

SUMMARY
Replication forks face multiple obstacles that slow their progression. By two-dimensional gel analysis, yeast forks pause at stable DNA protein complexes, and this pausing is greatly increased in the absence of the Rrm3 helicase. We used a genome wide approach to identify 96 sites of very high DNA polymerase binding in wild type cells. Most of these binding sites were not previously identified pause sites. Rather, the most highly represented genomic category among high DNA polymerase binding sites was the open reading frames (ORFs) of highly transcribed RNA polymerase II genes. Twice as many pause sites were identified in rrm3 compared to wild type cells as pausing in this strain occurred at both highly transcribed RNA polymerase II genes and the previously identified protein DNA complexes. ORFs of highly transcribed RNA polymerase II genes are the first class of natural pause sites that are not exacerbated in rrm3 cells.
Keywords: DNA replication, helicase, Rrm3, yeast, genome integrity, microarray, chromatin immunoprecipitation
INTRODUCTION
DNA replication in eukaryotic cells involves the coordinated assembly and bidirectional movement of the replisome from multiple origins per chromosome. Replication fork progression must contend with nucleosomal chromatin structure and site specific non-nucleosomal protein DNA complexes that assemble at genes and structural elements. As a result, the rate of DNA replication is not uniform throughout the genome (Raghuraman et al., 2001).
In Saccharomyces cerevisiae, sites that impede replication fork progression in vivo have been identified by two-dimensional (2D) gel analysis of replication intermediates. In wild type (WT) cells, the replication fork barrier (RFB), a site found in every ribosomal DNA (rDNA) repeat, is a highly efficient, unidirectional block to fork progression (Brewer and Fangman, 1988; Brewer et al., 1992; Linskens and Huberman, 1988). Modest pausing also occurs at centromeres (Greenfeder and Newlon, 1992), telomeres (Ivessa et al., 2002), and tRNA genes, but only when transcription and replication are oriented in opposite directions through these genes (Deshpande and Newlon, 1996). At each of these sites, replication pausing or arrest depends on a non-nucleosomal protein complex assembled at the affected site (Deshpande and Newlon, 1996; Greenfeder and Newlon, 1992; Ivessa et al., 2003; Kobayashi and Horiuchi, 1996). Many of the sites that impede fork progression exhibit elevated recombination, which can lead to gross chromosomal rearrangements, particularly when replication and checkpoints are compromised (Admire et al., 2006; Cha and Kleckner, 2002; Lemoine et al., 2005; Raveendranathan et al., 2006).
In the absence of the Rrm3 DNA helicase, pausing is substantially increased at all of the previously characterized pause sites (Ivessa et al., 2003; Ivessa et al., 2002; Ivessa et al., 2000). For example, pausing at a tRNAALA gene on chromosome VI is increased approximately 50-fold in rrm3 cells (Ivessa et al., 2003). Pausing is also detected at sites that do not slow fork progression in WT cells, such as the silent mating type loci, the 5S rDNA genes, and tRNA genes in which replication and transcription proceed in the same direction through the gene (Ivessa et al., 2003). We define Rrm3-sensitive sites as those that exhibit increased pausing in rrm3 compared to WT cells. Rrm3-sensitive replication sites are assembled into particularly stable non-nucleosomal protein complexes whose disassembly obviates the need for Rrm3 at the site during DNA replication (Ivessa et al., 2003; Torres et al., 2004a). Although Rrm3 affects replication only at discrete loci, it moves with the replication fork throughout the genome and interacts with the catalytic subunit of DNA polymerase epsilon, DNA Pol2, the leading strand DNA polymerase (Azvolinsky et al., 2006). Thus, Rrm3 is a replisome component.
The goal of this paper was to identify the most significant sites of replication fork pausing within the yeast genome in WT and rrm3 cells. We reasoned that loci that impede fork progression will have longer association with the replisome and should thus exhibit elevated association with DNA polymerase compared to sites where pausing does not occur. We assessed the distribution of DNA Pol2 throughout the genome in asynchronous cells using chromatin immunoprecipitation (ChIP) in cells expressing epitope tagged DNA Pol2, followed by hybridization to a S. cerevisiae whole genome microarray. We consider sequences with high DNA Pol2 binding to be pause sites whether high binding occurs at discrete sites or in a more regional manner.
We identified 96 sites in WT cells and 192 in rrm3 cells that have particularly high levels of DNA Pol2 binding. Unexpectedly, pausing by the criterion of very high DNA Pol2 occupancy in both WT and rrm3 cells was observed within the ORFs of highly transcribed RNA polymerase II (RNA Pol II) genes, and this pausing was transcription dependent. However, pausing within these genes was not increased in the absence of Rrm3. Thus, highly transcribed RNA Pol II genes are the first examples of natural pause sites that are not exacerbated in rrm3 cells. The most significant and frequently identified pause sites in rrm3 cells were previously identified Rrm3-sensitive replication sites such as telomeres and tRNA genes. Thus, in WT cells, the strongest impediments to fork progression are within highly transcribed RNA Pol II genes and only rarely at Rrm3-sensitive pause sites that occur at stable protein DNA complexes.
RESULTS
Experimental Rationale
To determine sites of replication pausing, we tagged the catalytic subunit of the leading strand DNA polymerase epsilon, DNA Pol2, with 13 MYC epitopes at its carboxy-terminus (DNA Pol2-MYC) in WT and rrm3 cells (Azvolinsky et al., 2006). Rrm3 was similarly tagged (Rrm3-MYC). Asynchronously growing log phase cultures were formaldehyde cross-linked in vivo and processed for ChIP. The immuno-precipitated DNA and corresponding input DNA were fluorescently labeled and hybridized to a whole genome DNA microarray (Lieb et al., 2001). We used asynchronous rather than synchronous cultures because replication fork pausing is more likely to be captured with asynchronous cells. At 26°C, yeast replication forks move at 3 kb/min (Raghuraman et al., 2001; Yabuki et al., 2002). At this speed, it takes only ~two seconds for the fork to move through a tRNA gene and ~20 seconds to move through a one kb ORF. Thus, it is difficult to detect pausing using synchronized cells.
Sites of high DNA Pol2-MYC and Rrm3-MYC association were determined using ChIPOTle, a software tool that uses a sliding window approach to identify significant binding sites (Buck et al., 2005). We used a stringent cutoff of p<10−4 for ChIPOTle, as this maximizes the number of sites identified in the tagged strain without increasing the number of sites identified in the untagged strain. We also used a ChIPOTLe-independent metric, the normalized enrichment spot values, to determine and rank high occupancy binding sites.
