

Abstract
Recent studies have shown that dendritic cells (DCs) regulate B cell functions. In this study, we report that bone marrow (BM)-derived immature DCs, but not mature DCs, can inhibit BCR-induced proliferation of B cells in a contact-dependent manner. This inhibition is overcome by treatment with BAFF and is dependent on the BCR coreceptor CD22; however, it is not dependent on expression of the CD22 glycan ligand(s) produced by ST6Gal-I sialyltransferase. We found that a second CD22 ligand (CD22L) is expressed on CD11c+ splenic and BM-derived DCs, which does not contain ST6Gal-I-generated sialic acids and which, unlike the B cell-associated CD22L, is resistant to neuraminidase treatment and sodium metaperiodate oxidation. Examination of splenic and BM B cell subsets in CD22 and ST6Gal-I knockout mice revealed that ST6Gal-I-generated B cell CD22L plays a role in splenic B cell development, whereas the maintenance of long-lived mature BM B cells depends only on CD22 and not on α2,6-sialic acids produced by ST6Gal-I. We propose that the two distinct CD22L have different functions. The α2,6-sialic acid-containing glycoprotein is important for splenic B cell subset development, whereas the DC-associated ST6Gal-I-independent CD22L may be required for the maintenance of long-lived mature B cells in the BM.
The role of dendritic cells (DCs)3 in Ag capture, processing, and presentation to T cells has been extensively studied (1-3). Upon entry into lymph nodes (LNs), DCs capture and process Ag and then migrate to T cell zones where they present the Ag to Th cells. The primed Th cells, in turn, induce B cell growth, differentiation, and Ab production through the release of cytokines and also through direct cell-cell contact requiring CD40-CD40L (CD40L, CD154) interactions. Thus, DCs can regulate B cell responses indirectly through T cell priming. Several studies have also shown that DCs can directly influence B cell responses, e.g., through the production of soluble factors including cytokines. DCs can stimulate CD40-activated B cells to proliferate and produce Ab by secreting soluble factors (4). The differentiation of naive B cells into IgM-secreting plasma cells is dependent on IL-12 production by DCs (5).
Other soluble factors produced by DCs that can regulate B cells include the TNF family members B cell activating factor BAFF (BLyS/TALL-1/THANK/zTNF4) and a proliferation-inducing ligand (APRIL) (6-10). BAFF and APRIL produced by DCs greatly enhance BCR-induced proliferation of B cells (9-10). Balazs et al. (6) also found that BAFF and APRIL play a role in the differentiation of naive marginal zone (MZ) B cells into plasmablasts. These TNF family members also induce IgG1 secretion in B cells (10). Furthermore, DC-generated BAFF and APRIL affect CD40-independent class switching (7). Type I IFNs and IL-6 produced by a subset of DCs also influence B cells to differentiate into Ig-secreting plasmablasts (11).
DCs can also exert their effects on B cells by direct cell-cell interactions. CD40L expressed on DCs can induce CD40-positive B cells to secrete IgG and IgA (12). DC-mediated CD40 triggering can also provide B cells with a survival signal (13). Ag-specific B cells can physically interact with DCs in vitro that have been pulsed with the cognate Ag (14). This direct interaction induces the release of intracellular Ca2+ by the B cells. DCs and B cells can form clusters in vitro, resulting in the induction of B cell proliferation, differentiation, and isotype class switching (15). In vivo, B cells and DCs interact in a few distinct locations. DCs can cluster with B cells in the lymph in an LFA-1 (CD11a/18)-dependent manner (16). Both VLA-4 (CD49d/29) and LFA-1 play important roles in enabling B cells to recognize membrane-bound Ags expressed on target cells (17). When LFA-1 and VLA-4 on B cells bind to their ligands (CD54/ICAM-1 and CD106/VCAM-1, respectively) on the surface of target cells also expressing membrane-bound Ag, the threshold for A-specific BCR signaling required for activation is lowered. The binding of both the integrin and BCR facilitates synapse formation between the B cell and target cell and allows the B cell to effectively spread over the membrane expressing the Ag. This subsequently results in B cell activation and allows the B cell to acquire Ag from the target cell (18, 19).
DCs and B cells also interact in the red pulp of the spleen (20). This interaction is thought to support plasmablast survival and their differentiation into Ab-secreting plasma cells. Balazs et al. (6) demonstrated that association between DCs and B cells can occur at the T cell-B cell borders in the spleen. Blood DCs capture bacteria and deliver them to MZ B cells, after which the DC-B cell-Ag complexes move to the T cell-B cell borders and the bridging channels connecting the white and red pulp, where plasmablast production subsequently takes place (6). In LNs, B cells and DCs associate in follicles (21, 22). B cells can also interact with DCs after DCs enter LNs via high endothelial venules, but before they enter follicles (23). This interaction results in the activation of the Ag-specific B cells by the DCs expressing cognate Ag and in the transfer of Ag on the surface of DCs to the Ag-specific B cells (23, 24).
In this study, we report that bone marrow (BM)-derived immature DCs (iDCs) and splenic DCs, but not mature DCs (mDCs), can inhibit BCR-induced proliferation of B cells in a contact-dependent manner. This inhibition is also dependent on the BCR coreceptor CD22, which contains an ITIM. We also report the novel finding that a second ligand for CD22, lacking ST6Gal-I-dependent α2,6-sialic acids, is expressed on the surface of DCs. This DC-associated CD22 ligand (CD22L) is resistant to removal of sialic acids and thus appears to be distinct from the ligands expressed on the surface of B cells, implicating differential roles for cis- vs trans-interactions between CD22 and its ligands.
Materials and Methods
Reagents
Recombinant mouse GM-CSF was purchased from Fitzgerald. LPS was obtained from Sigma-Aldrich. Goat anti-mouse IgM F(ab’)2 and PE-conjugated goat anti-human IgG were purchased from Jackson ImmunoResearch Laboratories. Recombinant mouse BAFF was purchased from Axxora. COS cells used for generation of CD22 fusion proteins were obtained from the American Type Culture Collection. All flow cytometry reagents were purchased from BD Biosciences. Arthrobacter ureafaciens neuraminidase was purchased from Roche Diagnostics and the biotinylated Sambucus nigra lectin (SNA) was obtained from Vector Laboratories.
Mice
Wild-type (WT; C57BL/6) and CD22 knockout (KO; C57BL/6) mice were housed under specific pathogen-free conditions, and all experiments were performed in compliance with the University of Washington Institutional Animal Care and Use Committee. ST6Gal-I KO mice were housed under specific pathogen-free conditions at the University of California, San Diego facility. The BM and splenocytes were collected at the University of California, San Diego and shipped overnight to the University of Washington as single-cell suspensions in complete RPMI 1640 medium.
DC culture
BM cells from the femurs and tibiae of mice were collected under sterile conditions and cultured in complete RPMI 1640 medium supplemented with 20 ng/ml recombinant mouse GM-CSF. The cells were fed every other day for 8 days to generate iDCs. To obtain mDCs, day 7-cultured iDCs were stimulated with 1 μg/ml LPS for 24 h. Before coculturing with the CFSE-labeled B cells, the DCs were extensively washed with complete RPMI 1640. The purity of the DCs was examined on day 8 and found to be between 85 and 95% CD11c+ cells.
