

Abstract
Chemokines are a superfamily of chemotactic cytokines that play an important role in leukocyte trafficking and have been implicated as functional mediators of immunopathology in experimental autoimmune encephalomyelitis (EAE). In the present study, we investigated the role of the CCL20 receptor, CCR6, in chronic EAE. After immunization with myelin oligodendrocyte glycoprotein 35–55 in CFA, CCR6-/- mice developed a significantly more severe chronic EAE as compared to wild type immunized animals. CCR6 expression was not required by T cells to induce EAE. Measurement of peripheral T cell responses showed differences in IFN-γ and IL-17 responses between CCR6-/- and wild type mice. At the time when CCR6-/- mice showed significantly more severe chronic EAE there was a significant decrease in PD-L1-expressing mDC in the spleens and no differences in Foxp3 Treg. Furthermore, add back of mDC with increased PD-L1 expression to CCR6-/- mice reduced the severe chronic EAE disease phase to that of wild type controls. The results suggest a role for CCR6-expressing PD-L1+ mDC in regulating EAE progression.
Keywords: multiple sclerosis, chemokine, CCL20, CCR6, EAE, central nervous system
1. Introduction
Experimental autoimmune encephalomyelitis (EAE)4 is a CD4+ T cell–mediated demyelinating disease of the CNS that serves as a model for MS (Hohlfeld et al., 2000). The pathogenic mechanisms of disease development include antigen-specific T cell activation and Th1 differentiation (Krakowski and Owens, 1997) followed by T cell (Hickey et al., 1991) and macrophage migration into CNS (Cross et al., 1990; Hickey, 1991). More recently a role for IL-17 producing T cells (Th17) in disease pathogenesis has been demonstrated (Bettelli et al., 2006). EAE can be induced in mice by immunization with whole myelin proteins or peptide emulsified in CFA (Tuohy et al., 1992) or by adoptive transfer of autoreactive T cells to normal recipient mice (Kuchroo et al., 1992; Whitham et al., 1991). Macrophages activated by the effector T cell response appear to be the end stage disease effector cells inducing damage in the CNS (Brosnan et al., 1981; Dogan et al., 2008; Huitinga et al., 1990).
Chemokines are leukocyte chemoattractants that play a crucial role during pathogenesis of tissue specific inflammatory diseases by directing leukocyte recruitment and/or accumulation at sites of inflammation (Murphy et al., 2000; Zlotnik and Yoshie, 2000). Chemokines and their receptors have been implicated as functional mediators of immunopathology in EAE (Karpus et al., 2003; Karpus and Ransohoff, 1998). CCL20 (MIP-3α) is a CC chemokine that has only one known receptor, CCR6 (Baba et al., 1997; Varona et al., 1998), which is expressed on a variety of cell types including lymphocytes (Liao et al., 1999; Schutyser et al., 2003), dendritic cells (DC) (Greaves et al., 1997) and Langerhans cells (LC) (Dieu-Nosjean et al., 2000). CCL20 has been shown to be expressed by astrocytes in the CNS after EAE induction (Ambrosini et al., 2003) and in the periphery after immunization with spinal cord homogenate (Kohler et al., 2003). Anti-CCL20 antibody treatments reduced clinical disease severity but failed to prevent the induction of disease (Kohler et al., 2003). Similarly, CCR6-/- mice also have been shown to have a delay in the development of acute EAE (Liston et al., 2009). These studies suggested a role for CCL20 and its receptor in priming peripheral immune responses. More recently it was shown that EAE-inducing Th17 cells expressed CCR6 (Yamazaki et al., 2008). We sought to address the mechanism by which the CCL20 receptor, CCR6, regulated chronic EAE pathogenesis. In the present report, we demonstrate that CCR6 plays a role in the control of chronic EAE by modulating the migration of regulatory DC.
2. Materials and Methods
2.1. Animals
Female C57BL/6 (H-2b) mice were purchased from Harlan Sprague Dawley (Indianapolis, IN). CCR6-/- (H-2b) homozygous knockout mice were previously described (Cook et al., 2000). CCR6-/- mice were backcrossed 8× onto the C57BL/6 background. All mice were maintained in the Center for Comparative Medicine at Northwestern University. Mice were 6-7 wk old at the initiation of the experiment and were maintained on standard laboratory chow and water ad libitum. Animal care was provided in accordance with Northwestern University and Public Health Service policies.
2.2. Peptides, Antibodies and Flow cytometry
MOG 35–55 (MEVGWYRSPFSRVVHLYRNGK) and OVA 323-339 (LSGAVHAAHAGLAGAGR) were purchased from Peptides International. Amino acid composition was verified by mass spectrometry, and purity was >98%. mAbs to CD3 (500A2), CD4 (RM4-5), CD8a (Ly-2), CD11b (M1/70), CD11c (HL3), CD45 (Ly-5), CD274 (B7-H1 or PD-L1; MIH5), CCR6 (140706), and purified Fc Block (2.4G2), (BD PharMingen, San Diego, CA). CNS mononuclear cells were isolated by centrifugation (500 g) at 24°C from the interface of a 30–70% discontinuous Percoll (Amersham Pharmacia Biotech, Piscataway, NJ) gradient and 1×106 cells were pre-incubated with Fc Block for 15 min at 4°C in PBS containing 0.1% NaN3 and 2% BSA (Sigma-Aldrich, St. Louis, MO), followed by specific Ab staining. Data collection was performed on a FACSCalibur™ and LSR II flow cytometers using CELLQuest™ and FACs Diva software (Becton Dickinson, Franklin Lakes, NJ) according to the setup recommendations of Maecker and Trotter (Holden T. Maecker, 2006). Analysis was performed on a PC workstation using FCS Express software (DeNovo Software, Thornhill, Ontario, Canada).
2.3. Cell Culture
Single cell suspensions from spleens were isolated from mice by mechanical disruption through mesh stainless steel screens. Red blood cells in the spleen samples were lysed by hypotonic shock in Tris-NH4Cl (pH 7.3) and cells were resuspended in HBSS. The cells were cultured in 96 well microtiter plates (Corning-Costar, Acton, MA) at a density of 5×106 viable cells/ml in DMEM (Life Technologies) containing 5% FCS (HyClone, Logan UT), 2 mM L-glutamine (Life Technologies, Carlsbad, CA), 100 U/ml penicillin (Life Technologies), 100μg/ml streptomycin (Life Technologies), 0.1 M nonessential amino acids (Life Technologies) and 5×10-5M 2-ME) in the presence of 0, 0.5, 50 μM MOG35-55. Cells were incubated at 37°C in a humidified atmosphere containing 7.5% CO2.
2.4. Disease Induction and Clinical Evaluation
For actively induced EAE, mice were immunized with 200 μg of MOG35–55 emulsified in CFA containing 4 mg/ml Mycobacterium tuberculosis (Difco, Kansas City, MO) subcutaneously and given 200 ng of pertussis toxin (List Biological Laboratories) intraperitoneally on the day of and 2 days after immunization. For adoptively induced EAE, donor wild-type and CCR6-/- mice were immunized with MOG35–55 in CFA. Ten days after immunization, draining LN were harvested and cultured. Cells were cultured at 1×106 cells/ml in DMEM for 72 h with 50 μg/ml MOG35–55, 20 ng/ml rmIL-12 (R&D Systems, Minneapolis, MN) (Segal et al., 1998). 5 × 106 CD4+ T cells were transferred intravenously to recipients who received 100 ng of pertussis toxin i.p. on the day of and two days after cell transfer. Individual animals were graded according to clinical severity as follows: grade 0, no abnormality; grade 1, limp tail; grade 2, limp tail and hind limb weakness; grade 3, partial hind limb paralysis; grade 4, complete hind limb paralysis.