Identification of high DNA polymerase occupancy sites in WT cells
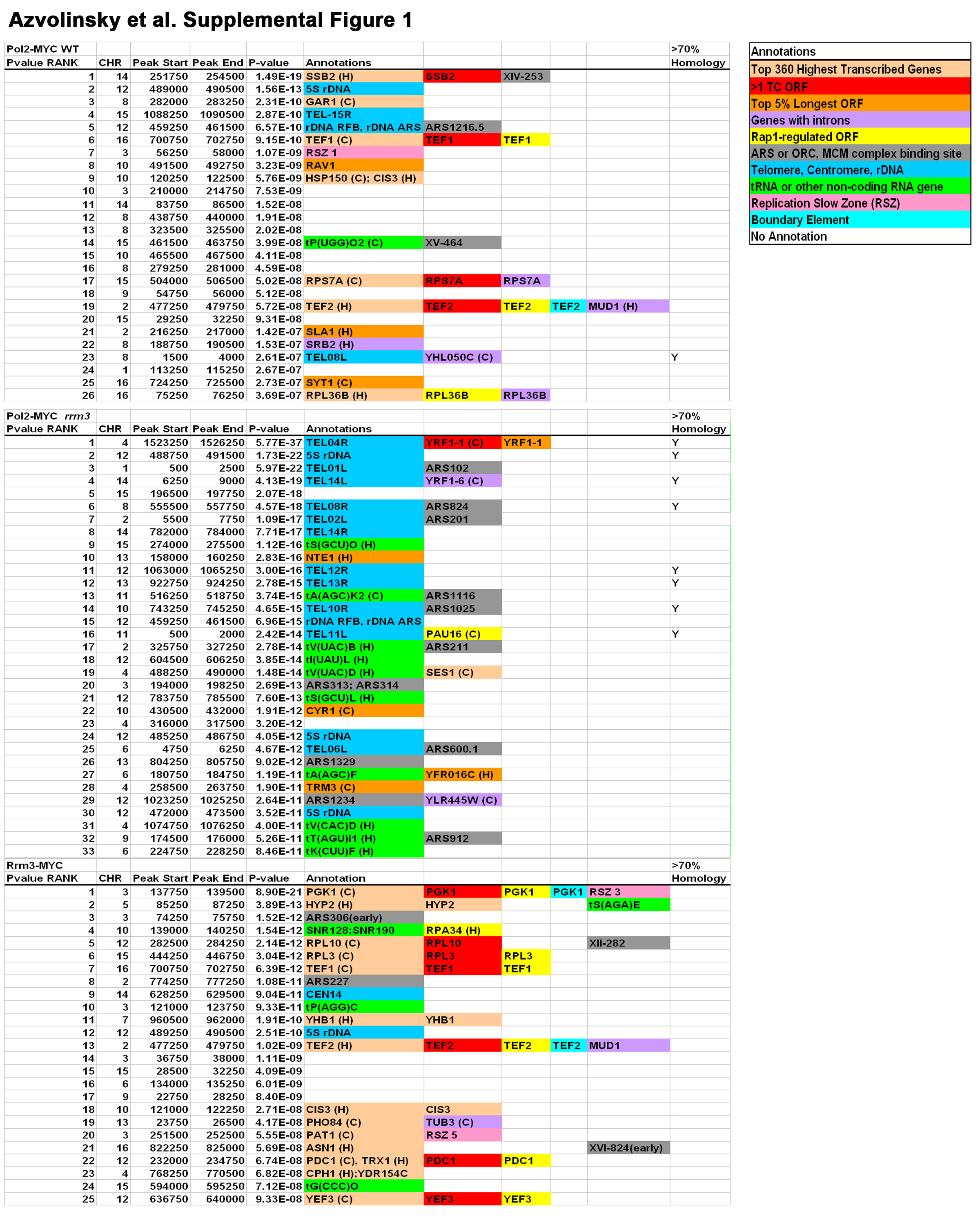
We identified 96 sites in the S. cerevisiae genome that were associated with unusually high levels of DNA Pol2-MYC, including several previously identified sites of replication fork pausing (see Fig. 1A for classes of sites with highly significant DNA Pol2-MYC association; see Supplemental Fig. 1 for the 25 most significant DNA Pol2-MYC binding sites). By 2D gels, the greatest impediment to fork progression is the RFB within the rDNA. Forks arrest at the RFB in the 10–20% of repeats with active origins (Brewer and Fangman, 1988; Brewer et al., 1992; Linskens and Huberman, 1988). Consistent with these data, the rDNA RFB was among the most significant DNA Pol2-MYC high occupancy sites in WT cells (see supplemental methods for p values). However, only ten other previously identified pause sites, (four telomeres, three tRNA genes, three inactive origins) were high occupancy DNA Pol2-MYC binding sites in WT cells.
Figure 1. High occupancy DNA Pol2-MYC and Rrm3-MYC sites by genomic class in WT and rrm3 cells.

A. The proportion of the 96 DNA Pol2-MYC WT, 192 DNA Pol2-MYC rrm3, 115 Rrm3-MYC, and 99 untagged high occupancy sites that overlap with listed genomic features (white bars) and expected proportions (black bars) are plotted. Because high occupancy sites may overlap with more than one genomic feature, numbers do not add up to the total number of high occupancy sites. Numbers in parentheses are the actual number of high occupancy sites that overlap with the indicated feature. Asterisks denote class enrichments that were significantly different from both the expected proportion and the number of sites that overlapped with the particular genomic class in the untagged control. See supplemental methods for p values. B. Rrm3-MYC and DNA Pol2-MYC both WT and rrm3 cells are highly associated with highly transcribed genes while Rrm3-sensitive sites are enriched only in DNA Pol2-MYC rrm3 cells. The plots are DNA Pol2-MYC normalized log(2) values with 250 bp sliding window averages of arrayed elements using ChIPOTle from untagged (black broken line), DNA Pol2-MYC WT (black), DNA Pol2-MYC rrm3 (gray), and Rrm3-MYC (dashed black). Enrichment values are plotted along 12 kb of chromosome VII (Fig 1B, top; includes highly transcribed TDH3) and 17 kb of chromosome XVI (Fig. 1B, bottom; includes highly transcribed TEF1). Annotations corresponding to the coordinates are shown below the plot (dark gray, characterized ORF; light gray, uncharacterized ORF, tRNA, or origin of replication). tW(CCA)G2 adjacent to TDH3 and inactive ARS1625 adjacent to TEF1 were significant DNA Pol2-MYC binding sites in rrm3 cells only.
DNA polymerase is enriched at the majority of Rrm3-sensitive pause sites only in an rrm3 background
We also determined high occupancy DNA Pol2-MYC sites in an rrm3 strain (see Fig. 1A and Supplemental Fig. 1). 192 sites were significantly associated with DNA Pol2-MYC in rrm3 cells, twice the number in WT cells. In contrast to WT cells, over half (61%) of the high occupancy DNA Pol2-MYC sites in rrm3 cells were known Rrm3-sensitive sites. All classes of Rrm3-sensitive replication sites, except centromeres and the silent mating type loci, were over-represented (Fig. 1A). Moreover, the number of Rrm3-sensitive sites is an underestimate as 19 of the DNA Pol2 high occupancy sites contained at least two Rrm3-sensitive sites but were counted as one site. tRNA genes were particularly enriched in rrm3 cells, comprising 38% (73 out of 192) of the DNA Pol2-MYC high occupancy sites compared with only three tRNA genes among high occupancy sites in WT cells. This number is also likely an under-estimate as six of the high occupancy sites contain two or three tRNA genes. Consistent with 2D gel data (Ivessa et al., 2003), tRNA genes in rrm3 cells were highly associated with DNA Pol2-MYC whether they were replicated in the same (27 genes, p=10−23) or opposite direction (53 genes, p=10−68) with respect to transcription (Supplemental Fig. 2A). Other RNA Pol III transcribed genes, the known Rrm3-sensitive 5S rDNA genes and the highly transcribed SCR1 and RPR1 genes (Dieci et al., 2002; Felici et al., 1989) were also high occupancy DNA Pol2-MYC sites in rrm3 cells.
