

Abstract
We have previously shown that activation of protein kinase C δ (PKCδ) is required for angiotensin II (AngII)-induced migration of vascular smooth muscle cells (VSMCs). Here, we have hypothesized that PKCδ phosphorylation at Tyr311 plays a critical role in VSMC hypertrophy induced by AngII. Immunoblotting was used to monitor PKCδ phosphorylation at Tyr311, and cell size and protein measurements were used to detect hypertrophy in VSMCs. PKCδ was rapidly (0.5–10 min) phosphorylated at Tyr311 by AngII. This phosphorylation was markedly blocked by a Src family kinase inhibitor and dominant-negative Src, but not by an epidermal growth factor receptor kinase inhibitor. AngII-induced Akt phosphorylation and hypertrophic responses were significantly enhanced in VSMCs expressing PKCδ wild type compared with VSMCs expressing control vector, whereas the enhancements were markedly diminished in VSMCs expressing PKCδ Y311F mutant. Also, these responses were significantly inhibited in VSMCs expressing kinase-inactive PKCδ K376A compared with VSMCs expressing control vector. From these data, we conclude that not only PKCδ kinase activation but also the Src-dependent Tyr311 phosphorylation contributes to Akt activation and subsequent VSMC hypertrophy induced by AngII, thus signifying a novel molecular mechanism for enhancement of cardiovascular diseases induced by AngII.
Keywords: angiotensin II, AT1 receptor, signal transduction, protein kinase C δ, Src, hypertrophy, vascular smooth muscle cells
Introduction
Angiotensin II (AngII) plays a major role in vascular remodeling outside of its hemodynamic effects. In cultured vascular smooth muscle cells (VSMCs), cardiac myocytes, and cardiac fibroblasts, AngII has been shown to promote hypertrophy and/or hyperplasia. There are two subtypes of AngII receptors, AT1 and AT2, although the major physiological and pathophysiological actions of AngII are facilitated through the AT1 receptor. In VSMCs, activation of the AT1 receptor coupled to Gq increases intracellular Ca2+ and activates protein kinase C (PKC) 1,2. In this regard, several PKC isoforms including PKCδ are believed to be activated by AngII in VSMCs 3–5. In addition, various tyrosine kinases and serine/threonine kinases are rapidly activated by AngII and likely play important roles in mediating vascular remodeling induced by AngII 6,7. However, the detailed role of each PKC isoform in mediating AngII-induced vascular remodeling as well as the possible signal crosstalk with other kinases has been insufficiently characterized.
Increasing evidence suggest that PKCδ is involved in many mechanisms promoting VSMC remodeling and dysfunction 8–11. It was reported that PKCδ is activated by mechanical stress and VSMCs from PKCδ–null mice migrate slower than control VSMCs 12. Previously, we have shown that PKCδ kinase activity is required for activation of several tyrosine kinases by AngII or reactive oxygen species in VSMCs 4,13,14. Moreover we have recently reported that PKCδ is required for activation of Rho, Rho-kinase and c-Jun NH2-terminal kinase, and subsequent migration of VSMCs by using kinase-inactive PKCδ over expression 15. These data suggest an important role of PKCδ in mediating vascular remodeling induced by AngII.
PKCδ is also phosphorylated on tyrosine residues in many cells including VSMCs and cardiac myocytes 13,16–18. Although there are multiple tyrosine phosphorylation sites on PKCδ, Tyr311 located between the regulatory and catalytic domains is of particular interest. This is because the Tyr311 phosphorylation has been linked to increased kinase activity in cells treated with H2O2 19. PKCδ phosphorylation at Tyr311 may also affect the selectivity of substrates 17. Taken together with the above information, we have tested the hypothesis that PKCδ Tyr311 phosphorylation plays a major role in AngII-induced vascular hypertrophy. We found that PKCδ phosphorylation at Tyr311 was induced by AngII through a Src family kinase and that this phosphorylation was involved in Akt activation and subsequent VSMC hypertrophy.
Methods
An expanded Methods section describing reagents, primary antibodies, cell culture and statistical analysis is available at http://hyper.ahajournals.org.
Retrovirus infection
Wild type or Y311F PKCδ containing enhanced green fluorescent protein (GFP) at the C-terminus 20 was cloned into the pBM-IRES-PURO vector and high titer retroviral supernatants were generated 21. VSMCs were infected with retrovirus and the infected VSMCs were selected as previously described 22,23. To assess complete viral transformation after an antibiotic selection, in addition to the detection of the over-expression by immunoblotting, we routinely confirmed more than 99% infection efficiency of our retrovirus vectors by the GFP tagged to the mutants and detected under a fluorescent microscope.
Adenovirus infection
The generation of adenovirus encoding wild type and a kinase-inactive K376A PKCδ mutant construct and dominant negative K295M+Y527F Src was described previously 24,25. The titer (pfu/ml) of adenovirus was determined by Adeno-XTM Rapid Titer Kit (BD Biosciences, Palo Alto, CA). VSMCs were infected with adenovirus for 2 days as previously described 14. To assess complete viral transformation, we routinely confirmed more than 99% infection efficiency of our adenovirus vectors by GFP encoded by these vectors separately and detected under a fluorescent microscope.
Immunoblotting
Cell lysates were subjected to SDS-PAGE and transferred to a nitrocellulose membrane as previously described 26. The membranes were then exposed to primary antibodies overnight at 4 °C. After incubation with the peroxidase-linked secondary antibody (Amersham Biosciences, Piscataway, NJ) with dilution between 1:1,000 – 1:10,000 (depending on the primary antibody) for 1 hour at room temperature, the immunoreactive proteins were visualized by a chemiluminescence reaction kit (Pierce, Rockford, IL).