2.5. Histology
Evaluation of CNS inflammation was performed using standard H&E methodology as previously described (Karpus et al., 1995).
2.6. ELISPOT assay
Spleen cells were obtained from mice cultured in 96-well microtiter ELISPOT plates (Whatman Polyfiltronics, Clifton, NJ) that were coated overnight with capture antibodies to IFN-γ (BD Pharmingen, Mountain) or IL-17 (eBioscience). Total cell numbers recovered were determined by use of a hemocytometer. ELISPOT assay was performed as previously described (Karpus et al., 1996). Plates were washed and spots were counted using an ELISPOT plate reader and software (Cellular Technologies Inc., Cleveland, OH). At least three wells per sample were counted, and presented as a mean value±standard deviation.
2.7. mDC isolation and adoptive transfer
Wild type mice were immunized with MOG35-55 in CFA as described above. The control mDC population was obtained from spleens of mice 3 days after MOG33-55 priming. The PD-L1-enriched mDC population was obtained similarly from spleens of mice 21 days after priming. Enrichment for PD-L1 expression was determined by flow cytometry prior to cell transfer. A total of 5 × 107 control or PD-L1-enriched spleen cells were transferred intravenously to recipient mice prior to showing peak signs of clinical EAE.
2.8. Statistical analysis
Comparisons of disease incidence were analyzed by the χ2 analysis, using Fisher's exact probability test. Statistical significance of cytokine levels, disease onset, and disease severity was analyzed using the Student's t test for comparisons of two means. Values of P ≤0.05 were considered significant.
3 Results
To understand the role of CCR6 in EAE, C57BL/6 wild type control and CCR6-/- mice were primed with 200 μg MOG35-55 peptide emulsified in CFA and monitored for clinical disease. The results shown in Figure 1A demonstrate that wild type and CCR6-/- mice developed severe ascending hind limb paralysis. Peak disease severity and disease incidence was comparable between the wild type and CCR6-/- groups at 18 days post-immunization. However, during the chronic phase of disease, between days 21-35, the CCR6-/- mice developed a significantly more severe disease (p<0.05). This result initially suggested that CCR6 was involved in the regulation of chronic disease progression.
Figure 1. Increased chronic disease severity in CCR6-/- mice.
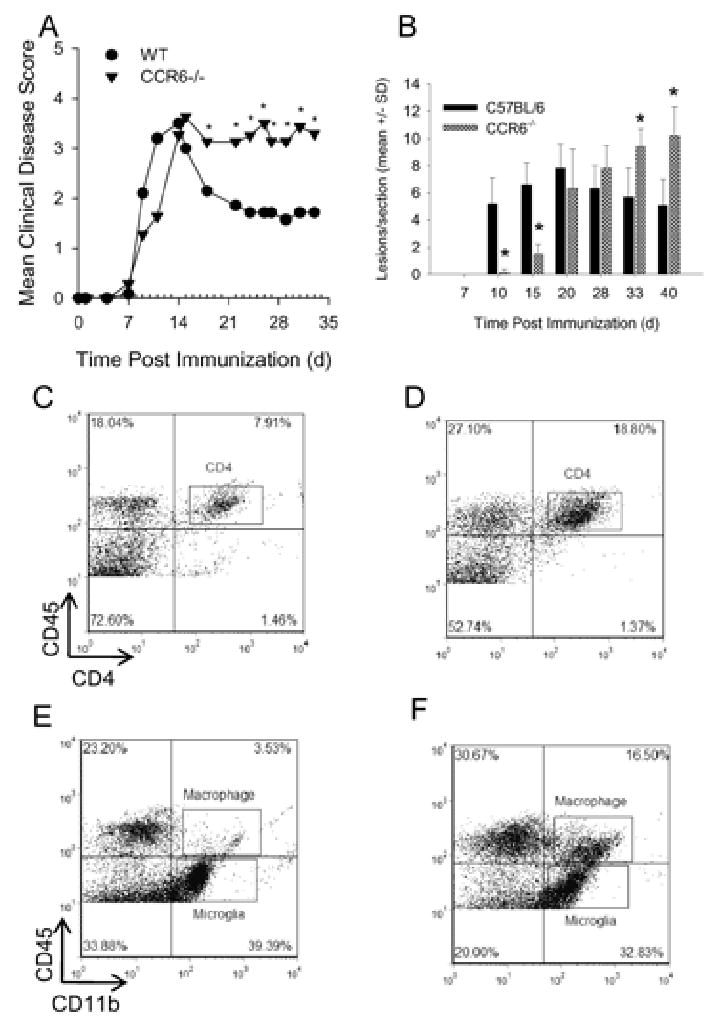
Wild type and CCR6-/- mice were immunized with MOG35-55 emulsified in CFA and monitored for clinical disease. A) CCR6-/- mice show increased chronic clinical disease severity. The data are expressed as the mean clinical disease score and are representative of three separate experiments. N=10 mice per group. *, p < 0.05 when mean clinical score was compared between CCR6-/- and wild type mice for that particular day. B) CCR6-/- mice show significantly (*, p < 0.05) increased chronic histological disease severity. The data are expressed as the mean (±SD) histologic disease score as determined from the microscopic analysis of H&E stained spinal cord sections. The data are representative of three separate experiments. N=3 mice per group.
Using CNS tissue from the previously described experiment, we examined the spinal cord histology by H&E staining. Paraffin-embedded sections were analyzed for mononuclear cell infiltrates and the number of perivascular lesions was quantified. We found that wild type mice demonstrated infiltrating mononuclear cells as early as day 10 post disease induction (Figure 1B). CCR6-/- mice had significantly (P<0.05) less infiltrate and fewer lesions at days 10 and 15, but by day 20 the spinal cords appeared identical to wild type mice with significant spinal cord white matter infiltrate (Figure 1B). During the chronic phase of disease, the CCR6-/- mice demonstrated a significantly (p<0.05) larger number of CNS white matter lesions. During chronic disease in wild type mice, a representative flow cytometric evaluation of CNS-infiltrating mononuclear cells revealed approximately 8% CD4+ T cells (Figure 1C) and 3.5% macrophages (Figure 1E) compared to 19% CD4+ T cells and 16.5% macrophages in the CCR6-/- mice that had more severe disease, while the percentages of microglia remained equivalent. These data further suggested that CCR6 expression played a role in the regulation of EAE progression.
Previous studies have demonstrated that CCR6-/- mice may have a defect in developing DTH responses (Varona et al., 2001) and a second group demonstrated that anti-CCL20 treatment had an effect on peripheral T cell priming which affected acute EAE development (Kohler et al., 2003). Moreover, a recent study suggested that CCR6 is predominantly expressed on Th17 encephalitogenic T cells (Yamazaki et al., 2008) and might be required for EAE development. To examine the role of CCR6 in peripheral T cell responses in EAE, wild type and CCR6-/- mice were primed with MOG35-55 in CFA and during chronic disease, representative animals were killed and lymphocytes from the draining lymph nodes, spleen and CNS were assessed for recall IL-17 and IFN-γ responses using ELISpot analysis. The results indicate the CCR6-/- mice had significantly (p<0.05) enhanced numbers of IL-17- (Figure 2B) and IFN-γ-producing (Figure 2E) T cells in the spleen and as well as significantly (p<0.05) enhanced numbers of IFN-γ-producing T cells in the peripheral lymph node (Figure 2D) when compared to wild type control mice. There were no significant differences between CCR6-/- and wild type control mice in terms of IL-17- or IFN-γ-producing T cells in the CNS (Figure 2C &F). CCR6-/- mice did not show any increases in either IL-17- or IFN-γ-producing T cells in any tissue compartment at earlier time points (data not shown). These data suggest that there is dysregulation of Th17 and Th1 responses during the chronic stage of EAE in the CCR6-/- mice.
