

Abstract

The author reviews how his early love for theoretical organic chemistry led to experimental research and the extended search for quantitative correlations between experiment and quantum calculations. The experimental work led to ion pair acidities of alkali-organic compounds and most recently to equilibria and reactions of lithium and cesium enolates in THF. This chemistry is now being modeled by ab initio calculations. An important consideration is the treatment of solvation in which coordination of the alkali cation with the ether solvent plays a major role.
Introduction
In the 1950s physical organic chemistry had two principal parts, the determination of reaction mechanisms and the effect of structure on reactivity. A number of tools had been developed for the study of reaction mechanisms: reaction kinetics, substituent effects, isotope effects, stereochemistry. In those early days I too was active in developing such tools. One of my most notable achievements related to secondary deuterium isotope effects.1 I was also interested in optical activity derived from hydrogen – deuterium asymmetry and prepared a number of compounds whose optical activity resulted from H-D asymmetry.2 These compounds were useful for examining the stereochemistry of reactions at primary centers.
Substituent effects often were confined to benzene derivatives and made use of Hammett – like σρ relationships. When the original version of the sigma of a substituent effect did not give satisfactory results, new definitions were proposed giving rise to a whole panoply of substituent constants: σ0, σ+, σ−, σI, etc.3 I was involved in many of these varied activities that constituted the physical organic chemistry of that era.
Disproportionation of Alkylbenzenes
The study of reactions of carbocations, then known as “carbonium ions”, was popular at mid-century and my group was active in various aspects of this chemistry. An example of one of these applications relates to the disproportionation reaction of alkylbenzenes, a reaction of some importance in petroleum chemistry. Not long after the discovery of the Friedel-Crafts alkylation reaction, Anschütz and Immendorff4 found that aluminum chloride converts ethylbenzene to benzene and a mixture of diethylbenzenes. When the reaction was applied to n-propylbenzene, the product still had n-propyl groups with no rearrangement to isopropyl.5 This observation shows that free alkyl cations are not involved, since rearrangement of n-propyl cations to isopropyl is facile. McCaulay and Lien found that neopentylbenzene is inert under conditions where ethylbenzene reacts readily; since this reactivity order is characteristic of SN2 reactions, they proposed a corresponding mechanism for disproportionation6 (Fig. 1).
Fig 1.

SN2 displacement mechanism for disproportionation.
We thought to test this mechanism using the classic method of Hughes, et al,7 who showed in the reaction of optically active 2-iodooctane with radioactive iodide ion that the rate of racemization was twice the rate of radio-iodine exchange; thus, each act of displacement went with inversion of configuration. Eliel had prepared optically active (ethyl-1-d)benzene earlier.8 Consequently, we could apply the same principle to optically active (ethyl-1-d)benzene labeled in the ring with 14C (Fig 2).
Fig 2.

Proposed test of the SN2 displacement mechanism with labeled optically active (ethyl-1-d)benzene in excess benzene.
The experiment was carried out by my first female graduate student, Liane Reif (now Reif-Lehrer).9 She found, to our surprise, that the rate of racemization was equal to the rate radio-exchange. The results of two runs are shown in Fig. 3. Mass spectroscopy of the isolated kinetic samples showed progressive scrambling to ethylbenzene and (ethyl-1,1-d2)benzene at a rate somewhat less than that of exchange or racemization. These observations led to a new mechanism shown in Fig. 4.
Fig. 3.

Two runs showing the reaction of opically active (ethyl-1-d)benzene labeled in the ring with 14C with GaBr3-HBr in benzene. Blue squares show optical activity, red circles are radioactivity and black squares are d1.
Fig. 4.

New mechanism for trans-ethylation.
The sequence starts with a trace amount of phenethyl cation from styrene impurity or a small amount of oxidation. Alkylation and dealkylation of benzene is rapid and results in loss of radiolabel and optical activity. The rate-determining step is hydride transfer from ethylbenzene to phenethyl cation. This type of process was shown earlier to occur readily between carbonium ions and hydrocarbons.10 Only transfer of deuterium results in scrambling and is expected to involve a significant primary isotope effect; hence, the scrambling process is slower than racemization or radioexchange. This mechanism has since become accepted for such trans-alkylation processes with primary alkyl groups. The mechanism requires that 1,1-diphenylalkanes rapidly dealkylate under the reaction conditions. This corollary was proved later when we showed that 1,1-di-p-tolylethane is completely converted to ethylbenzene and toluene within seconds on treatment with GaBr3-HBr under comparable reaction conditions.11 This mechanism example is also notable for its use of deuterium in several roles: as a label, for kinetic isotope effects and for optical activity from H-D asymmetry.
Early Theory
Molecular orbital theory at mid-century was primarily Hückel theory (HMO). The theory is limited to π – electronic systems but despite this limitation and the gross approximations in the theory it had a number of successful applications in its day. These early results were summarized in my book “Molecular Orbital Theory for Organic Chemists”, published in 1961.12 In order to determine how well HMO theory could model organic reactions, my group and several others studied the reactivities of several π-electronic systems that could provide reasonable tests. A typical example was our study of the exchange reaction of ArCH2D with lithium cyclohexylamide in cyclohexylamine, in which the Ar groups are polycyclic aromatic rings.13,14 The transition state for the reaction has a high degree of carbanionic character.15 The π–system model that could be calculated by HMO theory was that of an arene going to an arylmethyl anion. For π-energies given in the usual HMO form, Eπ = nα + mβ, this energy difference is 2α + Δmβ. The results we found are summarized in Figure 5.
Fig. 5.

Relative exchange rates of ArCH2D with lithium cyclohexylamide in cyclohexylamine. The polycyclic hydrocarbons are: Ph, benzene; Na, naphthalene; Pn, phenanthrene; An, anthracene; Py, pyrene; Fl, fluoranthene.
My late colleague, Professor Joel Hildebrand, was fond of pointing out that to the ancients the perfect geometric figure is the circle but to a scientist the perfect figure is a straight line joining his theory with experimental data. Figure 5 is not such a figure. To a scientist this scattering of points is esthetically ugly. The points do divide roughly into positions of the 2-naphthyl and more hindered 1-naphthyl types, although the 2-pyrenyl and 3-fluoranthyl positions are conspicuous exceptions.
This example points up the limited utility of the simple HMO method. It and its all-valence-electron analog, Extended Hückel Theory, have had notable successes, primarily in the qualitative understanding of many reactions, particularly of the pericyclic type. These successes are based on the 4n+2 rule and perturbation theory applied to the frontier orbitals (the highest occupied and lowest vacant orbitals) and work because their nodal properties are given correctly even by simple theories. Such nodal properties led to one of my most important early achievements, the conception and synthesis of uranocene, a sandwich compound with a uranium centered between two planar 8-membered rings.16 I recognized that bonding interaction of the highest occupied e2 MOs of a 10-electron cycle with fxyz atomic orbitals would be analogous to the interaction of the highest occupied e1 MOs of a 6-electron cycle with atomic d orbitals so important in the ring-metal bonding of ferrocenes. The story of this discovery and of my work in organo-f-element chemistry has been told before, most notably in my chemical autobiography, “A Lifetime of Synergy with Theory and Experiment”,17 and has been reviewed recently.18
Nevertheless, quantitative correlations of simple MO theory with experiment were rare. We subsequently tried some of the various all-valence-electron methods that arose about that time, methods having the acronyms CNDO, MNDO, etc., but without notable success. Instead, we had to await the development of ab initio computer programs. Fortunately, we did not have wait long.
The first general programs for ab initio MO computations, such as John Pople’s GAUSSIAN program, became available in the 1960’s, although our initial work in the late 60’s and early 70’s was done with IBMOL, a program generously made available to us by Enrico Clementi and his group at IBM, and which we modified for use on the campus CDC6400 computer. Computers in those days were quite limited – the campus CDC had far less computational power than the smallest laptop available today. Accordingly, computations were limited to small molecules and relatively small basis
sets. In a commentary in 1970,19 I pointed out that even restricted to small systems, one could still test important concepts in chemistry. A typical example was the demonstration that double bonds of the pπ-pπ type with second row elements are quite weak; e.g., the dipolar structure of phosphine oxides is far more important than the P=O structure:20
The electron density is a physical observable that can be computed readily and visualized with computer graphics. We made early use of this new tool in various studies21–23 and in a book, “Orbital and Electron Density Diagrams: An Application of Computer Graphics.”24 In studying the electron density function of methyllithium (Fig. 6),25 we made the surprising observation that the electron density has a low value between Li and C, much lower than we had expected from conventional thinking about organolithium bonding. I had been taught, and in my courses I also taught, that the carbon-lithium bond has comparable degrees of covalent and ionic character; that is, both of the following resonance structures are important in its electronic structure:
Fig 6.