Protein Assay
VSMCs on 12 well culture plates were incubated with serum-free DMEM for 3 days in retrovirus infected VSMCs. For adenovirus infection, VSMCs were incubated with serum-free DMEM for 1 day and infected with adenovirus at 100 multiplicity of infection in serum-free DMEM for 2 days. The cells were further incubated with or without 100 nmol/L AngII for 3 days. After aspiration of the medium, cells were washed twice with ice-cold Hanks balanced salt solution, and the total amount of cellular protein was measured as previously described 27.
Cell Volume Assay
After the pretreatments described in the protein assay, VSMCs were washed with Hanks balanced salt solution and trypsinized. The cells were then suspended in PBS and the cell volume was measured by Z2 Coulter Particle Count and Size Analyzer (Beckman Coulter, Fullerton, CA) 27.
Proliferation Assay
After the pretreatments described in the protein assay, cell proliferation was measured using a CellTiter 96 Aqueous cell proliferation assay kit (Promega, Madison, WI) following the manufactures’ protocol as previously described 27. Basically, this assay measures cell viabilities upon the PKCδ manipulation with or without AngII stimulation for 3 days.
BrdU assay
After the adenovirus infection as described for the protein assay, the cells were pretreated with or without 100 nmol/L AngII for 24 hours, and BrdU incorporation was determined for an additional 24 hours by a BrdU incorporation kit (Calbiochem, La Jolla, CA) according to the manufactures’ protocol.
Results
Phosphorylation of PKCδ at Tyr311 by AngII through the Gq-coupled AT1 receptor
In 3 day serum-starved rat aortic VSMCs, AngII (100 nmol/L) stimulated phosphorylation of PKCδ at Tyr311 in a rapid (within 0.5 min) and transient manner with a peak of 2 to 5 min (Figure 1). The phosphorylation returned to the baseline level at 40 min (Figure S1A, please see http://hyper.ahajournals.org.). Thus, in subsequent experiments, unless otherwise stated, VSMCs were stimulated with AngII for 2 min for evaluation of the PKCδ phosphorylation. Pretreatment with an AT1 receptor antagonist, RNH6270, totally blocked PKCδ phosphorylation by AngII (Figure S1B, please see http://hyper.ahajournals.org.). The AT1 receptor is mainly coupled to the heterotrimeric G-protein Gq, whereas G protein-independent signal transduction by the AT1 has been reported 2. Thus, we determined whether Gq contributed to the AngII-induced PKCδ phosphorylation. Pretreatment with a selective Gq inhibitor, YM-254890 22,28, completely blocked PKCδ phosphorylation at Tyr311 by AngII (Figure S1C, please see http://hyper.ahajournals.org.), indicating that AngII-induced phosphorylation of PKCδ at Tyr311 was mediated through Gq activation.
Figure 1.

Phosphorylation of PKCδ at Tyr311 by AngII. VSMCs were stimulated with 100 nmol/L AngII for the indicated time periods. The cell lysates were immunoblotted with phospho-selective antibody which detects PKCδ Tyr311 phosphorylation and anti-PKCδ antibody. The PKCδ phosphorylation at Tyr311 was measured by densitometry, normalized to total PKCδ and shown as mean ± SEM (n=3). *P < 0.05 compared to the basal control.
Involvement of Src in PKCδ Tyr311 phosphorylation by AngII
Activation of the AT1 receptor by AngII leads to rapid transactivation of the epidermal growth factor (EGF) receptor which appears to mediate many key components of downstream signal transduction in VSMCs 29, whereas a Src family kinase has been implicated as a PKCδ Tyr311 kinase 17. To clarify the involvement of Src family kinase and/or EGF receptor transactivation in PKCδ phosphorylation, we pretreated VSMCs with PP2, a Src family kinase inhibitor, or AG1478, an EGF receptor family kinase inhibitor. Interestingly, AngII-induced PKCδ Tyr311 phosphorylation was markedly blocked by PP2 (5 μmol/L), whereas AG1478 (1 μmol/L) had no inhibitory effect. As expected, AG1478, but not PP2, inhibited AngII-induced EGF receptor transactivation as detected by its autophosphorylation at Tyr1068 (Figure 2A). Also, PP3 (5 μmol/L), the inactive control chemical for PP2, had no inhibitory effect on AngII-induced PKCδ Tyr311 phosphorylation (Figure S1D, please see http://hyper.ahajournals.org.). To support these pharmacological experiments, the effect of dominant negative Src was examined. Infection of adenovirus encoding dominant negative Src, but not control vector, markedly inhibited PKCδ Tyr311 phosphorylation induced by AngII, whereas neither virus affected the EGF receptor transactivation (Figure 2B). Sufficient over expression of the dominant negative Src mutant as compared to endogenous Src was confirmed (Figure 2B). These data suggest that Src but not EGF receptor mediates AngII-induced PKCδ Tyr311 phosphorylation.
Figure 2.

Involvement of Src in PKCδ Tyr311 phosphorylation induced by AngII. A, VSMCs were pretreated with a Src family kinase inhibitor, PP2 (5 μmol/L) or an EGF receptor kinase inhibitor, AG1478 (1 μmol/L) for 30 min and stimulated with 100 nmol/L AngII for 2 min. B, VSMCs were infected with adenovirus encoding dominant negative (dn) Src or control vector, and stimulated with 100 nmol/L AngII for 2 min. A and B, the cell lysates were immunoblotted with phospho-specific antibodies which detect PKCδ Tyr311 phosphorylation or EGF receptor autophosphorylation at Tyr1068 and with anti-PKCδ, anti-EGF receptor and anti-Src antibodies as indicated. The PKCδ phosphorylation at Tyr311 was measured by densitometry and shown as mean ± SEM (n=3). *P < 0.05 compared to the basal control. †P < 0.05 compared to the stimulated control.