DNA Pol2-MYC binding sites were also highly enriched for inactive and late firing/rarely active origins of replication in rrm3 cells. For example, DNA Pol2-MYC exhibited strong association with the always inactive ARS313 and the rarely active ARS314 (Fig. 2C). Pausing at ARS313 and 314 was also detectable by 2D gels in rrm3 but not WT cells (Fig. 2D). Using the origin classification scheme from the Replication Origin Database (Fig. 2A) and excluding the rDNA ARS, 49 (25%) of the 192 DNA Pol2-MYC high occupancy sites in rrm3 cells were either late firing/rarely active (21 sites) or inactive (28 sites) origins (p=10−43) (Fig.1A and 2B). The association of DNA Pol2-MYC with origins was confirmed by plotting a moving average of normalized spot values based on their distance from an origin. Using this metric, DNA Pol2-MYC was significantly associated with inactive and late firing/rarely active origins but not with early origins in rrm3 cells (Fig. 2E, left), while in WT cells, DNA Pol2-MYC was not highly associated with any origin class (Fig. 2E, right ).
Figure 2. DNA Pol2-MYC in rrm3 cells is associated with inactive and late/rarely active origins of replication.

A. Distribution of classes of origins of replication (see supplemental methods for description of origin classes). B. Distribution of origin classes among the 57 origins in the 192 high occupancy DNA Pol2-MYC binding sites in rrm3 cells. C. Pattern of binding within the ARS313/ARS314 region in Rrm3-MYC and DNA Pol2-MYC in WT and rrm3 cells. This region is a high occupancy DNA Pol2-MYC site in rrm3 but not WT cells. See Fig. 1B for details. D. ARS313 and 314 are Rrm3-sensitive replication sites. DNA from WT and rrm3 cells was EcoRV digested, separated on 2D gels, and analyzed by Southern blotting. Schematic indicates the locations of the inactive ARS313 and rarely active ARS314. Gray circles indicate inactive origins; open circle indicates the nearest efficient replication origin (ARS315) whose activation is usually responsible for replicating this region (Poloumienko et al., 2001). Arrows denote pauses. E. DNA Pol2-MYC is associated with late/inactive and inactive origins in rrm3 (left panel) but not WT cells (right panel). Sliding window average of enrichment values versus distance from replication origins within an origin class. The positions of all probes on the arrays were mapped relative to the positions of early, inactive/late, and inactive origins (Fig. 2A). A sliding window (30 bp, 1 bp step size) was used to calculate the average DNA Pol2-MYC enrichment from the origins (0 bp to 4.5 kb away) in both WT and rrm3 cells. Moving averages of DNA Pol2-MYC ChIP enrichments are plotted for inactive (black solid line), inactive/late (solid gray), and early origins (black broken line) in rrm3 (left) and WT (right) cells. Rrm3-MYC and untagged plots are similar to Pol2-MYC WT for all origins (data not shown).
DNA Pol2-MYC was much more highly associated with telomeres in rrm3 (p=10−10) compared to WT (p=10−3) cells. Eleven and four of the 26 telomeres that can be distinguished unambiguously in the arrays were significant DNA Pol2-MYC sites in, respectively, rrm3 and WT cells (Supplemental Table 3).
Highly transcribed genes are sites of replication fork pausing in WT and rrm3 cells
Of the ~5000 genes in the S. cerevisiae genome that have measurable transcripts in cells growing exponentially in complete medium, 360 are in the top 5% for either transcript abundance (mRNAs/cell), transcription rate (mRNAs/hr), or both (Holstege et al., 1998; Nagalakshmi et al., 2008). Of the 96 sites that are highly associated with DNA Pol2-MYC in WT cells, 25 (26%) overlapped with these highly transcribed genes (p=10−18; Fig. 1A). DNA Pol2 occupancy was high regardless of whether replication and transcription moved in the same (p = 10−8) or opposite direction (p = 10−5) through the genes (Supplemental Fig. 2A). DNA Pol2-MYC was also highly associated with genes in the top 5% by transcript level (p=10−71) or transcription rate (p=10−69) when monitored by normalized spot values. Highly transcribed RNA Pol2 transcribed genes were similarly enriched within rrm3 cells, comprising 27 of the DNA Pol2-MYC high occupancy sites (Fig. 1A).
Pausing can be seen by 2D gels in a subset of highly transcribed RNA polymerase II genes that are also high occupancy DNA Pol2-MYC binding sites
We used 2D gels to see if this technique revealed fork pausing in seven highly transcribed genes that were high occupancy DNA Pol2-MYC binding sites by genome wide analysis (Fig. 1B, 4A). Replication and transcription move in opposite directions through four of these genes (TEF2, PDC1, RPL10 and RPS3) and in the same direction through three (PGK1, TEF1, RPL3). Discrete pauses were seen within the ORFs of two of the seven genes, TEF2 and PDC1 and in a regional manner in TEF1 and RPS3, although the effects are subtle (Fig. 4B and data not shown; brackets denote region of ORF, arrows indicate discrete pauses). These replication patterns were not affected by deleting RRM3 (Fig. 4B).
Figure 4. Highly transcribed genes are high occupancy DNA Pol2-MYC sites in WT and rrm3 cells as well as high occupancy Rrm3-MYC sites.

A. Association of DNA Pol2-MYC in WT and rrm3 cells and Rrm3-MYC with 12 kb regions on chromosomes XII, II and III, as in Fig. 1B. These regions contain the highly transcribed PDC1, TEF2, and PGK1 genes. Each gene was significantly associated with DNA Pol2-MYC in WT and rrm3 cells and with Rrm3-MYC. B. Schematics of restriction enzyme digested DNA with replication intermediates as visualized by 2D gels: 1N, non-replicating fragment; 2N, almost fully replicated fragment; P, replication pause; dotted line, arc of linear DNA molecules. C. DNA from WT and rrm3 cells was restriction enzyme digested, separated on 2D gels, and analyzed by Southern blotting at the indicated gene (restriction enzymes: EcoRV, TEF1 and PGK1; PstI, TEF2 and PDC1; BglII, RPS3; NdeI, TEF2). Brackets mark ORFs; arrows mark pauses. Pausing within TEF2 was also detectable in NdeI digested DNA when the pause was ~80% through the arc of replication intermediates.
Another replisome component, Rrm3, is also enriched at highly transcribed RNA polymerase II genes
The paucity of previously identified pause sites and the prevalence of highly transcribed RNA polymerase II genes among high occupancy DNA Pol2-MYC sites were surprising. Therefore, we confirmed these results by studying the association of a second replisome component, Rrm3-MYC (Fig .1A). The pattern of Rrm3-MYC binding was very similar to that seen for DNA Pol2-MYC (Fig. 1B, Fig. 2C, Fig. 4A, Supplemental Fig. 4). Using the same high stringency criteria, we identified 115 high occupancy Rrm3-MYC sites (see Fig. 1A for classes of sites with highly significant Rrm3-MYC association; Supplemental Fig. 1 for 25 most significant Rrm3-MYC binding sites). Only seven of the 25 most significant Rrm3-MYC binding sites were known Rrm3-sensitive sites, including 5S rDNA, three tRNA genes, CEN14, and two inactive origins; tRNA genes were the only class of Rrm3-sensitive sites that were significantly associated with Rrm3-MYC (p=10−5; Fig. 1A). These data support the conclusion that most Rrm3-sensitive sites do not have major effects on fork progression when Rrm3 is present.
