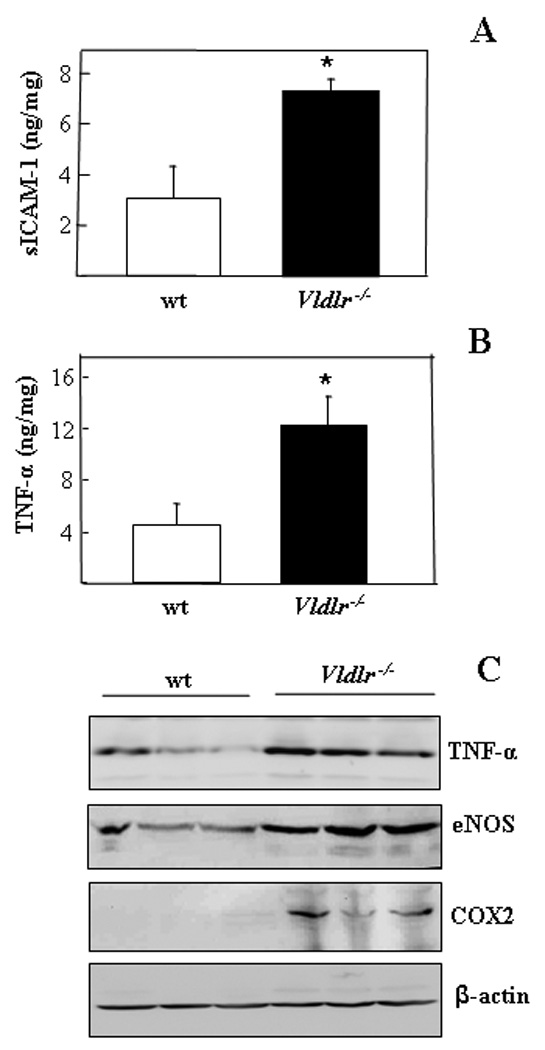
(A&B) The soluble ICAM-1 and TNF-α in the eyecup from 2 month-old wt and Vldlr−/− mice were measured by ELISA, and normalized by total protein concentrations in the eyecup, and expressed as ng/mg of total proteins (mean ± SD, n = 3). *P < 0.05. (C) Equal amounts (50 µg) of total proteins from the retina and RPE of Vldlr−/− mice and age-matched wt mice were separately blotted with antibodies against TNF-α, eNOS, COX2 and β-actin. Each lane represents an individual mouse.
Activated NF-κB and HIF-1α in the retina and RPE of Vldlr−/− mice
To further define the signaling pathways mediating the retinal inflammation in Vldlr−/− mice, we have measured the activation of HIF-1 and NF-κB, key transcription factors mediating inflammatory responses. As shown by Western blot analysis and immunohistochemistry, the VLDLR deficiency resulted in elevated levels of NF-κB and nuclear translocation in the RPE in Vldlr−/− mice (Fig. 7A–F). Similarly, the up-regulation of HIF-1α was detected in the RPE and sub-retinal space (Fig. 7G&H). Western blot analysis confirmed that both the NF-κB and HIF-1α levels were elevated in the eyecups of Vldlr−/− mice (Fig. 7I)
Figure 7. Up-regulation and activation of NF-κB and HIF-1α in Vldlr−/− mice.