Figure 2. Differences in Th17 and Th1 immune responses between CCR6-/- and wild type mice.
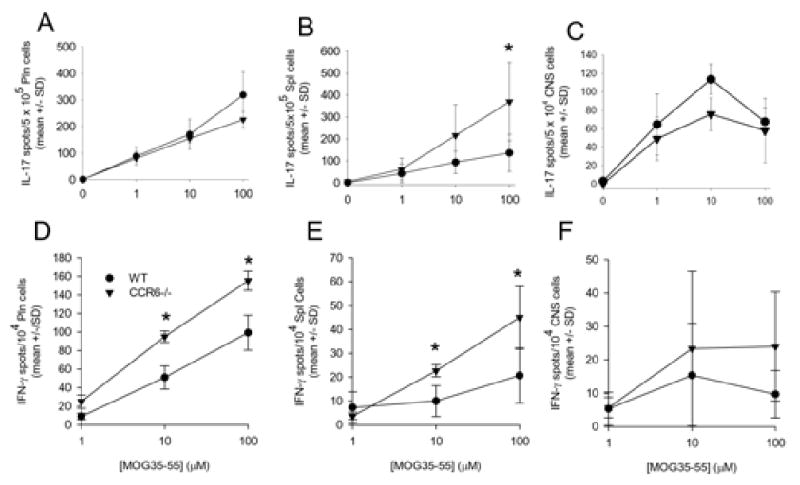
Peripheral draining lymph node cells (pln; A and D), splenocytes (spl; B and E) and CNS-infiltrating mononuclear cells (CNS; C and F) were harvested from wild type (WT) and CCR6-/- mice 21 days post immunization. Th17 and Th1 responses were determined by ELISpot. The data is shown as the mean number of cytokine-producing cells (±SD) per the number of input cells. There are statistically significant differences (*p<0.05) in IL-17- and IFN-γ-producing T cells in the spleen between wild type and CCR6-/- mice. The data is representative of two similar experiments.
To help understand if the difference in chronic disease seen in CCR6-/- mice was due to a defect in CD4+ T cell and/or macrophage/dendritic cell trafficking to the CNS we performed adoptive transfer experiments. Antigen re-stimulated CD4+ T cells from wild type donors were transferred intravenously to wild type or CCR6-/- recipients. Likewise, antigen re-stimulated CD4+ T cells from CCR6-/- donors were transferred intravenously to wild type or CCR6-/- recipients. The results in Figure 3 illustrate that wild type recipients of either MOG-specific wild type T cells or CCR6-/- T cells developed acute EAE with the same disease onset (13.6 ± 2.2 and 12.2 ± 2.7, respectively) and similar severity. These results suggest that encephalitogenic T cells do not need to express CCR6 in order to traffic to CNS and induce disease. Additionally, CCR6-/- recipients of MOG-specific wild type or CCR6-/- T cells also developed EAE with the same disease onset (13.4 ± 1.7 and 12 ± 1.5) and severity. These results suggest that host-derived T cells, macrophages and dendritic cells do not need to express CCR6 in order to traffic to CNS and induce disease.
Figure 3. CCR6 is not required for adoptively induced EAE.
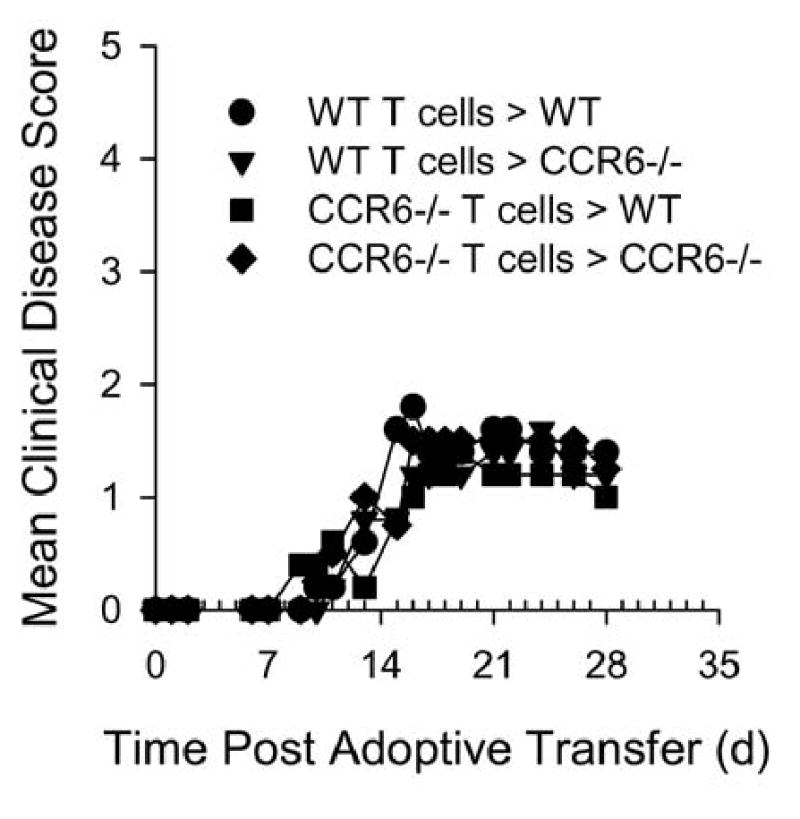
Wild type and CCR6-/- mice received primed, in vitro re-activated MOG35-55-specific CD4+ cells from wild type or CCR6-/- donors. The data are expressed as the mean clinical disease score for all mice in each group as a function of time post adoptive transfer. There are no statistical differences between any of the indicated groups. The data shown are representative of two similar experiments.
In order to explain the development of severe chronic EAE in the CCR6-/- mice, We hypothesized that CCR6 expression might affect trafficking of regulatory T cells to tissue-specific sites such as spleen or CNS where autoimmune responses could be down-modulated (Kleinewietfeld et al., 2005; Korn et al., 2007). To address this idea we examined wild type and CCR6-/- mice in the chronic phase of disease for the presence of Treg cells. At the time point when CCR6-/- mice were exhibiting significantly more severe clinical disease (c.f. Figure 1A, days 21-35 post disease induction) we assessed the spleen and CNS compartments for the presence of Tregs using flow cytometric analysis for CD4+Foxp3+ cells. The representative results shown in Figure 4 indicate no differences between wild type and CCR6-/- mice in terms of percentages or numbers (data not shown) of CD4+Foxp3+ cells in either the spleen or CNS. Likewise, there was no difference in regulatory cytokine (e.g. IL-10 and TGF-β) expression (data not shown). These data suggested that the failure to control chronic EAE was not due to the inability of CCR6+Foxp3+ Treg to traffic to either the spleen or the CNS.