Electron density function for methyllithium for the plane shown in units of e au−3. Compare the deep valley between C and Li with the high ridge between C and H. [Reproduced from ref. 25 Copyright 1976 American Chemical Society]
“Covalent” and “ionic” character are not physical observables and therefore have no exact definition, but, like many other such fuzzy concepts have a qualitative usefulness in understanding chemical properties. We interpreted this electron density result to mean that organolithium compounds are essentially tight ion pairs with little covalent bonding. We were also able to explain many of the properties of such compounds that seemed to require covalency, such as solubility in organic solvents, in terms of a wholly ionic model. Our interpretation was immediately controversial but has become generally accepted.26,27 The modeling of ion pairs has since become a major part of my computational research.
Carbon Acidity
My developing interest in carbanion chemistry that started in the late 1950’s with kinetic acidities, as measured by base-catalyzed hydrogen exchange reactions, extended in the late 1960’s to include equilibrium acidities. This work started originally in cyclohexylamine as solvent28,29 but later changed to tetrahydrofuran (THF),30 a much more important solvent for organic synthesis. Other groups were making measurements of carbon acidity at this same time. In a particularly important study, Ritchie and Uschold obtained equilibrium pKa values in dimethyl sulfoxide (DMSO).31–33 Bordwell later built on their work and determined the pKa’s in DMSO of hundreds of compounds.34 DMSO is a polar solvent with a relatively high dielectric constant, 46.6. Most salts are largely present as free ions. Nevertheless, any attempt to model such pKs’s with computed energies requires an evaluation of solvation energies of the carbanions. Figure 7 shows a comparison of gas phase proton affinities of anions with the corresponding pKa’s in DMSO. There is clearly a rough trend but much scatter because of solvation energies.
Fig. 7.

Gas phase acidities can be calculated with standard deviations of a few Kcal mol−1 using basis sets of reasonable size. Fu, et al,36 used a carefully calibrated polarized continuum model (PCM) to determine the solvation energies of ions in DMSO and were able to compute the pKa’s of over 100 compounds with an error of about ±2 pK-units. The solvation energies of anions in DMSO are an order of magnitude greater than that of neutral molecules. We considered, therefore, that solvation energies of ion pairs in THF would be of lower magnitude.
Ion pair acidities are based on the reaction in eq. 1, where M is an alkali metal which we chose generally to be either Li or Cs.
| eq. 1 |
The acidities measured, therefore, are acidity differences. For convenience we chose fluorene to be our standard and assigned its pK as 22.9, the pKa of fluorene in DMSO (per hydrogen). The measurements were made by UV-vis spectroscopy of dilute solutions in a glovebox in which the thermostated sample cell holder was connected to an external spectrometer with fiber-optic cables. A number of so-called indicator hydrocarbons were analyzed by this method using lithium37 and cesium30 as the counterions in eq. 1. These indicator hydrocarbons are relatively acidic polyarylmethanes or fluorenes whose conjugate bases are highly delocalized carbanions having absorption in the visible range. The cesium salts are contact ion pairs (CIP) in which the two ions are held in close juxtaposition. The lithium salts, however, are solvent-separated ion pairs (SSIP) in which the lithium is coordinated to four solvent molecules and this assembly forms an ion pair with the carbanion. This difference in the ion pair properties of lithium and cesium cations results from their relative size. The interaction of two ions falls off with the distance, R, as 1/R, whereas the electrostatic energy of charge-dipoles falls off as 1/R2. With delocalized carbanions the effective distance of negative charge to a cation is relatively large. With the small lithium cation, solvent dipoles can compete effectively with a delocalized carbanion in joining to the cation. With the much larger cesium cation, however, solvent dipoles cannot get close enough for 1/R2 to compete with the charge-charge 1/R.
No attempts have been made to model the SSIP theoretically. There is not much necessity to do so because the pK’s are generally quite similar to the ionic pKa’s in DMSO; that is, the SSIP lithium pK’s relative to fluorene in THF are numerically quite similar to the ionic pKa’s in DMSO. Localized carbanions, however, form CIP with lithium cations and these pK’s can differ substantially from the ionic pKa’s in DMSO. A number of the CIP organolithium compounds we have measured are summarized in Table 1. CIP lithium salts are frequently aggregated in THF solution. We have measured many of the aggregation constants as discussed below; the “lithium acidities” summarized in Table 1 pertain to the monomeric lithium compounds.
Table 1.
pK’s of lithium compounds in THF relative to fluorene = 22.90 (per hydrogen).
| Compound | Li pK THF | pKa DMSOo |
|---|---|---|
| Benzene | 39.5b,c | |
| Phenyldithiane | 29.4d | 30.65 |
| Adamantylacetylene | 23.7e | (28.7)p |
| 1,2,4,5-tetrafluorobenzene | 23.1c | |
| N-Methyl-p-phenylaniline | 22.09f | (30.9)q |
| p-Biphenylylmethyloxazoline | 21.55g | |
| Pentafluorobenzene | 21.5b,c | |
| DiphenylDMAa | 20.78g | |
| p-BiphenylylDMAa | 19.77g | |
| Diphenylamine | 19.05h | 24.95 |
| p-Phenylisobutyrophenone | 15.86i | (26.7)r |
| 6-Phenyltetralone | 14.22j | (23.3)s |
| Carbazole | 13.48k | 19.9 |
| Phenylcyclohexanone | 12.69l | |
| p-Biphenylylcyclohexanone | 12.31m | |
| Dibenzyl ketone | 11.62n | 19.3 |
| 2,6-Diphenyltetralone | 11.14j |
DMA = N,N-dimethylacetamide
Obtained by extrapolation
Ref 38
Ref 39
Ref 40
Unpublished results
Ref 41
Ref 42
Ref 43
Ref 44
Ref 45
Ref 46
Ref 47
Ref 48
Ref 34, per hydrogen
For phenylacetylene
For aniline
For cyclohexyl phenyl ketone
For 1-indanone
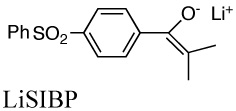
Not many compounds have been done in both systems. The most accurate measurements of the equilibrium constants of eq. 1 require distinctive uv-vis spectra for both carbanionic components. When only the indicator has a measurable spectrum, the equilibrium in eq. 1 can be determined by a decrease in the absorbance of the indicator when a known amount of substrate is added, a technique we have called the “single indicator method”. Many of our compounds have biphenylyl groups or other substituents that provide a useful chromophore. Most of Bordwell’s measurements in DMSO used the single indicator method. Nevertheless, in general, the CIP lithium pK’s are several units lower than the pKa’s in DMSO and is a measure of the effect of the stronger ionic bonding to the lithium compared to that in the SSIPs.
The experimental results can be compared with theoretical computations of eq. 1. Figure 8 presents the results of HF calculations at the 6–31+g(d) level. The use of electronic energies plus zero point energies relative to benzene in effect gives ΔE for eq 1 at absolute zero. The plot shows a general correlation but with much scatter. A regression line through these points has R2 = 0.85, a value too low for usefulness. The problem is undoubtedly the lack of consideration of solvation energies. Even though ion pairs are expected to have smaller solvation energies than free ions, they are still too large to be neglected. Much of this solvation energy is undoubtedly direct coordination of lithium to an ether solvent. Such coordination, after all, is strong enough to produce SSIPs with delocalized carbanions. A typical structure of a CIP RLi coordinated with three dimethyl ether molecules is shown in Fig. 9. In phenyllithium.3Me2O, two of the ether oxygens coordinate to lithium with essentially trigonal bonds (sum of bond angles = 358.5–358.6°) whereas the third is more highly pyramidal (sum of angles = 335.2°). The difference is probably a matter of space-filling. The Li-O bonds are almost certainly highly ionic.
Figure 8.