B cell purification
B cells from mouse spleens were purified using the EasySep Negative Selection Kit (Stem Cell Technologies) for mouse B cells according to the manufacturer’s protocol. Following the purification, the B cells were labeled by incubation with 5 μM CFSE for 10 min at 37°C, followed by quenching. For the B cell-DC cocultures, washed DCs and CFSE-labeled B cells were cultured in 24-well tissue culture dishes or 0.4-μm Transwells (Corning) at 37°C. Cell viability was monitored at 24, 48, and 72 h by trypan blue dye exclusion, forward/side scatter, and/or Δψm measured by FACS using a 50 nM Mitotracker Red CMXRos (Invitrogen Life Technologies). Following 72 h of incubation, the cells were collected and CFSE dilution was measured by flow cytometry. Data were analyzed using FlowJo software (Tree Star).
Splenic DC purification
Cells were liberated from the spleens using spleen dissociation medium (Stem Cell Technologies). The live and dead cells were separated using a Percoll gradient. DCs were subsequently purified using pan DC microbeads (Miltenyi Biotec). The cell purities were routinely checked and found to be ≥95% CD11c+.
Surface staining of cells
Generation of the CD22 Rg constructs, transfections, and protein purification were previously described (25). To stain with the CD22 Rg fusion proteins, cells were washed twice with PBS containing 0.2% BSA, incubated with the fusion proteins on ice, and then subsequently washed again. Next, the cells were stained with PE-conjugated goat anti-human IgG, incubated on ice, and then washed a final time before analyzing the CD22 Rg binding by flow cytometry. Data were analyzed using FlowJo software.
Sialic acid removal by neuraminidase and metaperiodate treatments
For neuraminidase treatment, cells were washed twice with PBS containing 2 mg/ml BSA and resuspended at a concentration of 5 × 106 cells/ml. A. ureafaciens neuraminidase was added to the cells at a final concentration of 400 mU/ml. The cells were then incubated at 37°C for 2 h, after which they were washed and stained for flow cytometry. For the sodium metaperiodate oxidation (26), cells were washed twice with cold PBS, diluted to a concentration of 1 × 106 cells/ml in 2 mM sodium metaperiodate, and then incubated on ice for 15 min in the dark, after which they were washed and stained.
Results
Immature DCs, but not mDCs, inhibit BCR-induced proliferation of B cells
DCs can present preserved Ag to B cells (27) and can activate B cells and transfer Ag to them in vivo (23). Similarly, we found that DCs can modulate BCR-dependent proliferative responses in splenic B cells. Mouse BM-derived iDCs (85-95% CD11c+) were cocultured with purified CFSE-labeled murine splenic B cells along with graded doses of anti-mouse IgM, and B cell division was determined 72 h later. As few as 1 iDC per 16 B cells inhibited BCR-induced proliferation (Fig. 1A) and, when more iDCs were cultured with B cells (4 B cells:1 iDC ratio), the inhibition was more pronounced. This inhibition was evident as early as 48 h after BCR cross-slinking (data not shown). Highly purified splenic DCs isolated ex vivo (≥95% CD11c+) from the spleen also inhibited B cell proliferation (Fig. 1B). We attribute the inhibition of B cells by both BM and splenic CD11c+-enriched populations to DCs.
FIGURE 1.
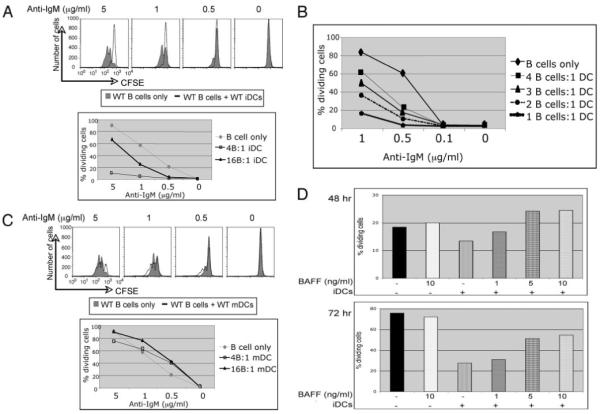
iDCs and splenic DCs, but not mDCs, inhibit BCR-induced proliferation of B cells. Purified CFSE-labeled B cells from WT mice were cocultured with either WT BM-derived iDCs (A), DCs purified ex vivo from spleens (B), or BM-derived mDCs (C) and the indicated graded doses of anti-IgM. CFSE dilution of the B cells was examined 72 h postincubation by flow cytometry. For the BM-derived iDCs, two different B cell:DC ratios were examined (4:1 and 16:1). The histograms shown in A and C are from cultures where the B cell:DC ratio was 4:1. Results from both ratios are shown in graph format. For the splenic DCs (B), four different B cell:DC ratios were used; the percentage of dividing cells in the cultures are indicated in the graph. These results with the BM-derived iDCs are representative of at least eight independent experiments and results with the splenic DCs are representative of three independent experiments. D, B cells and iDCs were cultured in the presence of the indicated doses of BAFF and 1 μg/ml anti-IgM at different B cell:DC ratios (32:1, 24:1, and 16:1). CFSE dilution was examined after 48 and 72 h of BCR cross-linking and the percentage of dividing cells in the cultures are shown. The results from the B cell:DC ratios of 24:1 are shown. These results are representative of three independent experiments.
We also examined the effects on B cell proliferation of mature BM-derived mDCs obtained after stimulating iDCs with LPS for 24 h. The mDCs were washed extensively before coculturing them with the B cells. Interestingly, BCR-induced proliferation was slightly enhanced by the presence of mDCs (Fig. 1C), and enhancement was most evident when lower doses of anti-IgM were used. Similar results were obtained using mDCs matured with peptidoglycan, another TLR agonist (data not shown).
Others and we have shown that DCs do not induce B cell proliferation unless the B cells receive both a signal from BCR cross-linking and a second signal from the DCs (9, 10). DCs can be induced by TLR signals or immune complexes to produce soluble factors, which augment B cell proliferation and, in particular, the TNF family member BAFF (9, 10, 28). Thus, production of BAFF by mDCs may explain the differential proliferation of B cells in the presence of iDCs and mDCs. To test whether the inhibition of B cell proliferation by iDCs could be overridden by BAFF, we added different doses of BAFF to B cell and iDC cocultures and examined B cell proliferation 48 or 72 h later (Fig. 1D). The DC-dependent inhibition of B cell proliferation was overridden by adding graded doses of BAFF to cultures. This was not simply due to BAFF promoting proliferation of anti-IgM-stimulated B cells, because BAFF did not increase BCR-induced proliferation of the B cells alone (Fig. 1D). Therefore, the inhibitory effect of iDCs on B cell proliferation can be overcome by BAFF.
One possibility was that the reduction of WT B cell proliferation by iDCs was due to an increase in B cell death. This was not case. In fact, B cell viability as measured by either trypan dye exclusion, forward and side scatter, or Mitotraker was consistently greater in the presence of iDCs than with B cells alone (data not shown) and was consistently greater at all time points evaluated in WT B cell-DC cultures than in CD22 KO B cell-DC cultures (n > 10 experiments). For example, in a representative experiment using a 4 B cell:1 DC ratio and stimulated with 5 μg/ml anti-IgM, WT B cells were 82, 81, and 86% viable by Mitotraker at 24, 48, and 72 h, while CD22 KO B cells at these time points were 58, 56, and 49% viable, respectively. Thus, decreased B cell proliferation was associated with increased viability.
iDC-mediated inhibition of BCR-induced proliferation is contact dependent
Because iDCs has been previously implicated in the induction of T cell tolerance (29), and because, analogous to T cells, the inhibition of B cells could be overcome with costimulation, we decided to focus on characterizing the inhibitory effects of iDCs on B cells. This inhibition effect could be the result of either direct contact between the two cell types and/or the production of soluble factors by the iDCs. To distinguish between these two possibilities, we performed the CFSE-based assay using Transwells with a pore size of 0.4 μm so that the cells could not traverse the barrier, but soluble factors could. When CFSE-labeled splenic B cells and iDCs were physically separated in the Transwells, iDCs did not inhibit the proliferation of WT B cells (Fig. 2); the amount of B cell division was similar to that in cultures containing only B cells. This was observed at both B cell:DC ratios tested. Thus, iDC-mediated inhibition of B cells requires direct contact between B cells and iDCs. These data suggest that surface molecule(s) on B cells may interact with structures on iDCs, which in turn trigger an inhibitory pathway within B cells, leading to inhibition of BCR-induced proliferation.