Involvement of PKCδ Tyr311 phosphorylation and PKCδ kinase activity in AngII-induced VSMC hypertrophy
To verify the functional significance of the Tyr311 phosphorylation, we established VSMCs which over-express wild type PKCδ or a PKCδ Y311F mutant containing GFP at the C terminus using retrovirus infection (Figure 3A). In wild type PKCδ expressing VSMCs, the AngII-induced increase in cellular protein was significantly enhanced compared to the control VSMCs. However, the enhancement was much less in Y311F expressing VSMCs (Figure 3B). Moreover, in wild type PKCδ VSMCs, AngII significantly increased the cell volume, whereas no enhancement was observed in Y311F mutant expressing cells (Figure S2A, please see http://hyper.ahajournals.org.). There was no significant change in cell number among these VSMCs stimulated by AngII (Figure S2B, please see http://hyper.ahajournals.org.). The confluency state of these VSMCs at the time of the measurements was less than 90% because without a mitogen the VSMC did not significantly proliferate after serum-starvation. Also, BrdU incorporation was not significantly changed by AngII regardless of wild type PKCδ over expression in VSMCs (Figure S2C, please see http://hyper.ahajournals.org.). In addition, there was no enhancement of an apoptotic marker, cleaved caspase-3, detected in both control and PKCδ over expressing VSMCs with 4 h AngII stimulation (Figure S3, please see http://hyper.ahajournals.org.).
Figure 3.

PKCδ phosphorylation at Tyr311 contributes to VSMC hypertrophy induced by AngII. A, VSMCs were infected with retrovirus encoding control vector, wild type PKCδ tagged with GFP, or PKCδ Y311F mutant tagged with GFP. The cell lysates were immunoblotted with antibodies as indicated. An arrow denotes exogenously introduced GFP-tagged PKCδ. B, VSMCs infected with the above retrovirus were stimulated with 100 nmol/L AngII for 3 days. Afterwards, cellular protein levels were measured by a protein assay kit. The data was presented as fold basal (mean ± SEM, n=3). *P < 0.05.
To investigate whether the kinase activity of PKCδ is also required for AngII-induced protein synthesis in VSMCs, we infected VSMCs with an adenovirus encoding a kinase-inactive PKCδ mutant (K376A). In VSMCs expressing K376A, both AngII-induced protein synthesis (Figure 4) as well as the increase in cell volume (Figure S4A, please see http://hyper.ahajournals.org.) was significantly inhibited compared with control VSMCs. Again, there was no significant change in cell number among these VSMCs stimulated by AngII (Figure S4B, please see http://hyper.ahajournals.org.). These data suggest that PKCδ Tyr311 phosphorylation and PKCδ kinase activity are both required for AngII-induced hypertrophy in VSMCs.
Figure 4.
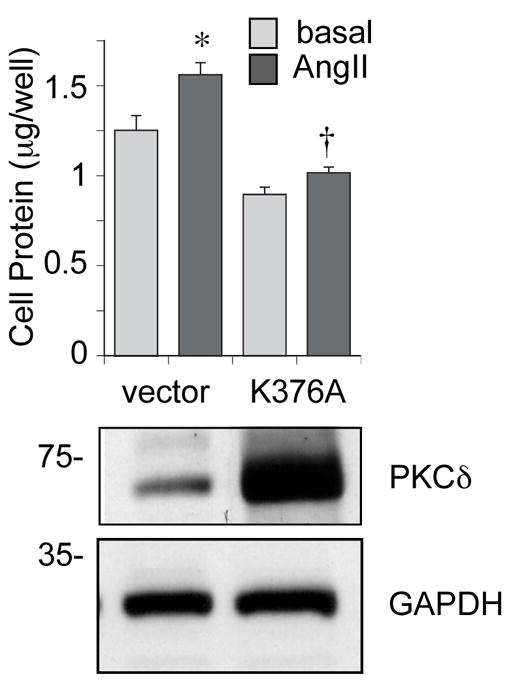
Kinase activity of PKCδ is required for AngII-induced protein synthesis in VSMCs. VSMCs were infected with adenovirus encoding a kinase-inactive PKCδ mutant (K376A) or control empty vector, and stimulated with 100 nmol/L AngII for 3 days. Afterwards, cellular protein levels were measured by a protein assay kit. The data was presented as fold basal (mean ± SEM, n=3). *P < 0.05 compared to the basal control. †P < 0.05 compared to the stimulated control. Also, immunoblotting of PKCδ and GAPDH to confirm K376A over-expression was performed.
It has been demonstrated that both Akt and extracellular signal-regulated kinase 1/2 (ERK1/2) are involved in AngII-induced VSMC hypertrophy 30–32. To assess the functional role of PKCδ Tyr311 phosphorylation and kinase activity in AngII-induced hypertrophic signaling, we examined Akt and ERK1/2 activation in the above treated cells. In wild-type PKCδ expressing VSMCs, AngII-induced Akt phosphorylation was markedly enhanced, whereas no enhancement of Akt phosphorylation by AngII was seen in Y311F expressing VSMCs (Figure 5A). In contrast, neither wild-type or Y311F expression altered AngII-induced ERK phosphorylation in VSMCs. Also, PKCδ Tyr311 phosphorylation by AngII precedes the Akt phosphorylation. Previously, we have demonstrated that K376A PKCδ had no inhibitory effect on ERK1/2 phosphorylation induced by AngII in VSMCs 15. In contrast, Akt phosphorylation induced by AngII was markedly inhibited in K376A expressing VSMCs (Figure 5B). These data suggest that AngII-induced VSMC hypertrophy is positively regulated by PKCδ kinase activation and Tyr311 phosphorylation through their involvement with Akt activation but not ERK activation.