The most frequent category of high occupancy Rrm3-MYC sites were highly transcribed RNA polymerase II genes (49 sites, 43%; p = 10−47). As with high occupancy DNA Pol2-MYC sites, highly transcribed genes that bound high levels of Rrm3-MYC were similarly distributed among genes where transcription and replication are co-directional (25 genes; p = 10−10) and those where replication and transcription collide (24 genes; p= 10−13). The results were the same when we considered only those highly transcribed genes that are within 10 kb of an early firing, efficient origin (Supplemental Fig. 2A).
DNA polymerase occupancy at highly transcribed genes is not increased in rrm3 cells
A striking aspect of the DNA Pol2-MYC binding to highly transcribed genes was that the level of binding was indistinguishable in WT and rrm3 cells. This similarity in DNA Pol2-MYC binding levels was evident from the traces of individual highly transcribed genes (Fig. 1B, 4). In the same traces, there was significantly higher DNA Pol2-MYC binding in rrm3 compared to WT cells at Rrm3-sensitive sites, such as at the tRNA genes to the left of TDH3 and the inactive ARS1625 to the left of TEF1 (Fig. 1B). The same pattern was evident from the validation experiments using ChIP and qPCR where enrichment of DNA Pol2-MYC within the ORFs of three highly transcribed genes was not higher in rrm3 compared to WT cells (Supplemental Figure 3). Thus, although highly transcribed RNA Pol II genes are high occupancy DNA Pol2-MYC binding sites in both WT and rrm3 cells, pausing is not increased at these genes in the absence of Rrm3.
DNA polymerase association is higher within the open reading frames compared to the promoters of highly transcribed genes
We used standard ChIP followed by qPCR to assess if DNA Pol2-MYC and Rrm3-MYC association in WT cells was higher at the promoters or ORFs of three highly transcribed RNA Pol II genes, PGK1, TEF1, and TEF2. Association of DNA Pol2-MYC and Rrm3-MYC were two to three-fold higher within the ORFs compared to the promoters of these genes (Fig. 5A, right panel). We further assessed whether DNA Pol2-MYC and Rrm3-MYC were associated with promoters or ORFs of highly transcribed genes in the genome wide data using a ChIPOTle independent metric (Fig. 5B). In the untagged strain, regardless of transcription rate, neither promoters (blue) nor ORFs (red) were enriched in the anti-MYC ChIP (Fig. 5B, left panel). In both WT and rrm3 cells (middle panels), DNA Pol2-MYC was associated at similarly low levels with ORFs and promoters of genes transcribed at rates below 25 mRNAs/hr (Fig. 5B, middle panel; arrows indicate 25 mRNAs/hr). However, DNA Pol2-MYC was associated with ORFs but not promoters of genes transcribed at ≥25 transcripts/hr in both WT and rrm3 cells. As all genes in the top 5% by transcription rate produce ≥25 transcripts/hr (Holstege et al., 1998), the transition to high DNA Pol2 enrichment at this value is consistent with the preferential association of DNA Pol2-MYC with highly transcribed genes in both strains (Fig. 1A). Likewise, Rrm3-MYC was preferentially associated with ORFs, not promoters, of highly transcribed genes (Fig. 5B, right panel).
Figure 5. DNA Pol2-MYC binding at highly transcribed genes is within ORFs, not promoters.

A. Diagrams (left) of the PGK1 region on chromosome III, TEF1 region on chromosome XVI, and TEF2 region on chromosome II including positions of nearest early firing replication origin. Forks moving from these origins are responsible for replication of the nearby genes in most cells. Gray arrows indicate ORFs and direction of transcription. Horizontal bars indicate positions of amplified segments (RT probe). Cells expressing DNA Pol2-MYC or Rrm3-MYC were processed for ChIP and qPCR. Results are plotted as % of input DNA in the immunoprecipitate (% IP’ed). Error bars are standard deviations from 3 to 8 biological replicates with two real-time PCR replicates per biological replicate. The sequence being amplified is indicated on the x-axis (ORFs, gray bars; promoters, white bars). The difference in DNA Pol2-MYC binding within the ORF versus its promoter was significant for PGK1, TEF1, and TEF2 (p=0.005, 0.03, and 0.01, respectively) and for Rrm3-MYC binding for PGK1, TEF1, and TEF2 (p= 0.007, 0.03, and 0.0006 respectively).
B. DNA Pol2-MYC is highly associated with ORFs, not promoters, of highly transcribed RNA Pol II genes. Enrichment values of ORFs (red) and promoters (blue) are plotted as a function of transcription rate (TR, mRNAs/hour) (Holstege et al., 1998) for untagged, Pol2-MYC WT, DNA Pol2-MYC rrm3, and Rrm3-MYC cells. Arrows indicate 25 mRNAs/hr.
C. DNA Pol2-MYC and Rrm3-MYC are more likely to be highly associated with ORFs whose TRs require more than one simultaneous transcription event. The enrichments arrayed ORFs are plotted as a function of the difference between the actual and theoretical number of mRNAs produced for the corresponding ORF in one hour for untagged, Pol2-MYC WT, DNA Pol2-MYC rrm3, and Rrm3-MYC cells. Actual number of mRNAs produced is based on measured transcription rates (Holstege et al., 1998).
DNA polymerase is more likely to be associated with RNA Pol II transcribed genes if they contain multiple transcription complexes
We reasoned that the transition to high DNA Pol2-MYC and Rrm3-MYC occupancy at genes transcribed at ≥25 mRNA molecules per hr may indicate that pausing is more likely if there are multiple RNA Pol II complexes per gene. For each gene, we calculated a theoretical maximum number of transcripts produced by a single polymerase based on an in vivo RNA Pol II synthesis rate of 20 nucleotides/sec and the length of each ORF (Uptain et al., 1997). We compared this theoretical maximum number of transcripts to the measured number of transcripts produced from each gene per hour. We then plotted the occupancy of DNA Pol2-MYC in WT and rrm3 cells (or Rrm3-MYC in WT cells) within ORFs versus the difference between the actual and the theoretical number of mRNAs produced (Fig. 5C). This analysis revealed that DNA Pol2-MYC and Rrm3-MYC were more likely to be bound at high levels within ORFs when their transcription requires two or more simultaneously active transcription complexes in order to achieve their measured transcription rates (Fig. 5C).