Plot of the experimental Li pK’s in Table 1 vs the electronic energy + ZPE at HF 6–31+G(d).
Figure 9.

Computed structure of PhLi.3Me2O.
If we consider all of the lithium compounds to be coordinated to three ethers, eq.1 becomes eq. 2 in which E is the ether.
| eq. 2 |
A plot of the lithium pK’s with the energies of eq. 2 relative to benzene is shown in Fig. 10. This simple change makes an enormous difference in the quality of the correlation; the regression line now has R2 = 0.96.
Figure 10.

Experimental Li pK’s in Table 1 for eq. 2 relative to benzene, HF 6–31+G(d). The regression line is pK = 40.25 ± 1.17 + (0.671 ± 0.036)ΔE, R2 = 0.96.
The only solvation energy term considered above is coordination of lithium cation with an ether. The dielectric solvation term of the coordinated lithium compounds is likely to be small because of the increased size of the molecules and the fact that one part of the molecule already looks like the solvent. Moreover, the differential solvation effect between R’Li.3E and RLi.3E is likely to be even smaller. Nevertheless, these solvation effects are not negligible and we should take into account the solvation energy differences between RH and R’H. Both of these solvation effects might well be responsible for much of the remaining scatter in Fig. 10. Various versions of PCM have generally been used for this purpose, and we have used these methods ourselves. But, when we tested various PCM methods with experimental results for typical organic compounds in solvents of low polarity like ethers we got disappointing results. Figure 11 shows a typical comparison of experimental and computed solvation energies in such solvents as ethyl ether and THF.49 A recent analysis of the popular IEF-PCM model gave mean errors of 5.66 Kcal mol−1 for the solvation energies of typical organic compounds in organic solvents, an error comparable to the magnitude of the solvation energies.50,51 Indeed, it was found that one does almost as well by assigning an average value to all compounds in a given solvent! This aspect of solvation remains a continuing problem. Quite recently, a new solvation model, “SM8”, became available from the Minnesota group of Truhlar and Cramer,50 but it was not successful in our application, perhaps because it is not calibrated for organolithium compounds.
Figure 11.

Experimental and computed free energies of solvation, Kcal mol−1, ref. 49
Aggregation of Lithium Enolates
Table 1 includes several ketones. Lithium enolates are important reagents in synthetic organic chemistry and have long been known to be frequently aggregated in ethereal solutions.52 A number of lithium enolate crystal structures have been established and show a common motif of cyclic dimers and cubic tetramers as symbolized by 1 and 2.

It has been suggested that the aggregates are involved in various synthetic reactions of such enolates,53–55 but the evidence for such involvement is not definitive. Establishing such a role is not simple. The system is that of a classic Curtin-Hammett equilibrium, Fig. 12.56 Each of the components has its own rate constant for reaction and the role of each depends on its concentration in the equilibrium and its rate constant. Thus, it is perfectly conceivable, for example, that the monomeric ion pair might be only a small component but have such a large rate constant that the major flux of reaction is through it. Indeed, that is what we have found for the alkylation of a number of enolates. A combination of careful equilibrium and kinetic studies was required to establish this generalization.
Fig. 12.

Curtin-Hammett equilibria for enolate aggregation and reaction. Each kn is the rate constant for the reaction of the corresponding RM system (e.g, (R− M+)n) with R’X.
In order to measure the equilibrium constants, K, one must work in a concentration regime in which all of the components are present in measurable amounts. For a monomer – dimer equilibrium, K1,2 = [D]/[M]2 and has units of M−1. To determine the appropriate concentration regime it is useful to consider that when [D] = [M], [M] = 1/K1,2. For K1,2 = 100 – 102 the concentration regime of about 0.01 – 1M is appropriate for NMR or infrared spectroscopy. Many of our enolate equilibria have K1,2 of the order of 103 – 105, which requires UV-vis spectroscopy for measurement. Dealing with such dilute solutions of air-sensitive organolithium compounds also required careful techniques using our glovebox –spectrometer apparatus. With this equipment we determined the aggregation equilibria by two different techniques: variation of the spectrum with concentration and by the “coupled equilibria” of aggregation and ion pair acidity. The studies were carried out principally with lithium and cesium as counterions in order to cover both ends of the alkali Periodic Table.
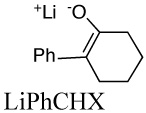
For a number of enolates, we found that different aggregates have significantly different spectra.57 An example is shown in Figure 13 for the lithium salt of 2-(p-biphenylyl)cyclohexanone (LiBiphCHX).47 As in other organolithium equilibria discussed above many of our examples required aryl groups, such as phenyl or biphenylyl, to provide an essential chromophore. It is clear in Figure 13 that λmax shifts to longer wavelength in more dilute solutions. James Krom42 showed that this data can be analyzed by the linear-algebraic method, “singular-value decomposition” (SVD). The method involves matrix diagonalization of the digitized data to produce a series of vectors and associated coefficients. The first three SVD vectors derived from the spectra in Fig. 13 are shown in Fig. 14. Only the first two vectors have significant magnitude. Thereafter, the vectors have small coefficients and just describe noise. These vectors have no physical significance but further matrix manipulation of the first two vectors does produce the spectra of the two significant components as shown in Fig. 15. That these are the spectra of the monomer and dimer will be shown in the discussion to follow.
Figure 13.