Figure 4. No difference in Foxp3+ Treg in CCR6-/- mice with severe chronic disease progression.
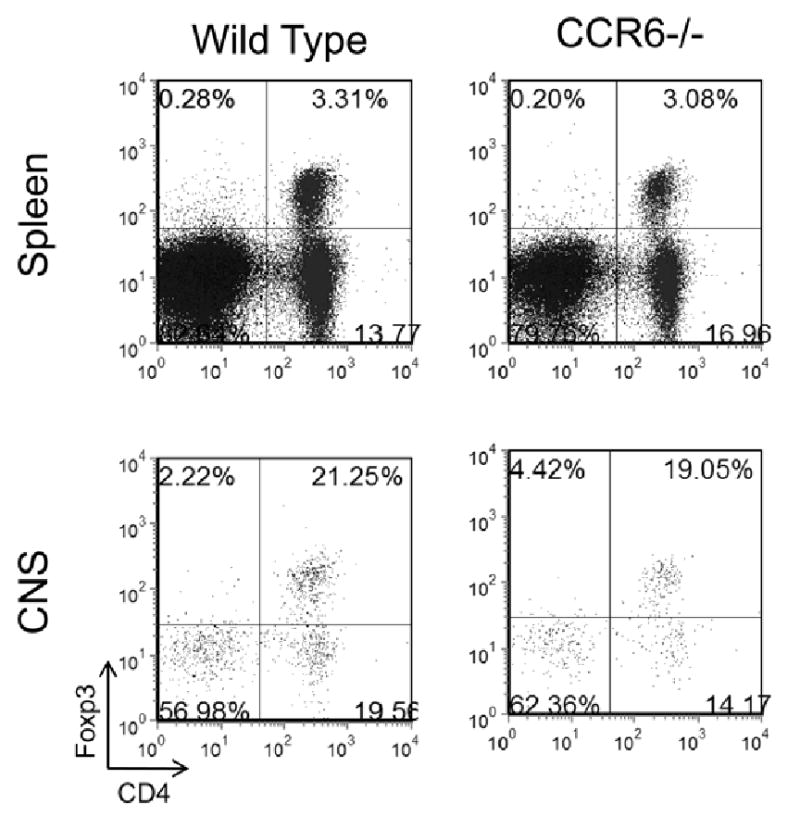
Wild type and CCR6-/- mice were immunized with 200 μg MOG35-55/CFA and monitored for clinical disease. Twenty eight days post immunization when CCR6-/- mice were showing increased clinical disease severity (cf. Figure 1A), lymphocytes from spleen and CNS were harvested and analyzed for the presence of CD4+Foxp3+ Treg by flow cytometry. The data is derived from a CD45+ cell gate and is representative of individually analyzed mice (N=5/group). The results indicate similar numbers of Treg in the spleen and CNS of wild type control and CCR6-/- mice.
Since we did not detect a difference in Treg cells between wild type and CCR6-/- mice, we assessed the possibility that peripheral lymphoid tissues and CNS from the CCR6-/- mice contained differences in B7-H1+ (PD-L1+) dendritic cells that have been shown to have regulatory function (Latchman et al., 2004). Groups of wild type and CCR6-/- mice were immunized with MOG35-55/CFA. During chronic disease when CCR6-/- mice were showing increased signs of clinical disease progression compared to wild type control mice, draining lymph node, spleen and CNS were harvested from both groups of mice and assessed for the presence of CD45+CD11c+PD-L1+ dendritic cells. The results shown in Figure 5A demonstrate no differences in the number of total mDC in either draining lymph node (pln) or CNS of wild type and CCR6-/- mice. Similarly, there were no differences in the number of PD-L1+ mDC in either draining lymph node (pln) or CNS of wild type and CCR6-/- mice (Figure 5B). However, there was a significant decrease in the number of total mDC (Figure 5A) and the number of PD-L1+ mDC (Figure 5B) in the spleens of CCR6-/- mice compared to wild type control mice.
Figure 5. Decreased splenic PD-L1+ mDC in CCR6-/- mice with severe chronic disease progression.
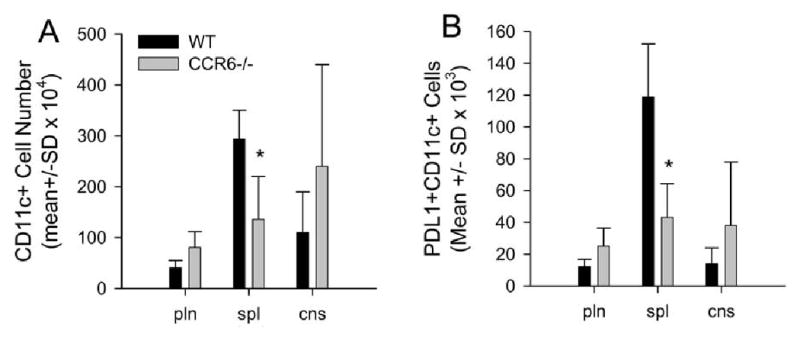
Wild type and CCR6-/- mice were immunized with 200 μg MOG35-55/CFA and monitored for clinical disease. Twenty eight days post immunization when CCR6-/- mice were showing increased clinical disease severity, peripheral lymph node cells (pln), splenocytes (spl) and CNS-infiltrating mononuclear cells (cns) were harvested and analyzed for the presence of CD11c+PD-L1(B7-H1)+ mDC by flow cytometry. Percentages of positive populations were obtained and total cell numbers were calculated based on the known input numbers. A) The data indicate similar numbers of mDC in the pln and cns of CCR6-/- mice compared to wild type controls. However, there is a significant (*, P<0.05) decrease in the number of mDC in the spleen of CCR6-/- mice compared to wild type controls. B) The data indicate similar numbers of PD-L1+ mDC in the pln and cns of CCR6-/- mice compared to wild type controls. However, there is a significant (*, p<0.05) decrease in the number of PD-L1+ mDC in the spleen of CCR6-/- mice compared to wild type controls. N=5 mice per group. Data is displayed as mean cell number ± SD.
Unprimed (naïve) and day 7 post-MOG35-55 immunized (primed) wild type and CCR6-/- mice were assessed for mDC PD-L1 expression in order to determine if there was an inherent difference between the two strains of mice in terms of the regulatory mDC population prior to disease development and progression. The data in Figure 6 demonstrate that in the peripheral lymph nodes and spleen there was no difference in the percentages of CD11c+PD-L1+ mDC in either the naïve or primed wild type (WT) and CCR6-/- mice (i.e. they contained similar low percentages of PD-L1+ mDC). Collectively, these results showed that in the absence of CCR6 expression there were fewer regulatory mDC in the spleen during the chronic EAE, suggesting that during normal disease pathogenesis CCR6 contributes to the regulation of disease progression by modulating the accumulation of regulatory mDC in the spleen.
Figure 6. PD-L1 expression by mDC in naïve and primed mice.