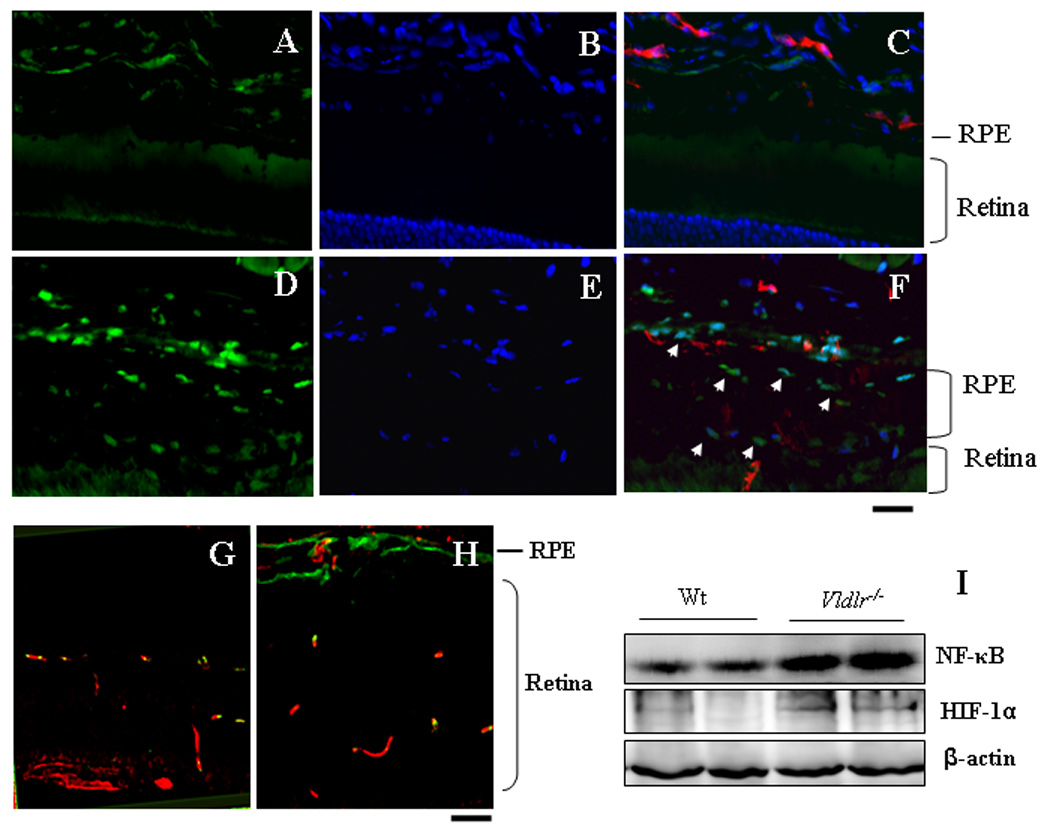
(A&B) The cross eye sections from 3 month-old wt (A–C) and Vldlr−/− mice (D–F) were double stained with antibodies for rabbit anti-NF-κB (green) and for rat anti-CD31 (red). The nuclei were counterstained with DAPI (blue). (A&D) NF-κB signal; (B&E) DAPI staining and (C&F) merged images. Note that increased NF-κB signal in the nuclei in the RPE and choroid of Vldlr−/− mice. (G&H) Cross eye sections from wt (G) and Vldlr−/− (H) mice were double stained with antibodies specific for CD31 (red) and HIF-1α (green), showing elevated HIF-1α levels in the sub-retinal space of Vldlr−/− mice. Scale bar, 50 µm. (I) Levels of NF- κB and HIF-1α in the retinae were measured by Western blot analysis and normalized by β-actin. Each lane represents an individual mouse.
DISCUSSION
It has been reported that Vldlr−/− mice develop abnormal subretinal NV (Heckenlively et al., 2003). Recently, we have shown that the subretinal NV in Vldlr−/− mice is mediated through LRP6 and activation of the canonical Wnt pathway (Chen et al., 2007). In the present study, we have demonstrated that the mice deficient of VLDLR, a negative regulator of the Wnt pathway, develop some pathological features of wet AMD such as declined cone ERG response and cone degeneration prior to rod loss, retinal vascular leakage and chronic inflammation in the retina and RPE.