Figure 5.

PKCδ kinase activity and Tyr311 phosphorylation are required for AngII-induced Akt activation of VSMCs expressing PKCδ mutants. A, the retrovirus-infected VSMCs (vector, wild type PKCδ, or Y311F mutant) were stimulated with 100 nmol/L AngII for the indicated time periods. An arrow indicates GFP-tagged PKCδ position. B, the adenovirus-infected VSMCs (vector or K376A mutant) were stimulated with 100 nmol/L AngII for the indicated time periods. A and B, cell lysates were immunoblotted with antibodies as indicated. The Akt Ser473 phosphorylation signal was measured by densitometry and shown as mean ± SEM (n=3). *P < 0.05 compared to the basal control. †P < 0.05 compared to the stimulated control.
Discussion
The major novel findings of this study revealed that PKCδ activation associated with Tyr311 phosphorylation mediates AngII-induced VSMC hypertrophy through a mechanism involving Akt. Also, AngII appears to induce PKCδ Tyr311 phosphorylation through the Gq-coupled AT1 receptor via Src. These findings provide a new signaling mechanism by which the AT1 receptor activation leads to PKCδ-mediated vascular remodeling and may serve as a potential therapeutic target toward cardiovascular diseases.
AngII-induced rapid PKCδ Tyr311 phosphorylation has been reported 18 with a slightly more sustained time course which may be due to a shorter serum starvation than the present study. However, the upstream regulators of PKCδ Tyr311 phosphorylation have not yet been identified. The present study using a selective Gq inhibitor indicates that the phosphorylation is mediated through Gq activation. This is in agreement with our recent observation suggesting that AngII-induced PKCδ Tyr311 phosphorylation in VSMCs requires intracellular Ca2+ elevation 33. Since the AT1 receptor is the dominant receptor expressed in our cultured VSMCs 34, we have not evaluated the possible confounding of these signal transductions by the AT2 receptor. Increasing evidence suggest the counter regulatory functions of the AT2 receptors toward the AT1 receptor-dependent functions including vascular hypertrophy as well as hyperplasia in vivo 35,36. Therefore, it will be interesting to further characterize a possible signal crosstalk of the PKCδ regulation between these subtype receptors in vivo.
Here, we report that PKCδ phosphorylation at Tyr311 by AngII is at least in part Src-dependent in VSMCs. Supporting this finding is the fact that several others have reported that PKCδ Tyr311 phosphorylation in select cell types was dependent on Src family kinases when stimulated with various non-GPCR agonists 19,37,38. Also, Src family kinases have been shown to be complexed with PKCδ in several cell types including VSMCs 13,38–40. However, possible contribution of other Src family kinases (Fyn and yes) expressed in VSMCs 41 in AngII-induced PKCδ Tyr311 phosphorylation remains to be determined. Although, we have not studied AngII-induced Src phosphorylation, such as at the positive regulatory Tyr416 residue, the Src inhibitor PP2 used in this study has been shown to block this phosphorylation effectively in VSMCs 42,43. In addition, our data suggest that the involvement of EGF receptor transactivation in the PKCδ phosphorylation by AngII is unlikely. However, the EGFR kinase inhibitor AG1478 if used at 10 times more concentration than in the present study, partially attenuated c-Src phosphorylation at Tyr416 induced by AngII in VSMCs 42. Thus, further careful evaluation may be necessary regarding the possible partial but minor involvement of the EGF receptor transactivation in this PKCδ cascade.
We have previously utilized a PKCδ inhibitor, rottlerin, to elucidate the role of this PKC isoform in signal transductions of the AT1 receptor in VSMCs 4,15. However, we have not utilized this inhibitor in the present study because of the reported off target effects 44, which would be inappropriate for our long term hypertrophic experiments. In PKCδ-deficient VSMCs, cytoskeletal signaling, reorganization and subsequent migration in response to mechanical stress was diminished 12. Also, an over-expression study using the wild type PKCδ suggested that PKCδ mediates p38 mitogen activated protein kinase (p38MAPK) activation induced by high glucose in VSMCs 45. However, by using the K376A mutant as well as rottlerin, our previous studies have shown that PKCδ kinase activity is essential for AngII-induced activation of a select set of protein kinases, which include JAK2, Rho-kinase, p21-activated kinase and c-Jun NH2-terminal kinase, but not ERK or p38MAPK 14,15,33. Thus, involvement of PKCδ in p38MAPK activation may be agonist-dependent.
It has been reported that PKCδ deficient mouse VSMCs are resistant to apoptotic responses compared to control VSMCs 46. Over expression of PKCδ in VSMC cell lines also results in G1 arrest and apoptosis 10,47. These apoptotic or necrotic changes if they occur could be associated with enlargement of cell volume 48. However, this scenario is quite unlikely in our present study because there was no difference in caspase-3 cleavage or cell viability with PKCδ over expression regardless of AngII stimulation as shown in the supplemental figures.
Here, we further revealed Akt as a PKCδ-dependent kinase in VSMCs stimulated by AngII which plays a significant role in VSMC hypertrophy 31. To support our notion, a similar link between PKCδ and Akt was observed in thrombin-induced NF-κB activation in endothelial cells 49. In addition, other mechanisms may coordinately regulate VSMC hypertrophy upon PKCδ activation by AngII such as expression of Smad3 and transforming growth factor β 11, and the Tyr311 phosphorylation of PKCδ.