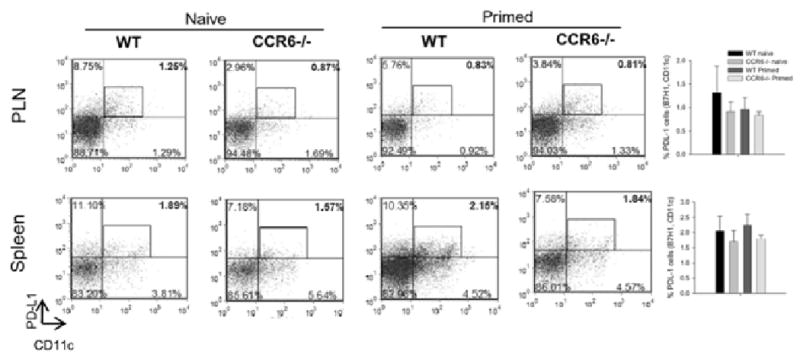
Wild type and CCR6-/- mice were either analyzed prior to immunization or immunized with 200 μg MOG35-55/CFA and analyzed for PD-L1+ mDC in peripheral lymph node (PLN) or spleen. In representative naïve mice, wild type peripheral lymph node and spleen PD-L1+ mDC were 1.26% and 1.89% compared to 0.87% and 1.57%, respectively, in CCR6-/- animals. In representative mice, seven days after MOG35-55 priming, wild type peripheral lymph node and spleen PD-L1+ mDC were 0.83% and 2.15% compared to 0.81% and 1.84%, respectively, in CCR6-/- animals. The bar graphs summarize the data from 5 individual mice in each group and indicate no significant differences between naïve or primed wild type and CCR6-/- animals (data is displayed as mean cell number ± SD).
The previous experiments indicated that enhanced chronic EAE in the CCR6-/- mice correlated with a decreased presence of PD-L1+ mDC in the spleen. In order to determine whether the mechanism of chronic disease regulation was a function of PD-L1+ mDC, we attempted to isolate a pure population of these cells and add them back to CCR6-/- mice that had shown increased chronic EAE severity (c.f.Figure 1). With this approach it has been difficult to obtain a large quantity of PD-L1+ mDC sufficient for the add-back experiments. Therefore, we utilized bulk populations of CD11c+ mDC isolated from the spleens of mice primed with MOG35-55/CFA that either had baseline (control DC) or upregulated (regulatory DC) levels of PD-L1 expression. Isolated cell populations were stained with antibodies specific for CD45, CCR6, CD11c and PD-L1 and analyzed by flow cytometry for the number of CD11c+PD-L1+ cells. Specifically, splenic CD45.1+CCR6+CD11c+ mDC harvested 3 days post MOG35-55 priming showed 1% PD-L1 expression (Figure 7A), similar to the baseline expression previously seen (Figure 6). In contrast, splenic CD45.1+CCR6+CD11c+ mDC harvested 21 days post MOG35-55 priming showed 8% PD-L1 expression (Figure 7B). The percentage of total CD11c+ mDC in the spleens of both groups of primed mice (day 3 and 21) was approximately the same (5.2% and 4.9%, respectively) and there were no differences in the other leukocyte constituents (i.e. T cells, B cells, macrophages and NK cells). Four groups of wild type CD45.2+ mice were primed with MOG35-55/CFA for the induction of EAE. At day 17 post immunization when the four groups showed similar EAE scores, the first wild type control group received a dose of 50×106 (containing approximately 29,000 PD-L1+ mDC) day 3 control spleen cells, the second wild type control group received a dose of 50×106 (containing approximately 200,000 PD-L1+ mDC) of the day 21 spleen cells, the first CCR6-/- group received 50×106 (containing approximately 29,000 PD-L1+ mDC) day 3 control spleen cells, and the second CCR6-/- group received 50×106 (containing approximately 200,000 PD-L1+ mDC) of the day 21 spleen cells. The results of this experiment shown in Figure 7C demonstrate that add back of the population of cells containing increased numbers of the PD-L1+ regulatory mDC reduced the severity of chronic EAE in the CCR6-/- mice compared to those mice that received the population of cells not enriched for regulatory mDC. Moreover, the CD45.1+ PD-L1+ regulatory mDC population could be detected in the spleens of wild type and CCR6-/- recipient mice (1.24±0.40% and 1.10±0.15%, respectively). These data further suggest that splenic CCR6+, PD-L1+ mDC function to regulate chronic EAE.
Figure 7. PD-L1+mDC administration decreases severe chronic EAE in CCR6 -/- mice.
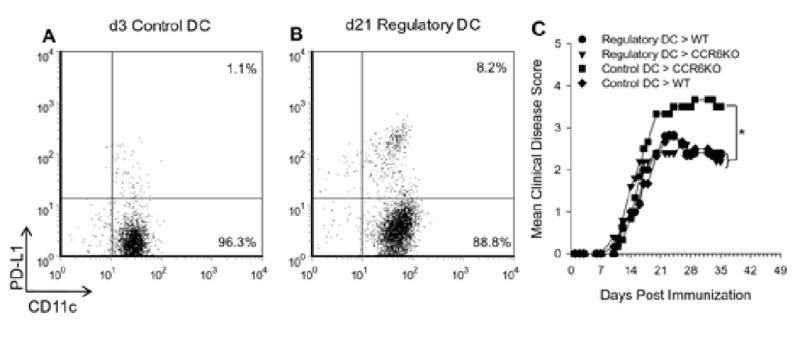
A) Splenocytes low in DC PD-L1 expression were harvested from mice 3 days after priming with MOG35-55/CFA. The data shown was gated on CD45 and CCR6 expression and plotted as PD-L1 vs. CD11c. This population of cells was termed control DC as there was only 1.1% PD-L1 expression. B) Splenocytes with elevated DC PD-L1 expression were harvested from mice 21 days after priming with MOG35-55/CFA. The data shown was gated on CD45 and CCR6 expression and plotted as PD-L1 vs. CD11c. This population of cells was termed regulatory DC as there was 8.2% PD-L1 expression. C) 50×106 splenocytes containing either regulatory or control DC were transferred i.v. to wild type or CCR6-/- (CCR6KO) mice primed with MOG35-55/CFA to induce EAE 14 days earlier. The data are shown as mean clinical disease score vs. days post immunization and indicate a significant (*, p<0.05) increase in chronic disease severity in the CCR6-/- group receiving control DC. The CCR6-/- group receiving regulatory DC showed a disease course similar to that of controls. N=5-6 mice/recipient group.
Since add-back of CCR6+PD-L1+ regulatory mDC to CCR6-/- mice with severe chronic EAE resulted in a reduction of EAE severity during disease progression, we sought to understand if the regulatory mDC functioned by reducing ongoing CNS inflammation. To that end we performed H&E histology on the CNS of the DC recipient mice and noted the CCR6-/- mice that had severe chronic EAE (c.f. Figure 7) and were recipients of control DC showed large focal CNS inflammatory lesions in the white matter (Figure 8). In contrast, wild type and CCR6-/- recipients of regulatory DC as well as wild type recipients of the control DC population showed a diffuse inflammatory cell infiltrate win the white matter (Figure 8). These data suggest that add-back of regulatory mDC to CCR6-/- mice downregulated CNS inflammation.
Figure 8. PD-L1+ DC add-back reduced chronic CNS inflammation.