FIGURE 2.
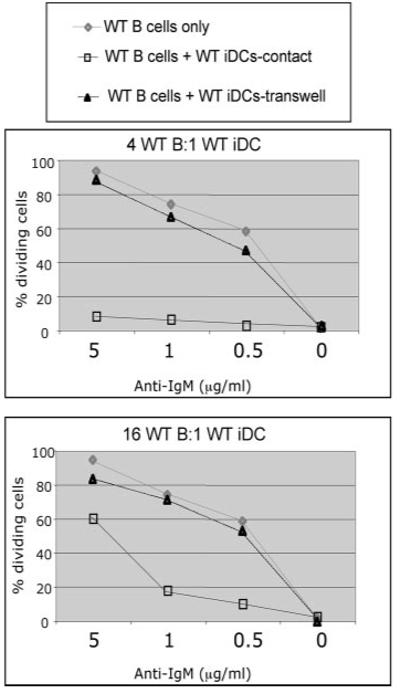
iDC-mediated inhibition of WT B cells is contact dependent. Purified CFSE-labeled B cells from WT mice were cocultured with WT BM-derived iDCs and the indicated concentrations of anti-IgM (“contact”). In some of the cocultures, the DCs and B cells were physically separated using Transwells with a pore size of 0.4 μm. CFSE dilution of the B cells was examined 72 h postincubation by flow cytometry. Two different ratios of B cells:DCs were examined (4:1 and 16:1). These results are representative of six independent experiments.
iDC-mediated inhibition of BCR-induced proliferation is dependent on the BCR coreceptor CD22
The results above indicate that the iDC-mediated inhibitory effects on B cell proliferation require cell-cell contact, possibly via inhibitory cell surface receptors on B cells. Two well-known inhibitory receptors expressed on B cells are CD22 and CD72 (30). Both receptors are known to activate inhibitory pathways within B cells because they both contain ITIMs within their cytoplasmic domains. CD22 was an attractive candidate because it is an adhesion molecule with CD22L expressed on hematopoietic cells and also because it can physically associate with BCR complexes and thereby inhibit BCR signaling (31). BCR cross-linking results in CD22 tyrosine phosphorylation and recruitment of the SHP-1 protein tyrosine phosphatase to the plasma membrane (31, 32). To determine whether CD22 might be involved in iDC-mediated regulation of B cells, we compared the ability of iDCs to inhibit WT and CD22 KO B cells. Unlike WT B cells, CD22 KO B cells proliferated equally well in the presence and absence of iDCs and at both the high and low B cell:DC ratios (Fig. 3A). Similar results were obtained when the CD22 KO B cells were cocultured with CD22 KO BM-derived iDCs (data not shown). Furthermore, physically separating the CD22 KO B cells and WT iDCs had no effect on the BCR-induced proliferation of the CD22 KO B cells (Fig. 3A). Thus, CD22 appears to be required for iDCs to inhibit BCR-induced cell division. We also examined the effects of iDCs on BCR-induced proliferation of B cells from Bam32 KO mice. B cells from these KO mice are deficient for the Bam32 adaptor protein, which plays a key role in signaling downstream of the BCR (33, 34). As was observed with the WT B cells, iDCs also inhibited BCR-induced proliferation of Bam32 KO B cells (data not shown).
FIGURE 3.
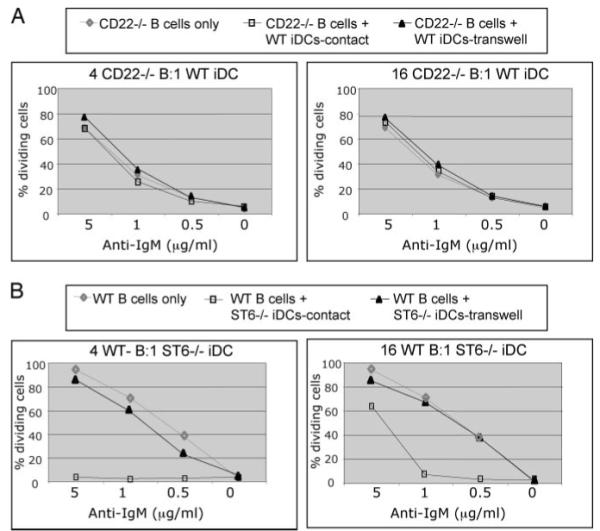
iDC-mediated inhibition of B cell proliferation requires CD22, but not ST6Gal-I sialyltransferase. A, Purified CFSE-labeled B cells from CD22-deficient mice were cocultured together (“contact”) or in Transwells with WT BM-derived iDCs and graded doses of anti-IgM (“contact”). B, Purified CFSE-labeled B cells from WT mice were cocultured with ST6Gal-I-deficient BM-derived iDCs and graded doses of anti-IgM. The same method described in Fig. 2 was used. These results are representative of at least three independent experiments.
ST6Gal-I sialyltransferase KO iDCs can inhibit BCR-induced proliferation
CD22 can mediate interactions with erythrocytes, neutrophils, T cells, B cells, and monocytes (25, 35, 36); thus, it is possible that CD22 on B cells binds to CD22L on iDCs. This binding might be required for iDCs to inhibit B cell proliferation in response to BCR cross-linking. To examine this possibility, we compared the effect on B cells of WT iDCs to iDCs derived from mice deficient for the sialyltransferase ST6Gal-I, which generates α2,6-sialic acid CD22L (37). Surprisingly, ST6Gal-I KO iDCs, like WT iDCs, inhibited BCR-induced proliferation of WT B cells (Fig. 3B). To determine whether the inhibition mediated by the ST6Gal-I KO iDCs is contact dependent, we used Transwells to provide a barrier between the iDCs and B cells. When the ST6Gal-I KO iDCs and WT B cells were physically separated, BCR-induced cell division was not inhibited (Fig. 3B), as was the case with WT iDCs (Fig. 2). Thus, physical interaction between WT B cells and ST6Gal-I KO iDCs is required for ST6Gal-I KO iDCs to inhibit BCR-induced B cell proliferation.