Progressive cone degeneration and impaired cone function have been observed in AMD patients (Eldred and Lasky, 1993). The ERG recording showed that the cone ERG a wave is depressed in Vldlr−/− mice at as early as 10 weeks of ages, suggesting impaired cone functions at early ages. In contrast, rod ERG is not significantly changed until 32 weeks of ages. Immunostaining of MWL cones showed decreased cone densities in the central retina and shortened cone outer segments, providing structural evidence of cone degeneration in Vldlr−/− mice. In older Vldlr−/− mice, retinoid analysis demonstrated decreased levels of 11-cis retinal, the chromophore for both rod and cone pigments, suggesting a decreased visual pigment formation in Vldlr−/− mice. Consistent with the decreased 11-cis retinal, rhodopsin levels were lower in Vldlr−/− mice than in wt mice of the same age. Further, Vldlr−/− mice showed decreased thickness of the photoreceptor layer at older ages. The decreased chromophore, rhodopsin and photoreceptors indicate progressive photoreceptor degeneration in Vldlr−/− mice. Although the pathogenesis of early cone degeneration and later both rod and cone loss in Vldlr−/− mice is uncertain at the present time, the retinal vascular leakage and inflammation, and consequent local ischemia may contribute to photoreceptor degeneration in Vldlr−/− mice.
Vascular leakage from the subretinal neovasculature is believed to be responsible for retinal edema in wet AMD (Witmer et al., 2003). Both fluorescein retinal angiography and retinal vascular permeability assay using FITC-BSA as a tracer showed significant vascular leakage in Vldlr−/− mice. The tight junction between endothelial cells is essential for the intact blood-retinal barrier. Our results showed that ZO-1, a tight junction protein, is significantly down-regulated in Vldlr−/− eyecups, providing a structural evidence of the blood-retinal barrier breakdown in Vldlr−/− mice. As shown in our previous studies, VEGF is over-expressed in Vldlr−/− retina (Chen et al., 2007). The VEGF over-expression may contribute to the retinal vascular leakage in Vldlr−/− mice.
Pericyte coverage is an important step in vascular maturation (Baffi et al., 2000; Dosso et al., 1999; Israely et al., 2003; Martin et al., 2005). Different from the mature retinal vasculature, in which endothelial cells are covered by mural cells (pericytes and vascular smooth muscle cells), the neovasculature in the sub-retinal space of Vldlr−/− mice lacks functional mural cells, which may also contribute to the immaturity and instability of blood vessels. Although the blood-retinal barrier primarily relies on the tight junction between endothelial cells, recent studies showed that functional pericytes also play an important role in maintaining the functional integrity of the blood-retinal barrier (Martin et al., 2005). Therefore, the lack of pericytes may contribute to the vascular leakage in Vldlr−/− mouse retina.
PDGF is known to play a crucial role in pericyte recruitment and survival during the vessel development and maturation (Marx et al., 1994; Risau et al., 1992). Through promoting the association of pericytes with endothelial cells, PDGF stabilizes the newly formed blood vessels, which in turn limits the proliferation of endothelial cells and inhibits angiogenesis. In the retina of early ages (2 weeks), PDGF levels are nearly undetectable in both wt and Vldlr−/− mice, suggesting pre-maturity of retinal vasculature at this age. At age of 6 weeks, PDGF levels increase significantly in wt mouse retina, consistent with vascular maturation and pericyte coverage of retinal vasculature. In Vldlr−/− mice, however, there is no detectable PDGF in the retina at both 2 and 6 weeks of age, suggesting that down-regulated expression of PDGF may be responsible for the lack of pericyte coverage in Vldlr−/− mice. These findings suggest that the maturation of vasculature is impaired or delayed in the Vldlr−/− retina.
Chronic inflammation is believed to play a pathogenic role in diabetic retinopathy and AMD (Green and Enger, 1993; Kern, 2007). Our results demonstrate that Vldlr−/− mice have significantly increased leukostasis and elevated levels of pro-inflammatory factors, indicating inflammation in the retina and RPE in Vldlr−/− mice. Increased leukostasis or adherent leukocytes in the retinal vasculature may impair endothelium and contribute to vascular leakage. Leukostasis may also cause capillary closure and lead to local ischemia, which subsequently induces VEGF expression and contributes to CNV. Increased expression of ICAM-1 and TNF-α in the eyecups of Vldlr−/− mice may be responsible for the increased leukostasis in Vldlr−/− mice.