There are 99 RNA Pol II transcribed genes whose transcript abundance and ORF length require more than one RNA Pol II complex per gene (Supplemental Table 4). Of these, 15 and 17 were high occupancy DNA Pol2-MYC sites in, respectively, WT and rrm3 cells, while 23 were high occupancy Rrm3-MYC sites (Fig 1A). 87 of these 99 genes were also among the 360 most highly transcribed genes. Two of the 12 genes that require more than one bound RNA Pol II complex but are not among the 360 most highly transcribed genes were high occupancy DNA Pol2 sites: YNL339C in WT and rrm3 cells and YGR296W in WT cells only. Thus, as a genomic class, genes whose transcription requires multiple transcription complexes per gene were significantly enriched among high occupancy binding sites for Rrm3-MYC (p=10−28) and DNA Pol2-MYC in WT (p=10−17) and rrm3 (p=10−15) cells (Fig. 1A).
Replication pausing at highly transcribed genes is transcription dependent
DNA Pol2-MYC was not enriched at GAL genes in our genome wide study, which was carried out with glucose grown cells, where GAL genes are not transcribed (Supplemental Fig. 4). To determine whether robust DNA Pol2-MYC or Rrm3-MYC binding to highly transcribed genes is transcription dependent, we used ChIP and qPCR to monitor association of these proteins within the GAL7 and GAL10 ORFs in galactose grown cells, when both genes are induced ~1000-fold and in raffinose grown cells, when neither gene is transcribed (Fig. 6A).
Figure 6. High association of DNA Pol2-MYC with highly transcribed genes is not Rap1 dependent.

A. Diagram of GAL7 and GAL10 region on chromosome II (see Fig 5A for symbols).
B. High DNA Pol2-MYC binding in WT and rrm3 cells is enriched within the GAL7 and GAL10 ORFs only in galactose grown cells (galactose, black bars; raffinose, white bars) (p=0.01 and 0.0006 for GAL7 in WT and rrm3, respectively. p=0.02 and 0.0073 for GAL10 in WT and rrm3, respectively). C. DNA Pol2-MYC enrichment at highly transcribed TEF2 and PGK1, whose transcription is carbon source independent, is not affected by carbon source (galactose, black bars; raffinose, white bars; p=0.73 and 0.07 for TEF2 and PGK1 in WT cells, and 0.27 and 0.85 for TEF2 and PGK1 in rrm3 cells, respectively). D. Diagram of modified RPL24A-RPL30 region integrated at URA3 and endogenous RPL24A-RPL30 genes on chromosome VII (see Fig 5A for symbols). RR, two Rap1 binding sites. E. mRNA abundance and transcription rate (Holstege et al., 1998) of the endogenous RPL30 and RPL24A genes and the predicted abundance and transcription rate of the mutated (rr) construct. F. Cultures expressing DNA Pol2-MYC with the integrated reporter construct (panel D) with either WT Rap1 binding sites (RR) or mutated Rap1 binding sites (rr) were processed for ChIP and qPCR. Amplifed sequences are indicated on x-axis.
G. Association of DNA Pol2-MYC with GFP ORF was indistinguishable in RR and rr strains (p=0.12).
In both WT and rrm3 cells, DNA Pol2-MYC binding was ~3-fold higher at both genes when they were transcribed (galactose) compared to when they were not (raffinose medium; Fig. 6B). We also examined DNA Pol2-MYC association in the same cultures with two highly transcribed genes, TEF2 and PGK1, whose transcription is carbon source independent. Both of these genes exhibited high DNA Pol2-MYC binding in our genome wide analysis, and both showed equivalently high DNA Pol2-MYC binding in raffinose and galactose medium by qPCR analysis in WT and rrm3 cells (Fig. 6B). Rrm3-MYC association with GAL genes was also transcription dependent while its high level of association with TEF2 and PGK1 was carbon source independent (data not shown).
Rap1 binding is not required for high DNA polymerase occupancy at highly transcribed genes
Almost half (89 of 217) of Rap1 regulated genes are highly transcribed. Of the 96 significant DNA Pol2-MYC associated loci in WT cells, 12 were the ORFs of Rap1 regulated genes (p=10−9) (Fig. 1A), and 10 of these were among the top 360 most highly transcribed genes (p=10−11). In addition to its role as a transcriptional activator, Rap1 is the major binding protein at telomeres, which are sites of fork pausing (Ivessa et al., 2002). Thus, we determined if replication pausing at highly transcribed genes was due to Rap1.
For these studies, we used an integrated construct that was designed to test the contributions of Rap1 to transcription of the two divergently transcribed RPL30 and RPL24A ribosomal protein genes that are both very highly transcribed and are known to be Rap1 regulated (Zhao et al., 2006). These genes are separated by a 609 bp region that contains two in vivo Rap1 binding sites. In this construct, the RPL30 and RPL24A ORFs are replaced, respectively, by G418r and GFP. The 609 bp intergenic region is either unchanged (RR construct) or the Rap1 sites are removed (rr construct) (Fig. 6D). Either the RR or rr RPL30/RPL24 cassette was introduced at the URA3 locus in the WT Pol2-MYC strain, leaving the endogenous RPL30/RPL24A locus unchanged. By northern analysis, removal of the Rap1 sites causes a 75% reduction in transcript levels from both RPL30 and RPL24 (Zhao et al., 2006). However, because the transcription rate of WT RPL30 is so high, even with a 75% reduction, its residual transcription still places it among the top 6% of transcribed genes (Fig. 6E).
Using ChIP and qPCR, we determined the association of DNA Pol2-MYC in WT asynchronous cells carrying the GFP ORF under the control of either the RR or rr promoter. As a control, we assayed DNA Pol2-MYC binding to the endogenous RPL30 ORF in the same cells. DNA Pol2-MYC binding to the endogenous RPL30 ORF was similarly high in cells carrying the RR or the rr construct (Fig. 6G). DNA Pol2-MYC association was also statistically indistinguishable within the GFP ORF when transcription levels were very high (RR construct) or when they were reduced by 75% (but still high) (rr construct; Fig 6F). Thus, high DNA Pol2-MYC association to the RPL30 ORF is not due to Rap1 binding.
The association of DNA polymerase with highly transcribed ORFs occurs mostly during their time of replication
DNA polymerases function not only in chromosome replication, but also in repair. To determine if the association of DNA Pol2 at highly transcribed genes occurred at their time of replication, we used alpha factor arrest to synchronize a strain expressing both DNA Pol2-HA and Rrm3-MYC, and examined association of both proteins with three highly expressed RNA Pol II transcribed genes as cells progressed through S phase at 17°C (Fig. 7). The highly transcribed PGK1 gene is replicated co-directionally from the early and efficiently firing ARS309 (Poloumienko et al., 2001) (diagram Fig. 5A). DNA Pol2-HA and Rrm3-MYC were both maximally associated with this origin 36 min after release from alpha factor. Both proteins were maximally associated with the PGK1 ORF, which is 6 kb from ARS309 at 48 and 60 min and with tRNAGLY, which is 10 kb from ARS309 at 60 min. Neither protein was PGK1 associated at earlier time points nor in G2 phase. Similarly, the association of DNA Pol2-HA and Rrm3-MYC peaked at ARS1625, an inactive origin 6 kb from TEF1 (diagram, Fig. 5A), at 48 and 60 min and with TEF1 at 60 and 72 min. TEF2 is maximally associated with the two replisome proteins at 48 and 60 min after release. Thus, the association of both DNA Pol2-HA and Rrm3-MYC with three of three highly transcribed RNA Pol II genes was limited to a discrete interval in S phase.