CD22 interacts with molecule(s) on DCs
To determine whether CD22 binds to molecules on the surface of DCs, we used chimeric CD22 fusion proteins to stain the DCs (25). One of the chimeric fusion proteins contains all seven extracellular Ig domains fused to the Fc region of human IgG (CD22 Rg1-7). The other fusion protein contained deletions of Ig domains 1 and 2 (CD22 Rg3-7), the regions previously shown to be required for binding to CD22L (see 25). These proteins, along with a human IgG (Fc fragment) control, were used to stain BM-derived iDCs from WT and ST6Gal-I KO mice. As expected, the CD22 Rg1-7 fusion protein bound to WT iDCs, but the CD22 Rg3-7 fusion protein did not (Fig. 4A). Surprisingly, CD22 Rg1-7 also bound well to ST6Gal-I KO iDCs (Fig. 4A). To ensure that the iDCs were correctly genotyped, we compared the binding of Sambucus nigra lectin (SNA), a lectin that specifically binds the Siaα2-6Galβ1-4GlcNac glycan ligand, to WT and ST6Gal-I KO iDCs. As expected, SNA bound to WT, but not to ST6Gal-I KO iDCs (data not shown and Ref. 37). We also compared the binding of the CD22 fusion proteins to WT and ST6Gal-I KO B cells. As expected, CD22 Rg1-7 binding was higher on the WT B cells compared with the ST6Gal-I KO B cells (Fig. 4B). However, some binding of CD22 Rg1-7 to ST6Gal-I KO B cells was also consistently detected.
FIGURE 4.
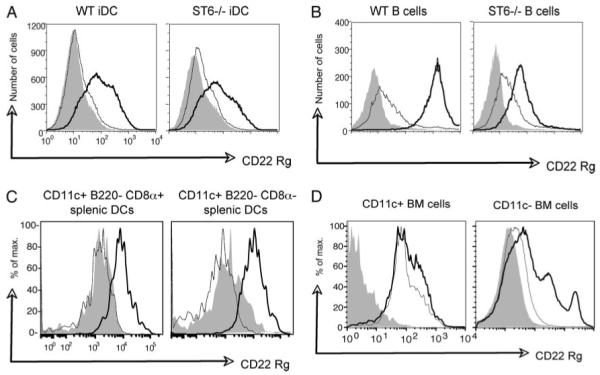
Soluble CD22 binds to WT and ST6GalI KO DCs. A-C, The binding of purified human IgG, Fc fragment (shaded), CD22 Rg1-7 (bold solid line), or CD22 Rg3-7 (dotted thin line) to the surface of WT and ST6Gal-I-deficient BM-derived iDCs (A), B cells (B), and WT splenic DCs (C) was quantified by flow cytometry. D, The binding of human IgG, Fc fragment (shaded), and CD22 Rg1-7 on WT (thick line) and ST6Gal-I KO (thin line) BM cells was examined by flow cytometry. The cells were stained with purified control or fusion proteins along with anti-human IgG-PE secondary and other surface markers. In the histograms shown, the cells were gated on CD11c+ cells (A), CD19+ cells (B), CD11c+B220-CD8α+ (C), or CD11c+B220-CD8α- cells, and CD11c+ or CD11c- cells (D). The results shown are representative of three independent experiments.
Because splenic DCs also inhibit B cell proliferation (Fig. 1B), we also tested whether the CD22 fusion proteins bound specifically to splenic DC subsets. CD22Ls were expressed on both CD11c+B220-CD8α+ and CD11c+B220-CD8α- DC subsets from the spleen (Fig. 4C). We then compared the binding of CD22 Rg1-7 to CD11c+ DCs and CD11c- cells in the BM of WT and ST6Gal-I KO mice. CD22 Rg1-7 bound to CD11c+ cells in the BM from both WT and ST6Gal-I KO mice (Fig. 4D, left histogram). As expected CD22 Rg1-7 bound to some CD11c- BM cells from WT mice, but it did not bind to any CD11c- BM cells from the ST6Gal-I KO mice (Fig. 4D, right histogram). Thus, CD22 binds to a ligand(s) on DCs that does not contain α2,6-sialic acids generated by ST6Gal-I. This CD22L on DCs is distinct from the major CD22L on B cells, which is comprised mostly of sialic acid-containing glycans produced by ST6Gal-I.
CD22 binding to DCs is not disrupted by cleavage of sialic acids by neuraminidase or sodium metaperiodate oxidation
To test further whether CD22 interacts with a CD22L on iDCs that is distinct from that produced by ST6Gal-I, we treated the cells with a neuraminidase from A. ureafaciens to remove cell surface sialic acid linkages (26). The A. ureafaciens neuraminidase can cleave sialic acids other than those with α2,6-linkages, however, we used conditions that tend to favor cleavage only of structures with α2,6-linkages (see Materials and Methods). Following treatment of iDCs with the A. ureafaciens neuraminidase, the cells were stained with the CD22 Rg fusion proteins, SNA, or human IgG control and analyzed by flow cytometry. Binding of CD22 Rg1-7 and CD22 Rg3-7 were largely unaffected by neuraminidase treatment of the iDCs (Fig. 5A, top panels). In contrast, CD22 Rg1-7 binding to B cells was dramatically reduced following neuraminidase treatment (Fig. 5A, bottom panels) as previously reported (37). To determine whether CD22 binding to DCs was affected by removal of sialic acids, we examined the binding of the fusion proteins to CD11c+ cells following treatment by sodium metaperiodate oxidation (26). CD22 fusion protein (both CD22 Rg1-7 and CD22 Rg3-7) and human IgG Fc binding was unaffected by removal of sialic acids in CD11c+ DCs from the BM (Fig. 5B) and spleen (data not shown). Thus, the requirements for CD22 to bind to B cells are different from the requirements for binding to DCs. The B cell CD22L is comprised mostly of α2,6-sialic acid linkages generated by ST6Gal-I, whereas the alternative DC CD22L is not dependent on ST6Gal-I and is not affected by A. ureafaciens neuraminidase or sodium metaperiodate treatment. Furthermore, the binding of CD22 to B cells requires Ig domains 1 and 2 of CD22 since binding of the deletion mutant (CD22 Rg3-7) was unaltered by neuraminidase treatment. In summary, B cells and DCs clearly express distinct sets of CD22Ls.
FIGURE 5.
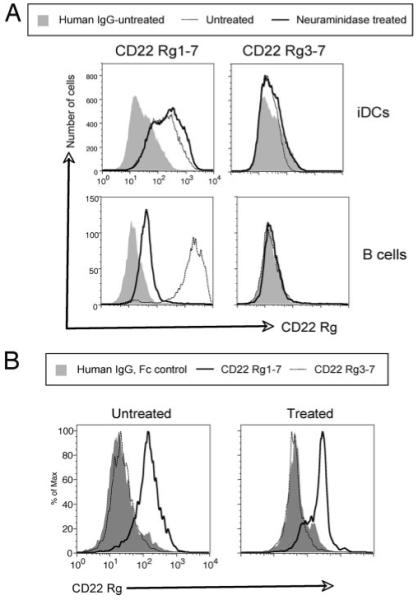
Cleavage of sialic acids does not disrupt CD22 binding on DCs. A, The binding of the purified chimeric CD22 fusion proteins to the surface of WT DCs and B cells following cleavage of α2,6-linked sialic acids (A) or to BM cells following sodium metaperiodate oxidation (B) was quantified by flow cytometry. A, Arthrobacter ureafaciens neuraminidase-treated cells were stained with the indicated fusion proteins or control human IgG (Fc fragment) along with anti-human IgG-PE secondary and anti-CD19-FITC (for the splenocytes) or anti-CD11c-FITC (for the iDCs). In the histograms shown, the cells were gated on CD19+ (B cells) or CD11c+ (iDCs). The neuraminidase-treated human IgG control was similar to the untreated sample shown. B, BM cells treated by sodium metaperiodate oxidation were stained with anti-CD11c-FITC and control or CD22 fusion proteins as indicated. In the histograms shown, the cells were gated on CD11c+ cells. The mean fluorescence intensity of the histograms for the experiment shown above (n = 4 mice) are as follow: untreated CD22 Rg1-7, 198 ± 4.2; untreated CD22 Rg3-7, 37 ± 3.5; treated CD22 Rg1-7, 243 ± 35; and treated CD22 Rg3-7, 50 ± 5.4. The results shown are representative of four experiments.