Our previous studies have shown that VLDLR functions like a negative regulator of the Wnt signaling pathway, as VLDLR knockout results in up-regulation of the Wnt co-receptors, LRP5/6, leading to activation of the canonical Wnt pathway (Chen et al., 2007). The VLDLR knockout-induced activation of the Wnt pathway is responsible, at least in part, for the VEGF over-expression and CNV in Vldlr−/− mice (Chen et al., 2007). As the Wnt pathway is known to mediate inflammatory responses (Lee et al., 2006), the activated Wnt signaling is likely to be responsible for the chronic inflammation in the retina of Vldlr−/− mice. It has been reported that NF-κB, a key regulator of inflammation (Gordon et al., 2005), is also regulated by the Wnt pathway (De Toni et al., 2006; Hoeflich et al., 2000). Our results demonstrate an increased nuclear translocation of NF-κB, a key step in its activation, in Vldlr−/− retina and RPE. The activation of NF-κB mediated by the Wnt pathway may play a key role in the inflammation in Vldlr−/− mice. The present study suggests that abnormal activation of the Wnt pathway induced by deficiency of VLDLR may be responsible for retinal inflammation in this mouse model.
Table 1.
A and B Wave Amplitudes of Rod ERG in Vldlr−/− Mice
| Age (Weeks) |
A wave |
p value (n=10) |
B wave |
p value (n=10) |
||
|---|---|---|---|---|---|---|
| wt | Vldlr−/− | wt | Vldlr−/− | |||
| 3 | 293.4 ± 38.8 | 269.8 ± 32.2 | p >0.05 | 604.4 ± 66.2 | 587.3 ± 54.2 | p >0.05 |
| 4 | 299.1 ± 42.6 | 278.8 ± 52.5 | p >0.05 | 597.5 ± 65.5 | 632.1± 61.8 | p >0.05 |
| 5 | 325.7 ± 54.1 | 305.2 ± 40.3 | p >0.05 | 632.6 ± 78.4.0 | 626.4 ± 82.1 | p >0.05 |
| 8 | 344.3 ± 32.5 | 298.8 ± 34.2 | p >0.05 | 600.0 ± 46.6 | 609.5± 59.6 | p >0.05 |
| 10 | 319.7 ± 43.0 | 279.6 ± 38.7 | p >0.05 | 656.4 ± 97.2 | 637.8 ± 71.2 | p >0.05 |
| 32 | 345.2 ± 47.5 | 252.6 ± 28.3 | p <0.05 | 670.5 ± 80.4 | 656.5 ± 53.4 | p >0.05 |
Table 2.
A and B Wave Amplitudes of Cone ERG in Vldlr−/− Mice
| Age (Weeks) |
A wave |
p value (n=10) |
B wave |
p value (n=10) |
||
|---|---|---|---|---|---|---|
| WT | Vldlr−/− | WT | Vldlr−/− | |||
| 3 | 22.4 ± 5.7 | 21.3 ± 5.6 | p >0.05 | 184.5 ± 12.3 | 169.6 ± 13.6 | p >0.05 |
| 4 | 23.9 ± 5.1 | 20.5 ± 4.8 | p >0.05 | 189.4 ± 22.2 | 181.5 ± 21.1 | p >0.05 |
| 5 | 26.1 ± 4.7 | 18.2 ± 3.4 | p >0.05 | 198.0 ± 13.6 | 201.9 ± 18.2 | p >0.05 |
| 8 | 25.5 ± 3.0 | 17. 9 ± 4.1 | p >0.05 | 226.4 ± 25.3 | 237.2 ± 23.6 | p >0.05 |
| 10 | 26.5 ± 4.1 | 16.2 ± 3.4 | P<0.05 | 192.5 ± 7.8 | 248.9 ± 37.8 | p >0.05 |
| 32 | 20.7 ± 3.3 | 15.2 ± 2.5 | P<0.05 | 192.0 ± 25.6 | 235.6 ± 42.5 | p >0.05 |
Acknowledgments
Supported by NIH grants EY012231 and EY015650, research award from ADA, a grant from OCAST and grant P20RR024215 from the National Center for Research Resources.