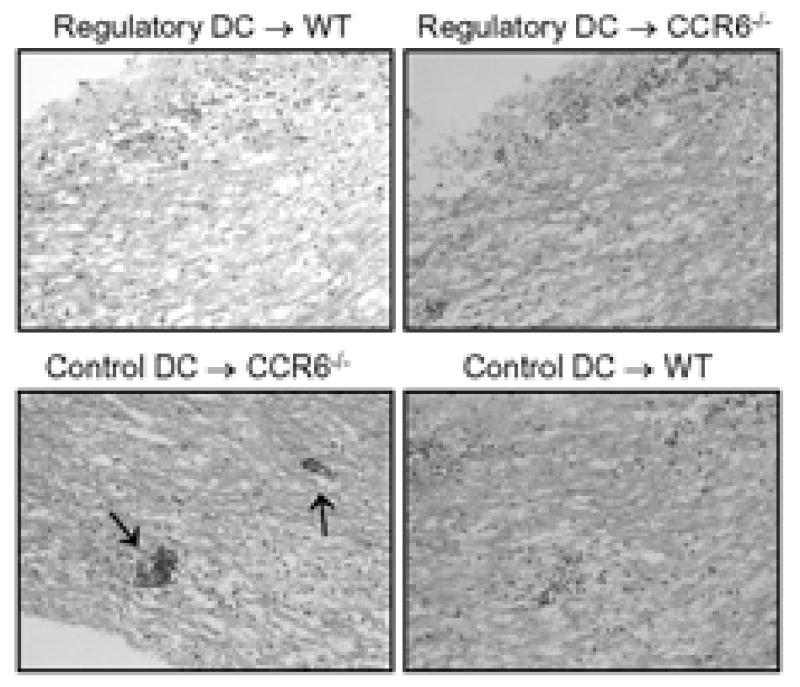
Spinal cords from the experiment in Figure 7 were analyzed by H&E histology for the presence of mononuclear cells. The data is a representative photomicrograph (200×) of the animals in each group. The following experimental groups showed diffuse mononuclear cell inflammation in the white matter tracts: Regulatory DC → WT, Regulatory DC → CCR6-/-, and Control DC → WT. The Control DC → CCR6-/- showed large focal perivascular inflammatory foci (arrows).
4. Discussion
The data in the present report suggests a role for CCR6 in the regulation of chronic EAE as CCR6-/- mice developed a significantly more severe clinical and histologic disease progression compared to wild type controls (Figure 1). The increased disease progression in CCR6-/- mice does not appear to be related to a requirement for T cell CCR6 expression (Figure 3), differences in Foxp3+ Treg accumulation (Figure 4), or ability to make suppressive cytokines (not shown). Rather, the fact that CCR6-/- mice develop a significantly more severe disease progression appears to be related to the lack of accumulation of PD-L1+ mDC in the spleen (Figure 5) that regulated ongoing effector T cells (Figure 2). PD-L1 has been shown to be expressed by both immature and mature DC and functions to down regulate T cells (Brown et al., 2003). That DC utilize PD-L1 as a mechanism to regulate immune responses and autoimmune disease was suggested by experiments showing PD-L1 expression by CNS-infiltrating cells in mice with EAE (Liang et al., 2003). The demonstration that PD-L1 was a potent effector of disease regulation was elucidated by experiments that showed decreased EAE in recipients of embryonic stem cell-derived DC that expressed PD-L1 (Hirata et al., 2005). Similarly, the severity of EAE progression was enhanced when mice were treated with a PD-L1-specific antibody (Zhu et al., 2006), further suggesting a down regulatory role for PD-L1. The role for PD-L1 as a negative regulator of disease development and progression in EAE was convincingly demonstrated by experiments where PD-L1 was genetically deleted and disease was more severe in mice deficient for PD-L1 expression (Carter et al., 2007; Latchman et al., 2004; Ortler et al., 2008). A role for PD-L1 in the immunoregulation of insulin-dependent diabetes has also been demonstrated (Fife et al., 2006), further supporting the idea that autoreactive T cell responses in general are modulated by PD-L1 DC with regulatory function. Our report is the first to demonstrate a role for CCR6 in the accumulation of these potent disease-regulating mDC. This line of investigation has therapeutic implications as treatment of MS patients with IFN-β resulted in enhanced expression of PD-L1+ DC, suggesting a role for that cell type in human disease regulation (Schreiner et al., 2004). This raises the possibility of CCR6 agonist development for the promotion of PD-L1+ mDC accumulation as a new therapeutic modality for autoimmune disease.
There are three possible explanations for increased severity in chronic EAE progression related to CCR6-dependent, PD-L1+ mDC: 1) lack of DC regulatory cytokine expression in tissue-specific sites (e.g., CNS); 2) lack of regulatory DC induction of Treg; and 3) lack of tissue-specific regulatory DC accumulation resulting in a lack of alteration of effector T cells leading to enhanced responses. We do not believe that the first of these possibilities plays a role in regulating chronic disease progression in our experiments because we did not detect a difference in production of inhibitory cytokines such as TGF-β or IL-10 (data not shown) related to PD-L1 mDC. Although PD-L1+ cells have been shown to influence Treg function (Tokita et al., 2008; Wang et al., 2008), we believe the possibility that CCR6-/- mice have decreases in PD-L1+ mDC-mediated Treg cells in tissue-specific compartments is also an unlikely explanation for the exacerbated chronic disease progression. This conclusion was reached primarily because the data showed no differences in CD4+Foxp3+ Treg cells in spleen or CNS between wild type or CCR6-/- mice (Figure 4).
We currently favor the explanation that the absence of CCR6 expression alters regulatory PD-L1 DC migration to the spleen where a direct regulation of encephalitogenic T cells does not occur thereby resulting in exacerbated chronic EAE progression. The data shown in Figure 5B supports this idea as there were no differences in numbers of PD-L1+ mDC in peripheral lymph node or CNS; however, there was a significant decrease of these cells in the spleen of CCR6-/- mice. Additionally, antigen-specific Th17 and Th1 responses in the spleen, but not CNS, were seen in CCR6-/- mice (Figure 2). Moreover, add back of wild type splenocytes containing an enriched CCR6+ PD-L1+ mDC population to CCR6-/- mice reduced the chronic disease progression severity to that of wild type mice (Figure 7C).
CCL20 expression was originally described for spleen, lymph nodes, liver, thymus, small intestine, colon and CNS (Ambrosini et al., 2003; Baba et al., 1997; Varona et al., 1998). We have observed that the spleen constitutively expresses CCL20, the only known chemokine ligand for CCR6, at higher levels than either the lymph node or CNS (data not shown). Therefore, it is reasonable to assume the added back regulatory PD-L1+ mDC (Figure 7) population could accumulate in the spleen in response to the constitutive CCL20 expression. In fact we have noted that the passively transferred PD-L1+ mDC could be found in the spleen. PD-L1 has been shown to be expressed in the spleen (Liang et al., 2003); the tissue site that we believe plays a role in the inhibition of autoreactive T cells. Indeed Flugel et al. (Flugel et al., 2001) have demonstrated the spleen to be a key regulatory checkpoint for development of EAE where T cells traffic prior to migration to the CNS.
Our results also indicate a delay in disease development between days 7 and 13 post immunization (Figure 1A) in CCR6-/- mice. This delay in clinical disease is not as substantial as observed by others (Liston et al., 2009; Reboldi et al., 2009). One possibility that might help to explain the differences in how CCR6 affects acute disease development might be related to differences between immunization protocols used by them and us, especially the amount of adjuvant. Regardless of the magnitude of CCR6 influence on acute EAE development, our results indicating T cell CCR6 expression is not necessary for adoptive EAE (Figure 3) are compatible with those of Liston et al (Liston et al., 2009). In aggregate, the evidence supports a role for antigen presenting cell rather than T cell expression of CCR6 in EAE development. We therefore speculate that CCR6 may contribute to EAE pathogenesis in two different ways: first to regulate disease development through modulation of APC migration and second to regulate disease progression through modulation of a regulatory DC population.