CD22-CD22L interactions play a role in the development of transitional 2 (T2) B cell precursors
To assess where in B cell development ST6Gal-I-generated CD22L vs alternative DC CD22L not containing ST6Gal-I-generated α2,6-sialic acids might be required, we compared BM and splenic B cell subsets from WT, CD22 KO, and ST6Gal-I KO mice. As we previously showed (38), CD22 KO mice had fewer long-lived mature BM B cells, which can be identified by their B220+IgMhigh CD24 (HSA)low phenotype (39, 40) compared with WT mice (Fig. 6A). However, the numbers of mature B cells in the BM of ST6Gal-I KO mice were comparable to those of WT mice as previously reported (Fig. 6A and Refs. 37 and 41). We also determined the numbers of the various splenic B cell subsets in the spleens, as described previously (42, 43), by separating T2 B cell populations into T2 follicular precursor (T2-FOP) and T2 MZ precursor (T2-MZP) subsets. Cells were stained for surface IgM, IgD, and CD21 and analyzed by flow cytometry. CD22 KO mice had a normal number of follicular (FO) mature B cells and slightly more T1 B cells (Fig. 6B). The MZ B cell population was reduced by one-half in the CD22 KO mice as previously reported (44). Interestingly, CD22 KO mice had dramatic deficiencies in the two T2 subsets. There were 65% fewer T2-FOP and 87% less T2-MZP in CD22 KO mice compared with WT mice. Interestingly, CD22 and ST6Gal-I KO mice had strikingly similar defects in splenic B cell development. The ST6Gal-I KO mice, like the CD22 KO mice, had normal numbers of T1 and FO B cells, but showed similar deficiencies in MZ and T2 precursor B cell populations (Fig. 6B).
FIGURE 6.
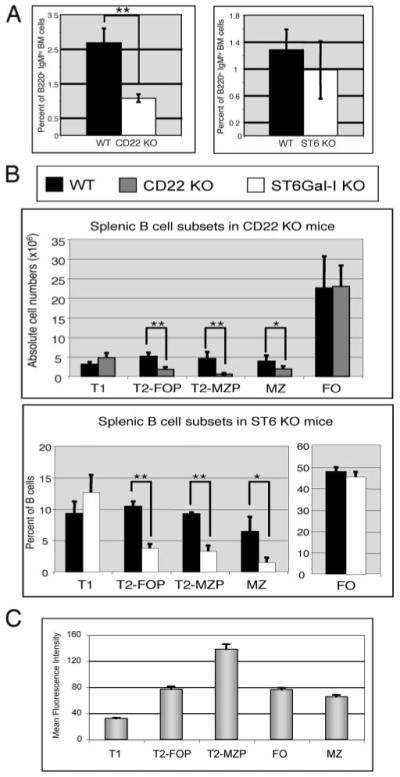
CD22 and ST6Gal-I sialyltransferase are required for development of MZ and T2 precursor B cells, but only CD22 is required for the maintenance of B cells in the BM. A, Total BM was collected from WT and ST6Gal-I KO mice and analyzed for surface expression of B220 and IgM by flow cytometry. The percentage of B220+IgMhigh cells is shown. B, Total splenocytes were isolated from WT, CD22, and ST6Gal-I KO mice. The splenocytes were analyzed for surface expression of IgM, IgD, and CD21 by flow cytometric analysis. The cell numbers or percentage of each B cell subset is shown in the graphs. Data represent mean ± SD. n, More than three mice per genotype. C, CD22 expression in the B cell subsets. WT splenocytes were stained with anti-IgM, anti-IgD, anti-CD21, and anti-CD22 and analyzed by flow cytometry. The graph shows the average mean fluorescence intensity of CD22 expression for each B cell subset. n = 9 mice. *, p < 0.05; and **, p < 0.01.
We then examined the levels of CD22 surface expression in the various splenic B cell subsets by flow cytometry. T1 cells expressed the lowest levels of CD22 while FO, MZ, and T2-FOP B cells expressed moderate levels (Fig. 6C). Interestingly, the cells that were most deficient in the CD22 KO mice (T2-MZP) also expressed the highest levels of surface CD22. These data suggest that CD22 may function to regulate the maturation of transitional B cells in the spleen.
Discussion
Our data show that iDCs and splenic DCs, but not mDCs, inhibit BCR-induced proliferation of WT B cells in a cell contact-dependent manner (Figs. 1 and 2). The inhibition induced by iDCs was not absolute, but rather dependent on the extent of BCR cross-linking because there was less inhibition with increasing BCR cross-linking. These results suggested that a negative inhibitory receptor on the surface of B cells may be binding to the iDCs, thereby activating a pathway raising the threshold of BCR stimulation required to induce proliferation. The inhibition of B cell division by iDCs but not mDCs is reminiscent of DC regulation of T cells (29). iDCs can inhibit and even tolerize T cells, while mDCs stimulate T cell proliferation. The ability of DCs to control the initiation of an immune response requires that iDCs undergo maturation by TLR or CD40 stimulation (45-47). An inhibitory role for DCs is more likely to occur in a steady state during induction of tolerance or anergy. Indeed, DCs can induce peripheral T cell anergy in vivo when Ag is delivered to them in the absence of CD40 or TLR costimulation (45, 47).
Previous studies suggested that DCs may inhibit B cells unless a costimulatory signal is provided. When a very low dose of Ag was delivered to DCIR2 (33D1)+ splenic DCs, mice became unresponsive to subsequent challenge with Ag and did not produce Ab (48). This B cell tolerance was overcome if a second signal, IL-1, was provided in vivo with the Ag. Similarly, suppressor of cytokine signaling 1 KO DCs, which are dysregulated and produce high levels of BAFF and APRIL, stimulate BCR-induced proliferation in vitro and autoantibody production in vivo (10). We found that the inhibition of B cell proliferation by iDCs could be overridden by BAFF (Fig. 1D). These results are consistent with a model (6) that Ag-induced proliferation does not occur unless a second signal such as BAFF is provided by DCs. In other words, B cells may require costimulation via DCs just like T cells do.
Because physical contact was necessary for iDCs to inhibit B cells, we speculated that an inhibitory receptor on B cells was involved. We discovered that CD22 is required for iDC-mediated inhibition of BCR-induced B cell proliferation (Fig. 3A). Interestingly, ST6Gal-I KO iDCs, which lack α2,6-sialic acid ligands for CD22, also inhibited WT B cell proliferation (Fig. 3B). Furthermore, CD22 bound equally well to both WT iDCs, CD11c+ BM DCs, and CD11c+ splenic DCs (Fig. 4), suggesting that a ligand for CD22 is expressed on all of these DCs. However, the ligand on these DCs was not dependent on the sialic acids modified by ST6Gal-I sialyltransferase because CD22 Rg1-7 still bound to ST6Gal-I KO iDCs (Fig. 4A), ST6Gal-I KO CD11c+ BM DCs (Fig. 4D), neuraminidase-treated WT iDCs (Fig. 5A), and sodium metaperiodate-treated BM and splenic DCs (Fig. 5B). In contrast, CD22 binding to ST6Gal-I KO B cells was lower compared with binding to WT B cells (Fig. 4B). Thus, DCs do not express ST6Gal-I-generated α2,6-sialic acid-containing glycoprotein ligands for CD22. DCs and B cells differ in their expression of CD22 ligands. Although DCs express just a ST6Gal-I-independent ligand, B cells express both an ST6Gal-I-generated ligands and a second ligand because some CD22 binding was evident in both ST6Gal-I KO B cells (Fig. 4B) and A. ureafaciens-treated WT B cells (Fig. 5A).