Spectra of LiBiphCHX, the lithium enolate of 2-(p-biphenylyl)cyclohexanone as a function of concentration in THF at 25° C.
Figure 14.

First three SVD vectors for the data in Fig. 11. The relative weights are S1=55.45, S2=2.32, S3=0.19.
Figure 15.

Spectra of the monomer and dimer of LiBiphCHX, the lithium enolate of 2-(p-biphenylyl)cyclohexanone, derived from the SVD analysis in Fig. 14.
From the spectra of the two components we can now go back to the original spectra in Fig. 13 and deconvolute to determine the concentration of each. Assuming these to be the monomer and dimer, we plot the concentration of the presumed dimer against the square of that of the presumed monomer and get a straight line confirming the assignments (Figure 16). The slope in Fig. 16 is directly the equilibrium constant, K1,2. Any other reasonable combination of aggregates would have given a curve in Figure 16.
Figure 16.

Plot of the concentration of the presumed dimer vs [monomer]2 to give a straight line whose slope is K1,2 = 4546 M−1.
The monomer has a longer wavelength λmax than the aggregate and we have found this generally to be the case. A simple rationalization is shown in Figure 17. The transition vector is assumed to involve transfer of electron density from the oxide ion to the other end of the double bond or aromatic system. The presence of additional cations nearby in the aggregate electrostatically impedes this process and requires higher energy (shorter wavelength).
Figure 17.

Monomer is at longer wavelength (lower energy).
An example of “coupled equilibria” is the equilibrium that defines lithium ion pair acidity together with the aggregation equilibrium, eq. 3.
| eq. 3 |
Formation of an aggregate pulls the first equilibrium to the right and makes the observed equilibrium constant, Kobs, as defined in eq. 4, dependent on concentration.
| eq. 4 |
In this equation {R− Li+} is the concentration of R− Li+ moieties as measured by spectroscopy whether present in monomer or aggregates. pKobs is defined in terms of the indicator pK, pK(Ind-H) by eq. 5.
| eq. 5 |
In a concentration regime where more than one aggregate species is present in measurable amounts, pKobs is concentration dependent. An example is shown in Fig. 18 for the lithium enolate of 2-(p-biphenylyl)cyclohexanone (LiBiphCHX). At higher concentrations more of the enolate is present as an aggregate and the effective pK is lower – more ketone is pulled towards enolate.
Fig. 18.

The observed pK of LiBiphCHX, the lithium enolate of 2-(p-biphenylyl)cyclohexanone, depends on the concentration. The black points are the theoretical pK’s for a monomer-dimer equilibrium with K1,2 = 4300 M−1.
An alternative way of treating the data makes use of eq. 6, which can be derived from the sum of all of the aggregation equilibria in eq. 3.
| eq. 6 |
For monomer-dimer and monomer-tetramer equilibria, eq. 6 reduces to eqs. 7 and eqs. 8, respectively.
| eq. 7 |
| eq. 8 |
In these equations, Kobs and {R− Li+} are experimental data. A plot of Kobs vs {R−Li+}/Kobs (for monomer-dimer) or ({R− Li+}/Kobs)3 (for monomer-tetramer) gives a straight line from which K0 and K1,n can be derived. An example for the data in Figure 18 is shown in Figure 19 in which the enolate concentration is given as {LiBPCH}. From the slope and intercept eq. 7 gives K1,2 = 4345 ± 130 M−1 in excellent agreement with the other approaches.
Fig. 19.

Plot of the data for the lithium enolate of 2-(p-biphenylyl)cyclohexanone, LiBPCH) according to eq. 7. The regression line shown is 1.029 ± 0.021 + (9202 ± 172)x; R2 = 0.995.
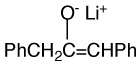
In this way we determined the aggregation equilibrium constants of a number of lithium and cesium enolates as summarized in Table 2. The enolate aggregates are generally either dimers or tetramers in equilibrium with monomers. In only a few cases were all three components present in measurable amounts. No examples of trimers were found.
Table 2.
Aggregation equilibrium constants of lithium and cesium enolates. The structure of the lithium enolate is shown. Values for cesium enolates are given in italics.
| Li Enolate | K1,2 M−1 | K1,4 M−3 | [M] in 1Ma | pK |
|---|---|---|---|---|
 |
5.0E+8e | 0.0047 | 15.9 | |
| 2.9E+4d | 7.8E+12 | 25.1 | ||
 |
5.0E+4b | 0.0032 | 14.7 | |
 |
4.2E+2c | 0.034 | 11.6 | |
| 3.5E+3 | 18.1 | |||
 |
2.8E+3f | 0.013 | 12.7 | |
| 1.8E+3 | 19.8 | |||
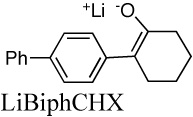 |
4.3E+3g | 0.011 | 12.6 | |
| 1.9E+3h | 19.3 | |||
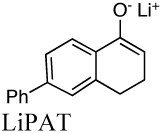 |
4.7E+10i | 0.0015 | 14.2 | |
| 2.3E+11k | 23.4 | |||
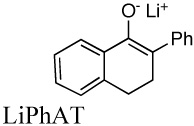 |
1.9E+3j | 0.016 | ||
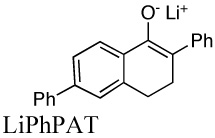 |
2.65E+3i | 0.014 | 11.1 | |
| 1.8E+3 | 17.8 | |||
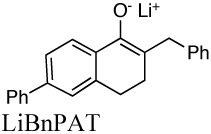 |
3.8E+3i | 0.011 | 14.0 | |
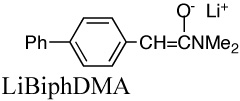 |
4.5E+2l | 0.033 | 19.8 | |
| 4.7E+2m | 24.9 | |||
Two dipoles arranged head-to-tail form an electrostatically stable orientation but Chabanel58 showed that for many salts in THF the principal driving force is entropic. The aggregate is frequently less solvated than the monomer and the release of solvent on aggregation provides a positive entropy change (eq. 9).
| eq. 9 |
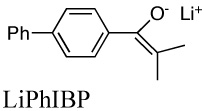
The same principle holds for phenolates65 and for enolate aggregation. The effect of temperature on the tetramerization of the lithium enolate pf p-phenylisobutyrophenone (LiPhIBP) gives ΔH° = 1.5 ± 1 Kcal mol−1 and ΔS° = 4 ± 5 eu.43,66 Thus, the change in equilibrium constant with temperature is relatively small.
Taking lithium as tetracoordinate, the dimerization of a lithium enolate can be formulated as in eq. 10.
| eq 10. |
The computed structure of a typical enolate dimer coordinated to four ethers is shown in Fig. 20 for the lithium enolate of cyclohexanone. The structure shows the iconic Li2O2 square. Li-O bond distances to the anionic enolates, 1.87–1.88Å, are significantly shorter than to the neutral oxygens of the ethers, 2.06Å. The structure is similar to that computed for the solvated dimer of PhCHLiCN that was also shown to be consistent with its IR spectrum.67
Figure 20.