Interestingly, our data suggest that AngII-induced PKCδ Tyr311 phosphorylation is also required for enhanced Akt activation and VSMC hypertrophy observed in VSMCs over-expressing wild-type PKCδ. However, the PKCδ Y311F mutant did not show a dominant-negative effect to inhibit Akt activation below the vector-transfected cells and one of the hypertrophic responses was still slightly greater than the control cells, demonstrating distinct characteristics of the PKCδ mutants. The kinase-inactive mutant, K376A, not only loses its wild-type ability to positively regulate Akt activation and subsequent hypertrophy, but also competes with endogenous PKCδ and thus acts as a dominant-negative PKCδ inhibitor. Y311F mutant also loses most of its own hypertrophic characteristics, whereas it is unable to interfere with the endogenous wild-type PKCδ. Although PKCδ Tyr311 phosphorylation has been proposed to enhance kinase activity, recent findings suggest additional roles of the Tyr311 phosphorylation in mediating unique functions of this PKC isoform 17,50. The Tyr311 phosphorylation may be the additional component required for the complex formation between PKCδ, Akt, and its upstream kinase, 3-phosphoinositide dependent kinase 1/PDK1, and subsequent Akt activation which appears to require the PKCδ kinase activity 51. Taken together, it is attractive to speculate that the PKCδ phosphorylation may contribute to Akt activation and subsequent hypertrophy independent from the kinase activity. To support this notion, we observed a comparable autophosphorylation of Y311F PKCδ at Ser643/676 phosphorylation to that of wild-type in AngII-stimulated VSMCs (unpublished observation), thus reflecting the kinase activity remains intact in Y311F mutant.
In the present study, we have not employed a standard protein synthesis assay measuring a radio-labeled leucine incorporation. However, we believe that our two distinct methods utilized here measure the hypertrophic effects of AngII just as sufficiently and perhaps more directly. Our data demonstrating hypertrophic responses by AngII stimulation in VSMCs are consistent with highly sited past articles using intact aortic segments 52 and cultured aortic VSMCs 53. Moreover, no significant enhancement of DNA synthesis was observed in AngII stimulated VSMCs regardless of PKCδ over expression. However, since our data rely on over expression strategies, a future study should be conducted by using specific RNA silencing to evaluate the overall roles of PKCδ in mediating VSMC hypertrophy induced by AngII. In addition, slight distinctions in control cell responses between Figure 3B and 4A may be caused by distinct control vectors utilized as well as by a selection of the permanently infected cells in the retroviral experiment. It is also unlikely that PKCδ Y311F mutant affects other PKC isoforms expressed in VSMCs non-specifically, because this residue is unique to PKCδ. Beside the data shown in Figure 3A, we and others have previously demonstrated the specificities of the PKCδ mutants utilized in the present study 14,54–57.
A recent study utilizing proteomic analysis of PKCδ-deficient VSMCs revealed that more than 30 proteins are altered including enzymes related to glucose and lipid metabolism, thus highlighting the critical role of PKCδ in the development of cardiovascular diseases 9. PKCδactivation increases O2 derived free radical generation from mitochondria and thereby promotes a pro-oxidant state 58. Therefore, it will be interesting to expand the present findings by evaluating the regulation of proteins such as pyruvate dehydrogenase and heat shock proteins which are likely involved in atherosclerosis as well as metabolic disorders 59,60.
Perspectives
We found that in addition to PKCδ kinase activity, PKCδ phosphorylation at Tyr311 appears to be required for Akt activation and subsequent VSMC hypertrophy induced by AngII, which is considered a potential mechanism of atherosclerosis and restenosis after vascular injury. However, our findings are limited within cell culture experiments. Therefore, further clarification of the signal transduction in vivo could contribute to a better understanding of the molecular mechanism of cardiovascular diseases as well as to the development of better strategies for their treatment.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by National Institute of Health Grants, HL076770 (S.E.), HL076575 (G.D.F.) and DE015648 (M.E.R.), by American Heart Association Established Investigator Award, 0740042N (S.E.), and by W. W. Smith Charitable Trust Grant, H0605 (S.E.).
Footnotes
Disclosures
None.
References
- 1.Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639–672. [PubMed] [Google Scholar]
- 2.Mehta PK, Griendling KK. Angiotensin II Cell Signaling: Physiological and Pathological Effects in the Cardiovascular System. Am J Physiol Cell Physiol. 2006;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 3.Liao DF, Monia B, Dean N, Berk BC. Protein kinase C-zeta mediates angiotensin II activation of ERK1/2 in vascular smooth muscle cells. J Biol Chem. 1997;272:6146–6150. doi: 10.1074/jbc.272.10.6146. [DOI] [PubMed] [Google Scholar]