The structure of the second CD22L is not yet clear. What is clear is that this ligand is not generated by ST6Gal-I and is not affected by treatment with A. ureafaciens neuraminidase or sodium metaperiodate oxidation (Figs. 4 and 5). However, it remains formally possible that the second CD22L contains α2,6-sialic acid linkages generated by another sialyltransferase, possibly ST6Gal-II (49, 50). ST6Gal-II prefers to act on glycolipids rather than glycoproteins (49), which may be resistant to treatment with A. ureafaciens neuraminidase. Another possibility is that the second DC CD22L contains α2,6-linked sialic acids on the GalNAc residue of O-glycans produced by one or more of the ST6GalNAc family of sialyltransferases or perhaps is composed in part of protein structure. These possibilities are under investigation.
CD22 can bind to its B cell CD22L in cis (on the same cellular surface) and to CD22L on non-B cells in trans (on a different cell) (51-53). The distinct CD22Ls on B cells and non-B cells may have different functions. We propose and discuss in detail elsewhere (54) that the CD22 binding to ST6Gal-I-dependent B cell CD22L in cis has a function distinct from CD22 binding to ST6Gal-I-independent DC CD22L in trans. The binding of CD22L in cis on B cells is very likely to be important for the regulation of BCR signaling thresholds (53, 55-57). In resting B cells, CD22 binds to CD22L in cis to form homomultimeric complexes, a process that does not require trans-interactions (58). These cis-interactions can control BCR signaling by preventing CD22 from interacting with BCR complexes. A number of studies suggest that weaker BCR signaling favors the development of MZ B cells and stronger signaling favor FO B cell production (59, 60). For instance, Cariappa et al. (42) found that Aiolos KO mice with enhanced BCR signaling have more FO B cells and fewer MZ B cells. Casola et al. (61) showed that reducing the strength of BCR signaling favors MZ B cell maturation. This model would explain why CD22 and ST6Gal-I KO mice, which lack cis-mediated B cell CD22L attenuation of BCR signaling, both have reduced numbers of MZ B cell precursors and MZ B cells.
trans-CD22L binding to CD22 has not been extensively studied, but on some cells binding to trans-ligands may be favored over binding to cis-ligands (62). Lanoue et al. (63) reported that the activation of B cells was attenuated if the Ag-bearing cells were also induced to express ST6Gal-I sialyltransferase. Our data are in agreement with these findings. Lanoue et al. (63) suggested that trans-CD22-CD22L interactions may be important for attenuating B cell responses to self-Ag. Thus, binding of CD22 to DC CD22L on iDCs may be an important mechanism to maintain tolerance to self-Ag, thereby preventing the occurrence of autoimmune diseases.
The binding of trans-DC CD22L to CD22 may directly signal a growth inhibitory signal to B cells so that B cell entry into the cell cycle is attenuated and B cell survival is promoted (54). Direct ligation of CD22 can induce BCR-independent tyrosine phosphorylation of CD22, the recruitment of SHP-1, and an inhibitory signaling cascade (32, 64, 65). The binding of CD22 in trans to DC CD22L may also disrupt the CD22 multimeric complexes on the B cell surface and shift the equilibrium of CD22 multimeric complexes so that more CD22-BCR complexes are formed, and BCR signaling is attenuated.
The fact that CD22 functions in trans via DC CD22L may explain some of the differences between CD22 KO mice, where both B cell and DC CD22L interactions are deficient. and ST6Gal-I KO mice, where the DC CD22L interactions still occur. CD22 KO mice have reduced numbers of recirculating B cells in the BM, whereas ST6Gal-I KO mice have normal numbers compared with WT animals (37, 41). The normal numbers of long-lived BM B cells in the ST6Gal-I KO mice may be due to the ability of DC CD22L on CD11c+ BM DCs to bind to CD22 on B cells and promote survival. The absence of this interaction in the CD22 KO mice may result in the dysregulation of BM B cells and contribute to the more rapid turnover and loss of B cells observed in CD22 KO mice (38, 54). This model is consistent with our finding that in the presence of DCs, WT B cells survive better than CD22 KO B cells even though CD22 KO B cells proliferate more. Although the cellular requirements for the generation and maintenance of long-lived BM B cells are still poorly understood, BM CD11c+ DCs may well be involved and interactions between CD22L on DCs and CD22 on B cells may be required. The inhibitory signal induced by this interaction may be important for the homeostatic maintenance of long-lived mature BM B cells.
In summary, our results strongly suggest that CD22 cis-interactions regulate BCR signaling thresholds and thus the development of splenic B cell subsets, but that the maintenance of mature B cells in the BM may be regulated by CD22 trans-interactions.
Acknowledgments
We thank Dr. Kevin Otipoby for critical reading of this manuscript and members of the Clark laboratory for helpful comments and suggestions during the course of this work. We thank Dr. Michel Nussenzweig for Bam32 KO mice.
Footnotes
This work was supported by National Institutes of Health Grants AI55203 and DE16381 (to E.A.C.) and P01HL57345 and the Howard Hughes Medical Institute (to J.D.M.).
- DC
- dendritic cell
- LN
- lymph node
- BM
- bone marrow
- Au
- Arthrobacter ureafaciens
- CD22L
- CD22 ligand
- FO
- follicular
- iDC
- immature DC
- mDC
- mature DC
- MZ
- marginal zone
- SNA
- Sambucus nigra lectin
- T1/2
- transitional 1/2
- FOP
- follicular precursor
- MZP
- MZ precursor
- WT
- wild type
- KO
- knockout
Disclosures
The authors have no financial conflict of interest.