Dimer of a lithium enolate coordinated to four dimethyl ethers, HF 6–31+g*.
Some structural effects on aggregation are readily rationalized. Simple enolates have high dimerization constants and readily form tetramers. With additional conjugation the electrostatic attraction of Li+ to the enolate is reduced and ether coordination is enhanced, leading to lower dimerization constants. A typical example is that of phenyl conjugation: lithium 6-phenylcyclohexene-1-oxide is a tetramer in THF, whereas the conjugated lithium 2-phenylcyclohexene-1-oxide (LiPhCHX in Table 2) forms a dimer.46 Substituents close to the enolate center sterically inhibit aggregation. The lithium enolate of 2-t-butyl-α-tetralone forms a monomer-dimer equilibrium compared to the tetramer of α-tetralone itself.68 Jackman also found 2–substituted tetralones to be less aggregated.52
A limited quantitative test of eq. 10 is available at this time. Figure 21 shows a plot of four points that give an excellent correlation considering the approximations required. K1,2 for isobutyrophenone (IBP) was estimated to be the same as that for the p-phenyl (PhIBP). Table 2 shows that phenyl substituents do have an effect but not a major one – about 0.2 in logK. Similarly, K1,2 for N,N-dimethylphenylacetamide was taken as that for its biphenylyl analog. Reich69 has reported dimerization constants of phenyllithium in THF at two temperatures that permits extrapolation to 117 M−1 at 25° C. This value fits the correlation in Fig. 21.
Figure 21.

Comparison of log(dimerization constant) vs computed energies for eq. 10, 6–31+G*. Regression line shown is −0.28 ± 0.39 – (0.309 ± 0.034)x; R2 = 0.98.
Further computations give qualitative agreement. Lithiothiophene is computed to have K1,2 = 27 M−1; experimentally, its dimerization is less than phenyllithium.70 Similarly, pentafluorophenyllithium is computed to have a small K1,2 (<1) and is known to be monomeric at normal concentrations. Note that only coordination solvation has been considered in this treatment. Moreover, some dimer examples might be less solvated than assumed in eq. 10 because of steric constraints. For some aggregates there is nmr evidence for other levels of solvation.52,71
Alkylation Reactions
For an equilibrium mixture of enolate aggregates the total rate of alkylation with RX is given by eq. 11 where [LiEn]n is the concentration of n-fold aggregate reacting with rate constant kn.
| eq. 11 |
For a simple monomer-dimer (M-D) equilibrium, eq. 11 reduces to eq. 12 and rearranges to eq. 13.
| eq. 12 |
| eq. 13 |
For the M-D enolates in Table 2, we know [M] and [D] for any nominal concentration of the enolate; thus, plotting the experimental quantity Rate/[RX][D] vs [M]/[D] gives a straight line of slope kM and intercept kD. For UV-concentrations of enolate of about 10−4 M, a concentration of RX of the order of 0.01–0.1M means that [RX] in these kinetic equations is essentially a constant. A typical example is shown in Fig. 22 for the reaction of LiBiphCHX with o-methylbenzyl bromide. The rate constant for the dimer is indistinguishable from 0. For such a case, the last term in eq. 12 drops out and the rate is simply pseudo-first order in [monomer] as shown in Fig. 23. Any contribution by dimer would show up as an upward curvature of the line; no such curvature is evident indicating that reaction is entirely with the monomer. These results suggest that kD is of the order of magnitude or less than kM/100. In one case a direct measurement was possible. For the reaction of LiSIBP with p-t-butylbenzyl bromide kM is about 3000 times kD.72
Fig 22.

Kinetic results for the reaction of LiBiphCHX with o-methylbenzyl bromide. The regression line shown is: Rate/[RX][D] = −0.006 ± 0.039 + (0.664 ± 0.023)[M]; R2 = 0.988
Fig. 23.

Plot of initial rate of reaction of 0.0205M o-methylbenzyl bromide vs monomer concn of LiBPCH. Slope of line through the origin is (13.4 ± 0.2) × 10−3; k2 = 0.655 M−1 s−1.
A reasonable model for the alkylation reaction involves the alkyl halide replacing one ether coordinated to lithium followed by a rearrangement to the SN2 transition state, eq. 14. EnLi.3E symbolizes a lithium enolate coordinated to 3 ethers; EnLi.RX.2Li symbolizes RX and two ethers coordinated to the lithium in EnLi.
| eq. 14 |
A similar process would apply to the enolate dimer.
Computed transition structures for the net reactions are shown in Fig. 24. The monomer and dimer transition structures show significant differences. The monomer reaction has a later TS with a shorter C-methyl bond and a long Li-Cl bond (4.23Å).
Fig. 24.

Computed transition structures (HF 6–31+g*) for reaction of lithium vinyloxide monomer (LiOV.3E) and dimer (2LiOV.4E) with methyl chloride.
The dimer reaction retains a greater Li-Cl interaction (2.34Å) but a strong Li-O bond in the original Li2O2 square is completely broken in order to have an almost linear SN2 reaction. It is this feature that is probably responsible for the higher reaction barrier of the dimer. The barrier difference of almost 4 Kcal mol−1 is consistent with the experimental rate difference of monomer and dimer.
These transition structures are those for C-alkylation. No O-alkylation was observed for the reactions we studied of lithium enolate monomers with alkyl halides. With sulfonate esters, however, some O-alkylation was found. This result can be rationalized by the extra oxygens on sulfur. In the computed transition structure for the O-alkylation reaction of lithium vinyloxide with methyl methanesulfonate (Fig. 25) the nucleophilic reactive oxygen is different from the oxygen coordinated with lithium. This arrangement permits a displacement reaction angle of 144° which is far better than the four-membered ring structures involved in O-alkylation with alkyl halides.
Fig. 25.

Computed TS for O-alkylation reaction of LiOV with methyl methanesulfonate.
In the reactions of several lithium enolates with benzyl bromides43,47,62,63 a m-Cl substituent give a rate increase of a factor of 2. This reactivity effect was modeled by applying the monomer TS in Fig. 24 to benzyl chloride as shown in Fig. 26. The calculated reaction barrier at MP2/HF631+G* for the reaction LiOV.3E + BnCl = TS + E is 26.67 Kcal mol−1; the barrier is 0.4 Kcal mol−1 lower with a m-chloro substituent, a result that agrees with experiment in direction and magnitude.
Fig. 26.