- 4.Frank GD, Saito S, Motley ED, Sasaki T, Ohba M, Kuroki T, Inagami T, Eguchi S. Requirement of Ca(2+) and PKCdelta for Janus kinase 2 activation by angiotensin II: involvement of PYK2. Mol Endocrinol. 2002;16:367–377. doi: 10.1210/mend.16.2.0768. [DOI] [PubMed] [Google Scholar]
- 5.Nakashima H, Suzuki H, Ohtsu H, Chao JY, Utsunomiya H, Frank GD, Eguchi S. Angiotensin II regulates vascular and endothelial dysfunction: recent topics of Angiotensin II type-1 receptor signaling in the vasculature. Curr Vasc Pharmacol. 2006;4:67–78. doi: 10.2174/157016106775203126. [DOI] [PubMed] [Google Scholar]
- 6.Yin G, Yan C, Berk BC. Angiotensin II signaling pathways mediated by tyrosine kinases. Int J Biochem Cell Biol. 2003;35:780–783. doi: 10.1016/s1357-2725(02)00300-x. [DOI] [PubMed] [Google Scholar]
- 7.Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci. 2007;112:417–428. doi: 10.1042/CS20060342. [DOI] [PubMed] [Google Scholar]
- 8.Wakino S, Kintscher U, Liu Z, Kim S, Yin F, Ohba M, Kuroki T, Schonthal AH, Hsueh WA, Law RE. Peroxisome proliferator-activated receptor gamma ligands inhibit mitogenic induction of p21(Cip1) by modulating the protein kinase Cdelta pathway in vascular smooth muscle cells. J Biol Chem. 2001;276:47650–47657. doi: 10.1074/jbc.M108719200. [DOI] [PubMed] [Google Scholar]
- 9.Mayr M, Siow R, Chung YL, Mayr U, Griffiths JR, Xu Q. Proteomic and metabolomic analysis of vascular smooth muscle cells: role of PKCdelta. Circ Res. 2004;94:e87–96. doi: 10.1161/01.RES.0000131496.49135.1d. [DOI] [PubMed] [Google Scholar]
- 10.Ryer EJ, Sakakibara K, Wang C, Sarkar D, Fisher PB, Faries PL, Kent KC, Liu B. Protein kinase C delta induces apoptosis of vascular smooth muscle cells through induction of the tumor suppressor p53 by both p38-dependent and p38-independent mechanisms. J Biol Chem. 2005;280:35310–35317. doi: 10.1074/jbc.M507187200. [DOI] [PubMed] [Google Scholar]
- 11.Ryer EJ, Hom RP, Sakakibara K, Nakayama KI, Nakayama K, Faries PL, Liu B, Kent KC. PKCdelta is necessary for Smad3 expression and transforming growth factor beta-induced fibronectin synthesis in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2006;26:780–786. doi: 10.1161/01.ATV.0000209517.00220.cd. [DOI] [PubMed] [Google Scholar]
- 12.Li C, Wernig F, Leitges M, Hu Y, Xu Q. Mechanical stress-activated PKCdelta regulates smooth muscle cell migration. FASEB J. 2003;17:2106–2108. doi: 10.1096/fj.03-0150fje. [DOI] [PubMed] [Google Scholar]
- 13.Saito S, Frank GD, Mifune M, Ohba M, Utsunomiya H, Motley ED, Inagami T, Eguchi S. Ligand-independent trans-activation of the platelet-derived growth factor receptor by reactive oxygen species requires protein kinase C-delta and c-Src. J Biol Chem. 2002;277:44695–44700. doi: 10.1074/jbc.M208332200. [DOI] [PubMed] [Google Scholar]
- 14.Frank GD, Mifune M, Inagami T, Ohba M, Sasaki T, Higashiyama S, Dempsey PJ, Eguchi S. Distinct mechanisms of receptor and nonreceptor tyrosine kinase activation by reactive oxygen species in vascular smooth muscle cells: role of metalloprotease and protein kinase C-delta. Mol Cell Biol. 2003;23:1581–1589. doi: 10.1128/MCB.23.5.1581-1589.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohtsu H, Mifune M, Frank GD, Saito S, Inagami T, Kim-Mitsuyama S, Takuwa Y, Sasaki T, Rothstein JD, Suzuki H, Nakashima H, Woolfolk EA, Motley ED, Eguchi S. Signal-crosstalk between Rho/ROCK and c-Jun NH2-terminal kinase mediates migration of vascular smooth muscle cells stimulated by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25:1831–1836. doi: 10.1161/01.ATV.0000175749.41799.9b. [DOI] [PubMed] [Google Scholar]
- 16.Jackson DN, Foster DA. The enigmatic protein kinase Cdelta: complex roles in cell proliferation and survival. FASEB J. 2004;18:627–636. doi: 10.1096/fj.03-0979rev. [DOI] [PubMed] [Google Scholar]
- 17.Steinberg SF. Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem J. 2004;384:449–459. doi: 10.1042/BJ20040704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan M, Xu X, Ohba M, Cui MZ. Angiotensin II-induced protein kinase D activation is regulated by protein kinase Cdelta and mediated via the angiotensin II type 1 receptor in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:2271–2276. doi: 10.1161/01.ATV.0000148449.92035.3a. [DOI] [PubMed] [Google Scholar]
- 19.Konishi H, Yamauchi E, Taniguchi H, Yamamoto T, Matsuzaki H, Takemura Y, Ohmae K, Kikkawa U, Nishizuka Y. Phosphorylation sites of protein kinase C delta in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc Natl Acad Sci U S A. 2001;98:6587–6592. doi: 10.1073/pnas.111158798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeVries TA, Neville MC, Reyland ME. Nuclear import of PKCdelta is required for apoptosis: identification of a novel nuclear import sequence. EMBO J. 2002;21:6050–6060. doi: 10.1093/emboj/cdf606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanderson MP, Erickson SN, Gough PJ, Garton KJ, Wille PT, Raines EW, Dunbar AJ, Dempsey PJ. ADAM10 mediates ectodomain shedding of the betacellulin precursor activated by p-aminophenylmercuric acetate and extracellular calcium influx. J Biol Chem. 2005;280:1826–1837. doi: 10.1074/jbc.M408804200. [DOI] [PubMed] [Google Scholar]