References
- 1.Clark EA, Ledbetter JA. How B and T cells talk to each other. Nature. 1994;367:425–428. doi: 10.1038/367425a0. [DOI] [PubMed] [Google Scholar]
- 2.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 3.Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 2003;3:984–993. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 4.Dubois B, Vanbervliet B, Fayette J, Massacrier C, Van Kooten C, Briere F, Banchereau J, Caux C. Dendritic cells enhance growth and differentiation of CD40-activated B lymphocytes. J. Exp. Med. 1997;185:941–951. doi: 10.1084/jem.185.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dubois B, Massacrier C, Vanbervliet B, Fayette J, Briere F, Banchereau J, Caux C. Critical role of IL-12 in dendritic cell-induced differentiation of naive B lymphocytes. J. Immunol. 1998;161:2223–2231. [PubMed] [Google Scholar]
- 6.Balazs M, Martin F, Zhou T, Kearney J. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity. 2002;17:341–352. doi: 10.1016/s1074-7613(02)00389-8. [DOI] [PubMed] [Google Scholar]
- 7.Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, Cerutti A. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat. Immunol. 2002;3:822–829. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, Brink R, Mackay F, Hodgkin PD, Tangye SG. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J. Clin. Invest. 2003;112:286–297. doi: 10.1172/JCI18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craxton A, Magaletti D, Ryan EJ, Clark EA. Macrophage- and dendritic cell-dependent regulation of human B-cell proliferation requires the TNF family ligand BAFF. Blood. 2003;101:4464–4471. doi: 10.1182/blood-2002-10-3123. [DOI] [PubMed] [Google Scholar]
- 10.Hanada T, Yoshida H, Kato S, Tanaka K, Masutani K, Tsukada J, Nomura Y, Mimata H, Kubo M, Yoshimura A. Suppressor of cytokine signaling-1 is essential for suppressing dendritic cell activation and systemic autoimmunity. Immunity. 2003;19:437–450. doi: 10.1016/s1074-7613(03)00240-1. [DOI] [PubMed] [Google Scholar]
- 11.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–234. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 12.Pinchuk LM, Klaus SJ, Magaletti DM, Pinchuk GV, Norsen JP, Clark EA. Functional CD40 ligand expressed by human blood dendritic cells is up-regulated by CD40 ligation. J. Immunol. 1996;157:4363–4370. [PubMed] [Google Scholar]
- 13.Wykes M, MacPherson G. Dendritic cell-B-cell interaction: dendritic cells provide B cells with CD40-independent proliferation signals and CD40-dependent survival signals. Immunology. 2000;100:1–3. doi: 10.1046/j.1365-2567.2000.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang NN, Han SB, Hwang IY, Kehrl JH. B cells productively engage soluble antigen-pulsed dendritic cells: visualization of live-cell dynamics of B cell-dendritic cell interactions. J. Immunol. 2005;175:7125–7134. doi: 10.4049/jimmunol.175.11.7125. [DOI] [PubMed] [Google Scholar]
- 15.Dubois B, Bridon JM, Fayette J, Barthelemy C, Banchereau J, Caux C, Briere F. Dendritic cells directly modulate B cell growth and differentiation. J. Leukocyte Biol. 1999;66:224–230. doi: 10.1002/jlb.66.2.224. [DOI] [PubMed] [Google Scholar]
- 16.Kushnir N, Liu L, MacPherson GG. Dendritic cells and resting B cells form clusters in vitro and in vivo: T cell independence, partial LFA-1 dependence, and regulation by cross-linking surface molecules. J. Immunol. 1998;160:1774–1781. [PubMed] [Google Scholar]
- 17.Carrasco YR, Fleire SJ, Cameron T, Dustin ML, Batista FD. LFA-1/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity. 2004;20:589–599. doi: 10.1016/s1074-7613(04)00105-0. [DOI] [PubMed] [Google Scholar]
- 18.Batista FD, Iber D, Neuberger MS. B cells acquire antigen from target cells after synapse formation. Nature. 2001;411:489–494. doi: 10.1038/35078099. [DOI] [PubMed] [Google Scholar]
- 19.Fleire SJ, Goldman JP, Carrasco YR, Weber M, Bray D, Batista FD. B cell ligand discrimination through a spreading and contraction response. Science. 2006;312:738–741. doi: 10.1126/science.1123940. [DOI] [PubMed] [Google Scholar]
- 20.Garcia De Vinuesa C, Gulbranson-Judge A, Khan M, O’Leary P, Cascalho M, Wabl M, Klaus GG, Owen MJ, MacLennan IC. Dendritic cells associated with plasmablast survival. Eur. J. Immunol. 1999;29:3712–3721. doi: 10.1002/(SICI)1521-4141(199911)29:11<3712::AID-IMMU3712>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 21.Berney C, Herren S, Power CA, Gordon S, Martinez-Pomares L, Kosco-Vilbois MH. A member of the dendritic cell family that enters B cell follicles and stimulates primary antibody responses identified by a mannose receptor fusion protein. J. Exp. Med. 1999;190:851–860. doi: 10.1084/jem.190.6.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindquist RL, Shakhar G, Dudziak D, Wardemann H, Eisenreich T, Dustin ML, Nussenzweig MC. Visualizing dendritic cell networks in vivo. Nat. Immunol. 2004;5:1243–1250. doi: 10.1038/ni1139. [DOI] [PubMed] [Google Scholar]
- 23.Qi H, Egen JG, Huang AY, Germain RN. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science. 2006;312:1672–1676. doi: 10.1126/science.1125703. [DOI] [PubMed] [Google Scholar]
- 24.Wykes M, Pombo A, Jenkins C, MacPherson GG. Dendritic cells interact directly with naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J. Immunol. 1998;161:1313–1319. [PubMed] [Google Scholar]
- 25.Law CL, Aruffo A, Chandran KA, Doty RT, Clark EA. Ig domains 1 and 2 of murine CD22 constitute the ligand-binding domain and bind multiple sialylated ligands expressed on B and T cells. J. Immunol. 1995;155:3368–3376. [PubMed] [Google Scholar]
- 26.Sgroi D, Varki A, Braesch-Andersen S, Stamenkovic I. CD22, a B cell-specific immunoglobulin superfamily member, is a sialic acid-binding lectin. J. Biol. Chem. 1993;268:7011–7018. [PubMed] [Google Scholar]
- 27.Bergtold A, Desai DD, Gavhane A, Clynes R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity. 2005;23:503–514. doi: 10.1016/j.immuni.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 28.Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin immunoglobulin G complexes. J. Exp. Med. 2004;199:1631–1640. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinman RM, Hawiger D, Nuusenzweig MC. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 30.Nitschke L, Tsubata T. Molecular interactions regulate BCR signal inhibition by CD22 and CD72. Trends Immunol. 2004;25:543–550. doi: 10.1016/j.it.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Cornall RJ, Goodnow CC, Cyster JG. Regulation of B cell antigen receptor signaling by the Lyn/CD22/SHP1 pathway. Curr. Top. Microbiol. Immunol. 1999;244:57–68. doi: 10.1007/978-3-642-58537-1_5. [DOI] [PubMed] [Google Scholar]
- 32.Law CL, Sidorenko SP, Chandran KA, Zhao Z, Shen SH, Fischer EH, Clark EA. CD22 associates with protein tyrosine phosphatase 1C, Syk, and phospholipase C-γ1 upon B cell activation. J. Exp. Med. 1996;183:547–560. doi: 10.1084/jem.183.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marshall AJ, Niiro H, Lerner CG, Yun TJ, Thomas S, Disteche CM, Clark EA. A novel B lymphocyte-associated adaptor protein, Bam32, regulates antigen receptor signaling downstream of phosphatidylinositol 3-kinase. J. Exp. Med. 2000;191:1319–1332. doi: 10.1084/jem.191.8.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han A, Saijo K, Mecklenbrauker I, Tarakhovsky A, Nussenzweig MC. Bam32 links the B cell receptor to ERK and JNK and mediates B cell proliferation but not survival. Immunity. 2003;19:621–632. doi: 10.1016/s1074-7613(03)00275-9. [DOI] [PubMed] [Google Scholar]