Our data suggests the mechanism of CCR6 regulation of chronic EAE is related to splenic accumulation of a PD-L1+ regulatory mDC population. Although we demonstrated an increase in antigen-specific Th responses in spleen at a time when there are fewer PD-L1+ mDC and no changes in either IL-10 or TGF-β, we do not know the precise mechanism of PD-L1 mDC-mediated effector T cell regulation. There is a suggestion that factors such as reactive oxygen species (ROS) produced by myeloid-derived suppressor cells may inhibit ongoing immune responses (reviewed in (Gabrilovich and Nagaraj, 2009)), a possibility that we cannot yet rule out. We are currently investigating the possibility that the lack of CCR6+PD-L1+ mDC in the spleen of CCR6-/- mice results in less direct ROS production by those DC resulting in enhanced autoreactive Th responses and exacerbated chronic EAE. Additionally, we are actively investigating whether PD-L1+ DC regulate disease progression by directly down modulating IL-17 and IFN-γ production by autoantigen-specific Th17 and Th1 cells, whether PD-L1+ DC regulate disease progression by altering chemokine and/or chemokine receptor expression and to what extent PD-L1+ DC affect the ability of APC to stimulate encephalitogenic T cells.
Acknowledgments
This work was supported in part by NIH grant R01 NS34510 (WJK). Sergio Lira is the Irene Diamond Professor of Immunology and this work was supported in part by The Irene Diamond Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ambrosini E, Columba-Cabezas S, Serafini B, Muscella A, Aloisi F. Astrocytes are the major intracerebral source of macrophage inflammatory protein-3α/CCL20 in relapsing experimental autoimmune encephalomyelitis and in vitro. Glia. 2003;41:290–300. doi: 10.1002/glia.10193. [DOI] [PubMed] [Google Scholar]
- Baba M, Imai T, Nishimura M, Kakizaki M, Takagi S, Hieshima K, Nomiyama, Yoshie O. Identification of CCR6, the specific receptor for a novel lymphocyte-directed CC chemokine LARC. Journal of Biological Chemistry. 1997;272:14893–14898. doi: 10.1074/jbc.272.23.14893. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Brosnan CF, Bornstein MB, Bloom BR. The effects of macrophage depletion on the clinical and pathologic expression of experimental allergic encephalomyelitis. Journal of Immunology. 1981;126:614–620. [PubMed] [Google Scholar]
- Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, Greenfield EA, Freeman GJ. Blockade of Programmed Death-1 Ligands on Dendritic Cells Enhances T Cell Activation and Cytokine Production. The Journal of Immunology. 2003;170:1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, Jussif J, Benoit S, Ireland G, Luxenberg D, Askew GR, Milarski KL, Groves C, Brown T, Carito BA, Percival K, Carreno BM, Collins M, Marusic S. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. Journal of Neuroimmunology. 2007;182:124–134. doi: 10.1016/j.jneuroim.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Cook DN, Prosser DM, Forster R, Zhang J, Kuklin NA, Abbondanzo SJ, Niu XD, Chen SC, Manfra DJ, Wiekowski MT, Sullivan LM, Smith SR, Greenberg HB, Narula SK, Lipp M, Lira SA. CCR6 mediates dendritic cell localization, lymphocyte homeostasis, and immune responses in mucosal tissue. Immunity. 2000;12:495–503. doi: 10.1016/s1074-7613(00)80201-0. [DOI] [PubMed] [Google Scholar]
- Cross AH, Cannella B, Brosnan CF, Raine CS. Homing to central nervous system vasculature by antigen-specific lymphocytes. I. Localization of 14C-labeled cells during acute, chronic, and relapsing experimental allergic encephalomyelitis. Laboratory Investigation. 1990;63:162–170. [PubMed] [Google Scholar]
- Dieu-Nosjean MC, Massacrier C, Homey B, Vanbervliet B, Pin JJ, Vicari A, Lebecque S, Dezutter-Dambuyant C, Schmitt D, Zlotnik A, Caux C. Macrophage Inflammatory Protein 3α Is Expressed at Inflamed Epithelial Surfaces and Is the Most Potent Chemokine Known in Attracting Langerhans Cell Precursors. The Journal of Experimental Medicine. 2000;192:705–718. doi: 10.1084/jem.192.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan RN, Elhofy A, Karpus WJ. Production of CCL2 by Central Nervous System Cells Regulates Development of Murine Experimental Autoimmune Encephalomyelitis through the Recruitment of TNF- and iNOS-Expressing Macrophages and Myeloid Dendritic Cells. J Immunol. 2008;180:7376–7384. doi: 10.4049/jimmunol.180.11.7376. [DOI] [PubMed] [Google Scholar]
- Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, Yagita H, Azuma M, Sayegh MH, Bluestone JA. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. The Journal of Experimental Medicine. 2006;203:2737–2747. doi: 10.1084/jem.20061577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flugel A, Berkowicz T, Ritter T, Labeur M, Jenne DE, Li Z, Ellwart JW, Willem M, Lassmann H, Wekerle H. Migratory Activity and Functional Changes of Green Fluorescent Effector Cells before and during Experimental Autoimmune Encephalomyelitis. Immunity. 2001;14:547–560. doi: 10.1016/s1074-7613(01)00143-1. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves DR, Wang W, Dairaghi DJ, Dieu MC, Saint-Vis B, Franz-Bacon K, Rossi D, Caux C, McClanahan T, Gordon S, Zlotnik A, Schall TJ. CCR6, a CC chemokine receptor that interacts with macrophage inflammatory protein 3α and is highly expressed in human dendritic cells. Journal of Experimental Medicine. 1997;186:837–844. doi: 10.1084/jem.186.6.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey WF. Migration of hematogenous cells through the blood-brain barrier and the initiation of CNS inflammation. Brain Pathology. 1991;1:97–105. doi: 10.1111/j.1750-3639.1991.tb00646.x. [DOI] [PubMed] [Google Scholar]
- Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. Journal of Neuroscience Research. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- Hirata S, Senju S, Matsuyoshi H, Fukuma D, Uemura Y, Nishimura Y. Prevention of Experimental Autoimmune Encephalomyelitis by Transfer of Embryonic Stem Cell-Derived Dendritic Cells Expressing Myelin Oligodendrocyte Glycoprotein Peptide along with TRAIL or Programmed Death-1 Ligand. The Journal of Immunology. 2005;174:1888–1897. doi: 10.4049/jimmunol.174.4.1888. [DOI] [PubMed] [Google Scholar]
- Hohlfeld R, Kerschensteiner M, Stadelmann C, Lassmann H, Wekerle H. The neuroprotective effect of inflammation: implications for the therapy of multiple sclerosis. Journal of Neuroimmunology. 2000;107:161–166. doi: 10.1016/s0165-5728(00)00233-2. [DOI] [PubMed] [Google Scholar]
- Holden T, Maecker JT. Flow cytometry controls, instrument setup, and the determination of positivity. Cytometry Part A. 2006;69A:1037–1042. doi: 10.1002/cyto.a.20333. [DOI] [PubMed] [Google Scholar]
- Huitinga I, Van Rooijen N, De Groot CJA, Uitdehaag BMJ, Dijkstra CD. Elimination of macrophages infiltrating into the central nervous system suppresses experimental allergic encephalomyelitis in Lewis rats. Ann N Y Acad Sci. 1990;594:458–460. [Google Scholar]