Computed TS (HF 6–31+G*) for reaction of lithium vinyloxide.2E with benzyl chloride.
Organocesium Compounds and Enolates
Many of our experimental results on ion pair acidity and enolate aggregation have dealt with cesium compounds. The intent was to compare the two extremes of the alkali elements with the expectation that the others would fall inbetween. Cesium is much larger and more polarizable than lithium. In organic solvents, cesium compounds are generally contact ion pairs; SSIP are rare. Although cesium is at the heavy end of the Periodic Table, computations are feasible using Effective Core Potentials (ECP) in which inner electron shells are not treated explicitly but rather as a core function. We had compared two popular cesium ECPs and found one (which will be referred to as Ross73) to be clearly superior.74 Despite its size, solvation effects are still important for cesium compounds. Table 3 summarizes some cesium ion pair pKs in THF and the calculated ΔE for eq. 1 at the HF 6–31+g(d,p) level using the Ross ECP for cesium and referred to fluorene and its reference pK of 22.90 (per H).
Table 3.
Results for equation 1 compared to experimental ion pair pKs in THF. For compounds known to be aggregated (eg cesium enolates) the quoted pKs refer to the monomers.
| RH Standard set | ΔEa Kcal mol−1 | pK (THF) |
|---|---|---|
| Fluorene | 0.0000 | 22.90b |
| 11 H-Benzo[a]fluorene | −4.6193 | 20.13b |
| 11 H-Benzo[b]fluorene | 3.6544 | 23.63b |
| 7H–Benzo[c]fluorene | −5.8302 | 19.47b |
| Benzo[def]fluorene | 1.8526 | 22.91b |
| 9-Methylfluorene | 1.6366 | 22.32b |
| 9-Phenylfluorene | −4.5744 | 18.15b |
| 9-tBuFluorene | 2.5349 | 24.39b |
| Triphenylmethane | 16.4247 | 31.26b |
| Phenalene | −4.7116 | 18.52c |
| Benzanthrene | 0.8934 | 21.48c |
| Benzo[cd]pyrene | −0.4154 | 19.95c |
| 1,3-Diphenylpropene | 12.0576 | 27.85b |
| 1,3-Diphenylindene | −13.6248 | 12.66d |
| Delocalized |
||
| Diphenylmethane | 11.5893 | 33.25b |
| Toluene | 24.6114 | 40.92b |
| p-PhToluene | 23.6189 | 38.73b |
| Indene | −7.8107 | 19.79e |
| Cyclopentadiene | −15.8297 | 16.11f |
| Aryl |
||
| Benzene | 35.2980 | 44.80g,h |
| p-fluorobenzene | 33.2329 | 43.10g,h |
| m-fluorobenzene | 31.2082 | 41.70g,h |
| 2,6-difluorobenzene | 11.6103 | 34.30h |
| Acetylenic |
||
| Adamantylacetylene | 6.2084 | 31.56i |
| Ketones |
||
| Bn2CO | −16.9726 | 18.07j |
| α-Tetralone | −0.7246 | 23.60k |
| 2-PhCyclohexanone | −8.9007 | 19.80l |
| PhIsobutyrophenone | −0.1831 | 25.08m |
| Acetophenone | −1.9777 | |
| Amines |
||
| Carbazole | −10.7763 | 19.24n |
| Ph2NH | −1.9869 | 24.20o |
| Sulfur Cpds |
||
| Dithiane | 14.3461 | 36.50p |
| Phenyldithiane | 10.6865 | 30.50p |
| MeDithiane | 19.0443 | 38.20p |
| Trithianer | 8.3652 | 30.20p |
| Dimethyl Sulfone | −3.9859 | 28.80q |
| Phenyl Methyl Sulfone | −2.1612 | 27.30q |
The plot of these data in Fig. 27 shows the expected scatter with one interesting feature. The compounds listed as “standard” in Table 3 form an excellent linear correlation of their own. The cesium salts of these compounds are ion pairs with highly delocalized carbanions such as those from fluorene, the benzofluorenes, triphenylmethane, phenalenes, etc. The relative cesium ion pair acidities of these compounds are similar to their pKa's in aqueous solutions82 and DMSO,34 and the relative ion pair acidities of SSIP lithium salts,37 and [2.2.1]cryptated lithium salts.83 Differential solvation within this group, accordingly, must be small. All of the other carbanions are thus more localized and solvation of the cesium cation is reduced, but differential effects lead to scatter. By this standard, benzylcesium, which lies far from the standard correlation, behaves as a rather localized carbanion. p-Phenylbenzylcesium is more delocalized and closer to the correlation. The salts of dithiane and methyldithiane behave as localized carbanions whereas the phenyl analog is clearly more delocalized. Somewhat surprisingly, triphenylmethane is in the standard set of delocalized systems whereas diphenylmethane is distinctly more localized; indeed, the gas phase calculation shows diphenylmethane to be more acidic than triphenylmethane.
Fig. 27.

Cesium ion pair pK’s in THF compared to calculated energy changes without solvation. The regression line through the blue “standard” points is 21.61 ± 0.26 + (0.584 ± 0.036)x; R2 = 0.956.
The group of Liu and Guo84 have recently shown that a carefully calibrated PCM can give a largely successful representation of the solvation of organocesium compounds in THF. Nevertheless, although cesium ion pair pKs are reproduced with good statistics, there are some disturbing chemical anomalies. For example, triphenylmethane is still predicted to be less acidic than diphenylmethane, in contrast to experiment. Thus, it was of interest to see how well the approach of direct coordination of solvent to cation, so successful with lithium, could be applied to cesium. One problem is that cesium, being so much larger than lithium, has higher coordination numbers. Depending on space available, coordination numbers of 6 and 8 are common. Moreover, the nature of coordination on cesium can be quite different from lithium. On successive coordinations of dimethyl ether with methylcesium, the first ether attaches not at the opposite end from the methyl group but at right angles to it (Fig. 28). The difference in coordination energies is almost 3 Kcal mol−1. This result can be rationalized by the high polarizability of cesium (Fig. 29). Polarization of the cesium by the methyl anion opposes coordination at the other polar (apical) position but has little effect on coordination at the equatorial position. Successive coordinations occur in an equatorial belt until this belt is fully occupied with four ethers. Only with the fifth ether does coordination occur at the apical position. Even then, the Me-Cs-O bond angle is only 152° and one of the equatorial ethers is pushed out to a Cs-O bond distance of 3.42Å. A sixth ether can also be located at a bent apical position but with a seventh ether, two ethers take up positions over 5Å from cesium; that is, five ethers form an inner solvation shell and two start a second solvation shell. Thus, to further explore this direct coordination approach, we considered organocesium compounds with five coordinated dimethyl ethers, in general with four forming an equatorial belt and the fifth directly opposite the organic moiety.
Fig. 28.

Successive coordinations of dimethyl ether (E) with methylcesium. Bond distances are given in Å, energy changes are Kcal mol−1 for HF 6–31+G** + ZPE.
Fig. 29.

The polarization of cesium cation by methyl anion encourages coordination at right angles rather than at the terminal position.
These calculations are computer-intensive and this work is still in progress. The preliminary results, however, are encouraging. Figure 30 shows some cesium ion pair pK’s in THF compared to MP2/HF6–31+G(d,p) computations for eq. 15 where FlH is fluorene.
| eq. 15 |
Figure 30.