Figure 7. DNA polymerase and Rrm3 are associated with highly transcribed RNA Pol II genes only during replication.

WT cells expressing both DNA Pol2-HA and Rrm3-MYC were arrested in G1 phase with alpha factor. After removal from alpha factor, cells were grown at 17° C and allowed to proceed synchronously through the cell cycle as monitored by FACS (panel B). At 12 minute intervals, samples were taken for FACS and cultures were split, processed for ChIP using either HA or MYC antibodies and then analyzed by qPCR. ChIP of Rrm3-MYC (black squares) and Pol2-HA (gray squares) from the same culture. See Fig. 5A for structures of the PGK1, TEF1, and TEF2 regions and positions of PCR primers. Real-time primers are indicated in top left corner.
DISCUSSION
Most studies on replication fork progression are carried out in cells subjected to exogenous DNA damage. However, even in unperturbed cells, replication forks must contend with obstacles that have the potential to slow fork progression in every S phase. In S. cerevisiae, our understanding of natural sites of fork slowing comes mainly from 2D gels, which reveal fork arrest at the RFB, which prevents collisions between transcription and replication within the rDNA and transient pauses at centromeres, telomeres, inactive replication origins, and tRNA genes. Replication fork pausing at all of these natural pause sites is increased in the absence of Rrm3 (Ivessa et al., 2003; Ivessa et al., 2002; Ivessa et al., 2000).
Here we used genome wide approaches to identify sites that slow fork progression in WT and rrm3 cells. To identify these loci, we mapped the level of association of an essential replisome component, DNA Pol2, throughout the genome. Although transient binding of DNA Pol2 occurs at all DNA sequences during every S phase, unusually high DNA Pol2 binding can be used to identify sites or regions where forks have a higher than average transit time (Azvolinsky et al., 2006). Results with DNA Pol2 were confirmed by studying the behavior of a second replisome component, Rrm3.
Using stringent criteria, we identified 96 natural pause sites in WT cells. Consistent with the observed fork arrest at this site, the RFB was one of the most significant DNA Pol2 binding sites (Supplemental Fig. 1A). However, only 11 of the high occupancy DNA Pol2 sites in WT cells were previously identified pause sites (Fig. 1A). Rather, the most significant class of pause sites in WT cells was highly transcribed RNA Pol II genes (Fig. 1A). The elevated association of DNA Pol2 with highly transcribed RNA Pol II genes was confirmed at a subset of these genes using standard ChIP plus qPCR (Fig. 5A). Similar results were seen with Rrm3, another replisome component. That is, high occupancy Rrm3 binding sites were only rarely previously identified pause sites. Rather the most robust and most frequent Rrm3 binding sites were the ORFs of RNA Pol II highly transcribed genes (Fig. 1A). In contrast, in rrm3 cells, where pausing at stable protein complexes by 2D gels is greatly increased (Ivessa et al., 2003; Ivessa et al., 2002; Ivessa et al., 2000), there were twice as many high occupancy DNA Pol2 sites, and over half (61%) of these were previously identified, Rrm3-sensitive sites. Thus, the relatively low levels of DNA Pol2 and Rrm3 binding to previously identified pause sites in WT cells is not due to some inherent difficulty identifying these sites using a ChIP-microarray approach.
Not only do the pauses identified by 2D gels rarely show high DNA Pol2 or Rrm3 binding in WT cells, only a subset of the RNA Pol II genes that were high occupancy DNA Pol2 sites by microarray analysis had discrete pauses in 2D gels (Fig. 4B). Why do 2D gels and ChIP-microarray analyses yield different views of the replication landscape in terms of fork progression? 2D gels are best at detecting pauses that are localized to discrete regions of 100-200 bps, such as the pauses at tRNA genes and centromeres and the arrest at the RFB. In contrast, fork slowing throughout the ORF of a highly transcribed RNA Pol II gene (Fig. 4B) is more difficult to detect by 2D gels. For example, in otherwise WT cells, replication pausing by 2D gels is not detected within the rDNA when the RFB is inactivated unless there are a very small number of rDNA repeats (Takeuchi et al., 2003). 2D gels are exquisitely sensitive at detecting pauses at discrete sites, even if the extent of pausing is quite modest, as it is at most previously identified pause sites in WT cells (Deshpande and Newlon, 1996; Greenfeder and Newlon, 1992; Ivessa et al., 2002). Indeed, only arrest at the RFB produces a robust 2D gel signal in WT cells. However, in rrm3 cells, 2D gel analysis reveals much stronger signals at previously identified pause sites (Ivessa et al., 2003; Ivessa et al., 2002; Ivessa et al., 2000). Consistent with results from 2D gels, many of these sites had high DNA Pol2 occupancy in rrm3 cells. These experiments emphasize the importance of Rrm3 for replication fork progression throughout the yeast genome since Rrm3-sensitive sites had very little impact on fork progression by the criteria of DNA Pol2 or Rrm3 occupancy in cells with a functional Rrm3 helicase.
There are hints from earlier studies that highly transcribed RNA Pol II genes slow fork progression. High RNA Pol II transcription from the strong yeast GAL promoter induces recombination (Thomas and Rothstein, 1989), which is replication dependent when the affected genes are on plasmids (Prado and Aguilera, 2005). The latter study also detected transcription dependent replication pausing by 2D gels within two of two plasmid-borne RNA Pol II transcribed genes. Likewise, transcription dependent fork pausing has been seen in E. coli both in vitro using phage components (Elias-Arnanz and Salas, 1997; Liu and Alberts, 1995) and in vivo on plasmids (Mirkin et al., 2006; Mirkin and Mirkin, 2005). However, our study is the first in any organism to document pausing within highly transcribed ORFs in a chromosomal context and to demonstrate the generality of this effect.
Our studies also document requirements for fork pausing at highly transcribed RNA Pol II genes. First, DNA Pol2 and Rrm3 association with highly transcribed genes was limited to a short period during S phase (Fig. 7), indicating that association was due to replication, not repair. Second, pausing was not only transcription dependent (Fig. 6B, C), it was also more frequent in genes having two or more active transcription complexes (Fig. 5C). DNA Pol2 binding occurred throughout the ORF, was higher within ORFs than at promoters (Fig. 5A, B; Supplemental Fig. 3) and was not due to Rap1 binding (Fig. 6F). Taken together, these results suggest that it is the transcribing polymerase complex itself and/or the nascent RNA, not trans-activators or other promoter associated complexes that impede fork progression. In support of this interpretation, ORFs of highly transcribed RNA Pol II genes were equally likely to be high DNA Pol2 and Rrm3 occupancy sites regardless of whether replication moved through the genes in the same or opposite direction as transcription (Supplemental Fig. 2A). In contrast, in rrm3 cells, tRNA genes were less likely to be high occupancy DNA Pol2 sites if they were replicated and transcribed in the same direction (27 genes, p=10−23) than when replication and transcription collide (53 genes, p=10−68). Since the average RNA Pol II ORF is about ten times larger than a tRNA gene, its orientation independent effects on replication fork progression may reflect increased torsional constraints due to the presence of multiple active transcription complexes along the ORF.