- 22.Mifune M, Ohtsu H, Suzuki H, Nakashima H, Brailoiu E, Dun NJ, Frank GD, Inagami T, Higashiyama S, Thomas WG, Eckhart AD, Dempsey PJ, Eguchi S. G protein coupling and second messenger generation are indispensable for metalloprotease-dependent, heparin-binding epidermal growth factor shedding through angiotensin II type-1 receptor. J Biol Chem. 2005;280:26592–26599. doi: 10.1074/jbc.M502906200. [DOI] [PubMed] [Google Scholar]
- 23.Ohtsu H, Dempsey PJ, Frank GD, Brailoiu E, Higuchi S, Suzuki H, Nakashima H, Eguchi K, Eguchi S. ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler Thromb Vasc Biol. 2006;26:e133–137. doi: 10.1161/01.ATV.0000236203.90331.d0. [DOI] [PubMed] [Google Scholar]
- 24.Ohba M, Ishino K, Kashiwagi M, Kawabe S, Chida K, Huh NH, Kuroki T. Induction of differentiation in normal human keratinocytes by adenovirus-mediated introduction of the eta and delta isoforms of protein kinase C. Mol Cell Biol. 1998;18:5199–5207. doi: 10.1128/mcb.18.9.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyazaki T, Sanjay A, Neff L, Tanaka S, Horne WC, Baron R. Src kinase activity is essential for osteoclast function. J Biol Chem. 2004;279:17660–17666. doi: 10.1074/jbc.M311032200. [DOI] [PubMed] [Google Scholar]
- 26.Eguchi S, Numaguchi K, Iwasaki H, Matsumoto T, Yamakawa T, Utsunomiya H, Motley ED, Kawakatsu H, Owada KM, Hirata Y, Marumo F, Inagami T. Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J Biol Chem. 1998;273:8890–8896. doi: 10.1074/jbc.273.15.8890. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki H, Eguchi K, Ohtsu H, Higuchi S, Dhobale S, Frank GD, Motley ED, Eguchi S. Activation of Endothelial Nitric Oxide Synthase by the Angiotensin II Type 1 Receptor. Endocrinology. 2006;147:5914–5920. doi: 10.1210/en.2006-0834. [DOI] [PubMed] [Google Scholar]
- 28.Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, Kobori M. A novel Gαq/11-selective inhibitor. J Biol Chem. 2004;279:47438–47445. doi: 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- 29.Eguchi S, Frank GD, Mifune M, Inagami T. Metalloprotease-dependent ErbB ligand shedding in mediating EGFR transactivation and vascular remodelling. Biochem Soc Trans. 2003;31:1198–1202. doi: 10.1042/bst0311198. [DOI] [PubMed] [Google Scholar]
- 30.Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022–15029. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- 31.Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, Griendling KK. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274:22699–22704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 32.Ohtsu H, Suzuki H, Nakashima H, Dhobale S, Frank GD, Motley ED, Eguchi S. Angiotensin II signal transduction through small GTP-binding proteins: mechanism and significance in vascular smooth muscle cells. Hypertension. 2006;48:534–540. doi: 10.1161/01.HYP.0000237975.90870.eb. [DOI] [PubMed] [Google Scholar]
- 33.Woolfolk EA, Eguchi S, Ohtsu H, Nakashima H, Ueno H, Gerthoffer WT, Motley ED. Angiotensin II-Induced Activation of P21-Activated Kinase 1 Requires Ca2+ and Protein Kinase C {delta} in Vascular Smooth Muscle Cells. Am J Physiol Cell Physiol. 2005;289:C1286–C1294. doi: 10.1152/ajpcell.00448.2004. [DOI] [PubMed] [Google Scholar]
- 34.Eguchi S, Matsumoto T, Motley ED, Utsunomiya H, Inagami T. Identification of an essential signaling cascade for mitogen-activated protein kinase activation by angiotensin II in cultured rat vascular smooth muscle cells. Possible requirement of Gq-mediated p21ras activation coupled to a Ca2+/calmodulin-sensitive tyrosine kinase. J Biol Chem. 1996;271:14169–14175. doi: 10.1074/jbc.271.24.14169. [DOI] [PubMed] [Google Scholar]
- 35.Carey RM. Cardiovascular and renal regulation by the angiotensin type 2 receptor: the AT2 receptor comes of age. Hypertension. 2005;45:840–844. doi: 10.1161/01.HYP.0000159192.93968.8f. [DOI] [PubMed] [Google Scholar]
- 36.Johren O, Dendorfer A, Dominiak P. Cardiovascular and renal function of angiotensin II type-2 receptors. Cardiovasc Res. 2004;62:460–467. doi: 10.1016/j.cardiores.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Blake RA, Garcia-Paramio P, Parker PJ, Courtneidge SA. Src promotes PKCdelta degradation. Cell Growth Differ. 1999;10:231–241. [PubMed] [Google Scholar]
- 38.Rybin VO, Guo J, Sabri A, Elouardighi H, Schaefer E, Steinberg SF. Stimulus-specific differences in protein kinase C delta localization and activation mechanisms in cardiomyocytes. J Biol Chem. 2004;279:19350–19361. doi: 10.1074/jbc.M311096200. [DOI] [PubMed] [Google Scholar]
- 39.Song JS, Swann PG, Szallasi Z, Blank U, Blumberg PM, Rivera J. Tyrosine phosphorylation-dependent and -independent associations of protein kinase C-delta with Src family kinases in the RBL-2H3 mast cell line: regulation of Src family kinase activity by protein kinase C-delta. Oncogene. 1998;16:3357–3368. doi: 10.1038/sj.onc.1201886. [DOI] [PubMed] [Google Scholar]
- 40.Brandt DT, Goerke A, Heuer M, Gimona M, Leitges M, Kremmer E, Lammers R, Haller H, Mischak H. Protein kinase C delta induces Src kinase activity via activation of the protein tyrosine phosphatase PTP alpha. J Biol Chem. 2003;278:34073–34078. doi: 10.1074/jbc.M211650200. [DOI] [PubMed] [Google Scholar]