REFERENCES
- Ambati J, et al. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48:257–293. doi: 10.1016/s0039-6257(03)00030-4. [DOI] [PubMed] [Google Scholar]
- Baffi J, et al. Choroidal neovascularization in the rat induced by adenovirus mediated expression of vascular endothelial growth factor. Invest Ophthalmol Vis Sci. 2000;41:3582–3589. [PubMed] [Google Scholar]
- Bok D. Evidence for an inflammatory process in age-related macular degeneration gains new support. Proc Natl Acad Sci U S A. 2005;102:7053–7054. doi: 10.1073/pnas.0502819102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylsma G, Guymer RH. Treatment of age-related macular degeneration. Clin. Exp. Optom. 2005;88:322–334. doi: 10.1111/j.1444-0938.2005.tb06716.x. [DOI] [PubMed] [Google Scholar]
- Chen Y, et al. VLDL receptor, a negative regulator of the WNT signaling pathway in choroidal neovascularization. J Biol Chem. 2007 doi: 10.1074/jbc.M611289200. [DOI] [PubMed] [Google Scholar]
- Croll SD, et al. VEGF-mediated inflammation precedes angiogenesis in adult brain. Exp Neurol. 2004;187:388–402. doi: 10.1016/j.expneurol.2004.02.010. [DOI] [PubMed] [Google Scholar]
- De Toni F, et al. A crosstalk between the Wnt and the adhesion-dependent signaling pathways governs the chemosensitivity of acute myeloid leukemia. Oncogene. 2006;25:3113–3122. doi: 10.1038/sj.onc.1209346. [DOI] [PubMed] [Google Scholar]
- Dosso AA, et al. Remodeling of retinal capillaries in the diabetic hypertensive rat. Invest Ophthalmol Vis Sci. 1999;40:2405–2410. [PubMed] [Google Scholar]
- Dvorak HF. VPF/VEGF and the angiogenic response. Semin Perinatol. 2000;24:75–78. doi: 10.1016/s0146-0005(00)80061-0. [DOI] [PubMed] [Google Scholar]
- Edwards AO, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- Eldred GE, Lasky MR. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature. 1993;361:724–726. doi: 10.1038/361724a0. [DOI] [PubMed] [Google Scholar]
- Gong Y, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- Gordon MD, et al. WntD is a feedback inhibitor of Dorsal/NF-kappaB in Drosophila development and immunity. Nature. 2005;437:746–749. doi: 10.1038/nature04073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman Lecture. Ophthalmology. 1993;100:1519–1535. doi: 10.1016/s0161-6420(93)31466-1. [DOI] [PubMed] [Google Scholar]
- Haines JL, et al. Functional candidate genes in age-related macular degeneration: significant association with VEGF, VLDLR, and LRP6. Invest Ophthalmol Vis Sci. 2006;47:329–335. doi: 10.1167/iovs.05-0116. [DOI] [PubMed] [Google Scholar]
- Heckenlively JR, et al. Mouse model of subretinal neovascularization with choroidal anastomosis. Retina. 2003;23:518–522. doi: 10.1097/00006982-200308000-00012. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, et al. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Hu W, et al. Expression of VLDLR in the retina and evolution of subretinal neovascularization in the knockout mouse model's retinal angiomatous proliferation. Invest Ophthalmol Vis Sci. 2008;49:407–415. doi: 10.1167/iovs.07-0870. [DOI] [PubMed] [Google Scholar]
- Ishida S, et al. VEGF164 is proinflammatory in the diabetic retina. Invest Ophthalmol Vis Sci. 2003;44:2155–2162. doi: 10.1167/iovs.02-0807. [DOI] [PubMed] [Google Scholar]
- Israely T, et al. Vascular remodeling and angiogenesis in ectopic ovarian transplants: a crucial role of pericytes and vascular smooth muscle cells in maintenance of ovarian grafts. Biol Reprod. 2003;68:2055–2064. doi: 10.1095/biolreprod.102.011734. [DOI] [PubMed] [Google Scholar]
- Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RJ, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeima K, et al. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J Cell Physiol. 2007;213:809–815. doi: 10.1002/jcp.21152. [DOI] [PubMed] [Google Scholar]
- Lee DK, et al. Activation of the canonical Wnt/beta-catenin pathway enhances monocyte adhesion to endothelial cells. Biochem Biophys Res Commun. 2006;347:109–116. doi: 10.1016/j.bbrc.2006.06.082. [DOI] [PubMed] [Google Scholar]
- Li C, et al. Biochemical alterations in the retinas of very low-density lipoprotein receptor knockout mice: an animal model of retinal angiomatous proliferation. Arch Ophthalmol. 2007;125:795–803. doi: 10.1001/archopht.125.6.795. [DOI] [PubMed] [Google Scholar]
- Lip PL, et al. Age-related macular degeneration is associated with increased vascular endothelial growth factor, hemorheology and endothelial dysfunction. Ophthalmology. 2001;108:705–710. doi: 10.1016/s0161-6420(00)00663-1. [DOI] [PubMed] [Google Scholar]
- Martin SL, et al. Retinal vascular integrity following correction of diabetic ketoacidosis in children and adolescents. J Diabetes Complications. 2005;19:233–237. doi: 10.1016/j.jdiacomp.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Marx M, et al. Modulation of platelet-derived growth factor receptor expression in microvascular endothelial cells during in vitro angiogenesis. J Clin Invest. 1994;93:131–139. doi: 10.1172/JCI116936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto K, et al. Vascular endothelial growth factor (VEGF)-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1) Am J Pathol. 2000;156:1733–1739. doi: 10.1016/S0002-9440(10)65044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseyev G, et al. RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci U S A. 2005;102:12413–12418. doi: 10.1073/pnas.0503460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak JZ. Age-related macular degeneration (AMD): pathogenesis and therapy. Pharmacol Rep. 2006;58:353–363. [PubMed] [Google Scholar]
- Nozaki M, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103:2328–2333. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provis JM, et al. Anatomy and development of the macula: specialisation and the vulnerability to macular degeneration. Clin Exp Optom. 2005;88:269–281. doi: 10.1111/j.1444-0938.2005.tb06711.x. [DOI] [PubMed] [Google Scholar]
- Risau W, et al. Platelet-derived growth factor is angiogenic in vivo. Growth Factors. 1992;7:261–266. doi: 10.3109/08977199209046408. [DOI] [PubMed] [Google Scholar]
- Schlingemann RO. Role of growth factors and the wound healing response in age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2004;242:91–101. doi: 10.1007/s00417-003-0828-0. [DOI] [PubMed] [Google Scholar]
- Usui T, et al. VEGF164(165) as the pathological isoform: differential leukocyte and endothelial responses through VEGFR1 and VEGFR2. Invest Ophthalmol Vis Sci. 2004;45:368–374. doi: 10.1167/iovs.03-0106. [DOI] [PubMed] [Google Scholar]
- Vinores SA, et al. Vascular endothelial growth factor (VEGF), transforming growth factor-beta (TGFbeta), and interleukin-6 (IL-6) in experimental herpesvirus retinopathy: association with inflammation and viral infection. Histol Histopathol. 2001;16:1061–1071. doi: 10.14670/HH-16.1061. [DOI] [PubMed] [Google Scholar]
- Witmer AN, et al. Vascular endothelial growth factors and angiogenesis in eye disease. Prog Retin Eye Res. 2003;22:1–29. doi: 10.1016/s1350-9462(02)00043-5. [DOI] [PubMed] [Google Scholar]