Although highly transcribed RNA Pol II genes were equally represented among high occupancy WT and rrm3 sites, the level of DNA Pol2 binding to these genes was not increased in the absence of Rrm3 (Fig. 1A, B, Fig. 3B, Fig. 4, Supplemental Fig. 2B). Thus, RNA Pol II transcribed genes are the only known intrinsic pause sites that are not Rrm3-sensitive. We suggest that Rrm3 is either redundant with another activity or there is an Rrm3 independent pathway that promotes fork movement through highly transcribed RNA Pol II genes.
Figure 3. Rrm3-sensitive replication sites are often high occupancy DNA Pol2-MYC binding sites in rrm3 but not WT cells.

A. Venn diagram of overlapping high occupancy DNA Pol2-MYC WT and DNA Pol2-MYC rrm3 sites.
B. The number of sites in common between DNA Pol2-MYC WT and rrm3 cells (overlap, black bars), DNA Pol2-MYC WT only (white bars), and DNA Pol2-MYC rrm3 only (gray bars) are plotted by genomic classes (Fig 1A). As more than 1 site within a single telomere may be associated with DNA Pol2, the number of telomeric loci and number of telomeres associated with DNA Pol2 are not equal.
Although individual telomeres, tRNA genes, and highly transcribed RNA Pol II genes had high DNA Pol2 occupancy, other members of these classes did not. For example, only 29% of tRNA genes (80 of 275) in rrm3 cells and 15% of RNA Pol II genes with more than one transcription complex (15 of 99) in WT cells met our stringent criteria for high DNA Pol2 occupancy. Our analysis likely underestimates the fraction of each genomic class that causes replication fork pausing, owing to the stringent criteria used to identify sites of high DNA Pol2 association. For example, the highly transcribed RPL30 gene clearly bound high levels of DNA Pol2 by standard ChIP-qPCR analysis (Fig. 6F), yet this high binding did not satisfy our criteria for a high occupancy site. In addition, genomic context likely plays a role in determining fork progression rates. For example, proximity to an efficient replication origin increases the probability of being a pause site (our unpublished results).
In addition to known categories, regions that currently lack annotation in SGD were among high occupancy DNA Pol2 or Rrm3 sites (Fig. 1A). Many of these sites mapped to biologically interesting structures that are not yet annotated, such as boundary elements (Bi and Broach, 1999; Yu et al., 2003) (p=10−4, 10−5, and 10−9 in, respectively, Pol2 wild type, Pol2 rrm3, and Rrm3 cells), sites where replication forks converge (9, 13, and 10 sites in, respectively, DNA Pol2 wild type, DNA Pol2 rrm3, and Rrm3; D. Fachinetti, R. Bermejo, Y. Doksani, S. Minardi, , Y. Katou, Y. Kanoh, S. Katou, A. Azvolinsky, V. Zakian, and Marco Foiani, submitted), and sites of high H2A phosphorylation in undamaged cells (p = 10−8,10−19, 10−1 in, respectively, DNA Pol2 wild type, DNA Pol2 rrm3 and Rrm3 cells; M. Grunstein, personal communication)
Although T4 bacteriophage helicases promote fork progression past protein complexes in vitro (Bedinger et al., 1983; Liu and Alberts, 1995), Rrm3 is the only helicase known to promote fork progression through hard to replicate protein complexes in vivo. The dramatic effects of Rrm3 on replication suggest that other eukaryotes must also have mechanisms to bypass these complexes. Rrm3 is a member of the conserved Pif1 family of DNA helicases with homologs in yeasts to mammals (reviewed in Boule and Zakian, 2006). As Rrm3 can supply the essential nuclear replication function of the S. pombe Rrm3 homolog (Pinter et al., 2008), Pif1 family helicases as a class may function to promote fork progression through hard to replicate sites. Finally, the methods used in this paper can be easily adapted to detect fork pausing in more complex eukaryotes.
EXPERIMENTAL PROCEDURES
Detailed methods are in supplementary materials, including a list of all strains (Supplemental Table 1). Yeast strains were derivatives of W1588 (a RAD5+ version of W303). WT and rrm3 versions of Pol2-MYC and Rrm3-MYC strains were described previously except that the rad5-535 mutation was corrected to RAD5+ (Azvolinsky et al., 2006). All experiments except the ones in Fig. 7 used asynchronous cells grown at 30° C. For experiments using galactose, single colonies were grown in YEPD for 6 hours, diluted into YEP plus 2% raffinose and grown to an OD660 of 0.25. Cultures were diluted to an OD660 of 0.15, galactose was added to a final concentration of 2% and cells were grown for an additional 2 hours. Synchronization methods and FACS analysis were as described except cells were released at 17 °(Azvolinsky et al., 2006). 2D gel methods were described previously (Ivessa et al., 2000). ChIP analyses were performed using modifications of (Taggart et al., 2002). Quantitation of immunoprecipitated DNA was performed using real time PCR on an iCycler iQ Real Time PCR Detection System (Bio-Rad Laboratories) and normalized to input DNA; data are expressed as the relative recovery of immunoprecipitated DNA relative to the amount of input DNA based on the quantification of the respective PCR products (% IP’ed). All ChIP experiments were repeated at least three times. For microarray experiments, a PCR based S. cerevisiae array containing ~13,000 spots representing both coding and intergenic regions at ~800 bp resolution amplified from yeast (S288C) was used (Iyer et al. 2001). Three or four biologically independent ChIP experiments as well as two technical replicates were performed for each strain. Data processing is detailed in supplemental methods. Briefly, the median standardized value for each spot was determined across all biological replicates. These standardized values were input for the peak-finding algorithm ChIPOTle (V1.015) with the following parameters: Gaussian background distribution, step size 0.25 kb and window size 1 kb (Buck et al., 2005). R 2.5.0 (R Foundation for Statistical Computing 2007) was used for statistical analysis. Significance of enrichment of a particular genomic feature among high occupancy sites was determined using the hypergeometric distribution.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Availability of data
All raw microarray data and images are available to the public through the UNC microarray database (https://genome.unc.edu/). Data are also available through GEO (accession number GSE16218).