- 41.Ishida M, Ishida T, Thomas SM, Berk BC. Activation of extracellular signal-regulated kinases (ERK1/2) by angiotensin II is dependent on c-Src in vascular smooth muscle cells. Circ Res. 1998;82:7–12. doi: 10.1161/01.res.82.1.7. [DOI] [PubMed] [Google Scholar]
- 42.Touyz RM, Wu XH, He G, Salomon S, Schiffrin EL. Increased angiotensin II-mediated Src signaling via epidermal growth factor receptor transactivation is associated with decreased C-terminal Src kinase activity in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension. 2002;39:479–485. doi: 10.1161/hy02t2.102909. [DOI] [PubMed] [Google Scholar]
- 43.Che Q, Carmines PK. Src family kinase involvement in rat preglomerular microvascular contractile and [Ca2+]i responses to ANG II. Am J Physiol Renal Physiol. 2005;288:F658–664. doi: 10.1152/ajprenal.00392.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soltoff SP. Rottlerin: an inappropriate and ineffective inhibitor of PKCdelta. Trends Pharmacol Sci. 2007;28:453–458. doi: 10.1016/j.tips.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Igarashi M, Wakasaki H, Takahara N, Ishii H, Jiang ZY, Yamauchi T, Kuboki K, Meier M, Rhodes CJ, King GL. Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways. J Clin Invest. 1999;103:185–195. doi: 10.1172/JCI3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leitges M, Mayr M, Braun U, Mayr U, Li C, Pfister G, Ghaffari-Tabrizi N, Baier G, Hu Y, Xu Q. Exacerbated vein graft arteriosclerosis in protein kinase Cdelta-null mice. J Clin Invest. 2001;108:1505–1512. doi: 10.1172/JCI12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fukumoto S, Nishizawa Y, Hosoi M, Koyama H, Yamakawa K, Ohno S, Morii H. Protein kinase C delta inhibits the proliferation of vascular smooth muscle cells by suppressing G1 cyclin expression. J Biol Chem. 1997;272:13816–13822. doi: 10.1074/jbc.272.21.13816. [DOI] [PubMed] [Google Scholar]
- 48.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rahman A, True AL, Anwar KN, Ye RD, Voyno-Yasenetskaya TA, Malik AB. Galpha(q) and Gbetagamma regulate PAR-1 signaling of thrombin-induced NF-kappaB activation and ICAM-1 transcription in endothelial cells. Circ Res. 2002;91:398–405. doi: 10.1161/01.res.0000033520.95242.a2. [DOI] [PubMed] [Google Scholar]
- 50.Kajimoto T, Shirai Y, Sakai N, Yamamoto T, Matsuzaki H, Kikkawa U, Saito N. Ceramide-induced apoptosis by translocation, phosphorylation, and activation of protein kinase Cdelta in the Golgi complex. J Biol Chem. 2004;279:12668–12676. doi: 10.1074/jbc.M312350200. [DOI] [PubMed] [Google Scholar]
- 51.Brand C, Cipok M, Attali V, Bak A, Sampson SR. Protein kinase Cdelta participates in insulin-induced activation of PKB via PDK1. Biochem Biophys Res Commun. 2006;349:954–962. doi: 10.1016/j.bbrc.2006.08.100. [DOI] [PubMed] [Google Scholar]
- 52.Holycross BJ, Peach MJ, Owens GK. Angiotensin II stimulates increased protein synthesis, not increased DNA synthesis, in intact rat aortic segments, in vitro. J Vasc Res. 1993;30:80–86. doi: 10.1159/000158979. [DOI] [PubMed] [Google Scholar]
- 53.Geisterfer AA, Peach MJ, Owens GK. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ Res. 1988;62:749–756. doi: 10.1161/01.res.62.4.749. [DOI] [PubMed] [Google Scholar]
- 54.Fujii T, Garcia-Bermejo ML, Bernabo JL, Caamano J, Ohba M, Kuroki T, Li L, Yuspa SH, Kazanietz MG. Involvement of protein kinase C delta (PKCdelta) in phorbol ester-induced apoptosis in LNCaP prostate cancer cells. Lack of proteolytic cleavage of PKCdelta. J Biol Chem. 2000;275:7574–7582. doi: 10.1074/jbc.275.11.7574. [DOI] [PubMed] [Google Scholar]
- 55.Matassa AA, Carpenter L, Biden TJ, Humphries MJ, Reyland ME. PKCdelta is required for mitochondrial-dependent apoptosis in salivary epithelial cells. J Biol Chem. 2001;276:29719–29728. doi: 10.1074/jbc.M100273200. [DOI] [PubMed] [Google Scholar]
- 56.Kajimoto T, Shirai Y, Sakai N, Yamamoto T, Matsuzaki H, Kikkawa U, Saito N. Ceramide-induced apoptosis by translocation, phosphorylation, and activation of protein kinase Cdelta in the Golgi complex. J Biol Chem. 2004;279:12668–12676. doi: 10.1074/jbc.M312350200. [DOI] [PubMed] [Google Scholar]
- 57.Motley ED, Eguchi K, Patterson MM, Palmer PD, Suzuki H, Eguchi S. Mechanism of endothelial nitric oxide synthase phosphorylation and activation by thrombin. Hypertension. 2007;49:577–583. doi: 10.1161/01.HYP.0000255954.80025.34. [DOI] [PubMed] [Google Scholar]
- 58.Mayr M, Madhu B, Xu Q. Proteomics and metabolomics combined in cardiovascular research. Trends Cardiovasc Med. 2007;17:43–48. doi: 10.1016/j.tcm.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Kanwar RK, Kanwar JR, Wang D, Ormrod DJ, Krissansen GW. Temporal expression of heat shock proteins 60 and 70 at lesion-prone sites during atherogenesis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1991–1997. doi: 10.1161/hq1201.100263. [DOI] [PubMed] [Google Scholar]
- 60.Mayr M, Mayr U, Chung YL, Yin X, Griffiths JR, Xu Q. Vascular proteomics: linking proteomic and metabolomic changes. Proteomics. 2004;4:3751–3761. doi: 10.1002/pmic.200400947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.