Cesium ion pair pK’s in THF compared to eq. 14 with energy differences in Kcal mol−1, MP2/6–31+G**. The regression line shown is 23.07 ± 0.57 + (0.483 ± 0.027)x; R2 = 0.962.
Even without ZPE corrections or other solvent effects, the average deviation of points from the regression line is only 1.3 pK units. One noteworthy feature is that triphenylmethane and diphenylmethane are in the correct order.
Conclusion
Much of the chemistry of organoalkali compounds, and particularly enolates, in non-polar donor solvents, such as ethers, can be modeled successfully even at modest theory levels by specifically including coordination of solvent to the metal.
Acknowledgement
It is a real pleasure to acknowledge the almost 300 undergraduates, graduate students, postdoctoral coworkers and visiting professors who have worked with me over the past half-century. My research could not have been done without them. I could only refer to the work of a few in the references to this paper. Major funding for my research over the years has come from the Petroleum Research Fund of the American Chemical Society, the Air Force Office of Scientific Research, The Department of Energy through LBNL, the National Institutes of Health, and, of course, the National Science Foundation, most recently by grant no. CHE-0243410. Recent support for undergraduate participation has come from Senior Scientist Mentor grants from the Camille and Henry Dreyfus Foundation. Many of the computations reported in this paper were run at the Molecular Graphics and Computational Facility, UCB College of Chemistry, supported in part by NSF grant no. CHE-0233882.
References
- 1.Streitwieser A, Jr, Jagow RH, Fahey RC, Suzuki S. J. Am. Chem. Soc. 1958;80:2326–2332. [Google Scholar]
- 2.Streitwieser A., Jr J. Am. Chem. Soc. 1953;75:5014–5018. [Google Scholar]
- 3.Charton M. Org. Prog. Phys. Chem. 1981;13:119. [Google Scholar]
- 4.Anschütz R, Immendorff H. Ber. 1884;17:2816–2817. [Google Scholar]
- 5.Heise R, Tö A. Ann. 1892;270:155–171. [Google Scholar]
- 6.McCaulay DA, Lien AP. J. Am. Chem. Soc. 1953;75:2411–2413. [Google Scholar]
- 7.Hughes ED, Juliusburger F, Masterman S, Topley B, Weiss J. J. Chem. Soc. 1935:1525–1529. [Google Scholar]
- 8.Eliel EL. J. Am. Chem. Soc. 1949;71:3970–3972. [Google Scholar]
- 9.Streitwieser A, Reif L. J. Am. Chem. Soc. 1960;82:5003–5004. [Google Scholar]
- 10.Bartrlett PD, Condon FE, Schneider A. J. Am. Chem. Soc. 1944;66:1531–1539. [Google Scholar]
- 11.Streitwieseer A, Downs WJ. J. Org. Chem. 1962;27:625. [Google Scholar]
- 12.Streitwieser A., Jr . Molecular Orbital Theory for Organic Chemists. New York: John Wiley and Sons, Inc; 1961. [Google Scholar]
- 13.Streitwieser A, Jr, Langworthy WC. J. Am. Chem. Soc. 1963;85:1757–1761. [Google Scholar]
- 14.Streitwieser A, Jr, Langworthy WC, Brauman JI. J. Am. Chem. Soc. 1963;85:1761–1763. [Google Scholar]
- 15.Streitwieser A, Jr, Koch HF. J. Am. Chem. Soc. 1964;86:404–409. [Google Scholar]
- 16.Streitwieser A, Jr, Müller-Westerhoff U. J. Am. Chem.Soc. 1968;90:7364. [Google Scholar]
- 17.Streitwieser A. A Life with the Synergy of Theory and Experiment. Washington DC: American Chemical Society; 1996. [Google Scholar]
- 18.Seyferth D. Organometallics. 2004;23:3562–3583. [Google Scholar]
- 19.Streitwieser A., Jr . In: In Sigma Molecular Orbital Theory. Sinanoglu O, Wiberg K, editors. New Haven, CN: Yale University Press; 1970. p. 197. [Google Scholar]
- 20.Streitwieser A, Jr, Rajca A, McDowell RS, Glaser R. J. Am. Chem. Soc. 1987;109:4184–4188. [Google Scholar]
- 21.Cambray J, Gasteiger J, Streitwieser A, Jr, Bagus PS. J. Am. Chem. Soc. 1974;96:5978–5984. [Google Scholar]
- 22.Streitwieser A, Jr, Williams JE, Cambray J, Owens PH. Comput. Chem. Res. Educ., Proc. Int. Conf. 1973;4:1–4. [Google Scholar]
- 23.Streitwieser A, Jr, Grier DL, Kohler BAB, Vorpagel ER, Schriver GW. In: In Electron Distributions and the Chemical Bond. Coppens P, Hall M, editors. New York: Plenum Press; 1982. [Google Scholar]
- 24.Streitwieser A, Jr, Owens PH. Orbital and Electron Density Diagrams: An Application of Computer Graphics. New York, NY: The Macmillan Co; 1973. [Google Scholar]
- 25.Streitwieser A, Jr, Williams JW, Alexandratos S, McKelvey JM. J. Am. Chem. Soc. 1976;98:4778–4784. [Google Scholar]
- 26.Streitwieser A, Bachrach SM, Dorigo A, Schleyer PvR. In: In Lithium Chemistry: A Theoretical and Experimental Overview. Sapse A-M, Schleyer PvR, editors. New York: John Wiley & Sons, Inc; 1995. pp. 12–43. [Google Scholar]
- 27.For a contrary view, see. Bickelhaupt FM, Sola M, Guerra CF. J. Chem. Theory Comput. 2006;2:965–980. doi: 10.1021/ct050333s. [DOI] [PubMed] [Google Scholar]
- 28.Streitwieser A, Jr, Nebenzahl LL. J. Syn. Org. Chem., Japan. 1975;33:889–902. [Google Scholar]
- 29.Streitwieser A, Jr, Juaristi E, Nebenzahl LL. Chap. 7. In: Buncel E, Durst T, editors. Comprehensive Carbanion Chemistry. Elsevier; 1980. In. [Google Scholar]
- 30.Streitwieser A, Jr, Bors DA, Kaufman MJ. J. Chem. Soc., Chem. Commun. 1983;23:1394–1395. [Google Scholar]
- 31.Ritchie CD. J. Am. Chem. Soc. 1969;91:6749–6753. [Google Scholar]
- 32.Ritchie CD, Uschold RE. J. Am. Chem. Soc. 1967;89:2752–2753. doi: 10.1021/ja00983a035. [DOI] [PubMed] [Google Scholar]
- 33.Ritchie CD, Uschold RE. J. Am. Chem. Soc. 1968;90:2821–2824. [Google Scholar]