- 35.Stamenkovic I, Seed B. The B-cell antigen CD22 mediates monocyte and erythrocyte adhesion. Nature. 1990;345:74–77. doi: 10.1038/345074a0. [DOI] [PubMed] [Google Scholar]
- 36.Engel P, Nojima Y, Rothstein D, Zhou LJ, Wilson GL, Kehrl JH, Tedder TF. The same epitope on CD22 of B lymphocytes mediates the adhesion of erythrocytes, T and B lymphocytes, neutrophils, and monocytes. J. Immunol. 1993;150:4719–4732. [PubMed] [Google Scholar]
- 37.Hennet T, Chui D, Paulson JC, Marth JD. Immune regulation by the ST6Gal sialyltransferase. Proc. Natl. Acad. Sci. USA. 1998;95:4504–4509. doi: 10.1073/pnas.95.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otipoby KL, Andersson KB, Draves KE, Klaus SJ, Farr AG, Kerner JD, Perlmutter RM, Law CL, Clark EA. CD22 regulates thymus-independent responses and the lifespan of B cells. Nature. 1996;384:634–637. doi: 10.1038/384634a0. [DOI] [PubMed] [Google Scholar]
- 39.Förster I, Vieira P, Rajewsky K. Flow cytometric analysis of cell proliferation dynamics in the B cell compartment of the mouse. Int. Immunol. 1989;1:321–331. doi: 10.1093/intimm/1.4.321. [DOI] [PubMed] [Google Scholar]
- 40.Allman DM, Ferguson SE, Lentz VM, Cancro MP. Peripheral B cell maturation: II. Heat-stable antigenhi splenic B cells are an immature developmental intermediate in the production of long-lived marrow-derived B cells. J. Immunol. 1993;151:4431–4444. [PubMed] [Google Scholar]
- 41.Collins BE, Smith BA, Bengtson P, Paulson JC. Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nat. Immunol. 2006;7:199–206. doi: 10.1038/ni1283. [DOI] [PubMed] [Google Scholar]
- 42.Cariappa A, Tang M, Parng C, Nebelitskiy E, Carroll M, Georgopoulos K, Pillai S. The follicular versus marginal zone B lymphocyte cell fate decision is regulated by Aiolos, Btk, and CD21. Immunity. 2001;14:603–615. doi: 10.1016/s1074-7613(01)00135-2. [DOI] [PubMed] [Google Scholar]
- 43.Saito T, Chiba S, Ichikawa M, Kunisato A, Asai I, Shimizu K, Yamaguchi T, Yamamoto G, Seo S, Kumano K, et al. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity. 2003;18:675–685. doi: 10.1016/s1074-7613(03)00111-0. [DOI] [PubMed] [Google Scholar]
- 44.Samardzic T, Marinkovic D, Danzer C-P, Gerlach J, Nitschke L, Wirth T. Reduction of marginal zone B cells in CD22-deficient mice. Eur. J. Immunol. 2002;32:561–567. doi: 10.1002/1521-4141(200202)32:2<561::AID-IMMU561>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 45.Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, Ravetch JV, Steinman RM, Nussenzweig MC. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001;194:769–779. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 2003;196:1627–1638. doi: 10.1084/jem.20021598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lui K, Iyoda T, Saternus M, Kimura Y, Inaba K, Steinman RM. Immune tolerance after delivery of dying cells to dendritic cells in situ. J. Exp. Med. 2002;196:1091–1097. doi: 10.1084/jem.20021215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finkelman FD, Lees A, Birnbaum R, Gause WC, Morris SC. Dendritic cells can present antigen in vivo in a tolerogenic or immunogenic fashion. J. Immunol. 1996;157:1406–1414. [PubMed] [Google Scholar]
- 49.Krzewinski-Recchi MA, Julien S, Juliant S, Teintenier-Lelievre M, Samyn-Petit B, Montiel MD, Mir AM, Cerutti M, Harduin-Lepers A, Delannoy P. Identification and functional expression of a second human β-galactoside α2,6-sialyltransferase, ST6Gal II. Eur. J. Biochem. 2003;270:950–961. doi: 10.1046/j.1432-1033.2003.03458.x. [DOI] [PubMed] [Google Scholar]
- 50.Takashima S, Tsuji S, Tsujimoto M. Comparison of the enzymatic properties of mouse β-galactoside α2,6-sialyltransferases, ST6Gal I and II. J. Biochem. 2003;134:287–296. doi: 10.1093/jb/mvg142. [DOI] [PubMed] [Google Scholar]
- 51.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 52.Grewal PK, Boton M, Ramirez K, Collins BE, Saito A, Green RS, Ohtsubo K, Chui D, Marth JD. ST6Gal-I restrains CD22-dependent antigen receptor endocytosis and Shp-1 recruitment in normal and pathogenic immune signaling. Mol. Cell. Biol. 2006;26:4970–4981. doi: 10.1128/MCB.00308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cyster JG, Goodnow CC. Tuning antigen receptor signaling by CD22: integrating cues from antigens and the microenvironment. Immunity. 1997;6:509–517. doi: 10.1016/s1074-7613(00)80339-8. [DOI] [PubMed] [Google Scholar]
- 54.Richards S, Santos L, Watanabe C, Clark EA. Regulation of B cell entry into the cell cycle. Immunol. Rev. 2008 doi: 10.1111/j.1600-065X.2008.00652.x. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poe JC, Fujimoto Y, Hasegawa M, Haas KM, Miller AS, Sanford IG, Bock CB, Fujimoto M, Tedder TF. CD22 regulates B lymphocyte function in vivo through both ligand-dependent and ligand-independent mechanisms. Nat. Immunol. 2004;5:1078–1087. doi: 10.1038/ni1121. [DOI] [PubMed] [Google Scholar]
- 56.Jin L, McLean PA, Neel BG, Wortis HH. Sialic acid binding domains of CD22 are required for negative regulation of B cell receptor signaling. J. Exp. Med. 2002;195:1199–1205. doi: 10.1084/jem.20011796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kelm S, Gerlach J, Brossmer R, Danzer CP, Nitschke L. The ligand-binding domain of CD22 is needed for inhibition of the B cell receptor signal, as demonstrated by a novel human CD22-specific inhibitor compound. J. Exp. Med. 2002;195:1207–1213. doi: 10.1084/jem.20011783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han S, Collins BE, Bengtson P, Paulson JC. Homomultimeric complexes of CD22 in B cells revealed by protein-glycan cross-linking. Nat. Chem. Biol. 2005;1:93–97. doi: 10.1038/nchembio713. [DOI] [PubMed] [Google Scholar]
- 59.Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat. Rev. Immunol. 2002;2:945–956. doi: 10.1038/nri955. [DOI] [PubMed] [Google Scholar]
- 60.Pillai S, Cariappa A, Moran ST. Positive selection and lineage commitment during peripheral B-lymphocyte development. Immunol. Rev. 2004;197:206–218. doi: 10.1111/j.0105-2896.2003.097.x. [DOI] [PubMed] [Google Scholar]
- 61.Casola S, Otipoby KL, Alimzhanov M, Humme S, Uyttersprot N, Kutok JL, Carroll MC, Rajewsky K. B cell receptor signal strength determines B cell fate. Nat. Immunol. 2004;5:317–327. doi: 10.1038/ni1036. [DOI] [PubMed] [Google Scholar]
- 62.Collins BE, Blixt O, DeSieno AR, Bovin N, Marth JD, Paulson JC. Masking of CD22 cis ligands does not prevent redistribution of CD22 to sites of cell contact. Proc. Natl. Acad. Sci. USA. 2004;101:6104–6109. doi: 10.1073/pnas.0400851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lanoue A, Batista FD, Stewart M, Neuberger MS. Interaction of CD22 with α2,6-linked sialoglycoconjugates: innate recognition of self to dampen B cell autoreactivity? Eur. J. Immunol. 2002;32:348–355. doi: 10.1002/1521-4141(200202)32:2<348::AID-IMMU348>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 64.Leprince C, Draves KE, Geahlen RL, Ledbetter JA, Clark EA. CD22 associates with the human surface IgM-B-cell antigen receptor complex. Proc. Natl. Acad. Sci. USA. 1993;90:3236–3240. doi: 10.1073/pnas.90.8.3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tuscano JM, Riva A, Toscano SN, Tedder TF, Kehrl JH. CD22 cross-linking generates B-cell antigen receptor-independent signals that activate the JNK/SAPK signaling cascade. Blood. 1999;94:1382–1392. [PubMed] [Google Scholar]