REFERENCES
- Admire A, Shanks L, Danzl N, Wang M, Weier U, Stevens W, Hunt E, Weinert T. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev. 2006;20:159–173. doi: 10.1101/gad.1392506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azvolinsky A, Dunaway S, Torres J, Bessler J, Zakian VA. The S.cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev. 2006;20:3104–3116. doi: 10.1101/gad.1478906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedinger P, Hochstrasser M, Jongeneel C, Alberts B. Properties of the T4 bacteriophage DNA replication apparatus: the T4 dda DNA helicase is required to pass a bound RNA polymerase molecule. Cell. 1983;34:115–123. doi: 10.1016/0092-8674(83)90141-1. [DOI] [PubMed] [Google Scholar]
- Bi X, Broach JR. UASrpg can function as a heterochromatin boundary element in yeast. Genes Dev. 1999;13:1089–1101. doi: 10.1101/gad.13.9.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boule JB, Zakian VA. Roles of Pif1-like helicases in the maintenance of genomic stability. Nucleic Acids Research. 2006;34:4147–4153. doi: 10.1093/nar/gkl561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer BJ, Fangman WL. A replication fork barrier at the 3' end of yeast ribosomal RNA genes. Cell. 1988;55:637–643. doi: 10.1016/0092-8674(88)90222-x. [DOI] [PubMed] [Google Scholar]
- Brewer BJ, Lockshon D, Fangman WL. The arrest of replication forks in the rDNA of yeast occurs independently of transcription. Cell. 1992;71:267–276. doi: 10.1016/0092-8674(92)90355-g. [DOI] [PubMed] [Google Scholar]
- Buck MJ, Nobel AB, Lieb JD. ChIPOTle: a user-friendly tool for the analysis of ChIP-chip data. Genome Biol. 2005;6:R97. doi: 10.1186/gb-2005-6-11-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha RS, Kleckner N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science. 2002;297:602–606. doi: 10.1126/science.1071398. [DOI] [PubMed] [Google Scholar]
- Deshpande AM, Newlon CS. DNA replication fork pause sites dependent on transcription. Science. 1996;272:1030–1033. doi: 10.1126/science.272.5264.1030. [DOI] [PubMed] [Google Scholar]
- Dieci G, Giuliodori S, Catellani M, Percudani R, Ottonello S. Intragenic promoter adaptation and facilitated RNA polymerase III recycling in the transcription of SCR1, the 7SL RNA gene of Saccharomyces cerevisiae. J Biol Chem. 2002;277:6903–6914. doi: 10.1074/jbc.M105036200. [DOI] [PubMed] [Google Scholar]
- Elias-Arnanz M, Salas M. Bacteriophage phi29 DNA replication arrest caused by codirectional collisions with the transcription machinery. EMBO J. 1997;16:5775–5783. doi: 10.1093/emboj/16.18.5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felici F, Cesareni G, Hughes JM. The most abundant small cytoplasmic RNA of Saccharomyces cerevisiae has an important function required for normal cell growth. Mol Cell Biol. 1989;9:3260–3268. doi: 10.1128/mcb.9.8.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfeder SA, Newlon CS. Replication forks pause at yeast centromeres. Mol Cell Biol. 1992;12:4056–4066. doi: 10.1128/mcb.12.9.4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell. 2003;12:1525–1536. doi: 10.1016/s1097-2765(03)00456-8. [DOI] [PubMed] [Google Scholar]
- Ivessa AS, Zhou J-Q, Schulz VP, Monson EM, Zakian VA. Saccharomyces Rrm3p, a 5' to 3' DNA helicase that promotes replication fork progression through telomeric and sub-telomeric DNA. Genes Dev. 2002;16:1383–1396. doi: 10.1101/gad.982902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivessa AS, Zhou J-Q, Zakian VA. The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell. 2000;100:479–489. doi: 10.1016/s0092-8674(00)80683-2. [DOI] [PubMed] [Google Scholar]
- Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- Keil RL, McWilliams AD. A gene with specific and global effects on recombination of sequences from tandemly repeated genes in Saccharomyces cerevisiae. Genetics. 1993;135:711–718. doi: 10.1093/genetics/135.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Horiuchi T. A yeast gene product, Fob1 protein, required for both replication fork blocking and recombinational hotspot activities. Genes Cells. 1996;1:465–474. doi: 10.1046/j.1365-2443.1996.d01-256.x. [DOI] [PubMed] [Google Scholar]
- Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell. 2005;120:587–598. doi: 10.1016/j.cell.2004.12.039. [DOI] [PubMed] [Google Scholar]
- Lieb JD, Liu X, Botstein D, Brown PO. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet. 2001;28:327–334. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- Linskens MHK, Huberman JA. Organization of replication of ribosomal DNA in Saccharomyces cerevisiae. Mol Cell Biol. 1988;8:4927–4935. doi: 10.1128/mcb.8.11.4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Alberts BM. Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science. 1995;267:1131–1137. doi: 10.1126/science.7855590. [DOI] [PubMed] [Google Scholar]
- Liu B, Wong ML, Tinker RL, Geiduschek EP, Alberts BM. The DNA replication fork can pass RNA polymerase without displacing the nascent transcript. Nature. 1993;366:33–39. doi: 10.1038/366033a0. [DOI] [PubMed] [Google Scholar]
- Mirkin EV, Castro Roa D, Nudler E, Mirkin SM. Transcription regulatory elements are punctuation marks for DNA replication. Proc Natl Acad Sci USA. 2006;103:7276–7281. doi: 10.1073/pnas.0601127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin EV, Mirkin SM. Mechanisms of transcription-replication collisions in bacteria. Mol Cell Biol. 2005;25:888–895. doi: 10.1128/MCB.25.3.888-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinter SF, Aubert SD, Zakian VA. The Schizosaccharomyces pombe Pfh1p DNA helicase is essential for the maintenance of nuclear and mitochondrial DNA. Mol Cell Biol. 2008;28:6594–6608. doi: 10.1128/MCB.00191-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poloumienko A, Dershowitz A, De J, Newlon CS. Completion of replication map of Saccharomyces cerevisiae chromosome III. Mol Biol Cell. 2001;12:3317–3327. doi: 10.1091/mbc.12.11.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F, Aguilera A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005;24:1267–1276. doi: 10.1038/sj.emboj.7600602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, Fangman WL. Replication dynamics of the yeast genome. Science. 2001;294:115–121. doi: 10.1126/science.294.5540.115. [DOI] [PubMed] [Google Scholar]
- Raveendranathan M, Chattopadhyay S, Bolon YT, Haworth J, Clarke DJ, Bielinsky AK. Genome-wide replication profiles of S-phase checkpoint mutants reveal fragile sites in yeast. EMBO J. 2006;25:3627–3639. doi: 10.1038/sj.emboj.7601251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart AKP, Teng S-C, Zakian VA. Est1p as a cell cycle-regulated activator of telomere-bound telomerase. Science. 2002;297:1023–1026. doi: 10.1126/science.1074968. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Horiuchi T, Kobayashi T. Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev. 2003;17:1497–1506. doi: 10.1101/gad.1085403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BJ, Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- Torres JZ, Bessler JB, Zakian VA. Local chromatin structure at the ribosomal DNA causes replication fork pausing and genome instability in the absence of the S. cerevisiae DNA helicase Rrm3p. Genes Dev. 2004a;18:498–503. doi: 10.1101/gad.1154704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uptain SM, Kane CM, Chamberlin MJ. Basic mechanisms of transcript elongation and its regulation. Annu Rev Biochem. 1997;66:117–172. doi: 10.1146/annurev.biochem.66.1.117. [DOI] [PubMed] [Google Scholar]
- Yabuki N, Terashima H, Kitada K. Mapping of early firing origins on a replication profile of budding yeast. Genes Cells. 2002;7:781–789. doi: 10.1046/j.1365-2443.2002.00559.x. [DOI] [PubMed] [Google Scholar]
- Yu Q, Qiu R, Foland TB, Griesen D, Galloway CS, Chiu YH, Sandmeier J, Broach JR, Bi X. Rap1p and other transcriptional regulators can function in defining distinct domains of gene expression. Nucleic Acids Res. 2003;31:1224–1233. doi: 10.1093/nar/gkg200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, McIntosh KB, Rudra D, Schawalder S, Shore D, Warner JR. Fine-structure analysis of ribosomal protein gene transcription. Mol Cell Biol. 2006;26:4853–4862. doi: 10.1128/MCB.02367-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.