- 34.Bordwell FG. Acc. Chem. Res. 1988;21:456–463. [Google Scholar]
- 35. http://webbook.nist.gov/chemistry.
- 36.Fu Y, Liu L, Li R-Q, Liu R, Guo Q-X. J. Am. Chem. Soc. 2004;126:814–822. doi: 10.1021/ja0378097. [DOI] [PubMed] [Google Scholar]
- 37.Streitwieser A, Wang DZ, Stratakis M, Facchetti A, Gareyev R, Abbotto A, Krom JA, Kilway KV. Canad. J. Chem. 1998;76:765–769. [Google Scholar]
- 38.Stratakis M, Wang PG, Streitwieser A. J. Org. Chem. 1996;61:3145–3150. doi: 10.1021/jo9516401. [DOI] [PubMed] [Google Scholar]
- 39.Xie L, Streitwieser A. J. Org. Chem. 1995;60:1339–1346. [Google Scholar]
- 40.Gareyev R, Streitwieser A. J. Org. Chem. 1996;61:1742–1747. doi: 10.1021/jo951243q. [DOI] [PubMed] [Google Scholar]
- 41.Facchetti A, Streitwieser A. J. Org. Chem. 1999;64:2281–2286. [Google Scholar]
- 42.Krom JA, Petty JT, Streitwieser A. J. Am. Chem. Soc. 1993;115:8024–8030. [Google Scholar]
- 43.Abbotto A, Leung SS-W, Streitwieser A, Kilway KV. J. Am. Chem. Soc. 1998;120:10807–10813. [Google Scholar]
- 44.Wang DZ, Streitwieser A. J. Org. Chem. 2003;68:8936–8942. doi: 10.1021/jo034543d. [DOI] [PubMed] [Google Scholar]
- 45.Abbotto A, Streitwieser A, Stratakis M, Krom JA. J. Am. Chem. Soc. 1998;120:1718–1723. [Google Scholar]
- 46.Wang DZ-R, Streitwieser A. Can. J. Chem. 1999;77:654–658. [Google Scholar]
- 47.Streitwieser A, Wang DZ-R. J. Am. Chem. Soc. 1999;121:6213–6219. [Google Scholar]
- 48.Gareyev R, Ciula JC, Streitwieser A. J. Org. Chem. 1996;61:4589–4593. doi: 10.1021/jo960013o. [DOI] [PubMed] [Google Scholar]
- 49.Reyes JR. unpublished results. [Google Scholar]
- 50.Cramer CJ, Truhlar DG. Acc. Chem. Res. 2008;41:760–768. doi: 10.1021/ar800019z. [DOI] [PubMed] [Google Scholar]
- 51.Marenich AV, Olson RM, Chamberlin AC, Cramer CJ, Truhlar DG. J. Chem. Theory Comput. 2007;3:2055–2067. doi: 10.1021/ct7001539. [DOI] [PubMed] [Google Scholar]
- 52.Jackman LM, Bortiatynski J. Adv Carbanion Chem. 1992;1:45–87. [Google Scholar]
- 53.Jackman LM, Lange BC. J. Am. Chem. Soc. 1981;103:4494–4499. [Google Scholar]
- 54.Seebach D. Angew. Chem. Int. Ed. Engl. 1988;27:1624–1654. [Google Scholar]
- 55.Seebach D, Amstutz R, Dunitz JD. Helv. Chim. Acta. 1981;64:2622–2626. [Google Scholar]
- 56.Seeman JI. Chem. Rev. 1983;83:83–134. [Google Scholar]
- 57.Ciula JC, Streitwieser A. J. Org. Chem. 1992;57:431–432. doi: 10.1021/jo960013o. correction p. 6686. [DOI] [PubMed] [Google Scholar]
- 58.Chabanel M. Pure & Appl. Chem. 1990;62:35–46. [Google Scholar]
- 59.Abu-Hasanayn F, Stratakis M, Streitwieser A. J. Org. Chem. 1995;60:4688–4689. [Google Scholar]
- 60.Streitwieser A, Krom JA, Kilway KA, Abbotto A. J. Am. Chem. Soc. 1998;120:10801–10806. [Google Scholar]
- 61.Streitwieser A, Wang DZ, Stratakis M. J. Org. Chem. 1999;64:4860–4864. doi: 10.1021/jo990251u. [DOI] [PubMed] [Google Scholar]
- 62.Streitwieser A, Kim Y-J, Wang DZ-R. Org. Lett. 2001;3:2599–2601. doi: 10.1021/ol0162872. [DOI] [PubMed] [Google Scholar]
- 63.Wang DZ-R, Kim Y-J, Streitwieser A. J. Am. Chem. Soc. 2000;122:10754–10760. [Google Scholar]
- 64.Krom JA, Streitwieser A. J. Org. Chem. 1996;61:6354–6359. doi: 10.1021/jo960687z. [DOI] [PubMed] [Google Scholar]
- 65.Jackman LM, DeBrosse CW. J. Am. Chem. Soc. 1983;105:4177–4184. [Google Scholar]
- 66.Streitwieser A. J. Mol. Modeling. 2006;12:673–680. doi: 10.1007/s00894-005-0045-3. [DOI] [PubMed] [Google Scholar]
- 67.Strzalko T, Seyden-Penne J, Wartski L, Corset J, Castella-Ventura M, Froment F. J. Org. Chem. 1998;63:3287–3294. [Google Scholar]
- 68.Streitwieser A, Wang DZ, McKeown AE. Paper in preparation. [Google Scholar]
- 69.Reich HJ, Green DP, Medina MA, Goldenberg WS, Gudmundsson Bö, Dykstra RR, Phillips NH. J. Am. Chem. Soc. 1998;120:7201–7210. [Google Scholar]
- 70.Jantzi KL, Puckett CL, Guzei IA, Reich HJ. J. Org. Chem. 2005;70:7520–7529. doi: 10.1021/jo050592+. [DOI] [PubMed] [Google Scholar]
- 71.DePue JS, Collum DB. J. Am. Chem. Soc. 1988;110:5518–5524. [Google Scholar]
- 72.Abu-Hasanayn F, Streitwieser A. J. Am. Chem. Soc. 1996;118:8136–8137. [Google Scholar]
- 73.Ross RB, Powers JM, Atashroo T, Ermler WC, LaJohn LA, Christiansen PA. J. Chem. Phys. 1990;93:6654–6670. [Google Scholar]
- 74.Streitwieser A, Liang JC-Y, Jayasree EG, Hasanayn FJ. Chem. Theory Comput. 2007;3:127–131. doi: 10.1021/ct600238v. [DOI] [PubMed] [Google Scholar]
- 75.Streitwieser A, Ciula JC, Krom JA, Thiele G. J. Org. Chem. 1991;56:1074–1076. [Google Scholar]
- 76.Streitwieser A, Kaufman MJ, Bors DA, MacArthur CA, Murphy JT, Guibe F. ARKIVOC. Gainesville, FL: United States; 2005. pp. 200–210. [Google Scholar]
- 77.Facchetti A, Kim Y-J, Streitwieser A. J. Org. Chem. 2000;65:4195–4197. doi: 10.1021/jo000054r. [DOI] [PubMed] [Google Scholar]
- 78.Streitwieser A, Jr, Nebenzahl LL. J. Am. Chem. Soc. 1976;98:2188–2190. [Google Scholar]
- 79.Xie L, Bors DA, Streitwieser A. J. Org. Chem. 1992;57:4986–4990. [Google Scholar]
- 80.Streitwieser A, Schriver GW. Heteroatom Chem. 1997;8:533–537. [Google Scholar]
- 81.Streitwieser A, Wang GP, Bors DA. Tetrahedron. 1997;53:10103–10112. [Google Scholar]
- 82.Cox RA, Stewart R. J. Am. Chem. Soc. 1976;98:488–494. [Google Scholar]
- 83.Antipin IS, Gareyev RF, Vedernikov AN, Konovalov AI. J. Phys. Org. Chem. 1994;7:181–191. [Google Scholar]
- 84.Fu Y, Shen K, Liu L, Guo Q-X. J. Am. Chem. Soc. 2007;129:13610–13619. doi: 10.1021/ja073548f. [DOI] [PubMed] [Google Scholar]