- Karpus WJ, Fife BT, Kennedy KJ. Immunoneutralization of chemokines for the prevention and treatment of central nervous system autoimmune disease. Methods. 2003;29:362–368. doi: 10.1016/s1046-2023(02)00360-2. [DOI] [PubMed] [Google Scholar]
- Karpus WJ, Kennedy KJ, Smith WS, Miller SD. Inhibition of relapsing experimental autoimmune encephalomyelitis in SJL mice by feeding the immunodominant PLP139-151 peptide. Journal of Neuroscience Research. 1996;45:410–423. doi: 10.1002/(SICI)1097-4547(19960815)45:4<410::AID-JNR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Karpus WJ, Lukacs NW, McRae BL, Strieter RM, Kunkel SL, Miller SD. An important role for the chemokine macrophage inflammatory protein-1α in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. Journal of Immunology. 1995;155:5003–5010. [PubMed] [Google Scholar]
- Karpus WJ, Ransohoff RM. Chemokine regulation of experimental autoimmune encephalomyelitis: temporal and spatial expression patterns govern disease pathogenesis. Journal of Immunology. 1998;161:2667–2671. [PubMed] [Google Scholar]
- Kleinewietfeld M, Puentes F, Borsellino G, Battistini L, Rotzschke O, Falk K. CCR6 expression defines regulatory effector/memory-like cells within the CD25+CD4+ T-cell subset. Blood. 2005;105:2877–2886. doi: 10.1182/blood-2004-07-2505. [DOI] [PubMed] [Google Scholar]
- Kohler RE, Caon AC, Willenborg DO, Clark-Lewis I, McColl SR. A Role for Macrophage Inflammatory Protein-3α/CC Chemokine Ligand 20 in Immune Priming During T Cell-Mediated Inflammation of the Central Nervous System. Journal of Immunology. 2003;170:6298–6306. doi: 10.4049/jimmunol.170.12.6298. [DOI] [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakowski ML, Owens T. The central nervous system environment controls effector CD4+ T cell cytokine profile in experimental allergic encephalomyelitis. European Journal of Immunology. 1997;27:2840–2847. doi: 10.1002/eji.1830271115. [DOI] [PubMed] [Google Scholar]
- Kuchroo VK, Sobel RA, Laning JC, Martin CA, Greenfield E, Dorf ME, Lees MB. Experimental allergic encephalomyelitis mediated by cloned T cells specific for a synthetic peptide of myelin proteolipid protein. Fine specificity and T cell receptor Vβ usage. Journal of Immunology. 1992;148:3776–3782. [PubMed] [Google Scholar]
- Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proceedings of the National Academy of Sciences (USA) 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, Sharpe AH. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33:2706–2716. doi: 10.1002/eji.200324228. [DOI] [PubMed] [Google Scholar]
- Liao F, Rabin RL, Smith CS, Sharma G, Nutman TB, Farber JM. CC-chemokine receptor 6 is expressed on diverse memory subsets of T cells and determines responsiveness to macrophage inflammatory protein 3α. Journal of Immunology. 1999;162:186–194. [PubMed] [Google Scholar]
- Liston A, Kohler RE, Townley S, Haylock-Jacobs S, Comerford I, Caon AC, Webster J, Harrison JM, Swann J, Clark-Lewis I, Korner H, McColl SR. Inhibition of CCR6 function reduces the severity of experimental autoimmune encephalomyelitis via effects on the priming phase of the immune response. J Immunol. 2009;182:3121–3130. doi: 10.4049/jimmunol.0713169. [DOI] [PubMed] [Google Scholar]
- Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- Ortler S, Leder C, Mittelbronn M, Zozulya AL, Knolle PA, Chen L, Kroner A, Wiendl H. B7-H1 restricts neuroantigen-specific T cell responses and confines inflammatory CNS damage: Implications for the lesion pathogenesis of multiple sclerosis. Eur J Immunol. 2008;38:1734–1744. doi: 10.1002/eji.200738071. [DOI] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F. C-C chemokine receptor 6-regulated entry of T(H)-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung HP, Weller M, Wiendl H. Interferon-β enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. Journal of Neuroimmunology. 2004;155:172–182. doi: 10.1016/j.jneuroim.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Schutyser E, Struyf S, Van Damme J. The CC chemokine CCL20 and its receptor CCR6. Cytokine & Growth Factor Reviews. 2003;14:409–426. doi: 10.1016/s1359-6101(03)00049-2. [DOI] [PubMed] [Google Scholar]
- Segal BM, Dwyer BK, Shevach EM. An Interleukin (IL)-10/IL-12 Immunoregulatory Circuit Controls Susceptibility to Autoimmune Disease. Journal of Experimental Medicine. 1998;187:537–546. doi: 10.1084/jem.187.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokita D, Mazariegos GV, Zahorchak AF, Chien N, Abe M, Raimondi G, Thomson AW. High PD-L1/CD86 ratio on plasmacytoid dendritic cells correlates with elevated T-regulatory cells in liver transplant tolerance. Transplantation. 2008;85:369–377. doi: 10.1097/TP.0b013e3181612ded. [DOI] [PubMed] [Google Scholar]
- Tuohy VK, Sobel RA, Lu Z, Laursen RA, Lees MB. Myelin proteolipid protein: minimum sequence requirements for active induction of autoimmune encephalomyelitis in SWR/J and SJL/J mice. Journal of Neuroimmunology. 1992;39:67–74. doi: 10.1016/0165-5728(92)90175-k. [DOI] [PubMed] [Google Scholar]
- Varona R, Villares R, Carramolino L, Goya I, Zaballos A, Gutierrez J, Torres M, Martinez A, Marquez G. CCR6-deficient mice have impaired leukocyte homeostasis and altered contact hypersensitivity and delayed-type hypersensitivity responses. Journal of Clinical Investigation. 2001;107:R37–R45. doi: 10.1172/JCI11297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varona R, Zaballos A, Gutierrez J, Martin P, Roncal F, Albar JP, Ardavin C, Marquez G. Molecular cloning, functional characterization and mRNA expression analysis of the murine chemokine receptor CCR6 and its specific ligand MIP-3α. FEBS Lett. 1998;440:188–194. doi: 10.1016/s0014-5793(98)01450-1. [DOI] [PubMed] [Google Scholar]
- Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105:9331–9336. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitham RH, Bourdette DN, Hashim GA, Herndon RM, Ilg RC, Vandenbark AA, Offner H. Lymphocytes from SJL/J mice immunized with spinal cord respond selectively to a peptide of proteolipid protein and transfer relapsing demyelinating experimental autoimmune encephalomyelitis. Journal of Immunology. 1991;146:101–107. [PubMed] [Google Scholar]
- Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, Martin-Orozco N, Kang HS, Ma L, Panopoulos AD, Craig S, Watowich SS, Jetten AM, Tian Q, Dong C. CCR6 regulates the migration of inflammatory and regulatory T cells. J Immunol. 2008;181:8391–8401. doi: 10.4049/jimmunol.181.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Guleria I, Khosroshahi A, Chitnis T, Imitola J, Azuma M, Yagita H, Sayegh MH, Khoury SJ. Differential Role of Programmed Death-1 Ligand and Programmed Death-2 Ligand in Regulating the Susceptibility and Chronic Progression of Experimental Autoimmune Encephalomyelitis. The Journal of Immunology. 2006;176:3480–3489. doi: 10.4049/jimmunol.176.6.3480. [DOI] [PubMed] [Google Scholar]
- Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]